
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical consequences of pyrazole orientation in RuII complexes of unsymmetric quinoline–pyrazole ligands†
Joachim
Hedberg Wallenstein
a,
Lisa A.
Fredin
b,
Martin
Jarenmark
c,
Maria
Abrahamsson
*a and
Petter
Persson
*b
aDepartment of Chemistry and Chemical Engineering, Chalmers University of Technology, SE-41296 Gothenburg, Sweden. E-mail: abmaria@chalmers.se
bTheoretical Chemistry Division, Department of Chemistry, Chemical Center, Lund University, Box 124, SE-22100 Lund, Sweden. E-mail: Petter.Persson@teokem.lu.se
cCentre for Analysis and Synthesis, Department of Chemistry Chemical Center, Lund University, Box 124, SE-22100 Lund, Sweden
First published on 23rd May 2016
Abstract
A series of homoleptic RuII complexes including the tris-bidentate complexes of a new bidentate ligand 8-(1-pyrazol)-quinoline (Q1Pz) and bidentate 8-(3-pyrazol)-quinoline (Q3PzH), as well as the bis-tridentate complex of bis(quinolinyl)-1,3-pyrazole (DQPz) was studied. Together these complexes explore the orientation of the pyrazole relative to the quinoline. By examining the complexes structurally, photophysically, photochemically, electrochemically, and computationally by DFT and TD-DFT, it is shown that the pyrazole orientation has a significant influence on key properties. In particular, its orientation has noticeable effects on oxidation and reduction potentials, photostability and proton sensitivity, indicating that [Ru(Q3PzH)3]2+ is a particularly good local environment acidity-probe candidate.
Introduction
Ruthenium polypyridine complexes have attracted much attention as sensors or probes in biology,1–3 as catalysts and sensitizers in photochemical applications,4–11 and as interesting model complexes in theoretical chemistry.12–15 The specific functions or properties of a complex can typically be understood in terms of electronic and steric factors.16,17 This kind of structure-function relationship is well established for the prototypical polypyridine complexes [Ru(bpy)3]2+ and [Ru(tpy)2]2+, (bpy is 2,2′-bipyridine and tpy is 2,2′:6′,2′′-terpyridine) and their derivatives.12,16–19 In comparison, Ru-complexes with other polyheteroaromatic ligands have received less attention, despite the fact that incorporation of non-pyridine type polyheteroaromatic ligands can substantially affect both structural and electronic properties of the complex.11,20,21One way of forming such polyheteroaromatic ligands is by the combinations of quinoline (Q) and pyridine (Py). Different orientations of the two subunits with respect to each other can yield both 8-(pyridine-2′-yl)quinoline (8-QPy) and 2-(pyridine-2′-yl)quinoline (2-QPy),22 which upon coordination to RuII form 6- or 5-membered chelates, respectively, Chart 1. The resulting heteroleptic complexes [Ru(bpy)2(8-QPy)]2+ and [Ru(bpy)2(2-QPy)]2+ display significant differences in photophysical and electrochemical properties; the Ru2+/3+ redox couple of [Ru(bpy)2(8-QPz)]2+ is negatively shifted 90 mV with respect to [Ru(bpy)2(2-QPy)]2+.22 In addition, the use of 8-QPy appears to preclude formation of homoleptic Ru2+ complexes due to steric interaction between the ligands; in the heteroleptic [Ru(bpy)2(8-QPy)]2+ Ru–N bond lengths and bite angles both increased compared to [Ru(bpy)3]2+ (from 2.05623 to 2.079 Å for Ru–NPy and from 78° to 88° for 8-QPy and bpy in the two complexes respectively). The distorted coordination geometry of [Ru(bpy)2(8-QPy)]2+ (compared to [Ru(bpy)3]2+) results in very short lived excited state and low emission quantum yields.22
 | ||
| Chart 1 (Top panel) Selected ligands under discussed, displayed for comparison, (bottom panel) the ligands used to form the homoleptic RuII-complexes reported in this paper. For PyPz R is –H, –CH3 or –CH2C6H5. | ||
When coordinating unsymmetric ligands to form heteroleptic complexes diastereomers can be formed, which may display significantly varying properties. Reports of cis/trans-isomers of heteroleptic transition metal complexes are abundant in the literature,16,24 commonly achieved by combining mono- and bidentate ligand motifs. However, reports of meridional/facial-homoleptic complexes with unsymmetrical ligands are less common. Notably, Metherell et al. has presented the X-ray structures of mer- and fac-isomers of a series of homoleptic Ru complexes with substituted 2-pyrazolyl-pyridine (PyPz) ligands, showing that mer- and fac-isomers of [Ru(PyPzH)3]2+ display different hydrogen bonding capability.25 Furthermore Tamayo et al. have reported 6 homoleptic iridium complexes with substituted 2-phenylpyridyl and 1-phenylpyrazolyl ligands where the oxidation of fac-isomers are shifted positively 50–100 mV compared to the mer-counterparts, and the isomers have substantially different photophysical properties.26 Dabb and Fletcher conclude, in a recent review of a large number of diimine complexes, that differences in electronic spectra between mer- and fac-isomers occur mainly when the difference in the diimine donors are substantial.27 These studies highlight the need to establish a detailed understanding of structure-function relationships in transition metal complexes with biheteroaromatic ligands, allowing for design of well-tailored complexes for specific applications. To this end, a combination of experimental and theoretical efforts can help to establish design-parameters and further understanding of the intricate interplay between complex parameters and properties.
