
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmbient pressure synthesis of MIL-100(Fe) MOF from homogeneous solution using a redox pathway†
Felix
Jeremias
ab,
Stefan K.
Henninger
*a and
Christoph
Janiak
*b
aDepartment of Thermally Active Materials and Solar Cooling, Fraunhofer Institute for Solar Energy Systems (ISE), Heidenhofstr. 2, D-79110 Freiburg, Germany. E-mail: stefan.henninger@ise.fraunhofer.de
bInstitut für Anorganische Chemie und Strukturchemie, Universität Düsseldorf, Universitätsstr. 1, D-40225 Düsseldorf, Germany. E-mail: janiak@uni-duesseldorf.de
First published on 26th April 2016
Abstract
Micro- to mesoporous iron(III) trimesate MIL-100(Fe) is a MOF of high interest for numerous applications. With regard to large-scale synthesis, e.g., by continuous flow or the in situ deposition of coatings, a replacement for the conventional, hydrothermal low-yield fluoride-containing synthesis is desirable. In this contribution, we present a method to synthesize crystalline fluoride-free MIL-100(Fe) from iron(III) nitrate and trimesic acid in zeotropic DMSO/water solution at normal ambient pressure involving a DMSO–nitrate redox pathway. Yields of 72%, surface areas of SBET = 1791 m2 g−1 and pore volumes of Vpore = 0.82 cm3 g−1 were achieved.
Introduction
Metal–organic frameworks (MOFs) are the subject of ongoing attention1,2 due to their high porosity which promises applications in, e.g., gas storage,3 gas4,5 and pollutant6 separation processes, drug delivery,7 heterogeneous catalysis,8,9 heat transformation10,11–13etc.14Among the about 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 MOFs known to date, the class of MIL compounds (MIL = Materials of Institute Lavoisier), which was pioneered by Férey and his group,5,16 sticks out due to their higher hydrothermal stability compared to many of the other MOFs.17
000 MOFs known to date, the class of MIL compounds (MIL = Materials of Institute Lavoisier), which was pioneered by Férey and his group,5,16 sticks out due to their higher hydrothermal stability compared to many of the other MOFs.17
Further, among the abundancy of known MIL-MOF structures, mesoporous MIL-100 has received special attention in the literature with respect e.g. to its catalytic, gas separation, gas storage, drug delivery7 and water sorption properties.2,11,18–20 MIL-100(M) compounds are three-dimensional chromium(III), iron(III) or aluminium(III) benzene-1,3,5-tricarboxylates of formula 3D-[M3(μ-O)(X)(H2O)2(btc)2·nH2O]n, (M = Cr,21,22 Fe,18,23 Mn,24 V25 or Al,26 X = OH, F, btc = benzene-1,3,5-tricarboxylate, trimesate). The porosity of MIL-100 originates from both 25 Å and 29 Å mesopores accessible via 5.5 Å and 8.6 Å windows, respectively (Fig. 1).18 MIL-100(Cr, Fe) have been proven to be hydrothermally very stable MOFs, even with regard to repeated adsorption/desorption cycles using water vapor.11,22 In particular, MIL-100(Fe) can be prepared from toxicologically and environmentally benign building blocks,27 – except for the use of HF required by the original synthesis procedure18 – with cost-efficient components. Despite these advantages, crystalline MIL-100(Fe) is still not commercially available. This can be attributed to the fact that its syntheses must be carried out in water, mostly at T > 100 °C, starting from a heterogeneous reaction mixture containing iron(III) chloride and trimesic acid or its trimethyl ester.18,20,28–30 The original synthesis procedure for MIL-100(Fe) was developed by Férey et al.18 In this original procedure, hydrofluoric acid, HF was used in strongly acidic solution (pH < 1, HNO3). The use of HF (a classified chemical toxicant) or acidic fluoride solutions is not desirable for large-scale or commercial syntheses.31 More important, overpressure and/or heterogeneous reaction mixtures render reaction control difficult and require expensive equipment, especially when it comes to scale-up. Continuous-flow synthesis has successfully been implemented for several MOFs as a promising method to their economic production.32 It can be expected that the space-time yield of a continuous process will exceed that of any batch process by far. However, in the case of MIL-100(Fe), the present demand for hydrothermal reaction conditions increases the technical complexity. Sub-hydrothermal reaction conditions, on the other hand, are known to lead to clogging.33In situ procedures, e.g., for the fabrication of coatings12,13 are also much more complicated to realize with the present hydrothermal procedures for MIL-100(Fe), be it with the addition of HF or without. Part of the commercial success of MOFs depends on the development of improved synthesis procedures.34 Recently, we reported a high-yield, fluoride-free and large-scale synthesis of related MIL-101(Cr).35
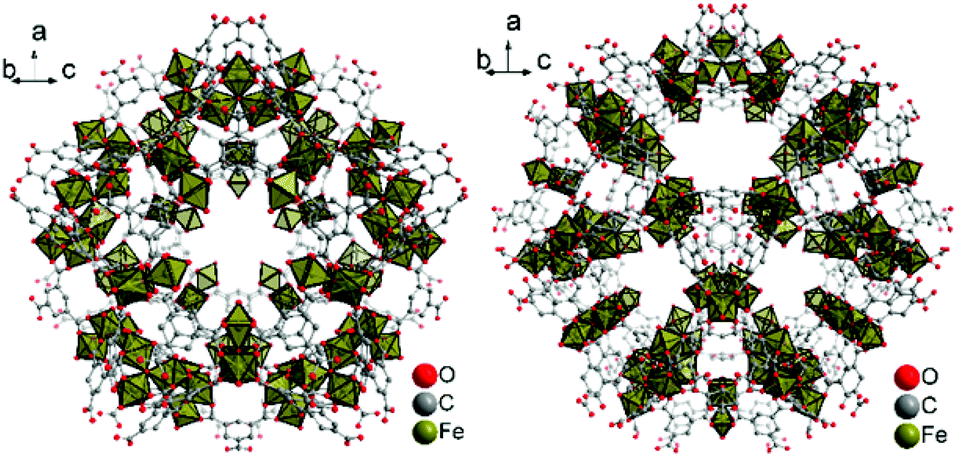 | ||
| Fig. 1 Small S cage (left) and large L cage (right) in MIL-100(Fe). Objects are not drawn to scale. Hydrogen atoms and solvent molecules of crystallization are not shown. Graphics produced by Diamond15 from cif-file for MIL-100(Fe) (CSD-Refcode CIGXIA, CCDC no. 640536).1 | ||
This contribution investigates the approach to overcome the overpressure and HF issue in replacing part of the water in the reaction mixture by a high-boiling, polar solvent forming a zeotropic mixture. This mixture should dissolve all starting materials and keep liquid water in the reaction-mixture even at T > 100 °C. Dimethyl sulfoxide, DMSO, (CH3)2S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O is an environmentally benign, aprotic and highly polar solvent forming zeotropic mixtures with water.36 In this contribution, we demonstrate the successful fabrication of MIL-100(Fe) from a homogeneous, fluoride-free solution at ambient pressure from a solvent where unreacted trimesic acid remains in solution.
