
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInduced europium CPL for the selective signalling of phosphorylated amino-acids and O-phosphorylated hexapeptides†
Emily R.
Neil
,
Mark A.
Fox
,
Robert
Pal
and
David
Parker
 *
*
Department of Chemistry, Durham University, South Road, Durham, DH1 3LE, UK. E-mail: david.parker@dur.ac.uk
First published on 20th April 2016
Abstract
Two bright, europium(III) complexes based on an achiral heptadentate triazacyclononane ligand bearing two strongly absorbing chromophores have been evaluated for the selective emission and CPL signalling of various chiral O-phosphono-anions. Binding of O-phosphono-Ser and Thr gives rise to a strong induced CPL signature and a favoured Δ complex configuration is adopted. A similarly large induced CPL signal arises when [Eu·L1]2+ binds to lysophosphatidic acid (LPA), where the strong binding (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K 5.25 (295 K)) in methanol allowed its detection over the range 5 to 40 μM. Strong and chemoselective binding to the phosphorylated amino-acid residues was also observed with a set of four structurally related hexapeptides: in one case, the sign of the gem value in the ΔJ = 1 transition allowed differentiation between the binding to O-P-Ser and O-P-Tyr residues.
K 5.25 (295 K)) in methanol allowed its detection over the range 5 to 40 μM. Strong and chemoselective binding to the phosphorylated amino-acid residues was also observed with a set of four structurally related hexapeptides: in one case, the sign of the gem value in the ΔJ = 1 transition allowed differentiation between the binding to O-P-Ser and O-P-Tyr residues.
Introduction
We introduce a dynamically racemic probe, [Eu·L1]Cl2, that can bind to a range of structurally different chiral O-phosphono anions, and signal the binding of the analyte via induction of circularly polarised luminescence.Rather erratic progress has been made in the design of lanthanide-based probes for O-phosphono oxy-anions, and many recent examples simply offer detection via emission quenching or via displacement assays of limited utility1 The parent phosphate di-anion is usually considered to bind to a lanthanide centre through a single negatively charged oxygen atom. The hypothesis of monodentate binding is based on the analysis of kinetic emission and NMR spectroscopic data for a wide range of complexes, based on 1,4,7,10-tetraazacyclododecane (12-N4) ligand systems.2,3 For these examples, which are less sterically demanding than the 1,4,7-triazacyclononane (9-N3) analogues described more recently, the available experimental data is in line with the preference of phosphate to act as a monodentate ligand to a single metal centre, as reflected in X-ray structural data (e.g. for Na+, Ca2+, Zn2+).4,5
The development of CPL probes has been dominated by strongly emissive lanthanide(III) complexes, as they serve as pure spherical emitters with large emission dissymmetry factors.6,7 Time-gating techniques allow the signal from the lanthanide(III) centre to be observed selectively over the millisecond timescale, long after ligand fluorescence has decayed.8–10 The emissive lanthanide excited state may be perturbed in a number of different ways.11 In particular, changes in the ligand field that take place following reversible binding to the metal or the ligand, cause significant variations in emission spectral form, lifetime and polarisation.1,12–15 The latter type of modulation is of particular use in signalling selectively the presence of chiral species in solution.
Emission spectra of LnIII complexes are well known to be sensitive to changes in the coordination environment determining the ligand field.16–18 With europium(III) emission spectra, the oscillator strength of the magnetic-dipole (MD) allowed ΔJ = 1 transition (ca. 590 nm) is normally independent of the ligand environment. The ΔJ = 2 and ΔJ = 4 transitions (ca. 615 and 700 nm respectively) are electric-dipole (ED)-allowed. In each case, they are hypersensitive to ligand perturbation. To a good first approximation, their intensities are directly proportional to the square of the ligand polarisabilities. Electric-quadrupole allowed transitions, e.g.5D0 to 7F2/4, gain ED strength via a quadrupole/induced dipole (Ln3+ion/ligand donor) mechanism of coupling. The induced dipoles on the ligands, in turn, are caused by direct coupling to the electric dipole components of the radiation field. In this way, the electric dipole strength of 4f–4f transitions is related to ligand dipolar polarisabilities and to the directional dependence (i.e. the anisotropy) of these polarisabilities.19,20 The perturbation of the ligand field has a major impact on the emission profile of Eu(III) complexes. Variation of the axial donor in mono-capped square-antiprismatic, 9-coordinate complexes, for example, has a major effect on the relative intensity of the 7F2 ← 5D0 transition,21 and has been exploited in signalling reversible anion binding to the metal centre. Examples of anion signalling and sensing have evolved, including practicable assays for lactate, bicarbonate and citrate, in complex bio-fluids.22–25
In an achiral environment, a racemic mixture of Eu(III) complexes does not exhibit CPL. Following addition of a chiral agent, a net CPL signal can be observed in principle. When a chiral anion binds reversibly to the metal centre of a racemic complex, diastereoisomeric complexes of differing relative stability are formed and may lead to an induced CPL signal, whose relative intensity is a function of the binding selectivity and conformational rigidity of the complex, on the emission timescale. Recently, a series of very bright Eu(III) complexes has been reported, in which 1,4,7-triazacyclononane (9-N3) acts as the core ligand structure. Up to three pyridyl-alkynylaryl groups can be introduced to serve as the chromophore, allowing sensitised emission to take place.26–32 In particular, coordinatively unsaturated complexes based on heptadentate ligands have been prepared33 that bind reversibly to anions in aqueous media.
In this work, we compare and contrast the behaviour of the Eu(III) complexes of the ligands L1 and L2, (Scheme 1) and examine the issues related to chemoselective signalling of binding for a range of chiral O-phosphono anions, allowing an evaluation of their scope and utility as chiral probes for CPL. At the outset, it was hypothesised that the greater steric demand at the europium centre would disfavour chelation of anions with a small bite angle, such as carbonate or a simple carboxylate. Such thinking was based on the experimental observation that [Eu·L2]+ does not bind a solvent molecule in water or methanol solution, consistent with the notion that the N-benzyl creates a significant steric demand as a result of free rotation about the N–C and C–phenyl bonds, thereby inhibiting hydrogen bonding to the second sphere of solvation that would otherwise stabilise the metal-coordinated solvent, with respect to dissociation.
 | ||
| Scheme 1 | ||
The ligand L2 has been reported earlier, and forms a mono-cationic complex with Eu(III). The introduction of a methylammonium group in the meta-position of the N-benzyl moiety, gives rise to a dicationic complex. It was hypothesised that the primary electrostatic binding interaction to any coordinated anion would be stronger with [Eu·L1]2+vs. [Eu·L2]+, aided in certain cases by directed hydrogen bonding between the ammonium group, serving as a hydrogen bond donor, and, for example, a metal-coordinated phosphate group acting as a hydrogen bond acceptor.