With a ligand similar to 8-QPy, wherein Py has been replaced with the smaller pyrazolyl (PzH) motif to form 8-(3-pyrazol)-quinoline (Q3PzH), Chart 1, we were recently able to decrease the steric hindrance, allowing for formation of homoleptic tris-bidentate complexes, [Ru(Q3PzH)3]2+.28 By changing the orientation of the pyrazole subunit, to instead form 8-(1-pyrazolyl)-quinoline (Q1Pz) we now report the synthesis of the analogous homoleptic tris-bidentate complex, [Ru(Q1Pz)3]2+. Furthermore, we recently reported the bistridentate analogue mer-[Ru(DQPz)2]2+ (DQPz is bis(quinolinyl)-1,3-pyrazole, Chart 1) which displays room temperature diastereomerization between two meridional isomers.29 The DQPz ligand is essentially a superposition of the Q1Pz and Q3PzH ligands, but with a higher relative number of quinolines. The complete pyrazole orientation series Q3PzH, Q1Pz, and DQPz, allows us to probe pyrazole orientation effects, as well as effects related to different quinoline–pyrazole ratios, on properties such as electronic absorption, electrochemical potentials and photostability by experimental and computational means.
Results and discussion
Synthesis of mer-[Ru(Q1Pz)3]2+
[Ru(Q3PzH)3]2+ and [Ru(DQPz)2]2+ were available from our previous studies and synthesized according to our reported procedures.28,29 The bidentate ligand Q1Pz was prepared through a copper catalyzed C–N cross-coupling reaction between pyrazole and 8-bromo-quinoline (Scheme 1). Using a high temperature procedure similar to that for Q3PzH and [Ru(Q3PzH)3](PF6)2,28 the tris-homoleptic ruthenium complex [Ru(Q1Pz)3](PF6)2 was synthesized, Scheme 1. | ||
| Scheme 1 Synthesis route to mer-[Ru(Q1Pz)3](PF6)2via Q1Pz and mer/fac-[Ru(Q1Pz)3](PF6)2. | ||
The 1H-NMR spectrum of [Ru(Q1Pz)3](PF6)2 displays two sets of peaks of non-equal intensities, indicating that two species are present (Fig. S1†). Although the spectrum is crowded, it can be readily explained by the presence of both the mer- and fac-isomers. The pure mer-[Ru(Q1Pz)3]2+ can be produced by visible light illumination of the mer/fac-mixture in solution, which leads to decomposition of the fac-[Ru(Q1Pz)3]2+ isomer, as monitored by 1H-NMR. The mer![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :fac-isomer ratio in [Ru(Q1Pz)3]2+ was 8:1, based on the relative integrals in the 1H-NMR. This is in contrast to the 3:1 ratio previously observed for [Ru(Q3PzH)3]2+.28
:fac-isomer ratio in [Ru(Q1Pz)3]2+ was 8:1, based on the relative integrals in the 1H-NMR. This is in contrast to the 3:1 ratio previously observed for [Ru(Q3PzH)3]2+.28
Geometric structure
