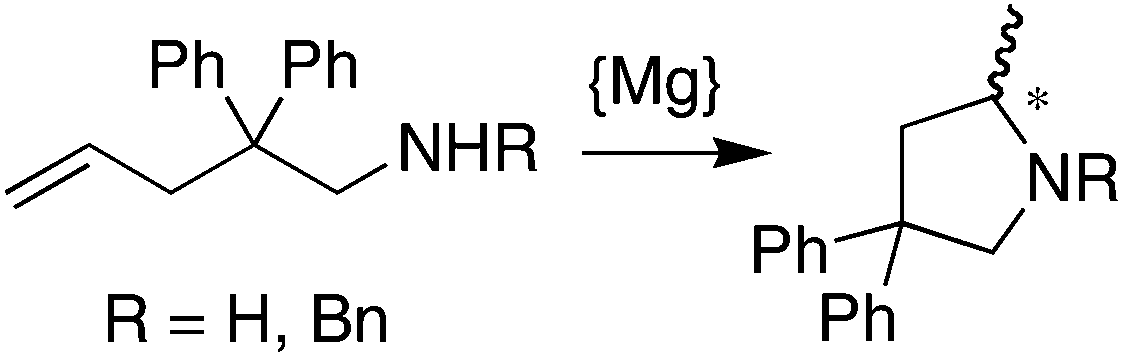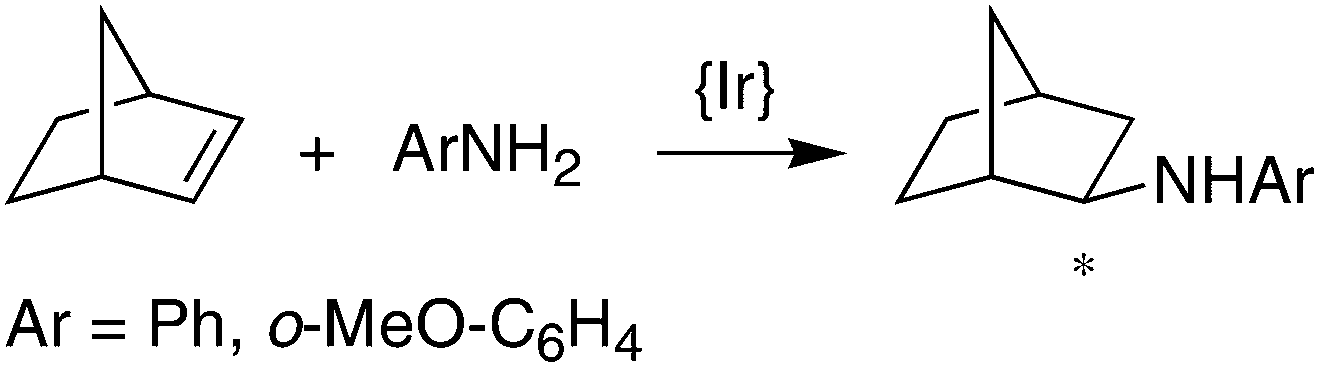DOI:
10.1039/C6DT01146B
(Paper)
Dalton Trans., 2016,
45, 12028-12040
Chiral amino-phosphine and amido-phosphine complexes of Ir and Mg. Catalytic applications in olefin hydroamination†
Received
23rd March 2016
, Accepted 30th June 2016
First published on 11th July 2016
Abstract
The reactions of rac- and (S,S)-trans-9,10-dihydro-9,10-ethanoanthracene-11,12-diamine (ANDEN) with PClPh2 in the presence of NEt3 yield the chiral amino-phosphine ligands rac-6 and (S,S)-6, respectively, on multi-gram scales. Both forms of 6 react quantitatively with MgPh2 to afford the C2-symmetric, N-bound Mg amidophosphine complexes rac-7 and (S,S)-7. The former crystallizes as a racemic conglomerate, which is a rare occurrence. Mixing (S,S)- or rac-6 with [IrCl(COE)2]2 leads in both cases to the homochiral dinuclear chloro-bridged P-ligated aminophosphine iridium complexes (S,S,S,S)-9 and rac-9 in excellent yields. X-ray quality single crystals only grow as the racemic compound (or ‘true racemate’) rac-9 thanks to its lowered solubility. In the coordinating solvent CH3CN, rac-9 transforms in high yield into mononuclear Ir-complex rac-10. The crystal structures of compounds rac-6, (S,S)-7, rac-9, and rac-10 reveal the ambidentate nature of the P–N function: amide-coordination in the Mg-complex (S,S)-7 and P-chelation of the softer Ir(I) centres in complexes rac-9 and rac-10. Furthermore, the crystal structures show flexible, symmetry lowering seven-membered P-chelate rings in the Ir complexes and a surprising amount of deformation within the ANDEN backbone. The simulation of this deformation by DFT and SCF calculations indicates low energy barriers. (S,S)-7 and (S,S,S,S)-9 catalyze the intra- and intermolecular hydroamination of alkenes, respectively: 5 mol% of (S,S)-7 affords 2-methyl-4,4′-diphenylcyclopentyl amine quantitatively (7% ee), and 2.5 mol% of (S,S,S,S)-9 in the presence of 5.0 mol% co-catalyst (LDA, PhLi, or MgPh2) gives exo-(2-arylamino)bornanes in up to 68% yield and up to 16% ee.
Introduction
Chiral amino-phosphines such as 1–3 are competent ligands for asymmetric hydrogenations of de-hydroaminoacids1 to the point where ligand 1 has been used industrially for the production of the artificial sweetener aspartame ((S,S)-aspartylphenylalanine).2 The N-atom in aminophosphines may strategically be functionalized either by bulky substituents for steric control3 or by leaving a hydrogen atom for reactivity control via H-bridging interactions4 and possibly η2-P,N coordination.5 An indication of the importance of the N-substitution is the observation that the optical yield of hydrogenation products obtained with the H-substituted ligand 2 is not only markedly higher than with its N-methylated analogue, but also leads to products with inverted configurations.6 Moreover, the deprotonation of the N–H function in an aminophosphine leads to a vicinal hard-amide/soft-phosphine donor system. The use of trans-9,10-dihydro-9,10-ethanoanthracene-11,12-diamine (termed as ANDEN in what follows) as a rigid 1,2-diamino scaffold7 for the construction of effective chiral ligands is exemplified by the success of Trost's ligand 4,8 which also remains the only example of an ANDEN-derived phosphine ligand. Here, we wish to communicate the synthesis of a new ANDEN-based amino-phosphine ligand and its Ir- and Mg-complexes, which are promising catalysts for the asymmetric hydroamination of olefins.9 Electron-rich Ir(I)-complexes figure among the first successful asymmetric intermolecular olefin hydroamination catalysts,10 and Hultzsch and co-workers recently showed that chiral Mg-complexes catalyze intramolecular hydroaminations with good enantioselectivities.11 Preliminary results on catalytic olefin hydroamination reactions employing the new chiral Ir- and Mg-complexes are also disclosed.
Results and discussion
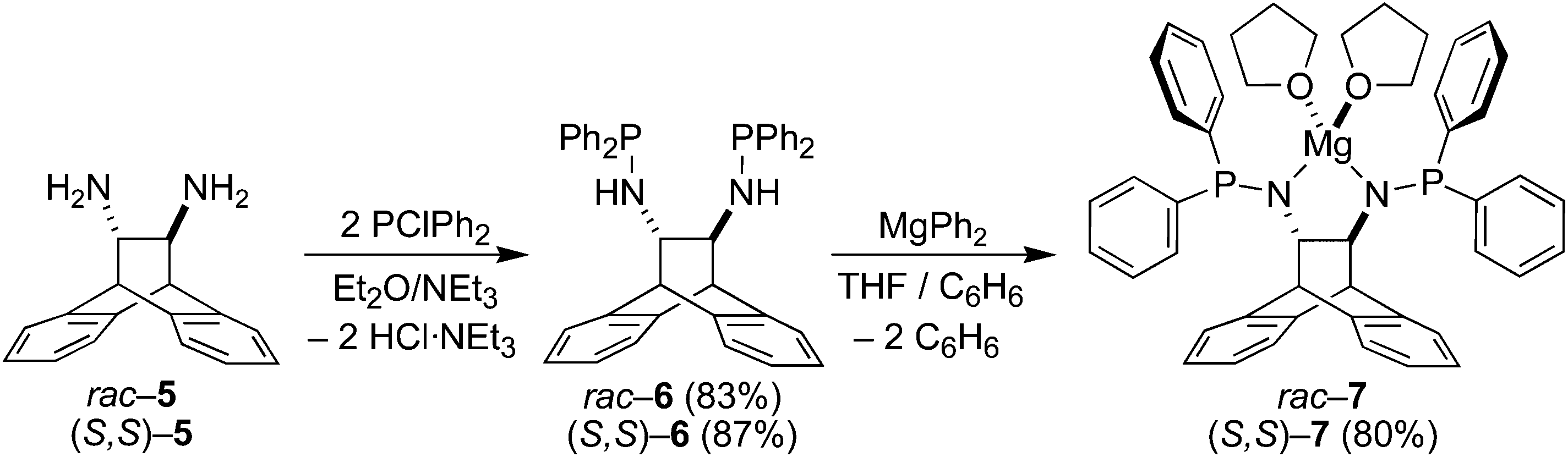
The amino phosphines (S,S)-6 and rac-6 are accessible in excellent yields and on multi-gram scales as crystalline materials according to Scheme 1. The solubility of (S,S)-6 is significantly higher than that of rac-6, and the 31P-NMR spectra of both products are characterized by a singlet resonance at 40.3 ppm. The protons of the N–H function resonate as dd (J values of 3.6 and 10.3 Hz) centered at 1.69 ppm, and the two protons of the connecting ethylene bridge appear as multiplets centered at 3.36 ppm. A single crystal X-ray diffraction study of rac-6 revealed that the molecule has approximate C2-symmetry (Fig. 1) and co-crystallizes with half an equivalent of n-pentane. The P–N–C bond angles of ca. 120° suggest sp2 hybridization of the N-atoms, while the sum of the angles about P1 and P2 are 308.27(12)° and 306.55(12)°, respectively. The N–C–C–N torsion angle of 107.88(14)° is close to the mean value of 111(5)° found in nine crystal structures of uncomplexed ANDEN-derived molecules (vide infra). The N–H groups are not involved in any hydrogen bonding interactions because they are sterically protected by the surrounding phenyl rings.