Results and discussion
Ligand and complex synthesis and characterisation
It was necessary first to synthesise an aromatic precursor containing a charged amino group that also contains an electrophilic benzylic substituent at the meta position. An N-methylbenzylamine group was selected, as it has an appropriate pKa value (∼9.7), ensuring that it is positively charged at ambient pH and is not too sterically bulky. It also offers scope for stabilising hydrogen-bonding interactions.The synthetic pathway began with formation of the mono-amide, 2: reaction of 1 with oxalyl chloride furnished the acid chloride, followed by addition of methylamine to generate the amide, 2. Simultaneous reduction of the amide and ester groups using LiAlH4 in THF gave the amino-alcohol, 3. A reasonable yield was achieved by using the classical ‘Fieser work-up’ procedure: the granular aluminium salts that formed were easily removed by filtration. Protection of the amine with a formamide group was achieved using 2,2,2-trifluoroethylformate (TFEF). The formamide group was selected as it is removed under basic conditions. Therefore, deprotection could occur at the same time as hydrolysis of the methyl ester groups of the ligand. The final step in the synthesis of the precursor was conversion of the alcohol, 4, to the mesylate, 5, under standard conditions, (Scheme 2).
 | ||
| Scheme 2 | ||
The synthesis of the ligand was completed by alkylation of the secondary amine, 6, with the mesylate, 5, in MeCN in the presence of potassium carbonate. Treatment with base followed by complexation afforded the cationic complex, [Eu·L1]2+, which was purified by reverse phase HPLC. The high absorbance of each complex in the range 332 to 352 nm arises from the strong ICT band.23,28 Several photophysical measurements were made to characterise the chloride salt of the complex [Eu·L1]2+ and data compared to the control N-benzyl complex, [Eu·L2]+, (Table 1). The complex was not fully soluble in H2O and the emission intensity was significantly quenched, probably due to self-aggregation. Therefore, in order to get an understanding of the number of metal bound solvent molecules, the rate of radiative decay of europium(III) emission was determined in methanol and d4-methanol. The solvation state, q, was estimated using literature methods, modified to allow for the presence of only one quenching OH oscillator. The number of metal-bound solvent molecules was estimated to be zero in each case, consistent with the data obtained for [Eu·L2]+ in water, reported earlier (Scheme 3).34
 | ||
| Scheme 3 | ||
| [Eu·L2]+ | [Eu·L1]2+ | |
|---|---|---|
| λ/nm | 348 | 352 |
| ε/mM−1 cm−1 | 36.0 | 35.0 |
| Φ | 0.18 | 0.18 |
| k (MeOH)/ms−1 | 0.83 | 0.77 |
| k (CD3OD)/ms−1 | 0.93 | 1.01 |
The two Eu(III) complexes do not possess a bound solvent molecule. In each case, the steric demand imposed by the benzylic substituent suppresses solvent coordination. The lack of variation of the emission spectrum in water compared to methanol is consistent with the rate data analysis.
Anion binding behaviour
The anion binding behaviour towards hydrogenphosphate, bicarbonate and lactate was examined first, prior to a study of the more complex phosphorylated amino acid and hexapeptide anions (Scheme 4). | ||
| Scheme 4 Structures of the three common O-phosphono amino acids, the non-phosphorylated hexapeptide Gly-Ala-Pro-Tyr-Lys-Phe (GAPYKF) and the phosphorylated hexapeptides Gly-Ala-Pro-O-P-Tyr-Lys-Phe (GAPY*KF), Gly-O-P-Ser-Pro-Phe-Lys-Phe (GS*PFKF) and Gly-O-P-Ser-Pro-O-P-Tyr-Lys-Phe (GS*PY*KF). | ||
Incremental addition of aqueous solutions containing a given anion, to each Eu complex in turn, was monitored by emission spectroscopy examining changes in the relative intensity and form of the Eu(III) emission spectrum. The relative intensity of the ΔJ = 2 and ΔJ = 1 emission bands was plotted as a function of anion concentration and the variation was fitted to a 1:1 binding model by non-linear least-squares fitting, to give an estimate of the association constant.
Each Eu(III) complex (5 μM, 295 K, 50/50 MeOH/water, apparent pH 7.4, 10 mM HEPES) exhibited no change in spectral form in aqueous methanol, following addition of hydrogencarbonate in one thousand fold excess, consistent with the absence of anion binding at the metal centre. The binding of lactate had earlier been examined with [Eu·L2]+ and a set of three analogues with differing sensitising groups and overall charge. Lactate bound reversibly to give a ternary adduct in each case, for which the estimated binding constants were 4.37 and 3.15(±0.07) for [Eu·L1]2+ and [Eu·L2]+ respectively. The higher affinity with the dicationic complex may simply be attributed to the greater attractive electrostatic term. With added hydrogenphosphate, whilst [Eu·L2]+ showed no change in spectral form with added anion, [Eu·L1]2+ exhibited a significant modulation, and a binding constant of 4.2 (±0.1) was estimated (ESI). Given that the analogue of [Eu·L2]+, where the N-benzyl group is replaced by NH, underwent decomplexation of the Eu(III) ion with added hydrogenphosphate, the origins of the enhanced stability of the phosphate adduct of [Eu·L1]2+ were assessed using DFT computational methods.
Stereochemistry of a model hydrogenphosphate adduct: a DFT study
As the paramagnetic europium(III) complexes are rather difficult to model computationally, optimised geometries of the analogous diamagnetic yttrium(III) complex was investigated. It has been shown earlier that they serve as suitable models for the Eu analogues;26,35–38 consistent with the fact that the Y(III) ion differs in ionic radius from the Eu(III) ion by only 0.05 Å. The functional/basis set of B3LYP/3-21G* gives model geometries of yttrium complexes with reasonable confidence34 and is arguably superior to alternative reported26,35–37,39,40 functionals and basis sets. The binding of one hydrogenphosphate molecule to the model geometry [Y·L1]2+ (Fig. 1) was examined here at B3LYP/3-21G* and compared to the behaviour of the aqua, acetate, lactate, hydrogencarbonate and carbonate adducts. The corresponding adducts of the model geometry [Y·L2]+ (Fig. 1) were also explored for comparison and Table 2 summarises the relative binding energies of [Y·L1]2+ and [Y·L2]+. | ||
| Fig. 1 Yttrium complexes [Y·L1]2+ and [Y·L2]+ explored here as model complexes in computations for [Eu·L1]2+ and [Eu·L2]+ respectively. | ||
| [Y·L1]2+ NH⋯O interaction | [Y·L1]2+ No NH⋯O interaction | [Y·L2]+ | |
|---|---|---|---|
| H2O | 27.2 | 46.4 | |
| HCO3− | 159.7 | 117.6 | 129.2 |
| CH3CH(OH)CO2− | 163.5 | 124.7 | 129.9 |
| CH3CO2− | 192.6 | 133.8 | 161.3 |
| HPO42− | 383.0 | 356.4 | 342.6 |
| CO32− | 406.1 | 327.9 |
Of the many minima located for the hydrogenphosphate complex [Y·L1(HOPO3)], the lowest energy minima are discussed in which hydrogenphosphate is a monodentate ligand (Fig. 2), resulting in an eight coordinate Y centre. There are no examples in the CCDC of hydrogenphosphate chelating to a Eu centre in a monomeric complex. Directed hydrogen bonds occur between the negatively charged phosphate oxygen and the ammonium NH proton that contribute to stronger hydrogenphosphate binding in [Y·L1]2+ compared to [Y·L2]+ observed experimentally for the europium analogues. The lowest energy minimum located without such an N-hydrogen interaction is 26.6 kJ mol−1 higher.