The optimized geometries of the complexes were calculated by DFT at the PBE0/SDD[Ru]6-31G(d,p)[N,C,H]/PCM(MeCN) level of theory, and the key structural parameters and energies of representative relaxed isomers are presented in Tables 1 and 2, respectively (additional isomers in Table S1†). RPz, RQ, and Rtot are the average Ru–N(pyrazole), Ru–N(quinoline), and overall Ru–N bond distances. O and P are the deviation of the Ru–ligand angles and the dihedral ligand angles (i.e. the angle between the planes of the heteroaromatic groups in each ligand) from an ideal octahedron or plane respectively (ideal ∼0°). Both, mer-[Ru(Q3PzH)3](PF6)2 and mer-[Ru(DQPz)2](PF6)2 geometries have been characterized previously.28,29 No crystals suitable for X-ray diffraction could be grown for mer-[Ru(Q1Pz)3](PF6)2, but since the previously reported X-ray structure of mer-[Ru(Q3PzH)3](PF6)2 was well matched by its DFT optimized ground state structure28 and since 1H-NMR and DFT-calculations between mer-[Ru(Q3PzH)3](PF6)2 and mer-[Ru(Q1Pz)3](PF6)2 are very similar it is safe to assume that the optimized ground state structure can serve as a reasonable representation of mer-[Ru(Q1PzH)3](PF6)2 (Fig. 1) in further structural comparisons. | ||
| Fig. 1 Optimized geometries of mer-[Ru(Q3PzH)3]2+ (top left), mer-[Ru(Q1Pz)3]2+ (top right), C-Ra-mer-[Ru(DQPz)2]2+ (bottom left) and C-Sa-mer-[Ru(DQPz)2]2+ (bottom right). | ||
|
R
Pzb |
R
Qb |
R
avgb |
O | P | |
|---|---|---|---|---|---|
| a All bond distances in Å and angles in °. b Average and standard deviation of the named Ru–ligand bond distances: Ru-pyrazole (RPz), Ru-quinoline (RQ), and the total average (Ravg) bond distances. c Deviation (rms) from ideal bond angles of all the N–Ru–N bonds (O, ideal of 90°) and the ligand dihedral angles (P, ideal 0°). | |||||
| mer-[Ru(Q3PzH)3]2+ (ref. 28) | 2.05 ± 0.01 | 2.13 ± 0.02 | 2.09 ± 0.05 | 3.41 | 13.46 |
| mer-[Ru(Q1Pz)3]2+ | 2.06 ± 0.02 | 2.12 ± 0.03 | 2.09 ± 0.04 | 2.64 | 17.41 |
| C-Ra-mer-[Ru(DQPz)2]2+ (ref. 29) | 2.004 ± 0.00 | 2.10 ± 0.00 | 2.07 ± 0.05 | 1.33 | 27.86 |
| C-Sa-mer-[Ru(DQPz)2]2+ (ref. 29) | 2.005 ± 0.00 | 2.10 ± 0.00 | 2.07 ± 0.05 | 1.18 | 27.51 |
The optimized geometries of both the tris-bidentate mer-isomers reveal that each ligand adopts a slightly different conformation, and that the differences are larger in mer-[Ru(Q3PzH)3]2+, Table 1. Bis-tridentate mer-[Ru(DQPz)2]2+ also exhibits non-planar ligands, i.e. twisting in the ligand back-bone, resulting in double chirality forming two diastereomers, that following IUPAC recommendation are denoted C-Ra and C-Sa, depending on the conformation of the ligand, Ra or Sa, Fig. 1.29 Their respective enantiomers, A-Ra and A-Sa, as well as trans-fac- and cis-fac-isomers are schematically illustrated in Fig. S2.† Both diastereomers of mer-[Ru(DQPz)2]2+ are calculated to be considerably more octahedral than either of the tris-bidentate complexes (O > 1.5°) and the Ru–N bond lengths are on average shorter for both Ru–NPz (∼0.05 Å) and Ru–NQ (∼0.02 Å). Interestingly, the ligands in mer-[Ru(Q3PzH)3]2+ are the most planar, but the complex is the least octahedral, indicating a tradeoff between flexibility in the ligand and coordination geometry.