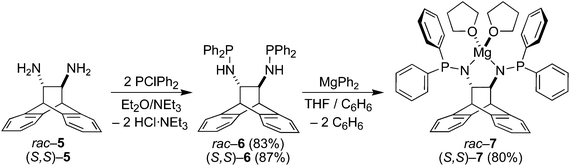 |
| | Scheme 1 Synthesis of diaminophosphines and their Mg–salts. | |
 |
| | Fig. 1 The molecular structure of the (R,R)-enantiomer in the crystal of rac-6 drawn with 50% probability displacement ellipsoids. Most H-atoms have been omitted for clarity. Selected bond lengths (Å) and angles (°) are: P1–N1 1.6840(14), P(2)–N(2) 1.6911(14), P(1)–C(17) 1.8363(17), N(1)–C(15) 1.4562(19), N(2)–C(16) 1.456(2), C(15)–C(16) 1.560(2), C(15)–N(1)–P(1) 122.20(11), C(16)–N(2)–P(2) 120.98(11), P1⋯P2 5.3045(15). | |
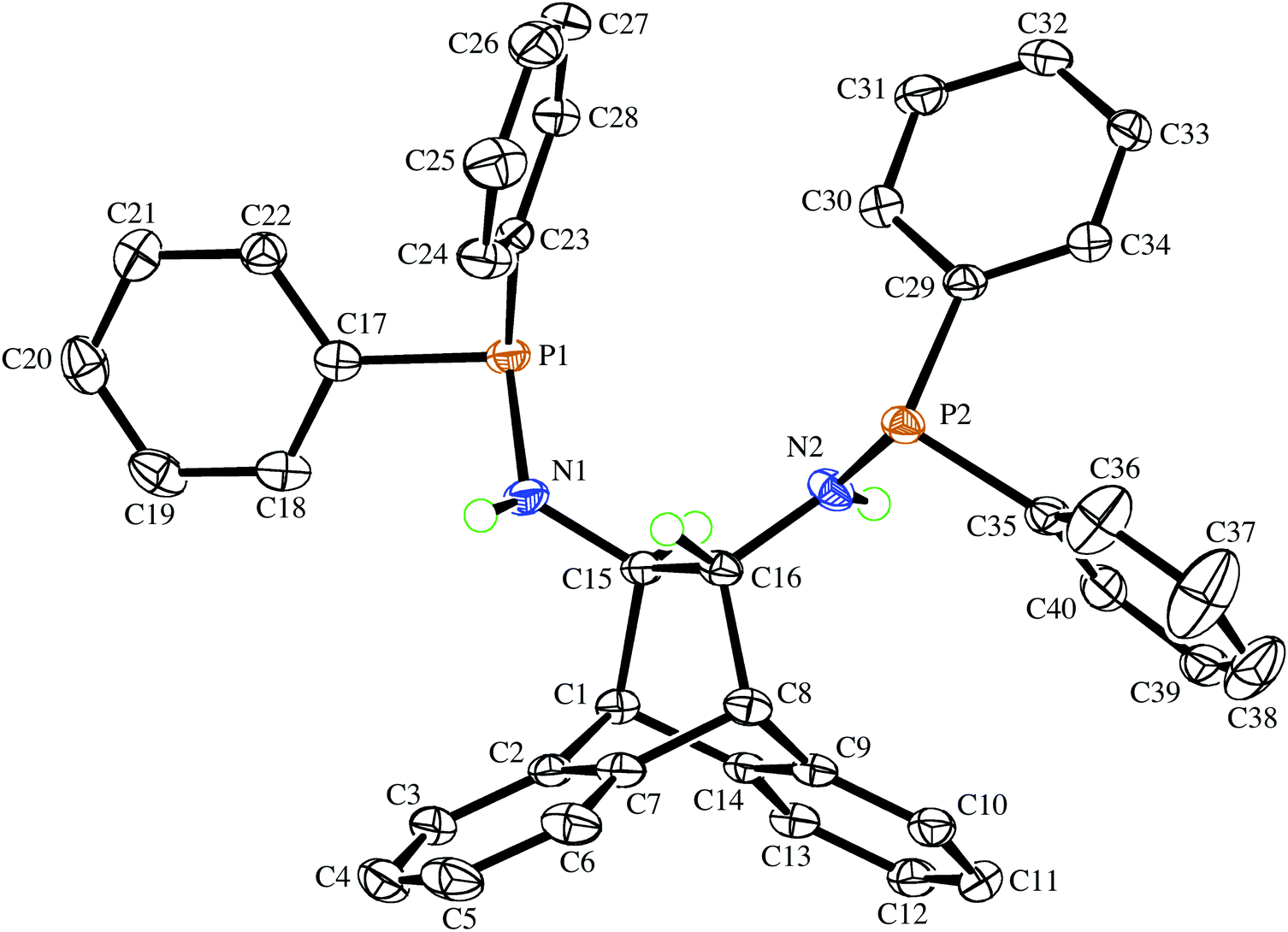
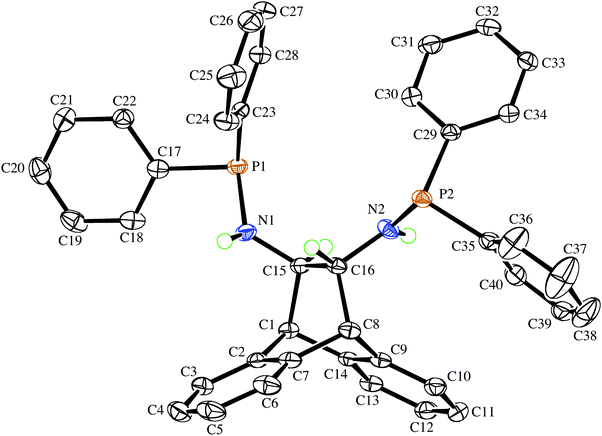
The deprotonation of (S,S)-6 with MgPh2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 12 in THF solution followed by addition of benzene affords the off-white crystalline solvento complex (S,S)-7 in good yield (Scheme 1). It has low solubility in benzene and toluene, and the 1H-NMR spectrum confirms the deprotonation of the amine functions (the dd resonance at 1.60 ppm is no longer present). The multiplet structure of the adjacent bridge-protons collapses to a singlet, which is shifted up-field to 3.17 ppm when compared to 3.36 ppm in 6, while the bridgehead protons exhibit a downfield shift from 3.95 in 6 to 4.55 ppm. The 31P{1H}-NMR spectrum of 7 in THF-d8 shows a singlet resonance at 46.5 ppm with only a moderate downfield shift compared to the free aminophosphine 6, which is consistent with the absence of P-coordination (as established by the crystal structure). Complex 7 is stable in the solid state, and a THF-d8 solution that was heated to 60 °C for 30 min did not show any signs of decomposition. Single crystals suitable for an X-ray diffraction analysis were obtained from the corresponding racemic complex rac-7, which crystallizes in the chiral space group P21 as a racemic conglomerate with one equivalent of benzene. The crystal that was picked contained exclusively the (S,S)-enantiomer, and its molecular structure is depicted in Fig. 2. The formation of a racemic conglomerate of enantiomorphous crystals is also termed ‘spontaneous resolution’13 and does not occur commonly.14 It means that rac-7 could in principle be mechanically separated into its enantiomers (Pasteur's method of triage). The formation of a conglomerate in the case of rac-7 is also consistent with the observed slower rate of crystallization due to its lower entropy of crystallization when compared to (S,S)-7. The coordination sphere around magnesium is distorted tetrahedral with quite a small N–Mg–N bite angle of 91.82(9)°, and the molecule possesses an approximate C2 symmetry axis passing through the Mg atom and the midpoint of C1–C2 (see insert of Fig. 2).15 While the P–C and P–N bond distances in rac-6 and (S,S)-7 are essentially unchanged, the N atoms in (S,S)-7 display a pyramidal rather than a trigonal planar geometry as observed in rac-6. Their considerable sp3 character is indicated by the sums of the bond angles around atoms N1 and N2, which are 340.6(3)° and 337.1(3)°, respectively. The sums of the bond angles around P1 and P2 in (S,S)-7 are 304.6(2)° and 304.73(2)°, respectively. The N–C–C–N torsion angle of the backbone is very small at 72.1(2)°,16 with a concomitant large deviation of −24.9(3)° from planarity of the C3–C2–C1–C6 bridge. These large distortions are caused by the five-membered magnesium chelate ring and also prove that the ANDEN backbone is surprisingly flexible (this observation will be discussed further below). The H⋯Mg distances of the hydrogen atoms pertaining to the stereogenic C atoms measure 2.6 and 2.7 Å and span Mg⋯H–C angles of 72.5° and 77.5°, respectively, and thus do not fulfill the criteria for agostic interactions.17 The co-crystallized benzene in the structure links a benzo-group of the backbone of one molecule of 7 with a phenyl substituent on a P atom of the other molecule of 7via two T-shaped C–H⋯π interactions.18 It should be noted that well characterized N-coordinated amido-phosphine metal complexes are rather rare,19 and to the best of our knowledge this is the first structurally authenticated chiral example of such a complex. Furthermore, the vicinity of the hard Lewis-acidic Mg2+ centre to the soft phosphine Lewis-bases creates a bifunctional complex that should facilitate the activation of polar bonds. Preliminary attempts to alkylate the amide functions in 7 with common organic electrophiles only led to inseparable mixtures.20
12 in THF solution followed by addition of benzene affords the off-white crystalline solvento complex (S,S)-7 in good yield (Scheme 1). It has low solubility in benzene and toluene, and the 1H-NMR spectrum confirms the deprotonation of the amine functions (the dd resonance at 1.60 ppm is no longer present). The multiplet structure of the adjacent bridge-protons collapses to a singlet, which is shifted up-field to 3.17 ppm when compared to 3.36 ppm in 6, while the bridgehead protons exhibit a downfield shift from 3.95 in 6 to 4.55 ppm. The 31P{1H}-NMR spectrum of 7 in THF-d8 shows a singlet resonance at 46.5 ppm with only a moderate downfield shift compared to the free aminophosphine 6, which is consistent with the absence of P-coordination (as established by the crystal structure). Complex 7 is stable in the solid state, and a THF-d8 solution that was heated to 60 °C for 30 min did not show any signs of decomposition. Single crystals suitable for an X-ray diffraction analysis were obtained from the corresponding racemic complex rac-7, which crystallizes in the chiral space group P21 as a racemic conglomerate with one equivalent of benzene. The crystal that was picked contained exclusively the (S,S)-enantiomer, and its molecular structure is depicted in Fig. 2. The formation