O is an environmentally benign, aprotic and highly polar solvent forming zeotropic mixtures with water.36 In this contribution, we demonstrate the successful fabrication of MIL-100(Fe) from a homogeneous, fluoride-free solution at ambient pressure from a solvent where unreacted trimesic acid remains in solution.
Experimental section
Note on DMSO
Dimethyl sulfoxide is less harmful than any other highly polar, aprotic, organic solvent. DMSO is a solvent with a median lethal dose higher than ethanol. DMSO is not listed as toxic or especially harmful. At high temperatures, it is flammable, and when brought into contact with skin, irritations may occur. DMSO is even used as an ingredient for pharmaceutical formulations (ointments). In medicine, DMSO is predominantly used as a topical analgesic, a vehicle for topical application of pharmaceuticals, as an anti-inflammatory, and an antioxidant substance.General synthesis
In a 250 mL three-necked round bottom flask equipped with a magnetic stirring bar, a reflux condenser and an internal thermometer, 1.12 g (5.3 mmol) of trimesic acid (≥98%, TCI) were dissolved in 40 mL of dimethyl sulfoxide. Solutions of 8 mmol (referred to Fe3+) of iron(III) nitrate, chloride, mesylate, hydrogen sulfate or sulfate in 8 mL of deionized water were prepared as described in the ESI.† The pH value was set according to Scheme 1 using the corresponding acids.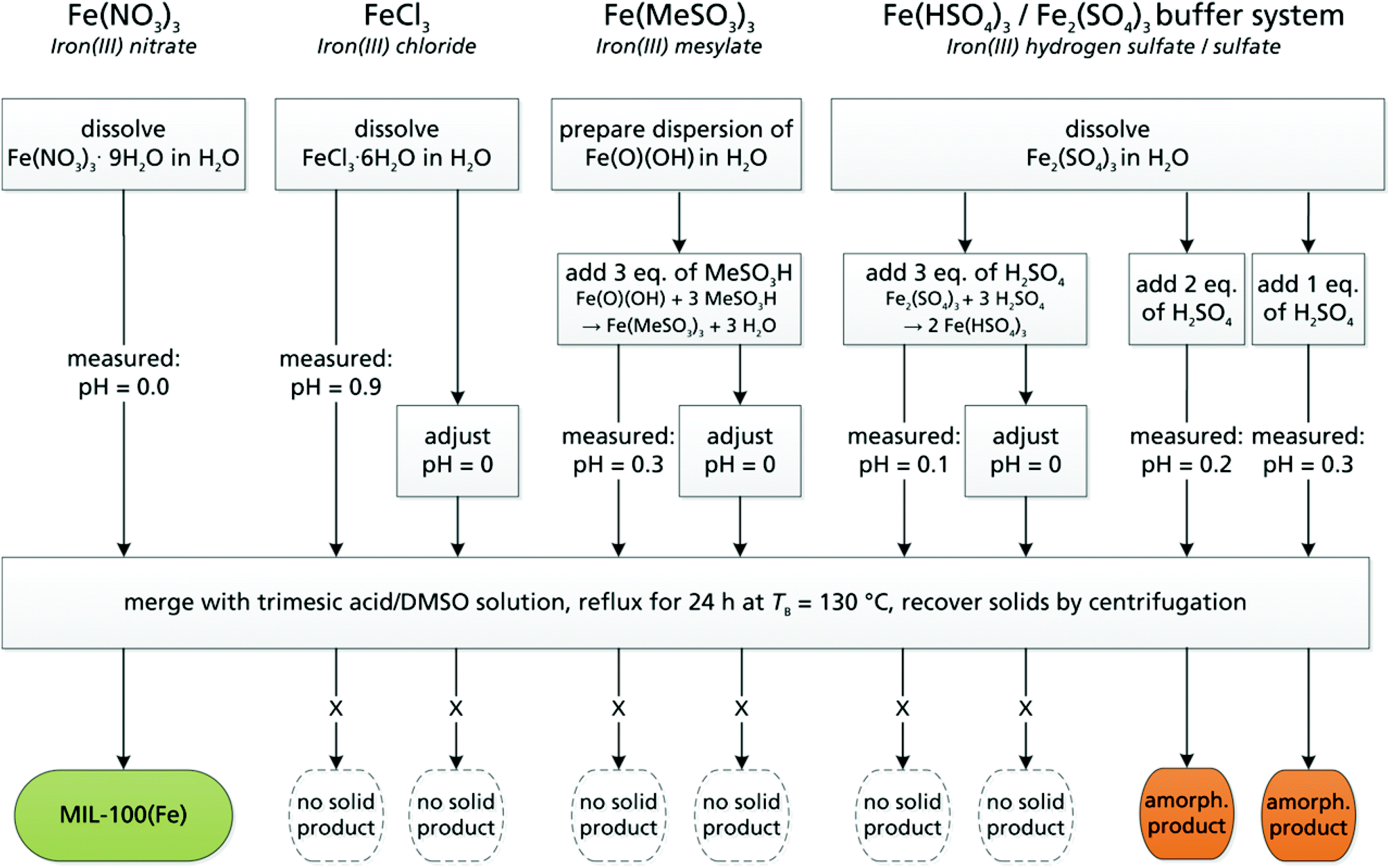 | ||
| Scheme 1 Experimental scheme for the synthesis of MIL-100(Fe) from DMSO/H2O under reflux employing different metal salts and pH values. | ||
Before merging the metal salt solution and the trimesic acid solution, the latter was heated to a temperature of 90 °C in order to avoid precipitation of [Fe(DMSO)6](NO3)3 (see ESI with Fig. S1†), upon addition of iron(III) nitrate into the DMSO solution of trimesic acid. Solid [Fe(DMSO)6](NO3)3 is a potential explosive but can be handled in the solution state.37 After merging, the clear, yellow mixture was heated further to its boiling point of 130 °C with gentle stirring. Within 30 min, the yellow-green, clear solution turned amber and then turbid when iron(III) nitrate was used. The solid product was separated by centrifugation of the still warm solution (80 °C) after 24 h.