The orientation of the pyrazole subunit with respect to the quinoline, in the tris-bidentate complexes, has three main effects: (i) the flexibility of the resulting ligand, where the Q3PzH ligand can adopt a wider range of conformations than Q1Pz, (ii) the coordination geometry of the complexes, where mer-[Ru(Q1Pz)3]2+ is more octahedral than mer-[Ru(Q3PzH)3]2+, and (iii) the energy separation between conformations, where the calculated energy difference between fac- and mer-ground states are, while still small, considerably larger for [Ru(Q3PzH)3]2+, 0.052 eV, compared to [Ru(Q1Pz)3]2+, 0.001 eV, Table 2. Resolving the pyrazole orientation effect between the bis-tridentate and the tris-bidentate complexes are not as straightforward. A comparison between the mer- and fac-isomers of the bis-tridentate complex display a considerably larger relative energy difference, where the fac-isomers are destabilized by ca. 0.6 eV compared to mer, much larger than that of the tris-bidentate complexes. C-Sa-mer-[Ru(DQPz)2]2+ is also the most octahedral complex, followed by C-Ra-mer-[Ru(DQPz)2]2+.
Electronic properties
The electronic structure of all three complexes was studied by computational and experimental means. The combination of ground state electronic structure, the redox reactivity of the complexes, and absorption spectra, gives ample information of the influence of pyrazole orientation on electronic properties. In particular, the oxidation and reduction potentials of the complexes are of critical importance to their use in catalysis.6Frontier molecular orbitals for each mer-isomer show that the three highest occupied molecular orbitals (HOMO, HOMO−1 and HOMO−2, Fig. S3†) all exhibit large density on the metal and can thus be assigned to the Ru t2g orbitals. The HOMO and the lowest unoccupied molecular orbital (LUMO) of the mer-isomers are presented in Fig. 2 (more isomers available in Fig. S4†). The LUMOs of each complex are instead mainly centered on the quinoline subunits. In the tris-bidentate complexes the contributions from the pyrazole is minor, however in the diastereomers of mer-[Ru(DQPz)2]2+ the LUMO density bleeds across the central pyrazole from the axial quinoline groups. All three complexes have similar HOMO energies, approximately −6 eV, and lowest lying LUMOs approximately −2.55 eV, with mer-[Ru(DQPz)2]2+ having the smallest HOMO–LUMO gap.
 | ||
| Fig. 2 Frontier molecular orbitals of the mer-isomers of three complexes. Energies in eV. | ||
Experimental Ru2+/3+ half wave redox potentials were collected by cyclic voltammetry (0.1 V s−1, using ferrocene/ferrocenium couple (Fc0/+) as an internal reference). Quasi-reversible oxidations were observed at +687 and +653 mV for mer-/fac-[Ru(Q1Pz)3](PF6)2 and mer-[Ru(DQPz)2](PF6)2 respectively, Table 3 (and Fig. S5†). The potential for mer-[Ru(Q3PzH)3](PF6)228 was previously reported to occur at +570 mV. It is typically assumed that, for Ru-polypyridyl complexes, it can be safely assumed that oxidation removes an electron from the metal dominated (HOMO) while the reduction is equivalent to reducing a ligand (LUMO).16 Thus, the potentials can be theoretically estimated using the free energies of the oxidized and ground state minima (Table 3).
| Experimental | Calc.c | Absorption datad | ||
|---|---|---|---|---|
|
E
oxa/V |
E Red,1/V | E ox/V | λ Max/nm (ε × 103/M−1 cm−1) | |
| ΔEb/V | E Red,2/V | |||
|
a
E
ox = taken as the midpoint of the reductive and oxidative waves for the Ru2+/3+ couple.
b ΔE = peak-to-peak separation of the reductive and oxidative waves.
c The values shown are for the lowest calculated value among possible stereoisomers.
d Sh = shoulder.
e Values from ref. 28, absorption spectra of mer-[Ru(Q3PzH)3]2+ recorded with 1.2 mM triflic acid in MeCN.
f Collected with cyclic voltammetry.
g Recorded in 8:1 mer:fac mixture of [Ru(Q1Pz)3]2+, Fig. S5.
h Collected by differential pulse voltammetry.
i Values calculated for C-Ra- isomers.
j Values calculated for C-Sa isomers.
|
||||
| mer-[Ru(Q3PzH)3]2+e | 0.57 | −1.46f | 0.528 | 243 (69.5), 291 (16.9), 315 (16.5), 444 (10.7), ∼530sh (4.5) |
| 0.060 | −1.89f | |||
| mer-[Ru(Q1Pz)3]2+ | 0.687g | −1.66h | 0.638 | 243 (82.8), 284sh (15.5), 318 (17.7), 434 (11.0), ∼530sh (3.1) |
| 0.064g | −1.84h | |||
| mer-[Ru(DQPz)2]2+ | 0.653 | −1.56h | 0.511i | 244 (70.9), 280 (29.4), 344 (25.6), ∼360sh (22,1), 486 (15.8) |
| 0.063 | −1.74h | 0.716j | ||
While the calculations provide formal potentials, and CV measurements provides the half-wave potential, one would not expect a complete agreement between calculated and measured potentials. The calculated oxidation potentials for both tris-bidentate complexes and the mean value of the two bis-tridentate isomers, +0.614 V, agree with the experimental values, although consistently negatively shifted ∼40 mV. The ∼120 mV difference between the mer-[Ru(Q3PzH)3]2+ and mer-/fac-[Ru(Q1Pz)3]2+ oxidation potentials shows that the pyrazole orientation has a large effect on the energy needed to remove an electron from the complex.