of a racemic conglomerate of enantiomorphous crystals is also termed ‘spontaneous resolution’13 and does not occur commonly.14 It means that rac-7 could in principle be mechanically separated into its enantiomers (Pasteur's method of triage). The formation of a conglomerate in the case of rac-7 is also consistent with the observed slower rate of crystallization due to its lower entropy of crystallization when compared to (S,S)-7. The coordination sphere around magnesium is distorted tetrahedral with quite a small N–Mg–N bite angle of 91.82(9)°, and the molecule possesses an approximate C2 symmetry axis passing through the Mg atom and the midpoint of C1–C2 (see insert of Fig. 2).15 While the P–C and P–N bond distances in rac-6 and (S,S)-7 are essentially unchanged, the N atoms in (S,S)-7 display a pyramidal rather than a trigonal planar geometry as observed in rac-6. Their considerable sp3 character is indicated by the sums of the bond angles around atoms N1 and N2, which are 340.6(3)° and 337.1(3)°, respectively. The sums of the bond angles around P1 and P2 in (S,S)-7 are 304.6(2)° and 304.73(2)°, respectively. The N–C–C–N torsion angle of the backbone is very small at 72.1(2)°,16 with a concomitant large deviation of −24.9(3)° from planarity of the C3–C2–C1–C6 bridge. These large distortions are caused by the five-membered magnesium chelate ring and also prove that the ANDEN backbone is surprisingly flexible (this observation will be discussed further below). The H⋯Mg distances of the hydrogen atoms pertaining to the stereogenic C atoms measure 2.6 and 2.7 Å and span Mg⋯H–C angles of 72.5° and 77.5°, respectively, and thus do not fulfill the criteria for agostic interactions.17 The co-crystallized benzene in the structure links a benzo-group of the backbone of one molecule of 7 with a phenyl substituent on a P atom of the other molecule of 7via two T-shaped C–H⋯π interactions.18 It should be noted that well characterized N-coordinated amido-phosphine metal complexes are rather rare,19 and to the best of our knowledge this is the first structurally authenticated chiral example of such a complex. Furthermore, the vicinity of the hard Lewis-acidic Mg2+ centre to the soft phosphine Lewis-bases creates a bifunctional complex that should facilitate the activation of polar bonds. Preliminary attempts to alkylate the amide functions in 7 with common organic electrophiles only led to inseparable mixtures.20
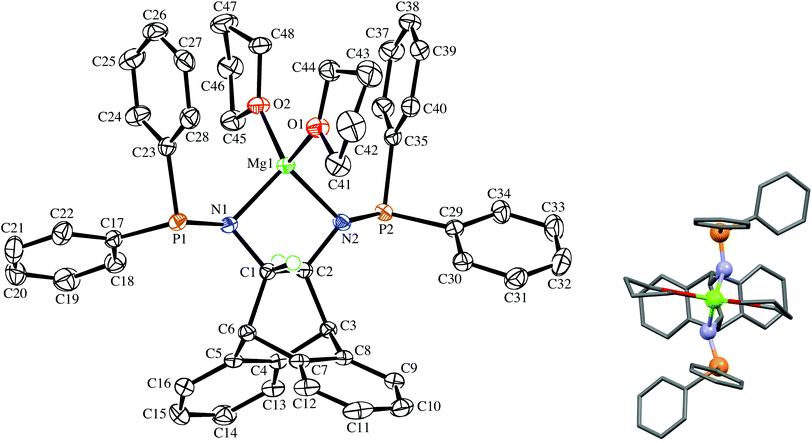 |
| | Fig. 2 The molecular structure of (S,S)-7 in the chiral crystal drawn with 50% probability displacement ellipsoids (the insert shows its approximate C2 symmetry). Most H-atoms have been omitted for clarity. Selected bond lengths (Å) and angles (°) are: Mg(1)–N(1) 2.026(2), Mg(1)–N(2) 2.035(2), Mg(1)–O(1) 2.020(2), Mg(1)–O(2) 1.988(2), P(1)–N(1) 1.677(3), P(2)–N(2) 1.676(3), P(1)–C(17) 1.847(3), C(1)–C(2) 1.539(4), N(1)–Mg(1)–N(2) 91.82(9), O(2)–Mg(1)–O(1) 106.08(9), O(2)–Mg(1)–N(1) 113.83(10), O(1)–Mg(1)–N(1) 115.41(11), O(2)–Mg(1)–N(2) 121.23(10), O(1)–Mg(1)–N(2) 108.44(10). | |
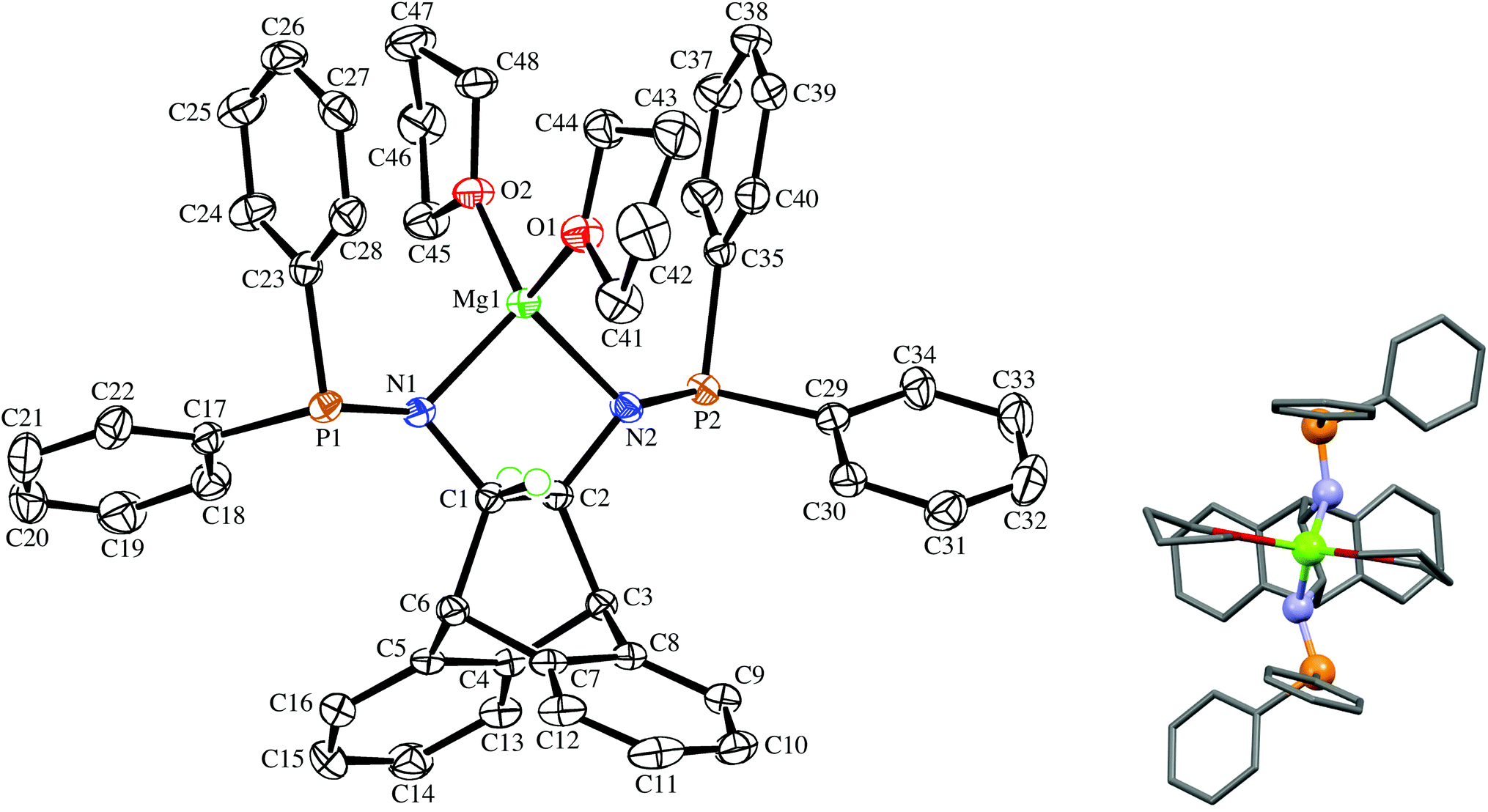
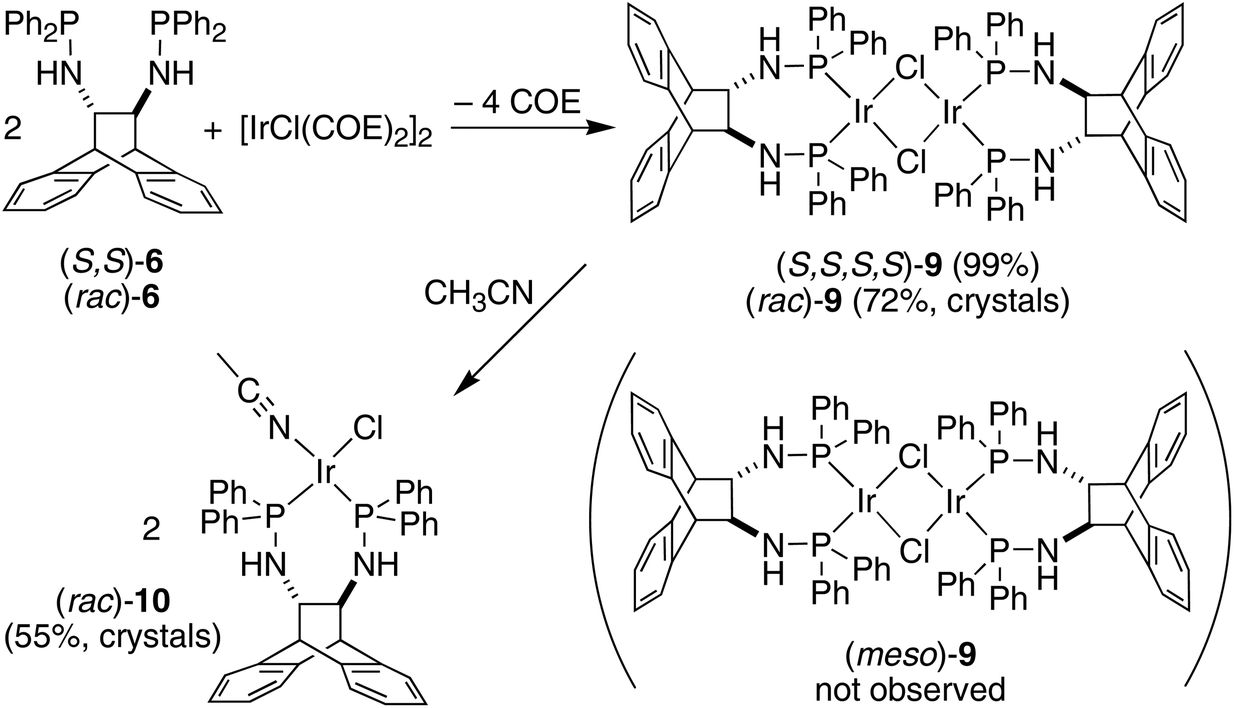
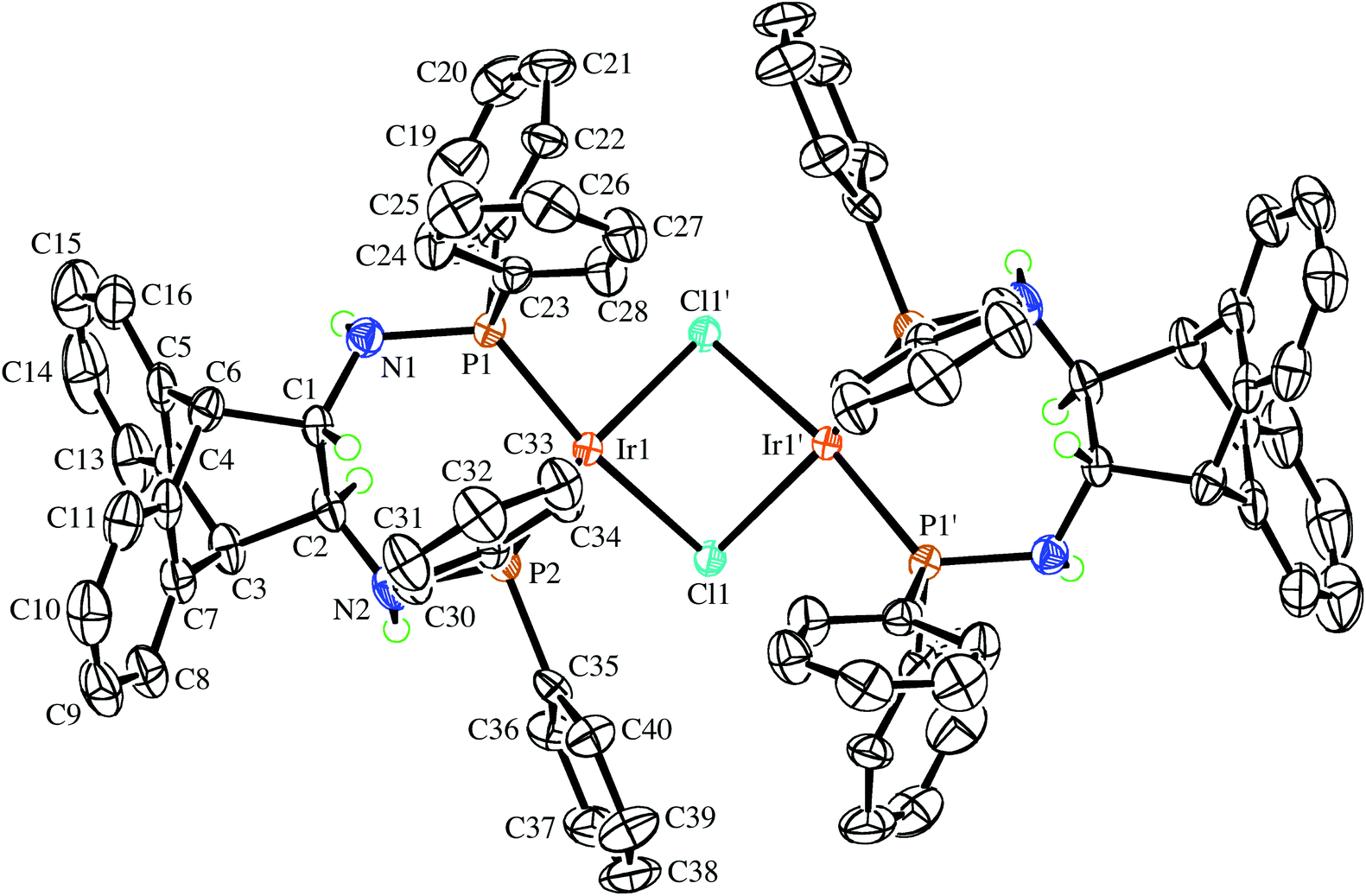
The enantiopure ligand (S,S)-6 reacts with [IrCl(COE)2]2 (COE = cyclooctene) in benzene solution to afford almost quantitatively the dimeric orange complex (S,S,S,S)-9 (see Scheme 2).21 The compound is very air-sensitive, turning green within seconds upon exposure to air. The NMR spectra show the complex to be symmetrical, containing no coordinated COE. Furthermore, no hydride resonances are observed even though similar Ir(I) complexes are known to readily activate N–H bonds.22 Attempts to grow crystals of the enantiopure complex (S,S,S,S)-9 were hampered by its high solubility. Therefore, the synthesis of the racemic analogue of homochiral (i.e. non-meso) dimers of 9 was attempted, because racemates often display better crystallinity than enantiopure compounds. Indeed, rac-6 reacts with [IrCl(COE)2]2 in toluene solution to afford copious amounts of analytically pure orange needles of rac-9. The X-ray diffraction analysis confirmed the exclusive formation of homochiral dimers, and the molecular structure of the (S,S,S,S)-enantiomer is depicted in Fig. 3.23 In the tetragonal unit cell four dimers are arranged around a large open channel, which runs parallel to the c-axis and is partially occupied by randomly oriented toluene molecules (for more details see the Experimental part). The space group, P42bc, is non-centrosymmetric, but the presence of glide planes requires the crystal to be a racemic compound (or ‘true racemate’) containing homochiral molecules with R,R,R,R- and S,S,S,S-configuration. The dinuclear complex resides on a crystallographic C2-axis. The butterfly-shaped chloro bridged Ir2Cl2 core is perpendicular to this axis and the butterfly wings subtend an angle of 123.68(6)°, which is in line with the conformation in similar chiral dinuclear Ir(I) phosphine complexes.10a,24 The two anthracenyl backbone units of each half-dimer are almost orthogonal to each other with a dihedral angle of 82.1(5)° between both C3–C2–C1–C6 mean planes. One side of the dimeric complex is sterically protected by four axial phenyl rings resembling a bowl, while the other side exposes the Ir2Cl2 core due to an equatorial arrangement of the remaining four phenyl rings. The seven-membered chelating rings have a twist-boat conformation with the Ir-atoms located approximately above the C1–C2 bridge, but the chelation is not C2-symmetrical, unlike that seen in the Mg complex (S,S)-7. The dihedral angle between the square coordination plane around Ir and the best plane fitted through the C3–C2–C1–C6 bridge is 51.9(4)°.