Influence of reaction time
The synthesis was performed as stated above. The precipitate was retrieved at set times (see Table 1) by centrifugation of the hot solution and the clear solution was continued to reflux.| Sample | Retrieved after | Yield, activated (mg) |
S
BETa (m2 g−1) |
V
poreb (cm3 g−1) |
|---|---|---|---|---|
| a Calculated in the pressure range 0.05 < p/p0 < 0.2 from N2 sorption isotherm at 77 K with an estimated standard deviation of ±50 m2 g−1. b Calculated from N2 sorption isotherm at 77 K (p/p0 = 0.95) for pores ≤20 nm. | ||||
| 1 | 24 h | 1250 | 1215 | 0.61 |
| 2a | 2 h | 115 | 1024 | 0.62 |
| 2b | 4 h | 109 | 1405 | 0.71 |
| 2c | 8 h | 147 | 1513 | 0.73 |
| 2d | 24 h | 286 | 1507 | 0.72 |
| 2e | 48 h | No further precipitation observed | ||
Influence of HNO3 addition
The influence of additional HNO3 on the synthesis was investigated by adding 0, 0.5, 1 or 2 HNO3 equivalents with respect to Fe (8 mmol).Activation
Activation was performed by stirring the solid, as synthesized MIL-100(Fe) product thrice in 25 mL each of a mixture of DMSO/H2O = 4:1 (v/v) for 1 h, 1 h and 24 h. Then, the solid product was stirred thrice in 25 mL each of ethanol for 1 h, 1 h and 24 h, and air-dried for 12 h at 80 °C. The washing solutions were separated by centrifugation and discarded. Yield: 1.25 g for the 24 h reflux synthesis using iron(III) nitrate (72%). Other yields are given in Tables 1 and 2.
| HNO3 equivalents to Fea | Initial HNO3 conc.b (mol L−1) |
S
BETc (m2 g−1) |
V
pored (cm3 g−1) |
|---|---|---|---|
|
a Molar ratio HNO3 to Fe. The Fe:H3btc ratio is always 3:2.
b Calculated from the amount of added HNO3 and the volume of the reaction solution.
c Calculated in the pressure range 0.05 < p/p0 < 0.2 from N2 sorption isotherm at 77 K with an estimated standard deviation of ±50 m2 g−1.
d Calculated from N2 sorption isotherm at 77 K (p/p0 = 0.95) for pores ≤20 nm.
|
|||
| 0 | 0 | 1412 | 0.71 |
| 0.5 | 0.083 | 1539 | 0.73 |
| 1 | 0.17 | 1791 | 0.82 |
| 2 | 0.33 | 1058 | 0.49 |
Analytical methods
Powder X-ray diffractograms were acquired on a Bruker® D8 Advance with DaVinci® design using Cu-Kα radiation, a Lynxeye detector, 0.02° and 1 s per step. Diffractograms were obtained on flat layer sample holders which led to the low relative intensities measured at low 2-theta angles (<5°) compared to the simulated pattern. Nitrogen adsorption isotherms were obtained at 77 K on a Quantachrome® Nova, after degassing for 24 h in fine vacuum at 120 °C.Results and discussion
Synthesis of MIL-100(Fe) under reflux conditions in DMSO/water solution using different metal salts
A mixture of DMSO:water = 4:1 (v:v) was selected as the solvent due to its ability to dissolve the starting materials already at room temperature, and because of the possible high water content. Furthermore, this mixture has an ambient pressure (101.3 kPa) boiling point of 130 °C (Fig. S2, ESI†). This boiling point matches the temperature given in a common hydrothermal procedure yielding highly porous MIL-100(Fe) without addition of HF.28,36 Iron(III) nitrate, chloride, mesylate and a sulfate/hydrogen sulfate system (pKa(HSO4−) = 1.9) were examined as Fe(III) sources with different anions. Metal salt precursors were selected according to the criteria of solubility in water/DMSO mixtures, the fact that MOFs have been successfully prepared using chloride, nitrate, sulfate anions and low (nitrate, sulfate, mesylate) anion coordination strength towards Fe3+. In order to find the optimal synthesis conditions for MIL-100(Fe) at ambient pressure, not only the iron salt, but also the pH was varied systematically (see Scheme 1 for an overview): first, all the metal salt solutions were used as prepared. In a second run, the pH of the Fe hydrogen sulfate, chloride and mesylate solutions was set to pH = 0, which is the initial, direct pH value of the 1 mol L−1 iron(III) nitrate solution (without acid addition).
Regardless of the pH, a color change of the solution (yellow to red), followed by the precipitation of MIL-100(Fe) could be observed only when using the nitrate salt (see Scheme 1). This indicates the generation of the Fe3(μ3-O) cluster forming the secondary building unit of MIL-100(Fe).38 At the same time, NOx fumes were observed inside the reflux condenser.
The fluoride-free synthesis of MIL-100(Fe) in DMSO/water under ambient pressure yielded a product in good yield (72%) with a BET surface area of SBET = 1215 m2 g−1 and pore volume Vpore = 0.61 cm3 g−1. This has to be compared with literature data for MIL-100(Fe) from hydrothermal synthesis with the addition of HF and SBET = 1917 m2 g−1, Vpore = 1.00 cm3 g−1 (ref. 11) or SLangmuir > 2800(100) m2 g−1.18 Under hydrothermal conditions but without the addition of HF, SBET = 1800 m2 g−1, and Vpore = 1.15 cm3 g−1 were achieved.29 From a reaction slurry containing trimesic acid, iron(III) nitrate and water, MIL-100 with SBET = 1836 m2 g−1 and Vpore = 1.1 cm3 g−1 was said to be obtained at 95 °C.20 The slightly lower porosity of MIL-100(Fe) produced by the DMSO/water approach can be attributed to the possible presence of an amorphous phase, as well as the overall slightly lower crystallinity of the product. Furthermore, the presence of hardly soluble dimethyl sulfone (DMSO2) in the pores cannot be ruled out.
The successful action of HF in terms of improved crystallinity and porosity has been documented,34,39 still its poisonous nature renders its replacement highly desirable even at the expense of slightly less surface area or pore volume.35
No solid products were obtained from the iron chloride, mesylate or hydrogen sulfate starting materials. From the hydrogen sulfate/sulfate system, only amorphous precipitates were formed, which we ascribe to the fact that the sulfate salt is the most basic among the investigated precursors, resulting in fast, non-crystalline precipitation.