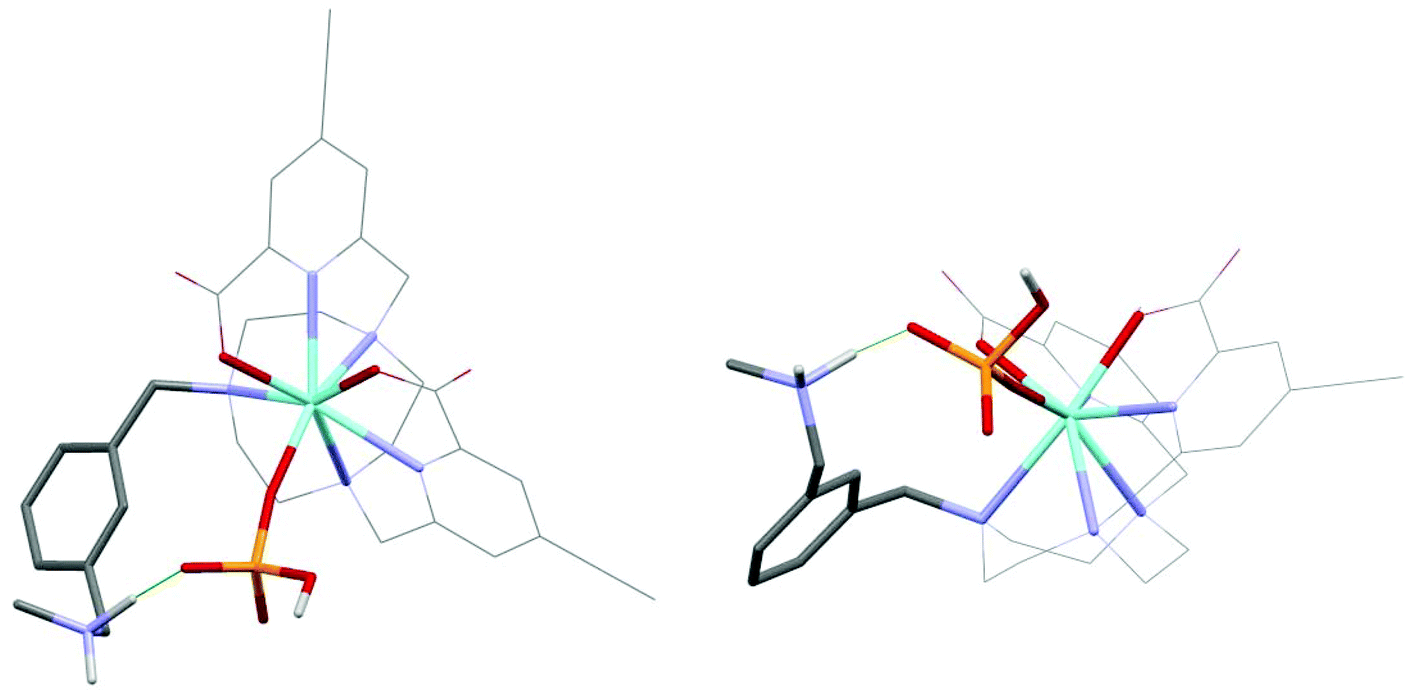 | ||
| Fig. 2 Top and side elevations showing the two lowest energy minima for [Y·L1(HOPO3)], in which the substituted phenyl groups are omitted for clarity; there is a directed hydrogen bond (green) between the negatively charged phosphate oxygen and the ammonium NH proton. | ||
The binding energy of [Y·L2]+ with hydrogenphosphate is 342.6 kJ mol−1 which is lower than the binding energy of [Y·L1]2+ with the phosphate by 40.4 kJ mol−1. This suggests that the stronger binding involving [Y·L1]2+ is a combination of the higher electrostatic binding interaction and the directed hydrogen bonding between the ammonium group and the phosphate group.
The binding energies listed (Table 2) follow the experimental observations of an absence of binding between [Eu·L1]2+ and [Eu·L2]+ with water molecules and are consistent with the spectral evidence for binding between [Eu·L1]2+ and [Eu·L2]+ and lactate anions. The higher affinity of [Eu·L1]2+ compared to [Eu·L2]+ with the lactate is in agreement with computed binding energies (Table 2). However, the spectral observations that [Eu·L1]2+ and [Eu·L2]+ bind neither with hydrogencarbonate nor with acetate anions, and, in the case of [Eu·L2]+, with hydrogenphosphate are not in agreement with DFT-computed binding energies. The absence of binding may be explained by two effects: first the fact the N-benzyl group is rotating quickly on the emission timescale creates a greater effective steric demand at the metal centre; secondly, the ligand hydrophobicity means there is a smaller second sphere of solvation which would otherwise stabilise the metal-coordinated solvent with respect to dissociation. Such solvation stabilisation models would require considerable computational efforts, and are beyond the scope of this study.
Binding behaviour of the chiral anions and CPL studies
Emission spectral changes for [Eu·L1]2+ were investigated following the addition of O-P-Ser, O-P-Thr and O-P-Tyr, in a MeOH–H2O solvent system (1:1, v/v), maintaining the apparent pH at 7.4 using a HEPES buffer (10 mM). The form of the total emission spectrum of [Eu·L1]2+ was similar to that following addition of inorganic phosphate, and the changes that did occur were relatively subtle, except in the hypersensitive ΔJ = 4 transitions, where isoemissive points could be distinguished (Fig. 2). Moreover, a broadening from one into three transitions in the ΔJ = 1 manifold was observed. In each case, it was possible to measure an estimated binding constant assuming a 1:1 stoichiometry for the ternary adduct. The stability constant for binding of each O-P-amino acid was calculated to be logK = 4.80 ± 0.05, revealing that there is no preference for a particular phosphorylated amino acid.
Such behaviour contrasts to previous work based on 12-N4 europium(III) complexes, where a preference for O-P-Tyr was observed in pure water, most likely as a result of the lower hydration energy of the tyrosine moiety.41 However, the study involving [Eu·L1]2+ was carried out in a 1:1 MeOH–H2O solvent system, to aid solubility. Selective solvation of the complex by a cluster of methanol molecules is likely to be occurring,42 and so the effect of the differential anion hydration energy is diminished. It is worth noting that no change in the emission spectrum was observed following addition of a 1000-fold excess of the non-phosphorylated amino acids. Binding must occur through the anionic phosphate group.