The calculated oxidation potentials for the two, C-Sa and C-Ra, mer-[Ru(DQPz)2]2+ diastereomers are +0.511 and +0.716 V vs. Fc0/+ respectively, while the measured Eox appear at +0.653 V vs. Fc0/+. Only one oxidation feature is observed in the cyclic voltammogram with near Nernstian (63 mV) separation between the anodic and cathodic peaks. While it is not expected that the two diastereomers act as one average entity, it is clear that oxidation on top of dynamical interconversion between the isomers C-Sa and C-Ra29 is not well represented by optimized individual conformation oxidation potentials. Additionally, it is possible that the redox couples of the mer-isomers are closer in energy than the calculations suggest due to e.g. solvation and ion pairing effects.
[Ru(Q1Pz)3]2+ and mer-[Ru(DQPz)2]2+ both displayed two irreversible reduction waves at more negative potentials than −1.5 V vs. Fc0/+ (Fig. S6†). The first and second reduction potentials of mer-[Ru(Q3PzH)3]2+, collected by cyclic voltammetry, were previously reported at −1.46 and −1.89 V vs. Fc0/+,28 while differential pulse voltammetry of mer-[Ru(DQPz)2]2+ yielded −1.56 and −1.74 V, and for [Ru(Q1Pz)3]2+ −1.66 and −1.84 V vs. Fc0/+, indicating that it is harder to reduce the Q1Pz ligand compared to Q3PzH. Since the main LUMO density resides on the quinolines, one might expect the quinolines to dominate the reduction potentials making the tris-bidentate complexes equivalent due to their equivalent number of quinolines; however, their first reduction potentials differ by 0.2 V. The main difference between Q1Pz and Q3PzH is the pyrazole orientation and the presence of the NH-group in Q3PzH, thus the reduction potentials are clearly influenced by inductive effects.
UV-Vis absorption spectra of mer-[Ru(DQPz)2]2+, mer-[Ru(Q1Pz)3]2+ and mer-[Ru(Q3PzH)3]2+ were collected in neat acetonitrile between 700 and 200 nm, Fig. 3. All of the complexes display a broad MLCT absorption in the visible and stronger absorption bands in the UV region, effectively reproduced by TD-DFT in shape and intensity (Fig. 3, Table 3 and details in ESI†). The absorption feature between 280–370 nm, typical for Ru-Pz motifs,28 is present in all three complexes. All complexes also exhibit ligand centered π–π*-transitions at 243–244 nm, consistent with the absorption spectra of the free ligands, which all displayed two absorption bands with λmax between 235–245 and 305–330 nm respectively (Fig. S7†). The extinction coefficient for mer-[Ru(DQPz)2]2+ is higher than for the bidentate complexes, likely due to the increased number of quinolines in the tridentate mer-[Ru(DQPz)2]2+, since the calculated LUMOs and first TD-DFT excited states are localized mainly on the quinolines for all three complexes.