Time-dependent MIL-100 formation
In order to investigate the influence of reaction time on the product formation, the precipitate was centrifuged off after fixed amounts of time, and the clear, red centrifugate was continued to reflux. The separated solids 2a to 2d were activated separately and it is apparent that both the crystallinity (Fig. 2) and the microporosity of the product increase with reaction time (although the formation rate does not). The sorption isotherms for 1 and 2a–2d exhibit a very similar curvature with the typical inflection point at p/p0 = 0.1, which is assigned to the two different cages (cf. Fig. 1). Surprisingly, the samples 2b, 2c and 2d separated after 4 h, 8 h and 24 h reaction time, respectively, yield a significantly increased BET surface area and porosity compared to the 24 h direct-sample 1. We explain the improved sample characteristics of 2b to 2d by a seeding effect of residual nano-MIL particles remaining in the “solution” after centrifugation. MIL nanoparticles of less than 0.2 μm can only be separated by high-speed ultracentrifugation.9 These nano-seeds can then lead to improved crystallization from a solution which has become more dilute compared to the rapidly formed crystallites of 2a from a more concentrated homogeneous solution. Sample 1 then presents an averaged BET surface area and porosity from rapidly precipitated low-quality product (cf. 2a) and slower formed high-quality products (cf. 2b to 2d) when the solution has become more dilute – a drawback of this otherwise attractive fractionated crystallization or synthesis of MIL-100(Fe).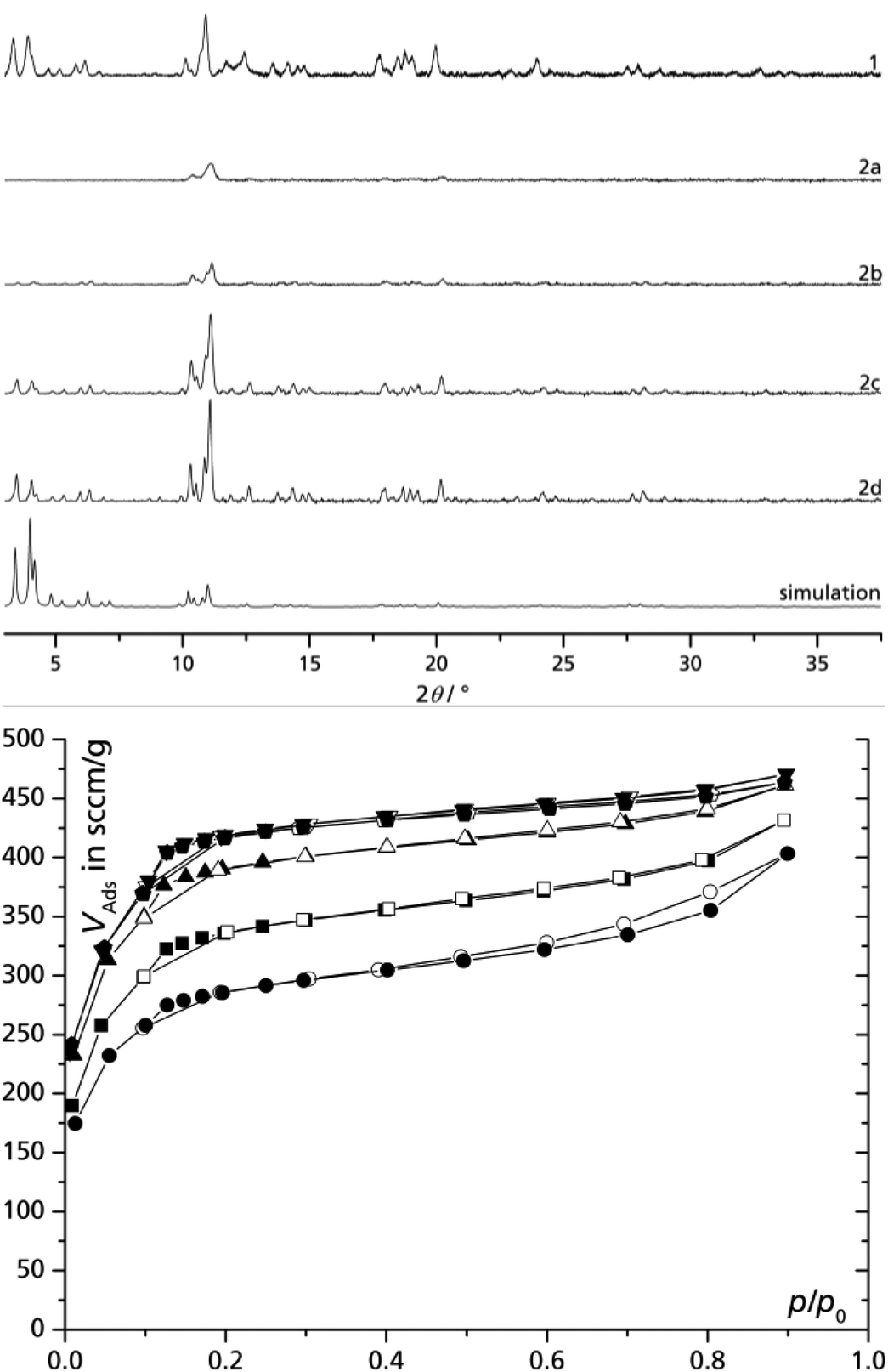 | ||
Fig. 2 Powder X-ray diffractograms (top) and nitrogen adsorption isotherms (77 K, bottom) of activated MIL-100(Fe) prepared by the DMSO/water route using iron(III) nitrate in a single batch with 24 h reaction time (1), and of products removed from the reaction solution by centrifugation after different reaction times (2a–2d): 1 – 24 h without interruption (■), 2a – 2 h (●), 2b – 4 h (▲); 2c – 8 h (▼), 2d – 24 h (![[pentagon filled]](https://www.rsc.org/images/entities/char_e13d.gif) ). Filled symbols = adsorption, empty symbols = desorption; see also Table 1). ). Filled symbols = adsorption, empty symbols = desorption; see also Table 1). | ||
Influence of HNO3 addition
It was recently shown in the fluoride-free synthesis of MIL-101(Cr) that an increased HNO3 content (up to one equivalent versus Cr) in the reaction mixture could substantially improve the yield, BET surface area and pore volume.35 In order to verify whether a higher initial content of nitric acid also improves the crystallization of MIL-100(Fe) syntheses 0, 0.5, 1 and 2 equivalents of nitric acid were added to the reaction mixture of iron(III) nitrate and trimesic acid in DMSO/water. The HNO3 addition is based on the perception that acidification should shift the equilibrium away from the formation of MIL-100(Fe) as nitric acid is released during the process. All products are of good crystallinity (Fig. 3) but improvement of product quality, judged by porosity, could be reached compared to experiments without addition of nitric acid (Table 2). The concentration of HNO3 determines the ligand deprotonation equilibrium in the reaction mixture and is therefore probably the key value rather than the ratio of