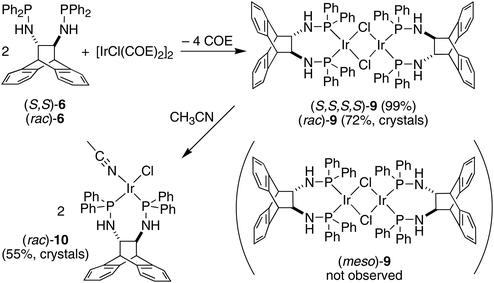 |
| | Scheme 2 Stereoselective syntheses of the Ir(I)–aminophosphine complexes 9 and 10. | |
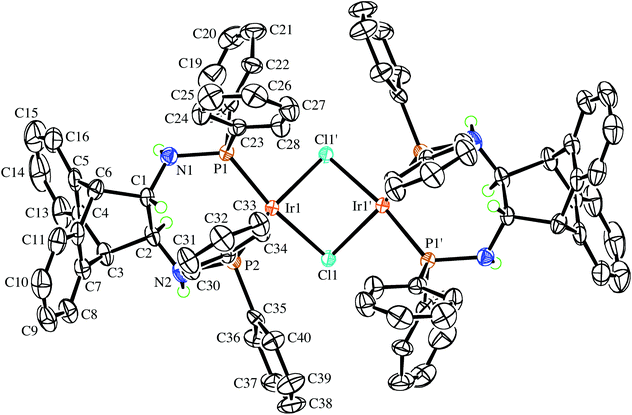 |
| | Fig. 3 View of the (S,S,S,S)-enantiomer in the crystal of rac-9 along the crystallographic C2-axis drawn with 50% probability displacement ellipsoids. Most H-atoms have been omitted for clarity. Selected bond lengths (Å) and angles (°) are: Ir(1)–P(1) 2.2024(16), Ir(1)–P(2) 2.1892(16), Ir(1)–Cl(1) 2.4200(15), Ir(1)–Cl(1′) 2.4167(15), P(1)–N(1) 1.709(6), P(2)–N(2) 1.701(5), N(1)–C(1) 1.446(8), N(2)–C(2) 1.440(9), C(1)–C(2) 1.538(11), Cl(1′)–Ir(1)–Cl(1) 79.48(6), Ir(1′)–Cl(1)–Ir(1) 85.37(5), P(2)–Ir(1)–P(1) 96.41(6), N(1)–P(1)–Ir(1) 124.3(2), N(2)–P(2)–Ir(1) 118.1(2), C(1)–N(1)–P(1) 120.5(5), C(2)–N(2)–P(2) 119.2(5), N(1)–C(1)–C(2) 112.8(6), N(2)–C(2)–C(1) 115.7(8); primed atom labels are for atoms related to the corresponding unprimed atoms by the symmetry operation −x, 1 − y, z. | |
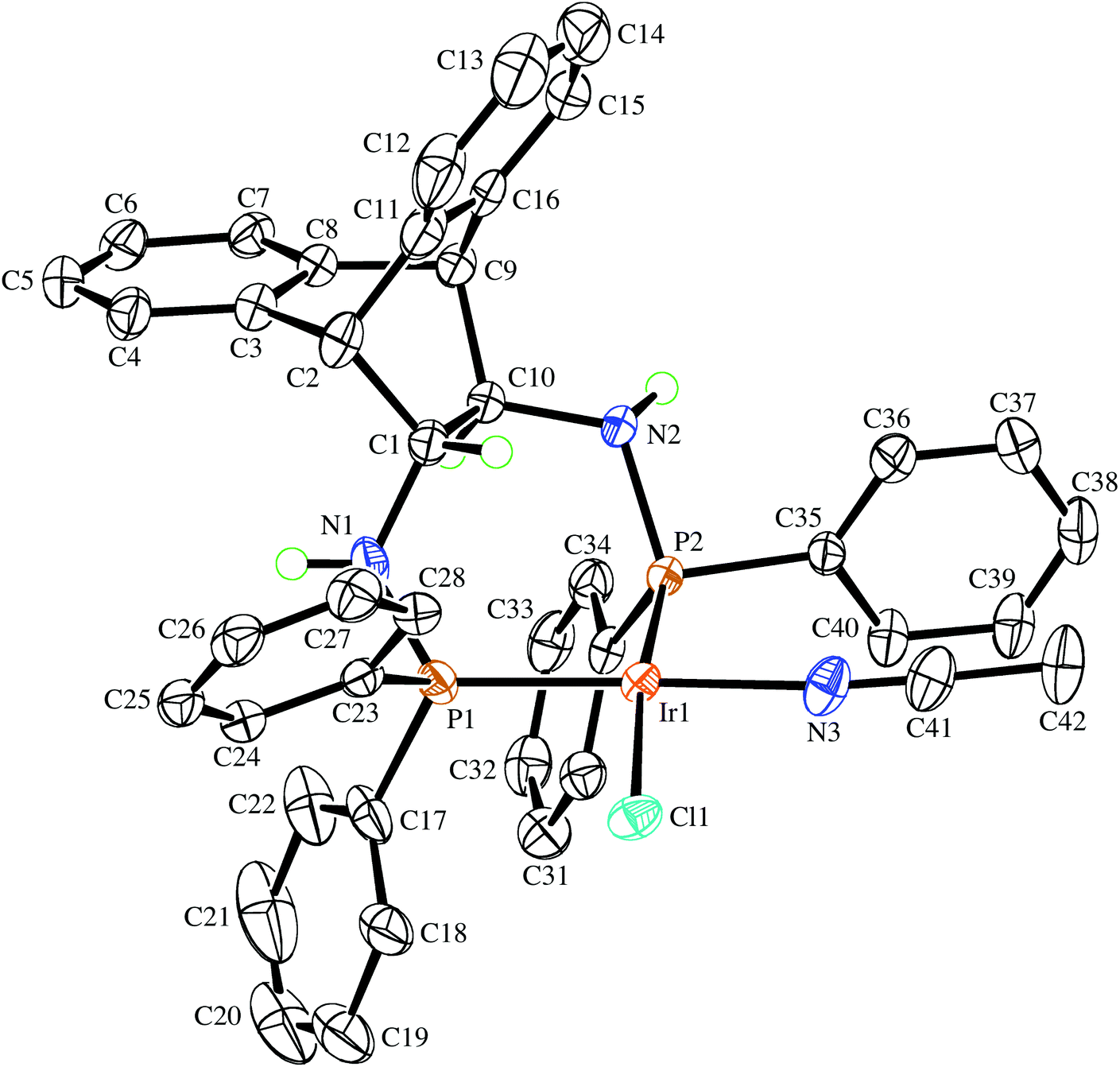
Even though the P donor atoms of the ligand are not symmetry-equivalent in the crystal structure, they display only one resonance in the 31P NMR spectrum and broadened 1H-NMR signals, which hints at a fast inversion – with respect to the NMR timescale – of either the chelate rings, or the Ir2Cl2-butterfly. rac-9 has very low solubility in benzene, toluene, and THF but readily dissolves in the presence of aniline forming an inseparable mixture of hydride species with 1H NMR resonances between −19.4 and −19.9 ppm, which are the result of N–H activation and a prerequisite for Ir(I) catalyzed hydroamination of norbornene (vide infra).25 Also acetonitrile initially dissolves rac-9 by splitting the chloro-bridge and forming the mononuclear adduct rac-10. The 31P NMR spectrum shows a pair of doublets and the proton spectrum is consistent with two diastereotopic half-sides of the ligand backbone.26 The single crystal X-ray diffraction analysis confirms the mononuclear nature of the acetonitrile adduct rac-10, which, along selected geometric parameters, is depicted in Fig. 4. The Ir-atom is in a square planar coordination environment and, as in rac-9, the conformation of the ligand is distorted considerably from the pseudo C2 symmetry it adopts in the Mg complex (S,S)-7. As in rac-9, the seven-membered chelate ring is boat-shaped,27 but in contrast to rac-9, where the Ir-atom is located centrally over the C–C bridge of the ligand backbone, it is tilted towards one of the benzo-groups due to more severe puckering of the chelate ring. Similar off-center coordination of the metal with respect to the anthracenyl moiety has been observed in di-NHC complexes with flexible seven-membered chelation.28 The dihedral angle between the mean coordination plane around the Ir atom and the mean plane defined by the C2–C1–C10–C9 bridge of the ligand backbone is 56.25(18)°. Two phenyl rings on each of atoms P1 and P2 are in an axial position and on the same side of the chelate ring, thereby shielding one coordination hemisphere, while the other two phenyl rings are equatorially arranged. The N1–C1–C10–N2 torsion angle in the ligand backbone of rac-10 decreased from 107.88(14)° to 100.1(3)° when compared to the free ligand in rac-6, showing once more the flexibility of the ANDEN backbone under the influence of strain caused by bidentate coordination.7aTable 1 lists the N–C–C–N torsion angles and shows their correlation with the C–C–C–C torsion angle of the ethynyl bridge of the ligand backbone. In 17 published crystal structures that contain the ANDEN substructure, the N–C–C–N torsion angles range between 61° and 119°, and in unstrained molecules (i.e. free ligands and protonated ANDEN) they are usually around 110°.7,29
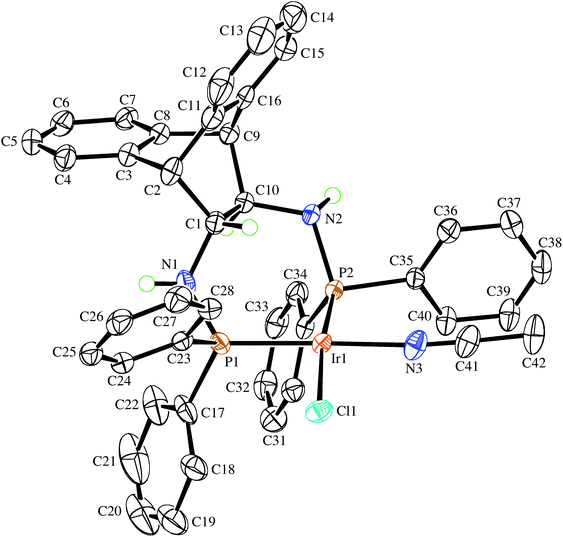 |
| | Fig. 4 The molecular structure of the (R,R)-enantiomer in the crystal of rac-10·1⅓CH3CN drawn with 50% probability displacement ellipsoids. Most H-atoms have been omitted for clarity. Selected bond lengths (Å) and angles (°) are: Ir1–N3 2.052(3), Ir1–P2 2.2123(7), Ir1–P1 2.2195(9), Ir1–Cl1 2.4015(7), P1–N1 1.690(3), P2–N2 1.718(2), P1–C17 1.820(3), P2–C35 1.835(3), N3–C41 1.148(5), N1–C1 1.478(4), N2–C10 1.477(4), N3–Ir1–P2 89.83(8), N3–Ir1–P1 174.02(8), P2–Ir1–P1 95.80(3), N3–Ir1–Cl1 85.01(8), P2–Ir1–Cl1 171.69(2), P1–Ir1–Cl1 89.62(3), C1–N1–P1 116.4(2), C10–N2–P2 117.38(19). | |
Table 1 Torsion angles within the ANDEN backbone as measured in the crystal structures of rac-6, (S,S)-7, rac-9 and rac-10
| Torsiona |
rac-6 |
(S,S)-7 |
rac-9 |
rac-10 |
|
By definition, negative and positive signs for the NCCN and CCCC angles, respectively, refer to the (R,R) enantiomers. The opposite applies to the (S,S) enantiomers.
|
| N–C–C–N |
−107.88(14) |
72.1(2) |
110.7(7) |
−100.1(3) |
| C–C–C–C |
0.28(16) |
−24.9(3) |
−1.2(9) |
9.2(4) |
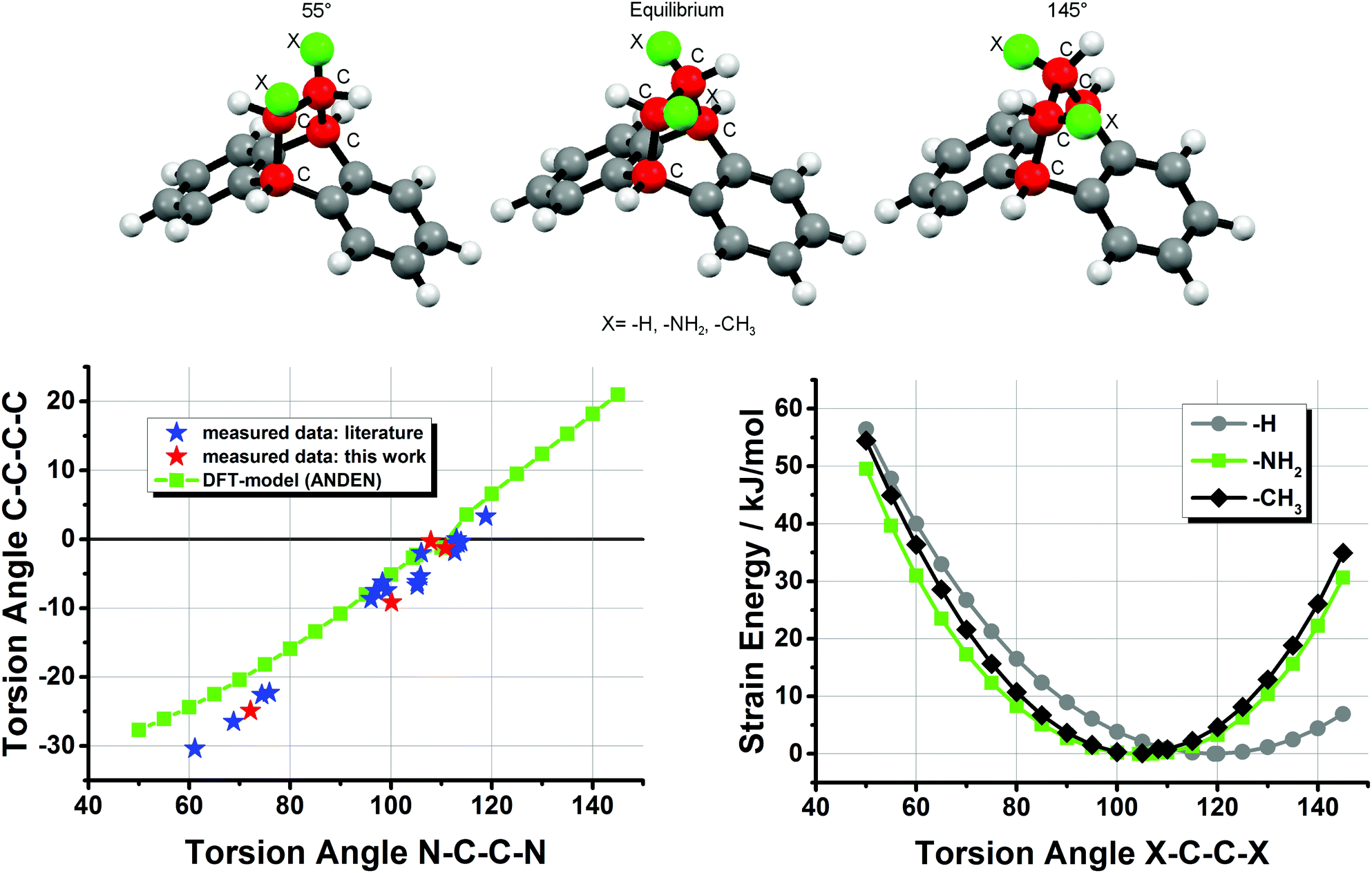
In order to model this type of flexibility in the ANDEN backbone we performed DFT calculations by varying the N–C–C–N torsion angle between 55° and 145°. The results shown in Fig. 5 agree well with the experimental data of an almost linear dependence of the two types of torsion angle, and for very strong deviations the linear trend between the angles is even more pronounced than predicted by the DFT-model. Furthermore, the calculations indicate an energy minimum at about 105° for the N–C–C–N angle in ANDEN, while the corresponding H–C–C–H torsion angle in the simple dibenzobicyclo[2,2,2]octane molecule approaches the expected ‘ideal’ value of 120°. The observation that substitution of the NH2 groups in ANDEN for CH3 groups also leads to a minimum at around 105° suggests that torsion angles smaller than 120° are the consequence of steric effects rather than H-bonding or electronegativity effects.30 The experimentally observed flexibility of the ANDEN backbone is consistent with the calculated low strain energies: the bulk of the X-ray crystal structures have N–C–C–N angles between 95° and 115° (i.e. 10° deviation from the ideal value), which amounts to less than 5 kJ mol−1 of strain.31 The strain energy in the highly distorted complex 7 is thus estimated to be around 20 kJ mol−1.