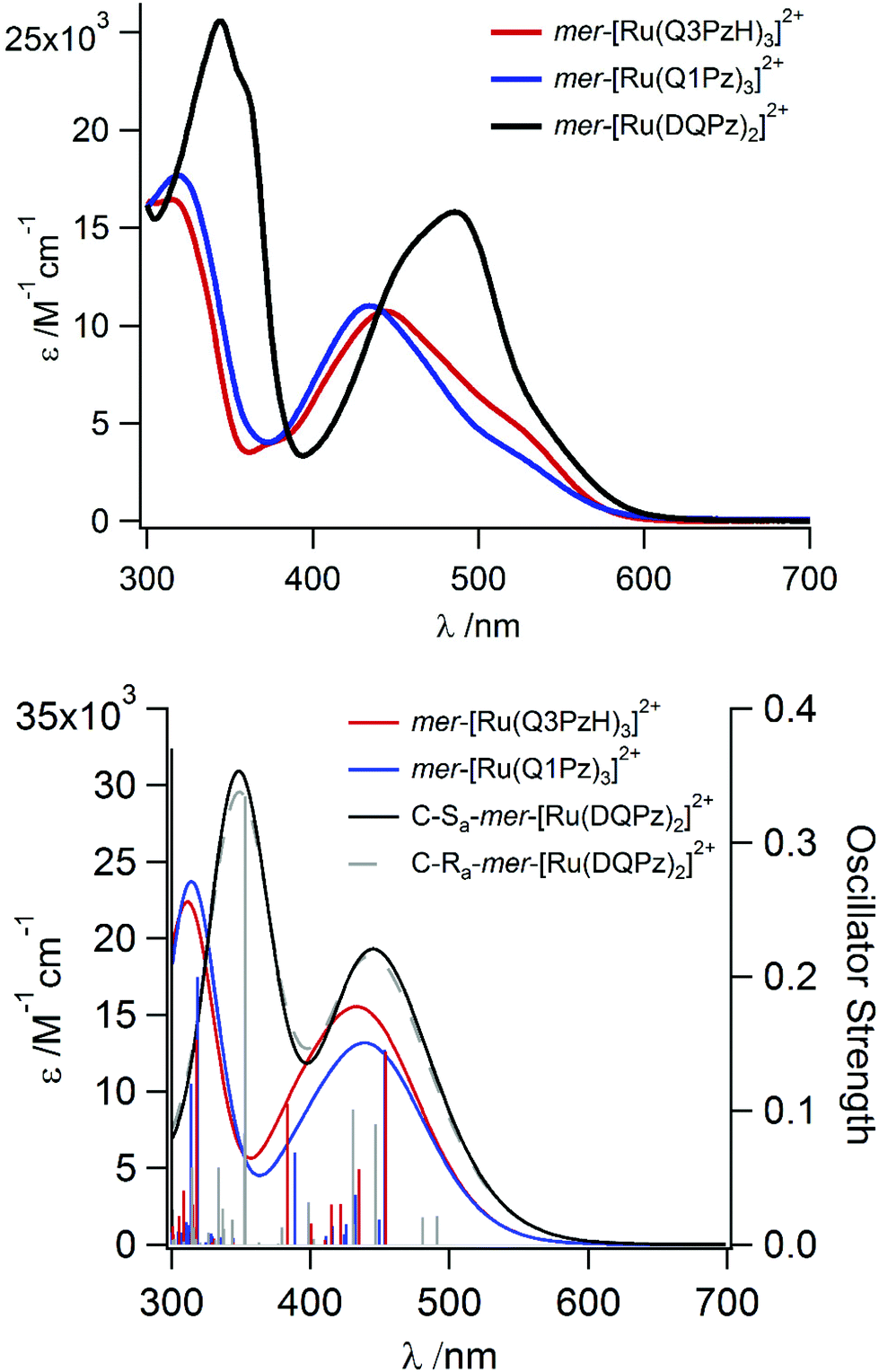 | ||
| Fig. 3 Experimental (top panel) and calculated (bottom panel) absorption spectra for mer-[Ru(Q3PzH)3]2+, mer-[Ru(Q1Pz)3]2+, C-Sa- and C-Ra-mer-[Ru(DQPz)2]2+. | ||
A detailed comparison of mer-[Ru(Q1Pz)3]2+ and mer-[Ru(Q3PzH)3]2+ reveals similar molar absorptivities and a slight red-shift of the maximum MLCT absorption for mer-[Ru(Q3PzH)3]2+, Table 3, qualitatively agreeing with the electrochemically determined oxidation potentials. Both tris-bidentate complexes display additional shoulders with lower intensity at ca. 530 nm, extending the absorption spectra up to almost 600 nm, not reproduced by the TD-DFT. The MLCT absorption bands observed for tridentate mer-[Ru(DQPz)2]2+ are red-shifted compared to the bidentate complexes, and display peak absorption at 486 (15 800 M−1 cm−1) and 344 nm (25 600 M−1 cm−1), in accordance with TD-DFT calculations.