HNO3 to Fe. Yet, it is likely that the most efficient HNO3 concentration will lie within the same order of magnitude as the Fe concentration, therefore, we gave and worked here with more illustrative HNO3 to Fe equivalents. At 1 eq. of HNO3 the highest BET surface area and pore volume was obtained (approaching the literature data from fluoride-containing syntheses, vide supra) which decreases considerably with higher amounts of HNO3. This finding matches the recent observation on the HNO3 effect in the synthesis of MIL-101(Cr). There, also the addition of 1.0 equivalent of nitric acid in synthesis produced the best porosity of MIL-101(Cr).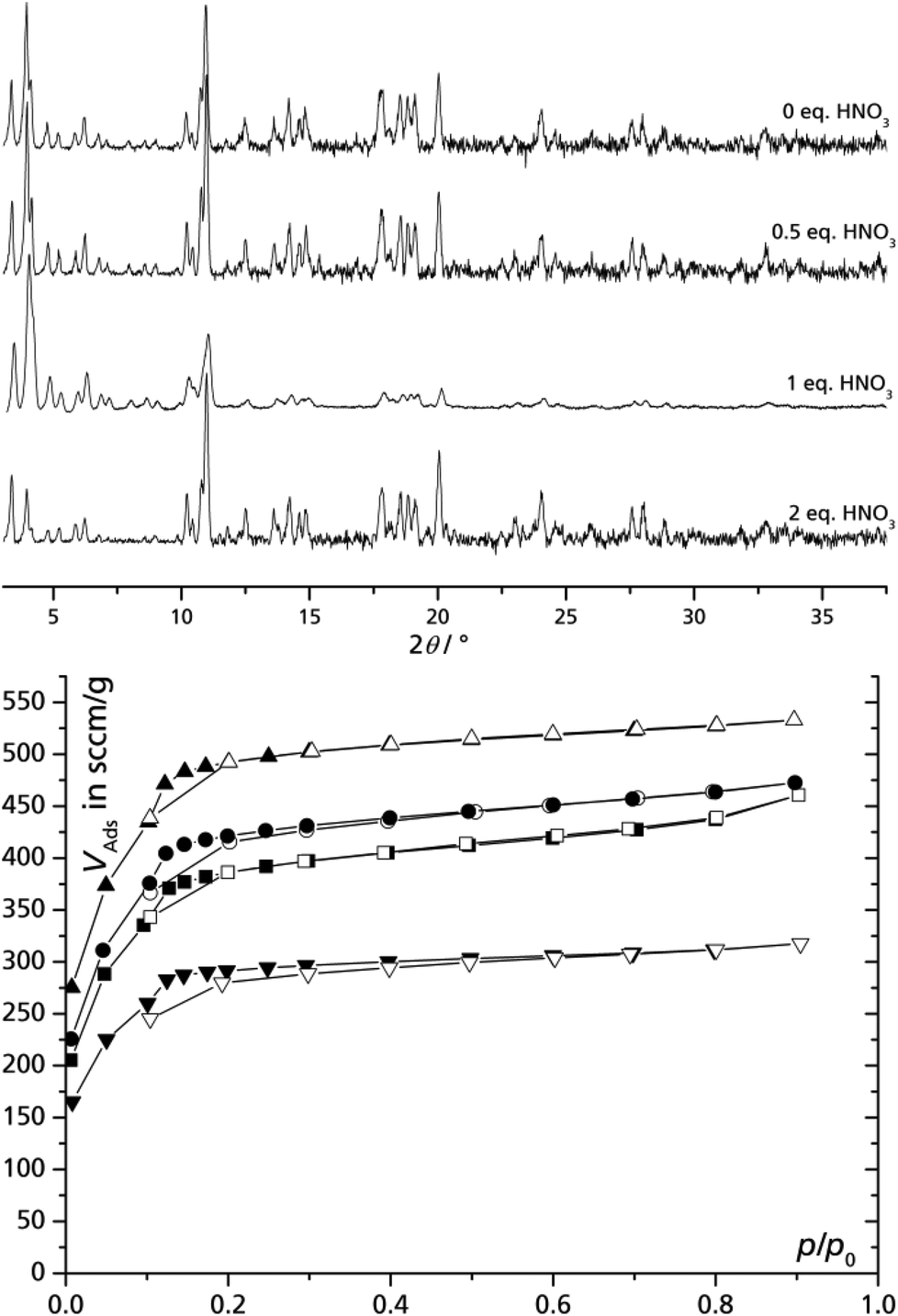 | ||
| Fig. 3 Powder diffractograms (top) and nitrogen adsorption isotherms (77 K, bottom) of MIL-100(Fe) prepared from iron nitrate and trimesic acid using a DMSO water solution with different equivalents of nitric acid (relative to Fe) added. Cu-Kα radiation. ■ – 0 eq., ● – 0.5 eq., ▲ – 1 eq., ▼ – 2 eq. Filled symbols = adsorption, empty symbols = desorption; see also Table 2). | ||
Mechanism of MIL-100 formation
To the best of our knowledge, MIL-100(Fe) has been synthesized up to date only using water as the solvent or dispersant. Under these conditions, formation of MIL-100(Fe) occurs most likely according to the general hydrothermal MOF formation mechanism: deprotonation of the linker due to increased temperature (Le Chatelier's principle) and withdrawal of the deprotonated linker from the equilibrium by precipitation, resulting in further deprotonation of the linker acid. Using the DMSO/water system, the formation mechanism appears to be fundamentally different:From the fact that MIL-100(Fe) is formed only when iron(III) nitrate is used as the Fe source, we deduce that there is a specific mechanism involved in the formation of MIL-100(Fe) in the DMSO/water solvent system. In DMSO/water there is an initial complexation of Fe3+ with formation of DMSO complexes [Fe(DMSO)6–n(H2O/OH)n]3+ instead of only aqua-hydroxido complexes, such as [Fe(H2O)5(OH)]2+. Dimethyl sulfoxide is known to form strong complexes with Fe3+ cations. [Fe(DMSO)6](NO3)3, where DMSO coordinates octahedrally to Fe3+via the oxygen atom (Fig. S3, ESI†), precipitates from solutions of iron(III) nitrate in DMSO (see ESI†).37,40 This has also been observed during our work when the solutions were allowed to sit at room temperature for longer than 5 min with no or too little water added, and the precipitate was hardly soluble in water. This insolubility is explained by the DMSO methyl groups forming the surface of the [Fe(DMSO)6]3+ cation and rendering it hydrophobic (Fig. S3, ESI†). Formation of Fe–DMSO complexes will occur with all iron salts used in Scheme 1 in DMSO solution. The special property of the nitrate anion among the used iron salts (Scheme 1) is its redox property. DMSO is known to decompose partially under experimental conditions generating e.g. formaldehyde and dimethyl sulfide (DMS).36 The distinct smell of DMS could be clearly noticed during all experiments. Especially DMS, but also DMSO reduces nitrate in an acidic environment converting it to nitrite (eqn (1) and (2)).41
| NO3− + DMS → NO2− + DMSO | (1) |
| NO3− + DMSO → NO2− + DMSO2 | (2) |
Nitrite in acidic solution (nitric acid) decomposes easily under disproportionation (eqn (3)).