The change in emission spectral form was accompanied by the induction of a strong CPL signal, upon adding O-P-Ser or O-P-Thr (Fig. 3). The emission dissymmetry values, gem (where gem = 2(IL − IR)/(IL + IR) for the ΔJ = 1 transition) were bigger for O-P-Thr (gem (592.5 nm) = +0.08 vs. +0.04 for O-P-Ser), perhaps as a result of the additional methyl group providing a slightly higher degree of conformational rigidity at the metal centre. No induced CPL signal could be recorded for the [Eu·L1·O-P-Tyr] adduct. From the total emission studies, it is clear that the phosphorylated amino acid binds to the lanthanide centre in a similar manner to O-P-Ser and O-P-Thr. Therefore, an alternative explanation must explain the lack of induced chiroptical activity: the chiral centre is more remote in the O-P-Tyr molecule, and simply explains the lack of CPL activity observed. Addition of R- and S-O-P-Ser resulted in an induced CPL spectrum of equal and opposite form, with identical gem values at 593 nm. Comparative analysis with the CPL spectrum of the control N-benzyl complex, [Eu·L2]+, following addition of R and S-lactate, allowed a tentative assignment of the absolute configuration of the complex adduct, as Δ-[Eu·L1·O-P-S-Ser] and Λ-[Eu·L1·O-P-R-Ser].
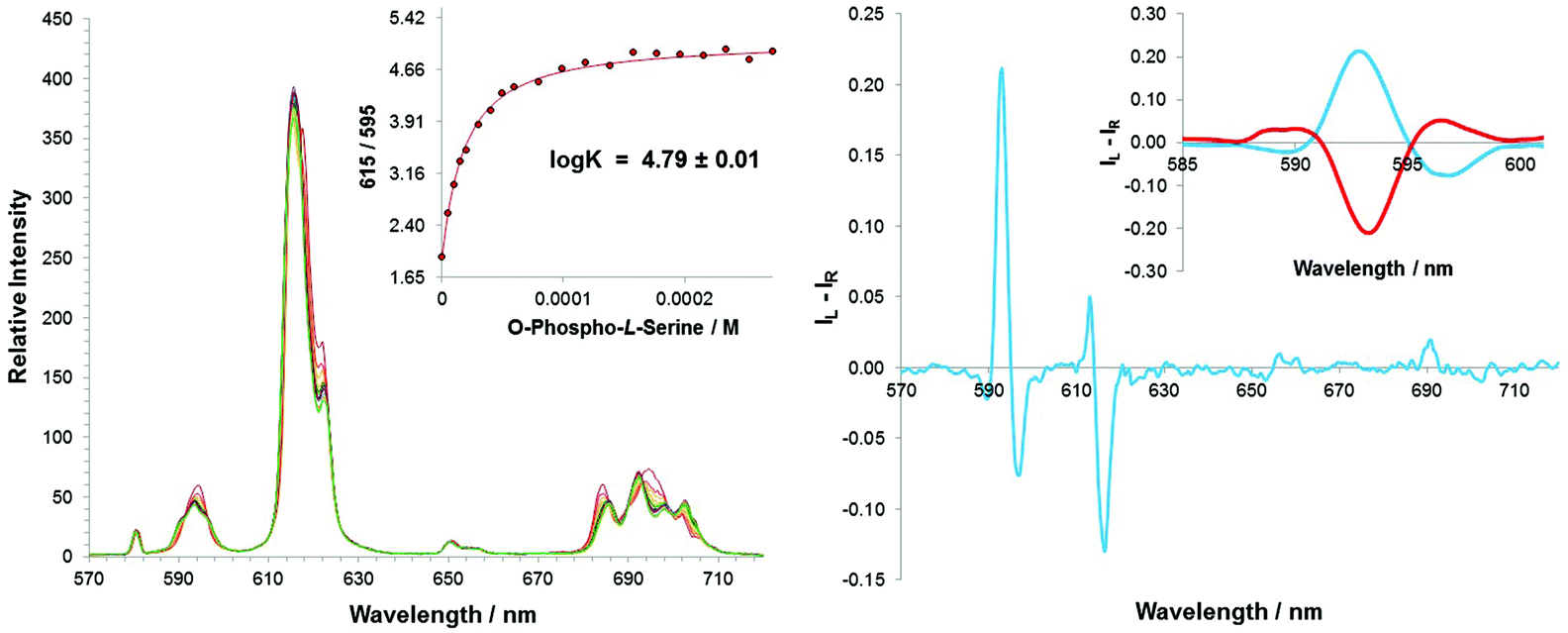 | ||
| Fig. 3 Variation of the Eu(III) total emission (left) as a function of added O-phosphono-Ser and the limiting CPL spectral profile for [Eu·L1] (right). The insets show the fit (line) to the experimental data points and an expansion of the ΔJ = 1 CPL transition for 50 μM added R-O-phosphono-Ser (red) and the S-enantiomer (blue) (λexc 352 nm, apparent pH 7.4; 50% H2O/MeOH, 10 mM HEPES, 5 μM complex). The CPL spectrum was nearly identical for addition of O-phosphono-Thr, but no CPL signal was observed with added O-phosphono-Tyr. | ||
Three phosphorylated peptides were selected for this study, varying in the phosphorylated residue (Ser or Tyr) and the number of phosphorylated sites (mono or di). The corresponding non-phosphorylated peptide, GAPYKF, was also studied as a control. These systems had been used earlier in related emission studies using 12-N4 based cationic complexes.41
In order to try and gain insight into the solution state structures of the three phosphorylated peptides, electronic circular dichroism (ECD) and 2D 1H NMR methods were used to assess peptide conformation. The hexapeptides each contain a proline residue, which can often induces a turn in peptide, particularly when followed by an aromatic residue.43 Weak ECD activity (around 200–230 nm) was observed for each of the three phosphorylated peptides. The hexapeptides GAPY*KF and GS*PY*KF exhibited ECD spectra that were very similar in form, consistent with the assignment of an open conformation peptide.44 In the case of GS*PFKF, the ECD spectrum was slightly more well-defined with a positive and negative component at 220 and 200 nm respectively, indicating that the mono-phosphorylated hexapeptide adopts a more structured preferred conformation in solution.
The induced CPL response of [Eu·L1]2+ was investigated, following addition of a 20-fold excess of each peptide. The addition of the non-phosphorylated control peptide, GAPYKF, resulted in no induced CPL, providing further evidence that the peptide binds to the europium(III) centre via the phosphate group, (Fig. 4). In the case of the O-P-Tyr peptide, GAPY*KF, the CPL spectral form resembles that of the O-P-amino acids, O-P-Ser and O-P-Thr). However, it is interesting to recall that the simple O-P-Tyr amino acid did not induce a CPL response. In contrast, the O-P-Tyr peptide induced significant CPL, with a measured emission dissymmetry value of gem (592.5 nm) = +0.10, which is amongst the highest induced gem values observed with a dynamically racemic europium(III) complex.12,13,45
 | ||
| Fig. 4 Limiting CPL spectra for [Eu·L1]2+, induced following addition of a 20-fold excess of peptide: (top left)-GAPY*KF; (top right)-GS*PFKF; (bottom left)-GS*PY*KF; (bottom right)-GAPYKF (10 μM complex, λexc 352 nm, 295 K, MeOH). | ||
Another interesting observation, was the induced CPL spectral response of the O-P-Ser peptide, GS*PFKF; this was opposite in sign and form to that of GAPY*KF. The gem value at the same transition, λ = 592.5 nm, was −0.04 and therefore much smaller than the O-P-Tyr peptide. Such behaviour, suggests that it may be the chiral structure of the entire peptide, when bound to the lanthanide centre that is associated with the induced CPL response.