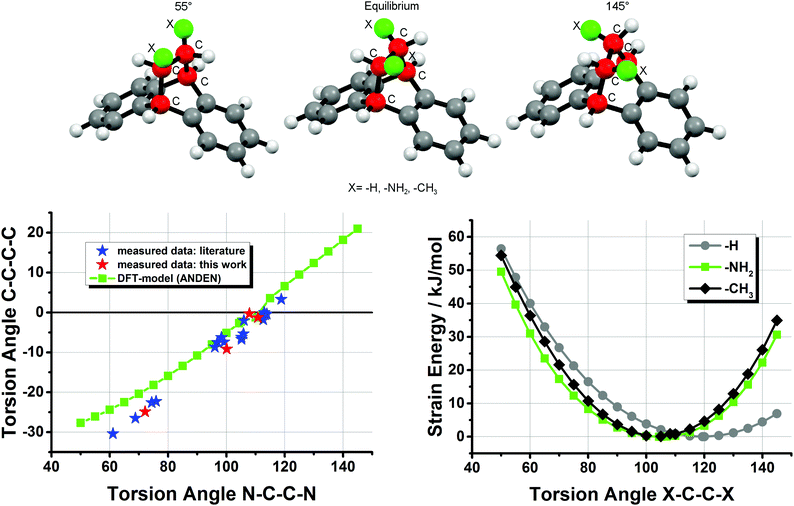 |
| | Fig. 5 DFT simulation (BP86/def2-TZVP) of N–C–C–N vs. C–C–C–C torsion angles in ANDEN compared with experimental data and strain SCF-energies (X–C–C–X torsion angles from 55° to 145°) of ANDEN, dibenzobicyclo[2,2,2]octane (the depicted molecule), and 2,3-dimethyl-dibenzobicyclo[2,2,2]octane. The torsion angles for the ANDEN-derivatives are taken from ref. 7 and 26. | |
Finally, complexes (S,S)-7 and (S,S,S,S)-9 were tested as catalysts for the hydroamination of olefins (see eqn (1) and (2) and Table 2 below). The Mg-complex (S,S)-7 turned out to be an active catalyst for the intramolecular hydroamination of 2,2′-diphenyl-pent-4-ene-amine to form 2-methyl-4,4′-diphenylcyclopentyl amine (eqn (1)). The reaction proceeds smoothly without the need for any additives and is surprisingly clean when monitored in situ by NMR, forming no side-products and reaching full conversion after only 4 h at room temperature with a catalyst loading of 5 mol% (Table 2, entry 1). However, at lower catalyst loadings, the reaction does not go to completion even after prolonged reaction times (entry 2), which is probably due to catalyst deactivation by trace amounts of water. Unfortunately, enantioselectivities are very low, and the slightly bulkier benzylated amine substrate yields the corresponding pyrrolidine in trace amounts only. For the reaction of eqn (2), the Ir-complex (S,S,S,S)-9 is a promising candidate due to its ability to activate the N–H bond of aniline (vide supra), and indeed, in the presence of co-catalytic amounts of MgPh2, LDA, or PhLi, the addition product exo-(2-phenylamino)bornane forms in good yields but low optical purity. Even the notoriously unreactive substrate o-anisidine32 affords the corresponding norbornylamine in 35% isolated yield. The precise role of Ph2Mg, LDA, and PhLi as co-catalysts in the addition of aniline to norbornene is still unclear. Separate experiments, however, showed that these co-catalysts on their own form the Mg- and Li-anilides in situ, which do not catalyze any of the reactions of eqn (1) and (2), even under prolonged heating at 75 °C. Not unexpectedly then, the hydroamination of norbornene with aniline is not catalyzed by (S,S)-7, because the magnesium complex reacts quantitatively with aniline to afford free ligand (S,S)-6 and catalytically inert Mg(NHPh)2. Conversely, (S,S,S,S)-9 is inactive in the intramolecular hydroamination of 2,2′-diphenyl-pent-4-ene-amine (eqn (1)).
| |  | (1) |
| | 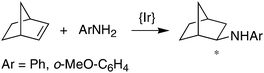 | (2) |
Table 2 Mg- and Ir-catalyzed intra- and inter-molecular olefin hydroaminations
| Entry |
Reaction |
R |
Catalyst (mol%) |
Co-catalyst (mol%) |
Temp. [°C] |
React. time [h] |
Isolated yield [%] |
ee [%] |
| 1 |
eqn (1)
|
H |
(S,S)-7 (5.0) |
— |
22 |
4 |
98 |
7 |
| 2 |
eqn (1)
|
H |
(S,S)-7 (1.0) |
— |
22 |
72 |
38 |
7 |
| 3 |
eqn (1)
|
Bn |
(S,S)-7 (5.0) |
— |
22 |
72 |
Traces |
— |
| 4 |
eqn (2)
|
Ph |
(S,S,S,S)-9 (2.5) |
MgPh2 (2.5) |
80 |
115 |
36 |
16 |
| 5 |
eqn (2)
|
Ph |
(S,S,S,S)-9 (2.5) |
LDA (5.0) |
80 |
115 |
68 |
7 |
| 6 |
eqn (2)
|
Ph |
(S,S,S,S)-9 (2.5) |
PhLi (5.0) |
80 |
115 |
67 |
7 |
| 7 |
eqn (2)
|
o-MeO-C6H4 |
(S,S,S,S)-9 (2.5) |
MgPh2 (2.5) |
110 |
115 |
35 |
5 |
In conclusion, the new ANDEN-based amino-phosphine ligand 6 is accessible in gram quantities and excellent yields as racemate and as optically pure compound. Its coordination chemistry with Mg and Ir highlights the ambidentate nature of the P–N function. The hard Mg(II) is coordinated by the hard amide functions in the C2-symmetric complex (S,S)-7, while the soft Ir(I) centres in complexes (rac)-9 and (rac)-10 are P-bound through seven-membered chelate rings of low symmetry. The crystal structures of (rac)-6, (S,S)-7, (rac)-9, and (rac)-10 reveal considerable flexibility within the bicyclic scaffold of the ANDEN backbone, and DFT calculations demonstrate that these distortions do not induce large amounts of strain energy. Thus, the ANDEN backbone is not as rigid as it appears, apart from the fact that it does not allow conformational isomerism. The Mg complex (S,S)-7 and the Ir complex (S,S,S,S)-9 are active catalysts for the intra- and intermolecular hydroamination of olefins, respectively. We are currently modifying ligand 6 by introducing different phosphine groups in an attempt to improve on enenantioselectivities and to expand the substrate scope, and results will be communicated in due course.
Experimental section
All reactions were carried out under anaerobic and anhydrous conditions, using standard Schlenk and glove box techniques, unless otherwise stated. THF, Et2O, and benzene were distilled from purple Na/Ph2CO solutions, toluene from Na, pentane, C6D6, and THF-D8 from Na2K alloy, CH3CN, CH2Cl2, and CD2Cl2 from CaH2, NEt3 and 1,4-dioxane from K. CD3CN and CDCl3 were degassed with three freeze–pump–thaw cycles and then kept in a glove box over activated molecular sieves (3 Å and 4 Å, respectively). 2-Norbornene was distilled from Na2K alloy, aniline from CaH2. ANDEN,33 [IrCl(COE)2]2,34 diphenylmagnesium,35 lithium diisopropylamide,36 and 2,2-diphenylpent-4-en-1-amine37 were prepared according to published procedures. Resolution of rac-ANDEN (5) was simplified by only using the cheaper (R)-enantiomer of the chiral auxiliary mandelic acid to obtain both enantiomers. Optically pure (S–S)-ANDENium-(R)-mandelate was obtained from a single crystallization in methanol, and the (S–S)-ANDEN was freed by treatment with KOH. The mother liquor containing mainly (R,R)-ANDENium-(R)-mandelate (as judged from its optical rotation) was likewise treated with KOH to obtain (R–R)-enriched ANDEN. Contrary to Lennon's protocol,33 this material was not treated with (S)-mandelic acid, but recrystallized from benzene to yield optically pure (R–R)-5. Elemental analysis samples of air sensitive compounds were handled and prepared in a glove box. NMR spectra were recorded on 270 MHz or 400 MHz Jeol Lambda/Eclipse spectrometers, and the solvent residual signals were used as the internal reference for the 1H-NMR-spectra.38 Optical rotations were measured on a Perkin-Elmer 241 polarimeter.
rac-ANDEN-PPh2 (rac-6)
rac-5 (3.00 g, 12.7 mmol) was suspended in Et2O (90 ml) and Et3N (6.43 g, 63.5 mmol) was added. Then a solution of Ph2PCl (5.61 g, 25.4 mmol) in Et2O (10 ml) was added dropwise affording a white precipitate. The mixture was stirred overnight and the solid removed by filtration over a glass fiber filter (GF/B). Evaporation of the filtrate under reduced pressure first formed a white voluminous solid and finally a paste, which was cooled to −32 °C for several hours to complete crystallization. The solid was washed with chilled Et2O and n-pentane by filtration and dried in vacuo to afford 6.77 g (88%) of a white solid. Elemental analysis found: C, 79.48; H, 5.39; N, 5.10. Calcd for C40H34N2P2: C, 79.45; H, 5.67; N, 4.63. 1H NMR (270 MHz, C6D6): δ 1.69 (m, 2H), 3.30 (m, 2H), 3.90 (s, 2H), 6.75–7.15 (m, 20H), 7.32 (m, 4H), 7.42 (m, 4H). 31P{1H} NMR (162 MHz, C6D6): δ 40.33 (s). 13C NMR (100 MHz, C6D6): δ 53.6(d, JCP = 4.9 Hz), 69.0 (dd, JCP = 29.1, 8.2 Hz), 124.5, 126.3, 126.56, 126.59, 128.5–128.8 (m), 130.9 (d, JCP = 4.9 Hz), 131.3 (d, JCP = 20.1 Hz), 139.4, 142.7, 142.9 (d, JCP = 10.4 Hz), 143.5 (d JCP = 14.8 Hz). 13C-DEPT-135 NMR (100 MHz, C6D6): δ 53.5 (d, JCP = 4.9 Hz), 69.0 (dd, JCP = 29.1, 8.2 Hz), 124.2, 126.0, 126.30, 126.33, 128.0–128.6 (m), 130.9 (d, JCP = 4.9 Hz), 131.3 (d, JCP = 20.1 Hz). Crystals suitable for an X-ray diffraction experiment were grown by vapor diffusion of pentane into an Et2O solution of rac-6.
(S,S)-ANDEN-PPh2 ((S,S)-6)
(S,S)-5 (4.00 g, 16.9 mmol) was suspended in Et2O (120 ml) and NEt3 (8.57 g, 84.7 mmol). A solution of Ph2PCl (7.48 g, 33.9 mmol) in Et2O (13 ml) was added dropwise affording a white precipitate. The mixture was stirred overnight and the solid removed by filtration over a glass fiber filter (GF/B). The filtrate was concentrated to approx. 10 ml under reduced pressure causing the formation of a white solid. Addition of n-pentane and cooling to −32 °C completed the crystallization. The white solid was washed with chilled pentane and dried in vacuo (8.86 g, 87%). Elemental analysis found: C, 79.65; H, 5.62; N, 4.56. Calcd for C40H34N2P2: C, 79.45; H, 5.67; N, 4.63. [α]21D +58.7 (c 1.00, THF). 1H NMR (400 MHz, C6D6): δ 1.75 (m, 2H), 3.36 (m, 2H), 3.95 (s, 2H), 6.82–7.19 (m, 20H), 7.37(m, 4H), 7.48 (m, 4H). 31P{1H} NMR (162 MHz, C6D6): δ 40.33 (s). 13C NMR (100 MHz, C6D6): δ 53.6 (d, JCP = 4.9 Hz), 69.0 (dd, JCP = 29.1, 8.2 Hz), 124.5, 126.3, 126.56, 126.59, 128.5–128.8 (m), 130.9 (d, JCP = 4.9 Hz), 131.3 (d, JCP = 20.1 Hz), 139.4, 142.7, 142.9 (d, JCP = 10.4 Hz), 143.5 (d JCP = 14.8 Hz). The ligand can be handled in air but should be kept under inert gas.