| 3NO2− + 2H+ → NO3− + 2NO + H2O | (3) |
Also formation of nitric oxide can occur either by a similar reductive pathway (eqn (4)) or by comproportionation of nitrate and nitrite (eqn (5)).42 NOx fumes were observed inside the reflux condenser.
| 2NO3− + DMS + 2H+ → 2NO2 + DMSO + H2O | (4) |
| NO3− + NO2− + 2H+ → 2NO2 + H2O | (5) |
Reactions (3)–(5) all consume protons thereby slowly increasing the pH, as required for the formation of the trinuclear {Fe3(μ3-O)(H2O)2(OH)} secondary building unit with its oxido and hydroxido anions and for the deprotonation of trimesic acid. Thus, for the formation of MIL-100(Fe) upon heating of iron(III) nitrate (with trimesic acid) in DMSO/water a special chemoreductive pathway is involved.
Dincă et al. recently showed that the reduction of nitrate is also key to the formation of MOF-5 using an electroreductive synthesis.43
Conclusions and outlook
The investigations presented in this contribution provide a method to produce highly porous and crystalline MIL-100(Fe) from iron(III) nitrate and trimesic acid in a homogeneous DMSO/water solution at ambient pressure as the main advantage over existing MIL-100(Fe) preparations. This facilitates potential industrial production as more expensive pressure vessels and concomitant precautions are not necessary. Further, the MIL product can be collected by simple solid/liquid separation, e.g., by filtration or centrifugation as unreacted trimesic acid remains in solution. Mechanistically, a specific redox pathway is claimed, based on the reaction of DMSO and its decomposition products with redox-active nitrate anions. This claim is strengthened by the negative results of comparative reaction systems using non-redox-active chloride, mesylate or sulfate anions. The overall procedure is especially promising not only for the deposition of coatings, but also for to the industrial production of this MOF, where heterogeneous, hydrothermal (pressure) reaction conditions should be avoided.Further research should be focused on increasing the reaction yield, solvent regeneration and product extraction. Further research could also be directed to combine the ambient pressure process with in situ coating procedures, e.g., the substrate heating method,12 or the cathodic electrodeposition method.43a
Acknowledgements
The work was supported by the Federal German Ministry of Economics (BMWi) under grant-# 0327851A/B and by the Federal German Ministry of Education and Research (BMBF) under grant-# 03SF0492A/C.Notes and references
- (a) G. Férey, Dalton Trans., 2009, 4400–4415 RSC; (b) C. Serre, S. Kitagawa and P. D. Dietzel, Microporous Mesoporous Mater., 2012, 157, 1–2 CrossRef CAS; (c) J. R. Long and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1213–1214 RSC; (d) H. C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef CAS PubMed; (e) C. Janiak and J. K. Vieth, New J. Chem., 2010, 34, 2366–2388 RSC; (f) S. H. Jhung, N. A. Khan and Z. Hasan, CrystEngComm, 2012, 14, 7099–7109 RSC; (g) M. Gaab, N. Trukhan, S. Maurer, R. Gummaraju and U. Müller, Microporous Mesoporous Mater., 2012, 157, 131–136 CrossRef CAS.
- G. Férey, Chem. Soc. Rev., 2008, 37, 191–214 RSC.
- (a) J. R. Li, R. J. Kuppler and H. C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC; (b) M. P. Suh, H. J. Park, T. K. Prasad and D. W. Lim, Chem. Rev., 2012, 112, 782–835 CrossRef CAS PubMed; (c) R. B. Getman, Y.-S. Bae, C. E. Wilmer and R. Q. Snurr, Chem. Rev., 2012, 112, 703–723 CrossRef CAS PubMed; (d) H. Wu, Q. Gong, D. H. Olson and J. Li, Chem. Rev., 2012, 112, 836–868 CrossRef CAS PubMed; (e) L. Wu, M. Xue, S.-L. Qiu, G. Chaplais, A. Simon-Masseron and J. Patarin, Microporous Mesoporous Mater., 2012, 157, 75–81 CrossRef CAS; (f) L. J. Murray, M. Dincă and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294–1314 RSC; (g) Z. Chen, S. Xiang, H. D. Arman, P. Li, S. Tidrow, D. Zhao and B. Chen, Eur. J. Inorg. Chem., 2010, 3745–3749 CrossRef CAS.
- (a) Z. Zhang, Y. Zhao, Q. Gong, Z. Li and J. Li, Chem. Commun., 2013, 49, 653–661 RSC; (b) J. R. Li, J. Sculley and H. C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS PubMed; (c) J.-R. Li, Y. Ma, M. C. McCarthy, J. Sculley, J. Yu, H.-K. Jeong, P. B. Balbuena and H.-C. Zhou, Coord. Chem. Rev., 2011, 255, 1791–1823 CrossRef CAS; (d) B. Zheng, H. Liu, Z. Wang, X. Yu, P. Yi and J. Bai, CrystEngComm, 2013, 15, 3517–3520 RSC; (e) T.-Z. Zhang, Z.-M. Zhang, Y. Lu, H. Fu and E.-B. Wang, CrystEngComm, 2013, 15, 459–462 RSC; (f) C. Hou, Q. Liu, T.-A. Okamura, P. Wang and W.-Y. Sun, CrystEngComm, 2012, 14, 8569–8576 RSC; (g) W.-Y. Gao, Y. Niu, Y. Chen, L. Wojtas, J. Cai, Y.-S. Chen and S. Ma, CrystEngComm, 2012, 14, 6115–6117 RSC; (h) Z. R. Herm, R. Krishna and J. R. Long, Microporous Mesoporous Mater., 2012, 157, 94–100 CrossRef CAS; (i) M. G. Plaza, A. M. Ribeiro, A. Ferreira, J. C. Santos, Y. K. Hwang, Y. K. Seo, U. H. Lee, J. S. Chang, J. M. Loureiro and A. E. Rodrigues, Microporous Mesoporous Mater., 2012, 153, 178–190 CrossRef CAS; (j) C. Li, W. Qiu, W. Shi, H. Song, G. Bai, H. He, J. Li and M. J. Zaworotko, CrystEngComm, 2012, 14, 1929–1932 RSC; (k) H. B. Tanh Jeazet, C. Staudt and C. Janiak, Dalton Trans., 2012, 41, 14003–14027 RSC; (l) B. Zornoza, C. Tellez, J. Coronas, J. Gascon and F. Kapteijn, Microporous Mesoporous Mater., 2013, 166, 67–78 CrossRef CAS; (m) G. Dong, H. Li and V. Chen, J. Mater. Chem. A, 2013, 1, 4610–4630 RSC; (n) B. Seoane, J. Coronas, I. Gascon, M. E. Benavides, O. Karvan, J. Caro, F. Kapteijn and J. Gascon, Chem. Soc. Rev., 2015, 44, 2421–2454 RSC.