The di-phosphorylated peptide, contains both a phosphorylated serine and tyrosine residue. The induced CPL is much weaker than for the other two peptides, and appears to be an additive sum of the two separate spectra. One possible explanation is that the complex has no preferential binding site, as the estimated affinity constants for the O-P-amino acids are the same (vide supra). Therefore, the emission spectrum observed is a combination of the lanthanide complex bound to the O-P-Ser of the peptide and the O-P-Tyr residue. The gem values for the induced CPL were bigger for the O-P-Tyr residue, which may explain why the major chiral emissive species for the GS*PY*KF adduct of [Eu·L1]2+, generated an induced CPL spectrum that more closely resembles that created by the O-P-Tyr hexapeptide.
Investigation of the binding affinities of the three hexapeptides with [Eu·L1]2+ was carried out by monitoring the emission and induced emission dissymmetry values, gem, as a function of peptide concentration. Emission spectral changes with [Eu·L1]2+ were very subtle, following addition of the phosphorylated peptide, GAPY*KF. It was much more informative to monitor the switching on of CPL with the system, particularly because the gem values were as large as +0.10 (Fig. 5). The change in the intensity of the ΔJ = 2 and ΔJ = 1 transitions of the emission spectrum was plotted as a function of added concentration of peptide, and the data was fitted to a 1:1 binding isotherm, following iterative least-squares fitting. The estimated binding affinity in methanol, logK = 5.9 (±0.1), was very large and consistent with the value obtained by plotting the induced gem values against concentration of peptide, logK = 6.1(±0.1). Such behaviour implies that the charged amino complex, [Eu·L1]2+, has a strong binding affinity with GAPY*KF, and that the major chiral species in solution is the most emissive species. A slightly higher binding affinity was calculated from the CPL data suggesting that it is associated with formation of the favoured isomer (Δ).
 | ||
| Fig. 5 (left): variation in the Eu(III) emission spectral profile for [Eu·L1]2+ as a function of added GAPY*KF showing the fit (line) to the experimental data points, assuming a 1:1 binding model; (right)- changes in the ΔJ = 1 manifold of the CPL spectrum for [Eu·L1]2+, following peptide addition, showing the fit to the data points (10 μM complex, λexc 352 nm, 295 K, MeOH). | ||
A similar experiment was carried out for the peptide GS*PFKF, however the data did not fit well to a 1:1 binding model, and a stability constant could not be reliably estimated. In the case of the di-phosphorylated peptide, GS*PY*KF, unusual behaviour was observed following the sequential addition of the peptide to the europium(III) complex. The induced CPL signal at 592.5 nm (ΔJ = 1) initially gave rise to increasingly negative values, until a critical concentration was reached. At this point, the sign of the CPL reversed and increasing positive gem values were recorded, until the limiting spectral response was achieved. Such behaviour provides further evidence that there is only a very small energy difference between the binding affinities of the two phosphorylated sites (O-P-Ser and O-P-Tyr). Nonetheless, at higher concentration of added peptide, the most emissive observed chiral species has a positive gem value at 592.5 nm, corresponding to binding at the O-P-Tyr residue.
[Eu·L1]2+ as a chiral probe for oleoyl-lysophosphatidic acid
Oleoyl-L-α-lysophosphatidic acid (LPA) is a phospholipid present at a physiological concentration of <0.1–6.3 μM and at significantly increased levels (up to about 40 μM) in ovarian cancer cells, (Scheme 5).46 | ||
| Scheme 5 Structures of lysophosphatidic acid (LPA) and a putative Eu(III)–salophene complex. | ||
Some attempts have been made to develop lanthanide probes able to detect LPA in MeOH, by monitoring an increase in the fluorescence intensity of the ligand.47 In a study using a coordinatively unsaturated europium(III) salophene complex of ill-defined speciation, an apparent selectivity of binding was demonstrated in methanol for a number of competitive analytes, such as serum albumin, phospholipids (phosphatidylserine, phosphatidylinositol, phosphatidylethanolamine) and selected carboxylates (phosphate and citrate). However, selectivity over bicarbonate and lactate (anions present at the highest physiological concentration) was not discussed. Furthermore, the chemical origin for the particularly high affinity of the europium(III) complex for LPA was neither apparent nor investigated. The study monitored the fluorescence response of the europium(III) complex in methanolic extracts of human serum, spiked with LPA. However, no information regarding the composition of the extract was given, making it difficult to determine the nature of the background medium in which LPA was ‘selectively’ detected. This particular lanthanide probe relied on short-lived ligand-centred emission to detect the biomolecule.
The anion LPA has a lower pKa2 than phosphatidic acid (PA), the related phospholipid with a simple phosphate head group, illustrated by the calculated values in a phosphatidylcholine bilayer (PA: pKa1 3.2 ± 0.3, pKa2 7.92 ± 0.03; LPA: pKa1 2.9 ± 0.3, pKa2 7.47 ± 0.03).48 Intramolecular hydrogen bonding is observed between the hydroxyl group on the glycerol backbone of the molecule and the phospho-monoester head group in the crystal structure, and is believed to persist at physiological pH (Fig. 6).48,49 Therefore, ionisation of the phosphate hydroxyl group of LPA occurs more easily than for PA (and other phosphate anions), generating a higher negative charge on the analyte, facilitating proposed binding to a positively charged probe, for example [Eu·L1]2+.
 | ||
| Fig. 6 Intramolecular hydrogen bonding of lysophosphatidic acid (LPA) and phosphatidic acid (PA), R = oleoyl acid chain. | ||
LPA must bind to the lanthanide centre via the phosphate head group, in the same mode discussed for the phosphorylated amino acids and peptides in the preceding sections.