(S,S)-[Mg(ANDEN-PPh2)(THF)2] ((S,S)-7)
A solution of Ph2Mg (178 mg, 0.841 mmol) in THF (3 mL) was added dropwise over 5 min to a well stirred solution of S,S-6 (505 mg, 0.836 mmol) in THF (3 mL). The resulting yellowish solution was stirred for 7 h, after which a white precipitate had formed. Benzene (6 mL) was added to complete precipitation. The solid was separated by centrifugation (1 h at 3500 rpm) and dried under HV to afford 544 mg (80%) of an off-white powder. Elemental analysis found: C, 74.83; H, 6.24; N, 3.54. Calcd for C40H32MgN2P2(THF)2.2(C6H6)0.2: C, 74.95; H, 6.39; N, 3.50. [α]21D +16.0 (c 1.00, THF). 1H NMR (400 MHz, THF-d8): δ 3.17 (s, 2H), 4.55 (s, 2H), 6.68–6.78 (m, 4H), 6.81–6.87 (m, 2H), 6.92–6.97 (m, 2H), 7.12–7.18 (m, 12H), 7.31–7.41 (m, 8H), coordinated THF is observed at 1.75 and 3.60 ppm. 31P{1H} NMR (162 MHz, THF-d8): δ 46.5 (s). 13C NMR (67 MHz, THF-d8): δ 51.7 (d, JCP = 21.7 Hz), 75.4 (dd, JCP = 22.0, 10.0 Hz), 121.8, 124.8 (d, JCP = 12.8 Hz), 126.7, 127.4, 127.7 (d, JCP = 4.8 Hz), 127.9, 128.4 (d, JCP = 5.8 Hz), 131.9 (d, JCP = 19.1 Hz), 132.1 (d, JCP = 20.7 Hz), 142.2, 149.9, 151.1, 151.5, 151.7, 152.0.
rac-[Mg(ANDEN-PPh2)]·Et2O and crystalline rac-7
A solution of MgPh2·0.4Et2O (1.10 g, 5.20 mmol) in Et2O (10 g) was added dropwise to a suspension of rac-6 (3.00 g, 4.73 mmol) in Et2O (20 g). The resulting solution was stirred overnight causing the precipitation of a white solid. Filtration, washing twice with n-pentane, and drying in HV afforded 2.95 g (91%) of an off-white powder. Elemental analysis found: C, 75.23; H, 5.98; N, 4.06. Calcd for C44H42MgN2OP2·C4H10O: C, 75.38; H, 6.04; N, 4.00. This material is very soluble in aromatic solvents, but NMR spectra in C6D6 solution are complicated, due to the presence of oligomeric species. Adding 2.2 equiv. of THF to such filtered (GF/B) benzene solutions affords the crystalline tetrahydrofuranate rac-7 in excellent yields (>85%). Single crystals obtained by this method were suitable for X-ray diffraction analysis. NMR spectra of these crystals are identical to those of (S,S)-7 (vide supra).
(S,S,S,S)-[IrCl(ANDEN-PPh2)]2 ((S,S,S,S)-9)
Benzene (4 g) was added to (S,S)-6 (500 mg, 0.827 mmol) and [IrCl(coe)2]2 (371 mg, 0.414 mmol) under stirring to afford a dark red solution that slowly turned orange. The solution was stirred for 3 d, and then the volatiles were removed under reduced pressure. The resulting bright yellow solid was slurried and washed in hexanes (3 × 10 mL, filtration over GF/B) and HV-dried to afford a pale yellow powder (688 mg, 99%). Elemental analysis found: C, 55.53; H, 4.02; N, 3.16. Calcd for C80H68Cl2Ir2N4P4·0.15C6H14: C, 57.91; H, 4.21; N, 3.34. Values for C were consistently low (5 measurements on two different batches), and we suspect formation of carbides during combustion. 1H NMR (400 MHz, C6D6): δ 1.96 (d, J = 7.5 Hz, 4H), 3.78–3.95 (m, 8H), 6.65–7.15 (m, 40H), 7.20–7.35 (m, 8H), 7.71–7.88 (m, 8H). 31P{1H} NMR (162 MHz, C6D6): δ 44.2 (s). 13C NMR (100 MHz, C6D6): δ 50.6, 61.9, 124.1, 125.3, 126.9, 127.0, 128.3, 128.8, 132.4, 138.6, 138.8, 139.0, 139.4, 139.6, 139.8, 140.0, 140.3, 140.6, 140.8, 141.4. 13C-DEPT-135 NMR (100 MHz, C6D6): δ 50.6, 61.9, 124.1, 125.3, 126.2, 126.9, 127.0, 128.3, 128.8, 132.4.
Crystalline rac-[IrCl(ANDEN-PPh2)]2 (rac-9)
Using toluene instead of benzene affords material of better crystallinity but in lower yield. Toluene (15 g) was added to rac-6 (708 mg, 1.12 mmol) and [IrCl(COE)2]2 (500 mg, 0.556) in a vial rapidly forming a clear, red solution, which after about 20 min turned orange. Within 2 d large amounts of needle-like orange crystals had grown and were collected by filtration, washed with chilled toluene (5 ml) and n-pentane (2 × 10 ml), and dried in vacuo (717 mg, 72%). The single crystals thus obtained were of X-ray diffraction quality. Elemental analysis found: C, 60.44; H, 4.50; N, 2.79. Calcd for C80H68Cl2Ir2N4P4·1½C7H8: C, 60.29; H, 4.47; N, 3.11. In common deuterated solvents the crystals are either almost insoluble (C6D6, toluene-d8, THF-d8) or they decompose readily (CD2Cl2). Therefore, NMR spectra had to be recorded in CD3CN, which leads to (rac)-10 (vide infra): 1H NMR (400 MHz): δ 2.17 (d, J = 4.8 Hz, 1H), 2.79 (m, 1H), 2.17 (m, 1H), 2.79 (m, 1H), 3.26 (m 1H), 3.64 (m 1H), 4.02 (m 1H), 4.36 (m 1H), 6.7–7–6 (m, 28H). The proton spectrum shows the presence of ca. 1.5 equiv. of toluene. 31P{1H} NMR (162 MHz, CD3CN): δ 41.32 (d, J = 24.7 Hz), 49.45 (d, J = 24.7 Hz).
Crystalline rac-[IrCl(ANDEN-PPh2)(CH3CN)]·1⅓CH3CN (rac-10)
rac-9 (239 mg, 0.144 mmol, used as its toluene solvate) was dissolved in acetonitrile (5 mL) and the solution left undisturbed for 5 d at −32 °C. This afforded copious amounts of yellow, X-ray quality crystals, which were separated from the orange mother liquor by decantation, washed with pentane (5 mL), and dried in HV (151 mg, 55%). Elemental analysis found: C, 58.49; H, 4.50; N, 6.26. Calcd for C42H37ClIrN3P2·1⅓CH3CN·¼C5H12: C, 58.29; H, 4.69; N, 6.41. Once crystals of (rac)-10 have formed, their solubility in CD3CN and other common solvents is too low for meaningful NMR-spectra to be recorded. For spectra of (rac)-10 that were recorded before crystallization took place, see the protocol for (rac)-9.
Mg-catalyzed cyclization of 2,2′-diphenyl-pent-4-eneamine
In a glovebox, 2,2-diphenylpent-4-en-1-amine (366.9 mg, 1.546 mmol) and (S,S)-7 (62.8 mg, 0.0775 mmol) were mixed in a Teflon-lined screw cap vial and dissolved in 1.5 mL of THF. The clear colorless solution was stirred for 4 h. The solvent was removed under high vacuum. Hexane (1 mL) was added and the white mixture was filtered through celite. After removing the solvent, the obtained oil was further purified by Kugelrohr distillation to afford 359 mg (98%) of product. 1H-NMR (400 MHz, CDCl3): δ 1.21 (d, 3H), 2.0–2.2 (m, 3H), 2.73 (dd, 1H), 3.38 (m, 1H), 3.46 (d, 1H), 3.67 (d, 1H), 7.1–7.3 (m, 10H). For HPLC analysis the product was derivatized with 1-naphthoyl chloride in an excess of NEt3 to give (2-methyl-4,4-diphenylpyrrolidin-1-yl)(naphthalen-1′-yl)methanone. The amide was obtained in 98% yield and was injected on a Daicel Chiralcel AD-H column n-hexane/i-PrOH = 85:15, 1.0 mL min−1, tR = 34.2 min (major), tR = 22.6 min (minor).
NMR-scale intramolecular hydroamination
(S,S)-7 (8 mg, 0.009 mmol) and 2,2-diphenylpent-4-en-1-amine (43 mg, 0.18 mmol) were added to an NMR tube and dissolved in 0.5 mL of THF-d8. The reaction was monitored by 1H-NMR and after 2 h, 87% conversion was achieved. Full conversion was observed after 4 h.
Representative Ir-catalyzed addition of aniline to 2-norbornene
(S,S,S,S)-9 (104 mg, 0.0625 mmol) and LDA (13.4 mg, 0.125 mmol) were dissolved in a solution of 2-norbornene (235 mg, 2.50 mmol) in aniline (233 mg, 2.50 mmol) that contained a few drops of benzene. The orange mixture was stirred at 80 °C for 115 h in a sealed vial (Teflon-lined screw cap) after which time the reaction was quenched by exposure to air. The product was isolated as a yellowish oil after flash chromatography (G60 silica, l = 10 cm, d = 2.5 cm, n-hexane/EtOAc = 9:1) and HV drying (320 mg, 68%). 1H-NMR (400 MHz, CDCl3): δ 1.12–1.31 (m, 4H), 1.43–1.69 (m, 4H), 1.78–1.91 (m, 1H), 2.29 (s, 2H), 3.20–3.35 (m, 1H), 3.61 (bs, 1H), 6.52–6.81 (m, 3H), 7.13–7.22 (m, 2H). HPLC analysis: Daicel Chiralcel OJ-H column, n-hexane/i-PrOH = 90:10, 0.5 mL min−1, 249 nm, tR (R) = 19.05 min (minor), tR (S) = 21.27 min (major). The addition of o-anisidine to norbornene gave a colorless oil after flash chromatography (G60 silica, l = 10 cm, d = 2.5 cm, n-hexane/EtOAc = 9:1) and HV drying. 1H–NMR (270 MHz, CDCl3): δ 1.12–1.42 (m, 4H), 1.48–1.67 (m, 3H), 1.78–1.99 (m, 1H), 2.26–2.48 (m, 2H), 3.13–3.42 (m, 1H), 3.86 (s, 1H), 4.14 (bs, 1H), 6.57–6.74 (m, 2H), 6.79 (d, J = 5.4 Hz, 1H), 6.90 (t, J = 5.4 Hz, 1H). HPLC analysis: Daicel Chiralcel OJ-H column, n-hexane/i-PrOH = 99.5:0.5, 0.5 mL min−1, 212 nm, tR = 19.95 min (major), tR = 21.66 min (minor).
Crystal structure determinations
The measurements for [Mg(C40H32N2P2)(C4H8O)2]·C6H6 ((S,S)-7) and [Ir2Cl2(C40H34N2P2)2]·2C7H8 (rac-9) were made on an Agilent Technologies SuperNova area-detector diffractometer39 using Mo Kα radiation (λ = 0.71073 Å) from a micro-focus X-ray source and an Oxford Instruments Cryojet XL cooler. That for C40H34N2P2·0.5C5H12 (rac-6) was conducted on a Bruker Smart APEX-II area-detector diffractometer40 using graphite-monochromated Mo Kα radiation, while the measurements for [Ir(C40H34N2P2)(C2H3N)Cl]·1.33C2H3N (rac-10) were made on a Bruker-Nonius KappaCCD area-detector diffractometer41 also using graphite-monochromated Mo Kα radiation. Data reduction was performed with CrysAlisPro,39 APEX240 or EvalCCD,42 respectively. The intensities were corrected for Lorentz and polarization effects, and empirical absorption corrections using spherical harmonics43 were applied. Equivalent reflections, other than Friedel pairs for (S,S)-7, were merged. The structures were solved by direct methods using SHELXS-2013,44 or SHELXTL45 and the non-hydrogen atoms were refined anisotropically. The data collection and refinement parameters are given in Table 3.