The pyrazole orientation not only affects the geometric structure of the resulting complexes but also the electronic structure. Although, the ground state electronic structures of all complexes are very similar with Ru t2g HOMOs and quinoline centered LUMOs, the oxidation and reduction potentials as well as the absorption spectra vary between mer-[Ru(Q3PzH)3]2+ and mer-[Ru(Q1Pz)3]2+ which differ mainly in the pyrazole orientation. Interestingly, Lever's electrochemical parametrization30 with combinations of pyrazole and quinoline subunits gives a Ru3+/2+ of +750 mV for the bidentate complexes, equal parts pyrazole and quinoline, and +810 mV for the tridentate one, 2:1 quinoline:pyrazole, vs. Fc0/+.30,31 The significant discrepancies between these parametrization based potentials and those measured indicate the importance of the pyrazole orientation, especially the ∼120 mV difference in the two bidentate complexes and that the bidentate mer-[Ru(Q1Pz)3]2+ potential is more positive than for the tridentate complex.
Proton sensitivity
If deprotonation has a distinct effect on the absorption spectra, the three protolyzable hydrogens in [Ru(Q3PzH)3]2+ can provide an environmental handle.25 This possibility was investigated here by UV-Vis spectroscopy and TD-DFT. The calculated absorption spectra of once, twice and three times deprotonated mer-[Ru(Q3PzH)3]2+ revealed consecutive redshifts of MLCT-band maxima for each removed hydrogen (Fig. 4) by, on average, 0.14, 0.38 and 0.55 eV respectively compared to the fully protonated complex. The negative deprotonated ligand causes destabilization of the metal HOMO, which in turn leads to lower energy HOMO–LUMO transitions manifested in the red-shifted absorption.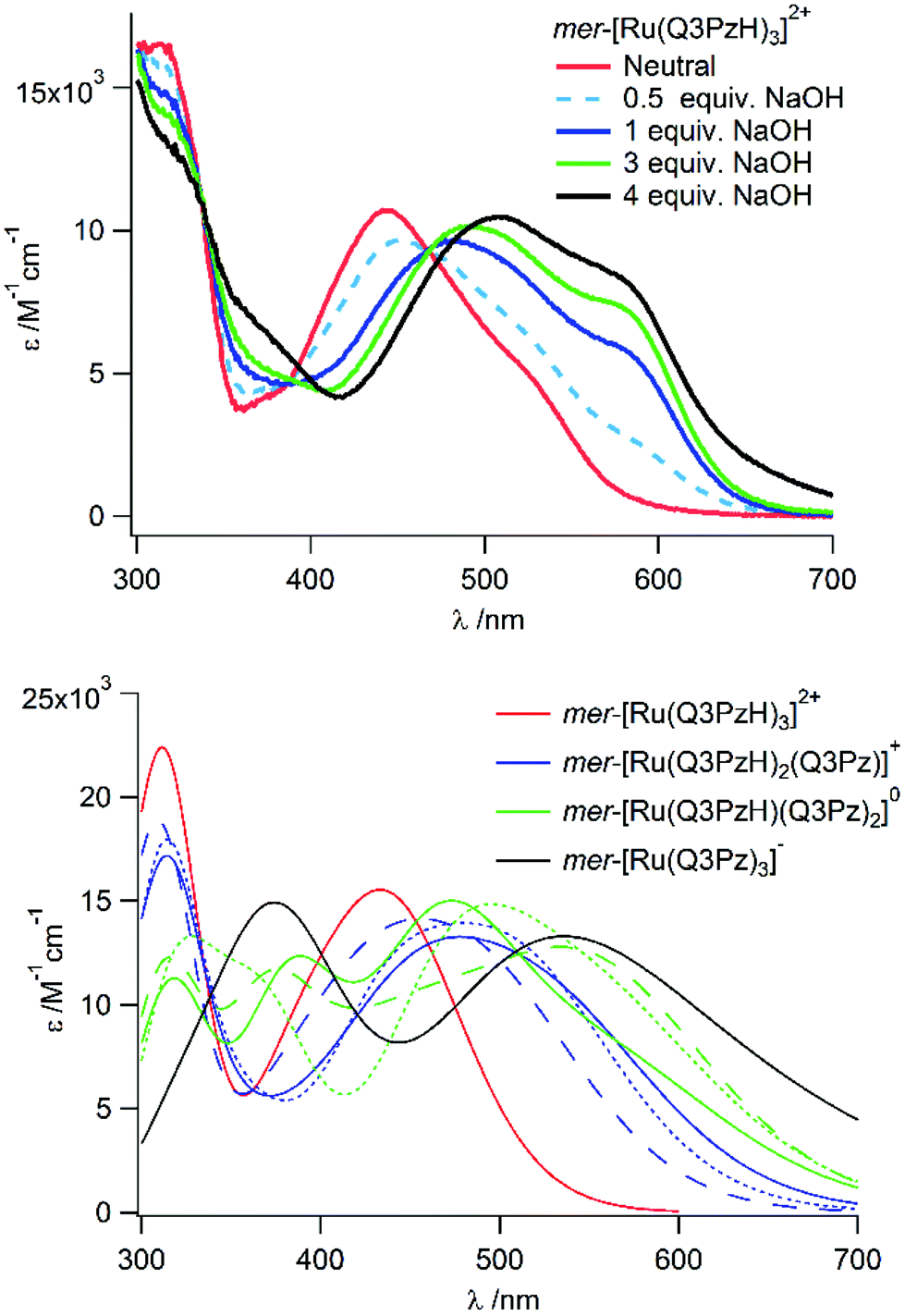 | ||
| Fig. 4 (Top panel) Titration of NaOH to mer-[Ru(Q3PzH)3]2+ in MeCN and (bottom panel) calculated absorption spectra with 0.25 eV broadening of all of the possible protonation states of mer-[Ru(Q3PzH)3−x(Q3Pz−)x](2−x)+ in acetonitrile (x = 0–3), where the lowest energy spectra of each protonation state is shown as a solid line. | ||
As previously reported, each ligand in mer-[Ru(Q3PzH)3]2+ adopts a different geometry, with significantly different ligand twists ranging from almost 0° to 20°.28 Hence, deprotonation of each unique ligand, with the same total number of deprotonations, affects the absorption profile differently, Fig. 4. UV-Vis spectra recorded upon titration of NaOH to mer-[Ru(Q3PzH)3]2+ in acetonitrile, Fig. 4, and aqueous solution (Fig. S8†) confirm the predicted redshift of the MLCT-band upon deprotonation.