Emission and induced CPL studies with LPA
The emission spectral form changes following addition of LPA to [Eu·L1]2+ were particularly apparent in the ΔJ = 1 transition, which broadened from a single manifold, into three distinct transitions, (Fig. 7). Such a change resembled the emission spectral response of [Eu·L1]2+ following addition of the other phosphate anions investigated (i.e. HPO42−, O-P-amino acids and O-P-peptides). Upon increasing the concentration of LPA, characteristic changes in the total emission spectrum were observed until a limiting spectrum was achieved, beyond which the total intensity continued to increase consistently across all transitions, with no further change to the emission spectral form. Such behaviour may be attributed to the long lipophilic chains in the molecule, which may aggregate to form a micelle, reducing the degree of quenching from second sphere solvent molecules. The changes in the local solvent permittivity may slightly change the ICT excited state energy level and favour the intramolecular energy transfer process, thereby significantly enhance the europium emission intensity. | ||
| Fig. 7 (left): variation of the Eu(III) spectral emission profile for [Eu·L1]2+ as a function of added LPA, showing the fit to the experimental data points for a 1:1 binding model (5 μM complex, MeOH, λexc 352 nm, 295 K). (right): increased emission intensity, without change of spectral form, in the range 100–600 μM LPA, consistent with micelle formation with an aggregation constant of 0.8 (±0.04) mM. | ||
The limit of detection for LPA was 5 μM; the limiting spectrum was reached at a concentration of 40 μM, which is in the desired range for LPA detection in ovarian cancer cells. The estimated stability constant in methanol, logK = 5.25, was high, demonstrating that there is indeed a strong binding affinity in the ternary adduct, [Eu·L1·LPA].
The induced CPL response (ESI) resembled that observed with O-P-Ser, O-P-Thr and the phosphorylated hexapeptides, with little activity observed in the ΔJ = 3 and ΔJ = 4 region. An emission dissymmetry value, gem, of +0.04 at 593 nm was measured. This value is lower than that measured for the O-P-Tyr phosphorylated peptide, suggesting that there is less conformational rigidity, consistent with the structure of LPA, which contains a long, aliphatic carbon chain.
Comparison of the CPL spectrum (ESI) with the parent N-benzyl complex, [Eu·L2]+, on addition of a number of chiral analytes, allowed a tentative assignment of the configuration of the complex adduct as Δ-[Eu·L2·L-LPA]. It is useful to recall that the O-P-S-amino acids also induced a similar CPL spectral response, resulting in the same Δ-assignment of helicity.
Following the promising results of the emission and CPL studies, further work set out to investigate the binding selectivity of the complex and LPA in a competitive environment. The nature of the ‘signature’ induced CPL response would provide further evidence for determination of the species bound to the europium centre.
No spectral evidence for an interaction was found for addition of other phospholipid species including phosphatidyl-serine, -inositol and -ethanolamine. The addition of HSA (human serum albumin) resulted in no change in the emission spectral form, although significant quenching of the emission intensity was observed at a protein concentration >0.2 mM, with no induced CPL. The binding affinity for LPA may be compared to other physiologically relevant anions, including lactate, bicarbonate and phosphate, (Table 3).
K, for analyte complexation with [Eu·L1]2+ and physiological concentrations of relevant biomolecules. (MeOH–H2O 1:1,10 mM HEPES, 295 K)
It is important to note that the affinity constant for LPA complexation was estimated in 100% MeOH and so is expected to be higher than the value measured in MeOH–H2O (1:1, v/v), due to the lower solvation energy of the anion. However, with that in mind, analysis of the affinity constants revealed that selective binding of LPA may be possible in a competitive environment. In aqueous methanolic solution (1:1, v/v; apparent ‘pH’ 7.4), simulating an extracellular fluid containing HSA (0.4 mM), sodium lactate (2.3 mM), hydrogen phosphate (0.9 mM), citrate (0.13 mM) and sodium bicarbonate (30 mM), the observed emission spectrum of [Eu·L1]2+ (5 μM) was the same as that observed for the complex alone. LPA was titrated into the solution in 5 μM aliquots until 100 μM was added. However, neither a change in the emission spectrum nor an induction of CPL was observed, under these conditions.
Summary and conclusions
This study has demonstrated that [Eu·L1]2+ binds selectively via the phosphate group to the O-phosphono amino acids and model phosphorylated peptides and a CPL response is induced. Accurate determination of stability constants can be achieved by monitoring the CPL spectral changes, as an alternative to europium(III) emission spectral modulation. Such an approach is particularly useful in cases where the changes in the total emission response are small or indistinct.The sign of the gem value in the ΔJ = 1 transition allows differentiation between O-P-Ser and O-P-Tyr residues. The magnitude of the induced CPL emission dissymmetry value for the O-P-Tyr hexapeptide, GAPY*KF was +0.10; such a value is amongst the highest that have been recorded for a dynamically racemic europium(III) system.12,13,45 The large gem value allows quick and easy detection of this peptide in solution when using CPL as the detection technique.
The detection of LPA using [Eu·L1]2+ as a lanthanide probe has also been demonstrated, within the range 5–40 μM. This result is the first example of induced CPL from an LPA adduct. The total emission and circularly polarised luminescence of the probe was monitored as a function of added lysophosphatidic acid. Due to the bright, strongly emissive nature of the lanthanide complex, detection of LPA was readily achieved. Analysis of the europium emission and CPL spectra revealed information about the structure of the ternary adduct, improving upon previous examples of lanthanide LPA probes. The absolute configuration of the [Eu·L1·L-LPA] adduct was tentatively assigned as Δ, consistent with the configuration assigned for the adducts with the O-P-L-amino acids.
Further work may seek to enhance the scope of the use of this, or analogous water-soluble complexes, in the detection and recognition of more complex and structurally diverse series of peptides, for example, those incorporating phosphorylated tyrosine residues.
Experimental
Details of the synthesis of the stated precursors 1–5, general experimental aspects and spectroscopic, analytical and computational methods are given in the ESI.† The synthesis of [Eu·L2]+ and compound 6 have been reported earlier.34Dimethyl 6,6′-((7-(3-((N-methylformamido)methyl)benzyl)-1,4,7-triazonane-1,4-diyl)bis(methylene))bis(4-((4-methoxy-2,6-dimethylphenyl)ethynyl)picolinate), 7
The bis-alkylated ligand, 6, (23 mg, 0.031 mmol) and K2CO3 (5 mg, 0.037 mmol) were dissolved in anhydrous CH3CN (2.5 mL) and bubbled with argon (20 minutes). The mesylate 5 (8 mg, 0.031 mmol) was added and the mixture was stirred under argon at 65 °C and monitored by LC-MS. After 24 h the reaction was cooled and filtered to remove excess potassium salts. The solvent was removed under reduced pressure and the crude material was purified by flash column chromatography (silica, gradient elution starting from 100% CH2Cl2 to 10% CH3OH in CH2Cl2 in 1% increments) to give 7 as a glassy solid (13 mg, 46%). TLC analysis Rf 0.32 (silica, 10% CH3OH in CH2Cl2); 1H NMR (295 K, 600 MHz, CDCl3) δH 8.25, 8.11 (1H, 2 × s, NCO

![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) , NCO′), 8.02 (2H, s, py-3), 7.62 (2H, s, py-5), 7.49–7.14 (4H, m, Ph-H), 6.62 (4H, s, Ar-2/2′), 4.49 (1H, s, NC2Ph), 4.39 (1H, s, NC′2Ph), 4.12 (4H, s, py-C2), 3.93 (6H, s, CO2C3), 3.80 (6H, s, OC3), 3.47–2.99 (12H, br m, rings Hs), 2.87, 2.74 (3H, 2 × s, NC3, NC′3), 2.47 (12H, s, Ar-C3); 13C NMR (295 K, 150 MHz, CDCl3) δC 165.3 (
, NCO′), 8.02 (2H, s, py-3), 7.62 (2H, s, py-5), 7.49–7.14 (4H, m, Ph-H), 6.62 (4H, s, Ar-2/2′), 4.49 (1H, s, NC2Ph), 4.39 (1H, s, NC′2Ph), 4.12 (4H, s, py-C2), 3.93 (6H, s, CO2C3), 3.80 (6H, s, OC3), 3.47–2.99 (12H, br m, rings Hs), 2.87, 2.74 (3H, 2 × s, NC3, NC′3), 2.47 (12H, s, Ar-C3); 13C NMR (295 K, 150 MHz, CDCl3) δC 165.3 (![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) O2CH3), 162.9 (NOH), 160.4 (Ar-1), 157.8 (py-6), 148.0 (Ar-4), 143.1 (Ar-3/3′), 137.4 (Ph-C), 134.6 (Ph-C), 130.3 (Ph-C), 129.6 (Ph-C), 129.5 (Ph-C), 128.8 (Ph-C), 128.0 (py-5), 125.9 (py-3), 113.8 (py-4), 112.9 (Ar-2/2′), 94.8 (alkyne 5), 93.0 (alkyne 6), 61.9 (ring Cs), 59.7 (ring Cs), 55.4 (OH3), 53.6 (CO2H3), 53.1 (py-H2), 51.7 (ring Cs), 47.7 (NH2Ph), 34.6, 29.7 (NH3), 21.5 (Ar-H3); m/z (HRMS+) 905.4599 [M + H]+ (C54H61N6O7 requires 905.4602).