Table 3 Crystallographic data for compounds rac-6, (S,S)-7, rac-9 and rac-10
| |
rac-6 |
(S,S)-7 |
rac-9 |
rac-10 |
| Empirical formula |
C40H34N2P2·½C5H12 |
C48H48MgN2O2P2·C6H6 |
C80H68Cl2Ir2N4P4·2C7H8 |
C42H37ClIrN3P2·1⅓C2H3N |
| Formula wt (g mol−1) |
640.71 |
849.28 |
1885.04 |
928.07 |
| Crystal size (mm) |
0.25 × 0.30 × 0.45 |
0.10 × 0.20 × 0.30 |
0.08 × 0.10 × 0.30 |
0.12 × 0.16 × 0.24 |
| Crystal colour, habit |
Colourless, block |
Pale yellow, prism |
Orange, needle |
Yellow, block |
| Temperature (K) |
100(2) |
160(1) |
160(1) |
100(2) |
| Crystal system |
Triclinic |
Monoclinic |
Tetragonal |
Trigonal |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21 |
P42bc |
R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) |
|
a (Å) |
10.016(2) |
10.4127(3) |
22.72621(15) |
27.620(5) |
|
b (Å) |
12.061(3) |
20.6503(3) |
22.72621(15) |
27.620(5) |
|
c (Å) |
16.103(4) |
11.3111(3) |
17.6278(2) |
28.5419(9) |
|
α (°) |
104.466(2) |
90 |
90 |
90 |
|
β (°) |
107.161(2) |
113.247(3) |
90 |
90 |
|
γ (°) |
102.209(2) |
90 |
90 |
120 |
|
V (Å3) |
1712.1(6) |
2234.72(10) |
9104.40(16) |
18856(5) |
|
Z
|
2 |
2 |
4 |
18 |
|
ρ
calcd (g cm−3) |
1.243 |
1.262 |
1.375 |
1.471 |
|
μ (mm−1) |
0.160 |
0.156 |
3.102 |
3.362 |
|
θ range (°) |
2.2–27.9 |
2.1–28.4 |
2.1–28.3 |
3.0–27.9 |
| Reflections measured |
42631 |
21935 |
44263 |
123409 |
| Independent reflns; Rint |
8109; 0.037 |
8599; 0.051 |
9564; 0.037 |
9993; 0.048 |
| Parameters; restraints |
476; 78 |
550; 1 |
418; 1 |
549; 180 |
|
R(F) [I > 2σ(I)] |
0.042 |
0.043 |
0.028 |
0.031 |
| wR(F2) (all data) |
0.109 |
0.105 |
0.068 |
0.084 |
| Goodness of fit (F2) |
1.029 |
1.064 |
1.113 |
1.239 |
| Δρmax, min (e Å−3) |
0.44, −0.59 |
0.64; −0.32 |
1.82; −0.87 |
3.37, −1.04 |
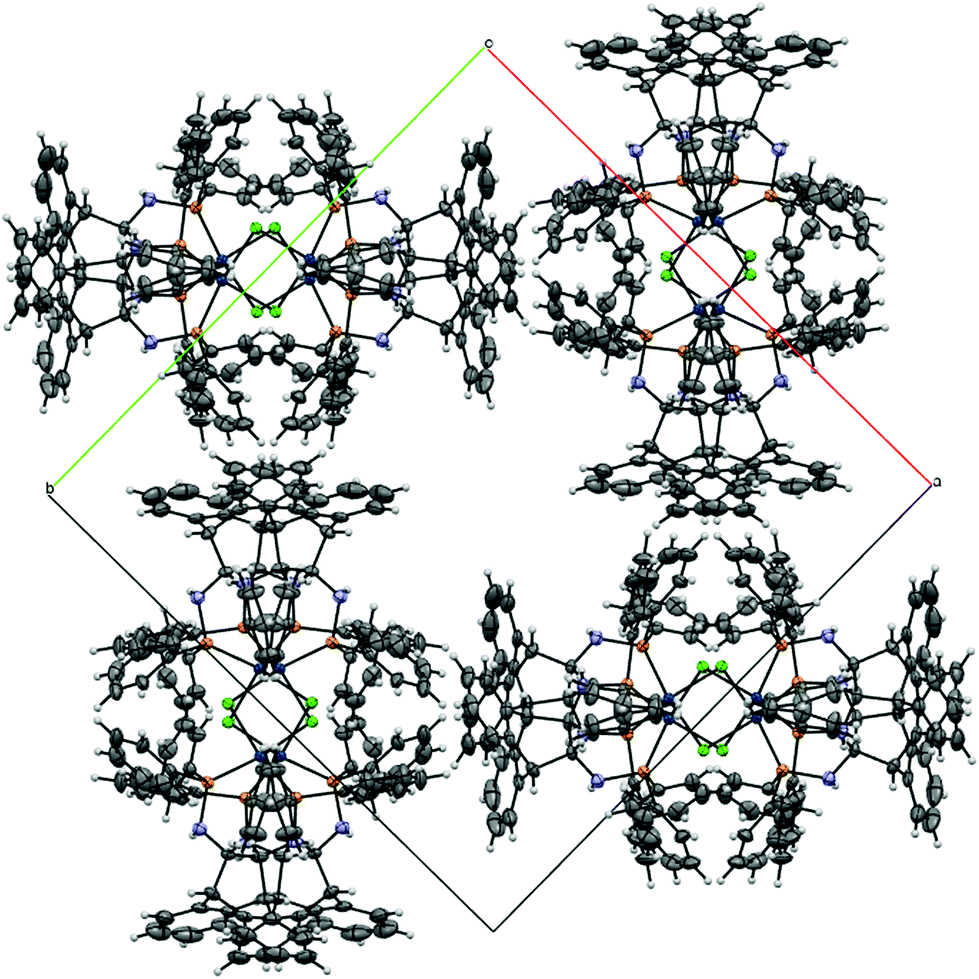
Compound rac-6 crystallized as its n-pentane hemisolvate. The solvent molecule is disordered about a crystallographic centre of inversion. Similarity and pseudo-isotropic restraints were applied to the anisotropic displacement ellipsoids of the disordered atoms. The asymmetric unit of (S,S)-7 contains one molecule of the Mg-complex plus one molecule of benzene. Refinement of the absolute structure parameter46 yielded a value of −0.01(5), which confidently confirms that the refined model represents the true absolute structure. The asymmetric unit of rac-9 contains half of a C2-symmetric dinuclear Ir-complex with the metal centres linked by a double Cl-bridge. The structure model, when defined just by the Ir-complex contains two large voids of 1567 Å3 per unit cell, in which no significant electron density peaks corresponding with obvious solvent molecules could be discerned (Fig. 6). However, based on NMR evidence and elemental analysis, it is clear that highly disordered non-stoichiometric amounts of toluene molecules occupy the cavities. As the solvent molecules could not be modelled, the SQUEEZE routine of the program PLATON47 was employed. The electron count in each void in the unit cell was calculated to be approximately 225 e. One toluene molecule has 50 e, so it has been assumed that four toluene molecules lie in each void. This approximation has been used in the subsequent calculation of the empirical formula, formula weight, density, linear absorption coefficient and F(000). Based on this assumption, the ratio of Ir-complex to toluene molecules in the structure is 1:2. Refinement of the absolute structure parameter46 yielded a value of −0.025(3), which confidently confirms that the refined model represents the true absolute structure. Although the space group for rac-9 is non-centrosymmetric, the presence of glide planes dictates that the compound in the crystal is racemic.
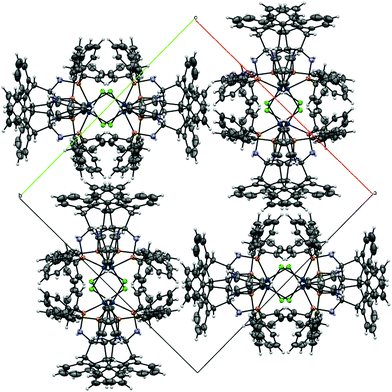 |
| | Fig. 6 View of the packing of rac-9 in the crystal showing the channel running parallel to the c-axis. An overlay of 8 molecules is shown, four of each (S,S,S,S)-9 and (R,R,R,R)-9. | |
For rac-10, the Ir-complex crystallized with 1.33 equivalents of MeCN. The solvent molecules are distributed over two crystallographic sites and are strongly disordered. One of the solvent molecules lies on a general position while the other is situated on a crystallographic three-fold axis. Two alternative orientations of the solvent molecules were refined at each site while applying bond length, bond angle and bond length similarity restraints. Similarity and pseudo-isotropic restraints were also applied to the anisotropic displacement ellipsoids of the disordered atoms. Three reflections, whose intensities were considered to be extreme outliers, were omitted from the final refinement.
For rac-9, the two unique amine H-atoms were placed in the positions indicated by a difference electron density map, then their positions were geometrically optimised and refined by using a riding model with refined isotropic displacement parameters. The H-atom of one amine group is disordered over the two possible positions on its parent N-atom. The amine H-atoms of rac-6 and rac-10 were placed in the positions indicated by difference electron density maps and their positions were allowed to refine together with a fixed isotropic displacement parameter with a value equal to 1.2Ueq of its parent atom. All remaining H-atoms in each structure were placed in geometrically calculated positions and refined using a riding model where each H-atom was assigned a fixed isotropic displacement parameter with a value equal to 1.2Ueq of its parent atom (1.5Ueq for any methyl groups). The refinement of each structure was carried out on F2 by using full-matrix least-squares procedures, which minimised the function ∑w(Fo2 − Fc2)2. The SHELXL-2014 program48 was used for the refinement of (S,S)-7 and rac-9, while SHELXTL45 was used for the other two structures.
Computational details
ANDEN and its modifications were drawn in a standard molecule editor and pre-optimized using the Merck Molecular Force Field MMFF94.49 Calculations were performed using the Turbomole 6.3 suite50 with the def2-TZVP51 basis set and the BP-86 exchange correlation functional.52 The calculations were accelerated using the RIJ approximation.53 The energy curves were obtained by iterative increments of a fixed internal redundant coordinate resembling the X–C–C–X torsion angle and starting from the equilibrium geometry. Energy differences were calculated with respect to the SCF-energies only. Gibbs free energies were not taken into consideration because the complex-valued vibrational modes of the non-equilibrium structures would be ignored in their calculation (this would generate an extra error due to the fact that the reference is an equilibrium structure).
Acknowledgements
We thank Dr Achim Zahl for performing NMR measurements and Ms. Christina Wronna for carrying out the elemental analyses. Financial support by Friedrich-Alexander University is acknowledged.
References
-
(a) M. Fiorini, F. Marcati and G. M. Giongo, J. Mol. Catal., 1978, 4, 125 CrossRef CAS;
(b) M. Fiorini and G. M. Giongo, J. Mol. Catal., 1979, 5, 303 CrossRef CAS;
(c) S. Miyano, M. Nawa and H. Hashimoto, Chem. Lett., 1980, 9, 729 CrossRef;
(d) S. Miyano, M. Nawa, A. Mori and H. Hashimoto, Bull. Chem. Soc. Jpn., 1984, 57, 2171 CrossRef CAS;
(e) F.-Y. Zhang, C.-C. Pai and A. S. C. J. Chan, Am. Chem. Soc., 1998, 120, 5808 CrossRef CAS;
(f) F.-T. Zhang, W. H. Kwok and A. S. C. Chan, Tetrahedron: Asymmetry, 2001, 12, 2337 CrossRef CAS;
(g) C.-J. Wang, Z.-P. Xu, X. Wang and H.-L. Teng, Tetrahedron, 2010, 66, 3702 CrossRef CAS.
-
G. M. Giongo, (Anic S.p.A.) EP77099, 1983 Search PubMed.
- The idea is to create a steric transportation chain of chiral information from the diamine backbone to the substituents on the P-atoms. Structural studies of Pt complexes of ligand 4 may serve as an illustration thereof: R. A. Swanson, R. S. Haywood, J. B. Gibbson, K. E. Cordova, B. O. Patrick, C. Moore, A. L. Rheingold and C. J. A. Daley, Inorg. Chim. Acta, 2011, 368, 74 CrossRef CAS.
- For a recent example, see: T. Chen, H. Li, S. Qu, B. Zheng, L. He, Z. Lai, Z.-X. Wang and K.-W. Huang, Organometallics, 2014, 33, 4152 CrossRef CAS.
- For a structurally authenticated η2-P,N(H)–Ru coordination mode, see: M. D. Palacios, M. C. Puerta, P. Valerga, A. Lledós and E. Vellly, Inorg. Chem., 2007, 17, 6958 CrossRef PubMed.
- M. Fiorini, F. Marcati and G. M. Giongo, J. Mol. Catal., 1979, 4, 125 CrossRef . These authors also observed the opposite effect by N-methylation of the corresponding 1,2-diaminocyclohexane derived ligands, affording the hydrogenation product with higher ee and inverted configuration.
- For ANDEN-derived racemic NHCs, see:
(a) R. J. Lowry, M. K. Veige, O. Clément, K. A. Abboud, I. Ghiviriga and A. S. Veige, Organometallics, 2008, 27, 5184 CrossRef CAS;
(b) R. J. Lowry, M. T. Jan, K. A. Abboud, I. Ghiviriga and A. S. Veige, Polyhedron, 2010, 29, 553 CrossRef CAS. For non-racemic NHCs:
(c) A. B. Mullick, M. S. Jeletic, A. R. Powers, K. A. Abboud, I. Ghiviriga and A. S. Veige, Polyhedron, 2013, 52, 810 CrossRef CAS. For tetradentate Schiff base ligands, see:
(d) M. Furutachi, S. Mouri, S. Matsunaga and M. Shibasaki, Chem. – Asian J., 2010, 5, 2351 CrossRef CAS PubMed. For an amino-thiophosphine ligand, see:
(e) F. Zhang, H. Song and G. J. Zi, Organomet. Chem., 2010, 695, 1993 CrossRef CAS. For sulfonamide derivatives, see:
(f) G. Huelgas, L. K. LaRochelle, L. Rivas, Y. Luchinina, R. A. Toscano, P. J. Carroll, P. J. Walsh and C. Anaya de Parodi, Tetrahedron, 2011, 67, 4467 CrossRef CAS.