- G. Férey, C. Serre, T. Devic, G. Maurin, H. Jobic, P. L. Llewellyn, G. de Weireld, A. Vimont, M. Daturi and J. S. Chang, Chem. Soc. Rev., 2011, 40, 550–562 RSC.
- (a) E. M. Dias and C. Petit, J. Mater. Chem. A, 2015, 3, 22484–22506 RSC; (b) A. J. Howarth, Y. Liu, J. T. Hupp and O. K. Farha, CrystEngComm, 2015, 17, 7245–7253 RSC; (c) K. A. Cychosz, R. Ahmad and A. J. Matzger, Chem. Sci., 2010, 1, 293–302 RSC.
- K. A. Cychosz and A. J. Matzger, Langmuir, 2010, 26, 17198–17202 CrossRef CAS PubMed.
- (a) M. Yoon, R. Srirambalaji and K. Kim, Chem. Rev., 2012, 112, 1196–1231 CrossRef CAS PubMed; (b) J. Guo, J. Yang, Y.-Y. Liu and J.-F. Ma, CrystEngComm, 2012, 14, 6609–6617 RSC; (c) J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC; (d) B. J. Burnett, P. M. Barron and W. Choe, CrystEngComm, 2012, 14, 3839–3846 RSC; (e) H.-J. Pang, H.-Y. Ma, J. Peng, C.-J. Zhang, P.-P. Zhang and Z.-M. Su, CrystEngComm, 2011, 13, 7079–7085 RSC.
- A. Herbst, A. Khutia and C. Janiak, Inorg. Chem., 2014, 53, 7319–7333 CrossRef CAS PubMed.
- (a) F. Jeremias, D. Fröhlich, C. Janiak and S. K. Henninger, New J. Chem., 2014, 38, 1846–1852 RSC; (b) C. Janiak and S. K. Henninger, Chimia, 2013, 67, 419–424 CrossRef CAS PubMed; (c) C. Janiak and S. K. Henninger, Nachr. Chem., 2013, 61, 520–523 CrossRef CAS; (d) S. K. Henninger, F. Jeremias, H. Kummer and C. Janiak, Eur. J. Inorg. Chem., 2012, 2625–2634 CrossRef CAS; (e) S. K. Henninger, H. A. Habib and C. Janiak, J. Am. Chem. Soc., 2009, 131, 2776–2777 CrossRef CAS PubMed; (f) J. Ehrenmann, S. K. Henninger and C. Janiak, Eur. J. Inorg. Chem., 2011, 471–474 CrossRef CAS; (g) F. Jeremias, V. Lozan, S. Henninger and C. Janiak, Dalton Trans., 2013, 42, 15967–15973 RSC; (h) D. Fröhlich, S. K. Henninger and C. Janiak, Dalton Trans., 2014, 43, 15300–15304 RSC.
- F. Jeremias, A. Khutia, S. K. Henninger and C. Janiak, J. Mater. Chem., 2012, 22, 10148–10151 RSC.
- F. Jeremias, S. K. Henninger and C. Janiak, Chem. Commun., 2012, 48, 9708–9710 RSC.
- F. Jeremias, D. Fröhlich, C. Janiak and S. K. Henninger, RSC Adv., 2014, 4, 24073–24082 RSC.
- (a) A. U. Czaja, N. Trukhan and U. Muller, Chem. Soc. Rev., 2009, 38, 1284–1293 RSC; (b) M. J. Prakash and M. S. Lah, Chem. Commun., 2009, 3326–3341 RSC.
- K. Brandenburg, Diamond 3.2, Crystal and Molecular Structure Visualization, Crystal Impact - K. Brandenburg & H. Putz GbR, Bonn, Germany, 2009 Search PubMed.
- (a) G. Férey, Chem. Mater., 2001, 13, 3084–3098 CrossRef; (b) G. Férey, C. Mellot-Draznieks, C. Serre and F. Millange, Acc. Chem. Res., 2005, 38, 217–225 CrossRef PubMed; (c) G. Férey and C. Serre, Chem. Soc. Rev., 2009, 38, 1380–1399 RSC.
- (a) J. J. Low, A. I. Benin, P. Jakubczak, J. F. Abrahamian, S. A. Faheem and R. R. Willis, J. Am. Chem. Soc., 2009, 131, 15834–15842 CrossRef CAS PubMed; (b) J. Canivet, J. Bonnefoy, C. Daniel, A. Legrand, B. Coasne and D. Farrusseng, New J. Chem., 2014, 38, 3102–3111 RSC; (c) J. Canivet, A. Fateeva, Y. Guo, B. Coasne and D. Farrusseng, Chem. Soc. Rev., 2014, 43, 5594–5617 RSC.
- P. Horcajada, S. Surble, C. Serre, D. Y. Hong, Y. K. Seo, J. S. Chang, J. M. Greneche, I. Margiolaki and G. Férey, Chem. Commun., 2007, 2820–2822 RSC.
- (a) C. Volkringer, T. Loiseau, M. Haouas, F. Taulelle, D. Popov, M. Burghammer, C. Riekel, C. Zlotea, F. Cuevas, M. Latroche, D. Phanon, C. Knöfelv, P. L. Llewellyn and G. Férey, Chem. Mater., 2009, 21, 5783–5791 CrossRef CAS; (b) P. L. Llewellyn, S. Bourrelly, C. Serre, A. Vimont, M. Daturi, L. Hamon, G. de Weireld, J. S. Chang, D. Y. Hong, Y. Kyu Hwang, S. Hwa Jhung and G. Férey, Langmuir, 2008, 24, 7245–7250 CrossRef CAS PubMed; (c) L. Hamon, C. Serre, T. Devic, T. Loiseau, F. Millange, G. Férey and G. de Weireld, J. Am. Chem. Soc., 2009, 131, 8775–8777 CrossRef CAS PubMed; (d) A. Vimont, J. M. Goupil, J. C. Lavalley, M. Daturi, S. Surble, C. Serre, F. Millange, G. Férey and N. Audebrand, J. Am. Chem. Soc., 2006, 128, 3218–3227 CrossRef CAS PubMed.