O2CH3), 162.9 (NOH), 160.4 (Ar-1), 157.8 (py-6), 148.0 (Ar-4), 143.1 (Ar-3/3′), 137.4 (Ph-C), 134.6 (Ph-C), 130.3 (Ph-C), 129.6 (Ph-C), 129.5 (Ph-C), 128.8 (Ph-C), 128.0 (py-5), 125.9 (py-3), 113.8 (py-4), 112.9 (Ar-2/2′), 94.8 (alkyne 5), 93.0 (alkyne 6), 61.9 (ring Cs), 59.7 (ring Cs), 55.4 (OH3), 53.6 (CO2H3), 53.1 (py-H2), 51.7 (ring Cs), 47.7 (NH2Ph), 34.6, 29.7 (NH3), 21.5 (Ar-H3); m/z (HRMS+) 905.4599 [M + H]+ (C54H61N6O7 requires 905.4602).
[Eu·L1]Cl2
An aqueous solution of sodium hydroxide 0.3 M (0.5 mL) was added to a solution of ligand 7 (13 mg, 14 μmol) in methanol (0.5 mL). The mixture was stirred at 70 °C for 48 h. The reaction was cooled to r.t. and aqueous hydrochloric acid (0.1 M) was added until pH 7 was achieved. The crude solid was purified by preparative HPLC, tR = 16.5 min. The solid was passed down an ion exchange column to remove the presence of formate. Europium chloride hexahydrate (3 mg, 8 μmol) was added and the pH was readjusted to 6.5 by addition of aqueous sodium hydroxide (0.1 M). The reaction was stirred at 65 °C for 24 h. The solvent was removed under reduced pressure to give the complex [Eu·L1]Cl2 as a white solid (5 mg, 68%). m/z ESI (MeOH no column) 1044 [M + H]+; λexc (MeOH) = 352 nm; Φem (MeOH) = 0.18; ε (MeOH) 36
 000 M−1 cm−1; τ (MeOH) = 0.77 ms.
000 M−1 cm−1; τ (MeOH) = 0.77 ms.
Acknowledgements
We thank the ERC (FCC 266804) and The Royal Society for support.Notes and references
- (a) S. J. Butler and D. Parker, Chem. Soc. Rev., 2013, 42, 1652–1666 RSC; (b) W. T. Xu, Y. F. Zhou, D. C. Huang, M. Y. Su, K. Wang, M. Xiang and M. C. Hong, J. Mater. Chem. C, 2015, 3, 2003–2015 RSC; (c) S. Nadella, J. Sahoo, P. S. Subramanian, A. Sahu, S. Mishra and M. Albrecht, Chem. – Eur. J., 2014, 20, 6047–6053 CrossRef CAS PubMed; (d) D. F. Caffrey and T. Gunnlaugsson, Dalton Trans., 2014, 43, 17964–17970 RSC.
- R. S. Dickins, S. Aime, A. S. Batsanov, A. Beeby, M. Botta, J. I. Bruce, J. A. K. Howard, C. S. Love, D. Parker, R. D. Peacock and H. Puschmann, J. Am. Chem. Soc., 2002, 124, 12697–12705 CrossRef CAS PubMed.
- J. I. Bruce, R. S. Dickins, L. J. Govenlock, T. Gunnlaugsson, S. Lopinski, M. P. Lowe, D. Parker, R. D. Peacock, J. J. B. Perry, S. Aime and M. Botta, J. Am. Chem. Soc., 2000, 122, 9674–9684 CrossRef CAS.
- R. S. Alexander, Z. F. Kanyo, L. E. Chirlian and D. W. Christianson, J. Am. Chem. Soc., 1990, 112, 933–937 CrossRef CAS.
- B. Schneider and M. Kabeláč, J. Am. Chem. Soc., 1998, 120, 161–165 CrossRef CAS.
- G. Muller, Dalton Trans., 2009, 9692–9707 RSC.
- R. Carr, N. H. Evans and D. Parker, Chem. Soc. Rev., 2012, 41, 7673–7686 RSC.
- F. Zinna and L. Di Bari, Chirality, 2015, 27, 1–13 CrossRef CAS PubMed.
- J.-C. G. Bunzli and S. V. Eliseeva, Chem. Sci., 2013, 4, 1939–1949 RSC.
- J. M. Zwier, H. Bazin, L. Lamarque and G. Mathis, Inorg. Chem., 2014, 53, 1854–1866 CrossRef CAS PubMed.
- F. Kielar, C. P. Montgomery, E. J. New, D. Parker, R. A. Poole, S. L. Richardson and P. A. Stenson, Org. Biomol. Chem., 2007, 5, 2975–2982 CAS.
- K. Okutani, K. Nozaki and M. Iwamura, Inorg. Chem., 2014, 53, 5527–5537 CrossRef CAS PubMed.
- M. Iwamura, Y. Kimura, R. Miyamoto and K. Nozaki, Inorg. Chem., 2012, 51, 4094–4098 CrossRef CAS PubMed.
- C. M. G. dos Santos, A. J. Harte, S. J. Quinn and T. Gunnlaugsson, Coord. Chem. Rev., 2008, 252, 2512–2527 CrossRef CAS.