- B. M. Trost, D. L. Van Vranken and C. Bingel, J. Am. Chem. Soc., 1992, 114, 9327 CrossRef CAS.
- For reviews on asymmetric olefin hydroamination, see:
(a) I. Aillaud, J. Collin, J. Hannedouche and E. Schulz, Dalton Trans., 2007, 5105 RSC;
(b) K. C. Hultzsch, Adv. Synth. Catal., 2005, 347, 367 CrossRef CAS;
(c) K. C. Hultzsch, Org.
Biomol. Chem., 2005, 3, 1819 CAS;
(d) P. W. Roesky and T. E. Müller, Angew. Chem., Int. Ed., 2003, 42, 2708 CrossRef CAS PubMed.
-
(a) R. Dorta, P. Egli, F. Zürcher and A. Togni, J. Am. Chem. Soc., 1997, 119, 10857 CrossRef CAS;
(b) D. Vasen, A. Salzer, F. Gerhards, H.-J. Gais, R. Stürmer, N. H. Bieler and A. Togni, Organometallics, 2000, 19, 539 CrossRef CAS;
(c) J. (S.) Zhou and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 12220 CrossRef CAS PubMed. For an achiral Ir-system for intramolecular reactions, see:
(d) K. D. Hesp, S. Tobisch and M. Stradiotto, J. Am. Chem. Soc., 2010, 132, 413 CrossRef CAS PubMed. For a review on Ir complexes that are relevant to hydroamination up to the year 2007, see:
(e)
R. Dorta, in Iridium Complexes in Organic Synthesis, ed. L. Oro and C. Claver, Wiley-VCH, Weinheim, 2008 Search PubMed.
- X. Zhang, T. J. Emge and K. C. Hultzsch, Angew. Chem., Int. Ed., 2012, 51, 394 CrossRef CAS.
- Attempts to deprotonate the aminophosphine with BuLi only led to inseparable yellow (Et2O) or dark red (THF) mixtures.
- For definitions and comments, see:
E. L. Eliel and S. H. Wilen, Stereochemistry of Organic Compounds, Wiley-Interscience, 1994, p. 298 ff Search PubMed.
- Only 5–10% of the racemic mixtures crystallize as racemic conglomerates: H. Lorenz, A. Perlberg, D. Sapoundijev, M. P. Elsner and A. Seidel-Morgenstern, Chem. Eng. Process., 2006, 45, 863 CrossRef CAS.
- For a very similar C2-symmetric coordination environment around Mg with an achiral ligand, see: F. Majoumo-Mbe, E. Smolensky, P. Lonnecke, D. Shpasser, M. S. Eisen and E. Hey-Hawkins, J. Mol. Catal. A: Chem., 2005, 240, 91 CAS.
- Even smaller torsion angles are seen in one of Zi's N-ligated Ti-complex, which also exhibits a five-membered chelate ring (61.1°, ref. 7e) and Veige's Rh-complex (69°, ref. 7a).
- M. Brookhart, M. L. H. Green and G. Parkin, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6908 CrossRef CAS PubMed.
- The distances are well within the range for π–σ interactions, and the shortest C⋯C distances are less than 4 Å. See: C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885 RSC.
- For structurally characterized amidophosphine complexes of group 2 metals, see:
(a)
ref. 14;;
(b) T. Chivers, M. C. Copsey, C. Fedorchuk, M. Parvez and M. Stubbs, Organometallics, 2005, 24, 1919 CrossRef CAS;
(c) P. W. Roesky, Inorg. Chem., 2006, 45, 798 CrossRef CAS PubMed;
(d) K. Panda, M. T. Gamer and P. W. Roesky, Inorg. Chim. Acta, 2006, 359, 4765 CrossRef;
(e) D. Olbert, A. Kalisch, N. Herzer, H. Gorls, P. Mayer, L. Yu, M. Reiher and M. Westerhausen, Z. Anorg. Allg. Chem., 2007, 633, 893 CrossRef CAS;
(f) S. Datta, M. T. Gamer and P. W. Roesky, Dalton Trans., 2008, 2830 Search PubMed;
(g) D. A. Dickie, K. B. Gislason and R. A. Kemp, Inorg. Chem., 2012, 51, 1162 CrossRef CAS PubMed. For structurally characterized amidophosphines complexes of transition metals, see:
(h) E. A. Gwynne and D. W. Stephan, Organometallics, 2011, 30, 4128 CrossRef CAS;
(i) S. Kuppuswamy, T. M. Powers, B. M. Johnson, M. W. Bezpalko, C. K. Brozek, B. M. Foxman, L. A. Berben and C. M. Thomas, Inorg. Chem., 2012, 52, 4802 CrossRef PubMed;
(j) B. Wu, R. Hernández Sánchez, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, Inorg.
Chem., 2014, 53, 10021 CAS.
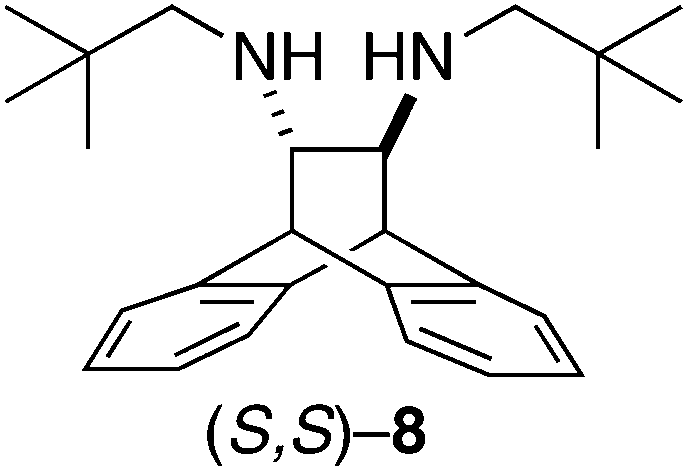
- Conversely, secondary diamines, such as (S,S)-8, did not react with PClPh2 under the conditions described for ANDEN. Attempts to first deprotonate 8 with strong bases (BuLi, MeLi, LDA, MgPh2, etc.) led to the rapid decomposition of 8 to anthracene and other unidentified products. Preparation of (S,S)-8: (S,S)-5 and pivaloyl chloride afforded the bis-amide, which then was reduced with NaBH4/I2 (M. J. McKennon, A. I. Meyers, J. Org. Chem., 1993, 58, 3568) to neo-ANDEN on a gram scale. 1H-NMR (300 MHz, CD3CN): δ 7.24–7.36 (m, 4H), 7.05–7.17 (m, 4H), 4.39 (s, 2H), 2.64 (d, 2H, J = 11.4), 2.38 (t, 2H, J = 1.5), 2.29 (d, 2H, J = 11.4), 0.81 (s, 18H), 0.55 (br, 2H). 1H-NMR (300 MHz C6D6): δ 7.20–7.31 (m, 4H), 7.04–7.15 (m, 4H), 4.38 (s, 2H), 2.68 (d, 2H, J = 11.5), 2.65 (s, 2H), 2.33 (d, 2H, J = 11.4), 0.91 (s, 18H), 0.53 (br, 2H). 13C-NMR (75 MHz, C6D6): δ 132.82 (s), 131.06 (s), 116.67 (s), 116.29 (s), 114.60 (s), 58.48 (s), 50.07 (s), 39.82 (s), 22.05 (s). Elemental analysis found: C, 82.65; H, 9.64; N, 8.25. Calcd for C26H36N2: C, 82.93; H, 9.64; N, 7.44. [α]21D +24.4 (c 1.00, THF).
.
- Reactions of (S,S)-7 with [IrCl(COE)2]2 under various conditions in an attempt to synthesize Ir(I)–amido complexes only led to inseparable mixtures. Precipitation of MgCl2 was not observed.
- For examples, see:
(a)
Ref. 10e, pp. 162–165; ;
(b) O. V. Ozerov, C. Guo, V. A. Papkov and B. M. Foxman, J. Am. Chem. Soc., 2004, 126, 4792 CrossRef CAS PubMed;
(c) H. Locke, A. Herrera, F. Heinemann, A. Linden, S. Frieß, B. Schmid and R. Dorta, Organometallics, 2015, 34, 1925 CrossRef CAS.
- As the yield of the crystals was around 70% their composition may be regarded as representative of the bulk material.
- T. Yamagata, A. Iseki and K. Tani, Chem. Lett., 1997, 26, 1215 CrossRef.
-
(a) A. L. Casalnuovo, J. C. Calabrese and D. Milstein, J. Am. Chem. Soc., 1988, 110, 6738 CrossRef CAS;
(b)
Ref. 10c
.
- Small peaks from rac-9 remain visible in the NMR-spectra and hint at an equilibrium with an apparent Keq of around 15. Unfortunately, VT NMR experiments to prove the equilibrium were hindered by the high tendency of rac-10 to form sparingly soluble crystalline precipitates.
- This conformation is common for cis-bis-(1,2-diamino)phosphine complexes.
(a) K. Onuma and A. Nakamura, Bull. Chem. Soc. Jpn., 1981, 761, 761 CrossRef;
(b) V. F. Kuznetsov, G. R. Jefferson, G. P. A. Yap and H. Alper, Organometallics, 2002, 21, 4241 CrossRef CAS;
(c) P. Bergamini, V. Bertolasi and F. Milani, Eur. J. Inorg. Chem., 2004, 1277 CrossRef CAS.
- For an NMR method to measure such ligand flexibility, see: M. S. Jeletic, C. E. Lower, I. Ghiviriga and A. S. Veige, Organometallics, 2011, 30, 6034 CrossRef CAS . Related X-ray crystal structures are found in ref. 7b.
- For crystal structures of protonated ANDEN, see: Y. Imai, K. Murata, K. Kamon, T. Kinuta, T. Sato, R. Kuroda and Y. Matsubara, Cryst. Growth Des., 2009, 9, 602 CAS.
- Fluorine substitution leads to an equilibrium F–C–C–F angle of 114°.
- From this point of view it is not surprising that the torsion angles from the X-ray crystallographic data vary over such a wide range, because hydrogen bonds and packing effects may easily overcome this barrier.
-
(a)
Ref. 10c;;
(b)
R. Dorta and A. Togni, unpublished results.
- M. E. Fox, A. Gerlach and I. C. Lennon, Synthesis, 2005, 3196 CAS.
- A. Van der Ent, A. L. Onderlinden and R. A. Schunn, Inorg. Synth., 1990, 28, 90 CrossRef CAS.
- H. Tang and H. G. J. Richey, Organometallics, 2001, 20, 1569 CrossRef CAS.
- N. Barnett and R. Mulvey, J. Am. Chem. Soc., 1991, 113, 8187 CrossRef CAS.
- G.-Q. Liu, Adv. Synth. Catal., 2014, 356, 2303 CrossRef CAS.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176 CrossRef CAS.
-
CrysAlisPro, Version 1.171.37.31d, Agilent Technologies, Yarnton, Oxfordshire, England, 2014 Search PubMed.
-
APEX2, Version 2014.9-0, Bruker AXS, Madison, WI, U.S.A., 2014 Search PubMed.
-
COLLECT, Bruker AXS BV, Delft, The Netherlands, 2002 Search PubMed.
- A. J. M. Duisenberg, L. M. J. Kroon-Batenburg and A. M. M. Schreurs, J. Appl. Crystallogr., 2003, 36, 220–229 CrossRef CAS.
-
(a) R. H. Blessing, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1995, 51, 33–38 CrossRef;
(b)
SADABS, Version 2008/1, Bruker AXS, Madison, WI, U.S.A. Search PubMed, 2008 for 10.
Version 2012/1, Bruker AXS, Madison, WI, U.S.A., 2012 Search PubMed for 6.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
-
SHELXTL Version 6.12, Bruker AXS, Madison, WI, U.S.A., 2002 Search PubMed.
-
(a) H. D. Flack and G. Bernardinelli, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1999, 55, 908–915 CrossRef;
(b) H. D. Flack and G. Bernardinelli, J. Appl. Crystallogr., 2000, 33, 1143–1148 CAS.
- A. L. Spek, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 9–18 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef PubMed.
- T. A. Halgren, J. Comput. Chem., 1996, 17, 490 CrossRef CAS.
-
TURBOMOLE V6.5 2013, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, available from http://www.turbomole.com Search PubMed.
-
(a) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC;
(b) A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571 CrossRef;
(c) F. Weigend, M. Häser and R. Ahlrichs, Chem. Phys. Lett., 1998, 294, 143 CrossRef CAS;
(d) F. Weigend, F. Furche and R. Ahlrichs, J. Chem. Phys., 2003, 119, 12753 CrossRef CAS.
-
(a) P. A. M. Dirac, Proc. R. Soc. A, 1929, 123, 714 CrossRef CAS;
(b) J. C. Slater, Phys. Rev., 1951, 81, 385 CrossRef CAS;
(c) S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200 CrossRef CAS;
(d) A. D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS;
(e) J. P. Perdew, Phys. Rev. B: Condens. Matter, 1983, 33, 8822 CrossRef.
- R. Ahlrichs, Phys. Chem. Chem. Phys., 2004, 6, 5119 RSC.
Footnotes |
| † CCDC 1463736–1463739. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt01146b |
| ‡ Authors contributed equally to this publication. |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a