- F. Zhang, J. Shi, Y. Jin, Y. Fu, Y. Zhong and W. Zhu, Chem. Eng. J., 2015, 259, 183–190 CrossRef CAS.
- G. Férey, C. Serre, C. Mellot-Draznieks, F. Millange, S. Surble, J. Dutour and I. Margiolaki, Angew. Chem., Int. Ed., 2004, 43, 6296–6301 CrossRef PubMed.
- G. Akiyama, R. Matsuda and S. Kitagawa, Chem. Lett., 2010, 39, 360–361 CrossRef CAS.
- (a) J. S. Lee, S. H. Jhung, J. W. Yoon, Y. K. Hwang and J.-S. Chang, J. Ind. Eng. Chem., 2009, 15, 674–676 CrossRef CAS; (b) L. Kurfiřtová, Y.-K. Seo, Y. K. Hwang, J.-S. Chang and J. Čejka, Catal. Today, 2012, 179, 85–90 CrossRef; (c) P. Küsgens, M. Rose, I. Senkovska, H. Fröde, A. Henschel, S. Siegle and S. Kaskel, Microporous Mesoporous Mater., 2009, 120, 325–330 CrossRef.
- H. Reinsch and N. Stock, CrystEngComm, 2013, 15, 544 RSC.
- A. Lieb, H. Leclerc, T. Devic, C. Serre, I. Margiolaki, F. Mahjoubi, J. S. Lee, A. Vimont, M. Daturi and J.-S. Chang, Microporous Mesoporous Mater., 2012, 157, 18–23 CrossRef CAS.
- C. Volkringer, D. Popov, T. Loiseau, G. Férey, M. Burghammer, C. Riekel, M. Haouas and F. Taulelle, Chem. Mater., 2009, 21, 5695–5697 CrossRef CAS.
- P. Horcajada, T. Chalati, C. Serre, B. Gillet, C. Sebrie, T. Baati, J. F. Eubank, D. Heurtaux, P. Clayette, C. Kreuz, J. S. Chang, Y. K. Hwang, V. Marsaud, P. N. Bories, L. Cynober, S. Gil, G. Férey, P. Couvreur and R. Gref, Nat. Mater., 2010, 9, 172–178 CrossRef CAS PubMed.
- R. Canioni, C. Roch-Marchal, F. Sécheresse, P. Horcajada, C. Serre, M. Hardi-Dan, G. Férey, J.-M. Grenèche, F. Lefebvre, J.-S. Chang, Y.-K. Hwang, O. Lebedev, S. Turner and G. van Tendeloo, J. Mater. Chem., 2011, 21, 1226–1233 RSC.
- Y.-K. Seo, J. W. Yoon, J. S. Lee, U. H. Lee, Y. K. Hwang, C.-H. Jun, P. Horcajada, C. Serre and J.-S. Chang, Microporous Mesoporous Mater., 2012, 157, 137–145 CrossRef CAS.
- J. Shi, S. Hei, H. Liu, Y. Fu, F. Zhang, Y. Zhong and W. Zhu, J. Chem., 2013, 2013, 1–4 Search PubMed.
- M. Yamashita, M. Yamashita, M. Suzuki, H. Hirai and H. Kajigaya, Crit. Care Med., 2001, 29, 1575–1578 CrossRef CAS PubMed.
- (a) M. Faustini, J. Kim, G.-Y. Jeong, J. Y. Kim, H. R. Moon, W.-S. Ahn and D.-P. Kim, J. Am. Chem. Soc., 2013, 135, 14619–14626 CrossRef CAS PubMed; (b) P. A. Bayliss, I. A. Ibarra, E. Pérez, S. Yang, C. C. Tang, M. Poliakoff and M. Schröder, Green Chem., 2014, 16, 3796 RSC; (c) M. P. Batten, M. Rubio-Martinez, T. Hadley, K.-C. Carey, K.-S. Lim, A. Polyzos and M. R. Hill, Curr. Opin. Chem. Eng., 2015, 8, 55–59 CrossRef.
- S. Waitschat, M. T. Wharmby and N. Stock, Dalton Trans., 2015, 44, 11235–11240 RSC.
- N. Stock and S. Biswas, Chem. Rev., 2012, 112, 933–969 CrossRef CAS PubMed.
- T. Zhao, F. Jeremias, I. Boldog, B. Nguyen, S. K. Henninger and C. Janiak, Dalton Trans., 2015, 44, 16791–16801 RSC.
- Gaylord Chemical, Dimethyl Sulfoxide (DMSO). Physical Properties - Bulletin # 101, http://www.gaylordchemical.com/65-2/literature/101b-dmso-physical-properties/, 2005.
- H. Dehn, US Pat, 3463684, 1969 Search PubMed.
- R. C. Paul, R. C. Narula and S. K. Vasisht, Transition Met. Chem., 1978, 3, 35–38 CrossRef CAS.
- E. A. Berdonosova, K. A. Kovalenko, E. V. Polyakova, S. N. Klyamkin and V. P. Fedin, J. Phys. Chem. C, 2015, 119, 13098–13104 CAS.
- (a) W. B. Moniz, C. F. Poranski and D. L. Venezky, U. S. Gov. Res. Dev. Rep., 1968, 1–38 Search PubMed; (b) J. R. Tzou, M. Mullaney, R. E. Norman and S. C. Chang, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1995, 51, 2249–2252 CrossRef.
- Gaylord Chemical, Dimethyl Sulfide Reaction Solvent Guide - Bulletin # 203B, http://www.gaylordchemical.com/?page=203b-dms-reaction-solvent-guide, 2007.
- A. Saytzeff, Liebigs Ann. Chem., 1867, 144, 148–156 CrossRef.
- (a) M. Li and M. Dincă, J. Am. Chem. Soc., 2011, 133, 12926–12929 CrossRef CAS PubMed; (b) M. Li and M. Dincă, Chem. Mater., 2015, 27, 3203–3206 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: PXRD of Fe(NO3)3·6DMSO, dew point/bubble point curves of DMSO/water. See DOI: 10.1039/c6dt01179a |
| This journal is © The Royal Society of Chemistry 2016 |