- G.-L. Law, D. Parker, S. L. Richardson and K.-L. Wong, Dalton Trans., 2009, 8481–8484 RSC.
- L. M. P. Lima and R. Tripier, Curr. Inorg. Chem., 2011, 1, 36 CrossRef CAS.
- A. M. Funk, K.-L. N. A. Finney, P. Harvey, A. M. Kenwright, E. R. Neil, N. J. Rogers, P. Kanthi Senanayake and D. Parker, Chem. Sci., 2015, 6, 1655–1662 RSC.
- A. M. Funk, P. Harvey, K.-L. N. A. Finney, M. A. Fox, A. M. Kenwright, N. J. Rogers, P. K. Senanayake and D. Parker, Phys. Chem. Chem. Phys., 2015, 17, 16507–16511 RSC.
- S. F. Mason, Struct. Bonding, 1980, 39, 43 CrossRef CAS.
- M. F. Reid and F. S. Richardson, Chem. Phys. Lett., 1983, 95, 501–506 CrossRef CAS.
- R. S. Dickins, D. Parker, J. I. Bruce and D. J. Tozer, Dalton Trans., 2003, 1264–1271 RSC.
- R. Pal, D. Parker and L. C. Costello, Org. Biomol. Chem., 2009, 7, 1525–1528 CAS.
- D. G. Smith, G. Law, B. S. Murray, R. Pal, D. Parker and K.-L. Wong, Chem. Commun., 2011, 47, 7347–7349 RSC.
- D. G. Smith, R. Pal and D. Parker, Chem. – Eur. J., 2012, 18, 11604–11613 CrossRef CAS PubMed.
- R. Pal, A. Beeby and D. Parker, J. Pharm. Biomed. Anal., 2011, 56, 352–358 CrossRef CAS PubMed.
- M. Soulié, F. Latzko, E. Bourrier, V. Placide, S. J. Butler, R. Pal, J. W. Walton, P. L. Baldeck, B. Le Guennic, C. Andraud, J. M. Zwier, L. Lamarque, D. Parker and O. Maury, Chem. – Eur. J., 2014, 20, 8636–8646 CrossRef PubMed.
- J. W. Walton, A. Bourdolle, S. J. Butler, M. Soulie, M. Delbianco, B. K. McMahon, R. Pal, H. Puschmann, J. M. Zwier, L. Lamarque, O. Maury, C. Andraud and D. Parker, Chem. Commun., 2013, 49, 1600–1602 RSC.
- N. H. Evans, R. Carr, M. Delbianco, R. Pal, D. S. Yufit and D. Parker, Dalton Trans., 2013, 42, 15610–15616 RSC.
- M. Delbianco, V. Sadovnikova, E. Bourrier, G. Mathis, L. Lamarque, J. M. Zwier and D. Parker, Angew. Chem., Int. Ed., 2014, 53, 10718–10722 CrossRef CAS PubMed.
- M. Delbianco, L. Lamarque and D. Parker, Org. Biomol. Chem., 2014, 12, 8061–8071 CAS.
- S. J. Butler, M. Delbianco, N. H. Evans, A. T. Frawley, R. Pal, D. Parker, R. S. Puckrin and D. S. Yufit, Dalton Trans., 2014, 43, 5721–5730 RSC.
- S. J. Butler, L. Lamarque, R. Pal and D. Parker, Chem. Sci., 2014, 5, 1750–1756 RSC.
- S. J. Butler, B. K. McMahon, R. Pal, D. Parker and J. W. Walton, Chem. – Eur. J., 2013, 19, 9511–9517 CrossRef CAS PubMed.
- E. R. Neil, M. A. Fox, R. Pal, L.-O. Palsson, B. A. O'Sullivan and D. Parker, Dalton Trans., 2015, 44, 14937–14951 RSC.
- T. T. da Cunha, J. Jung, M.-E. Boulon, G. Campo, F. Pointillart, C. L. M. Pereira, B. L. Guennic, O. Cador, K. Bernot, F. Pineider, S. Golhen and L. Ouahab, J. Am. Chem. Soc., 2013, 135, 16332–16335 CrossRef CAS PubMed.
- K. Sénéchal-David, A. Hemeryck, N. Tancrez, L. Toupet, J. A. G. Williams, I. Ledoux, J. Zyss, A. Boucekkine, J.-P. Guégan, H. Le Bozec and O. Maury, J. Am. Chem. Soc., 2006, 128, 12243–12255 CrossRef PubMed.
- F. Pointillart, B. L. Guennic, O. Maury, S. Golhen, O. Cador and L. Ouahab, Inorg. Chem., 2013, 52, 1398–1408 CrossRef CAS PubMed.
- R. S. Dickins, A. S. Batsanov, J. A. K. Howard, D. Parker, H. Puschmann and S. Salamano, Dalton Trans., 2004, 70–80 RSC.
- A. Bourdolle, M. Allali, J.-C. Mulatier, B. L. Guennic, J. M. Zwier, P. L. Baldeck, J.-C. G. Bünzli, C. Andraud, L. Lamarque and O. Maury, Inorg. Chem., 2011, 50, 4987–4999 CrossRef CAS PubMed.
- E. R. Neil, A. M. Funk, D. S. Yufit and D. Parker, Dalton Trans., 2014, 43, 5490–5504 RSC.
- P. Atkinson, Y. Bretonnière, D. Parker and G. Muller, Helv. Chim. Acta, 2005, 88, 391–405 CrossRef CAS.
- S. Dixit, J. Crain, W. C. K. Poon, J. L. Finney and A. K. Soper, Nature, 2002, 416, 829–832 CrossRef CAS PubMed.
- J. Yao, H. J. Dyson and P. E. Wright, J. Mol. Biol., 1994, 243, 754–766 CrossRef CAS PubMed.
- A. Perczel and G. D. Fasman, Protein Sci., 1992, 1, 378–395 CrossRef CAS PubMed.
- R. Carr, R. Puckrin, B. K. McMahon, R. Pal, D. Parker and L.-O. Pålsson, Methods Appl. Fluoresc., 2014, 2, 024007 CrossRef.
- Y. Xu, Z. Shen, D. W. Wiper, M. Wu, R. E. Morton, P. Elson, A. W. Kennedy, J. Belinson, M. Markman and G. Casey, JAMA, J. Am. Med. Assoc., 1998, 280, 719–723 CrossRef CAS PubMed.
- O. Alptürk, O. Rusin, S. O. Fakayode, W. Wang, J. O. Escobedo, I. M. Warner, W. E. Crowe, V. Král, J. M. Pruet and R. M. Strongin, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 9756–9760 CrossRef PubMed.
- E. E. Kooijman, K. M. Carter, E. G. van Laar, V. Chupin, K. N. J. Burger and B. de Kruijff, Biochemistry, 2005, 44, 17007–17015 CrossRef CAS PubMed.
- I. Pascher and S. Sundell, Chem. Phys. Lipids, 1985, 37, 241–250 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6dt01212d |
| This journal is © The Royal Society of Chemistry 2016 |