In acetonitrile, isosbestic points on the blue and red side of the MLCT-band were maintained up to 3 equivalences of base, suggesting complete deprotonation of one ligand in the complex. In the aqueous titration, the complex was fully protonated up to pH 6.38, and singly deprotonated at pH 9.46. Further addition of base caused a larger red-shift, but the second and third deprotonations could not be conclusively discerned in either acetonitrile or aqueous solution. In acetonitrile, consecutively recorded 1H-NMR spectra revealed a time dependent process at >3 equivalents of base in the dark, causing broadening of all the resonances and attempts to structurally elucidate the product(s) of this process were inconclusive (Fig. S9†). In aqueous solution, the second and third deprotonations occurred at similar pH (Fig. S8†). The first protonation equilibrium constant, pKa1, of mer-[Ru(Q3PzH)3]2+ was determined to be 8.7. Examination of isosbestic points and spectral shape of spectra resulting from the titration in aqueous solution, together with the TD-DFT calculations of the different protonation states of the complex, resulted in the estimation of pKa2 to be 10.7.
The theory models each deprotonation state as a separate species giving a picture of the otherwise hard-to-obtain individual spectra of mer-[Ru(Q3PzH)(Q3Pz−)2] and mer-[Ru(Q3Pz−)3]−. While the experimentally obtained spectra cannot provide an isolated protonation state in solution, the theory simplifies the assignment of the different experimentally acquired spectra, and provides understanding of the observed red-shifts. The strong pH-dependence of the UV-Vis spectra in mer-[Ru(Q3PzH)3]2+ makes it a good candidate as a local environment acidity-probe. With the different spectral signatures depending on which ligand becomes deprotonated, as displayed by the TD-DFT, one could also envision a scenario where this type of complex is applied for hydrogen bonding in guest-host chemistry.25
Photochemical reactivity
As the mer-complexes of both Q3PzH and Q1Pz are separable from the mer/fac-mixture via light-induced decomposition of the fac-isomer, we also investigated the response to light of all the three isolated mer-isomers. The effects of broad-band visible irradiation was followed by UV-Vis absorption, electro-spray ionization high resolution mass spectrometry (ESI-HRMS), and 1H-NMR. The bis-tridentate mer-[Ru(DQPz)2]2+ displayed no signs of degradation or transformation when irradiated with a Xe-arc lamp in neat acetonitrile or in acetonitrile with added triflic acid (TfOH), showing only minute changes in UV-Vis spectra even beyond 106 excitation cycles, Fig. 5.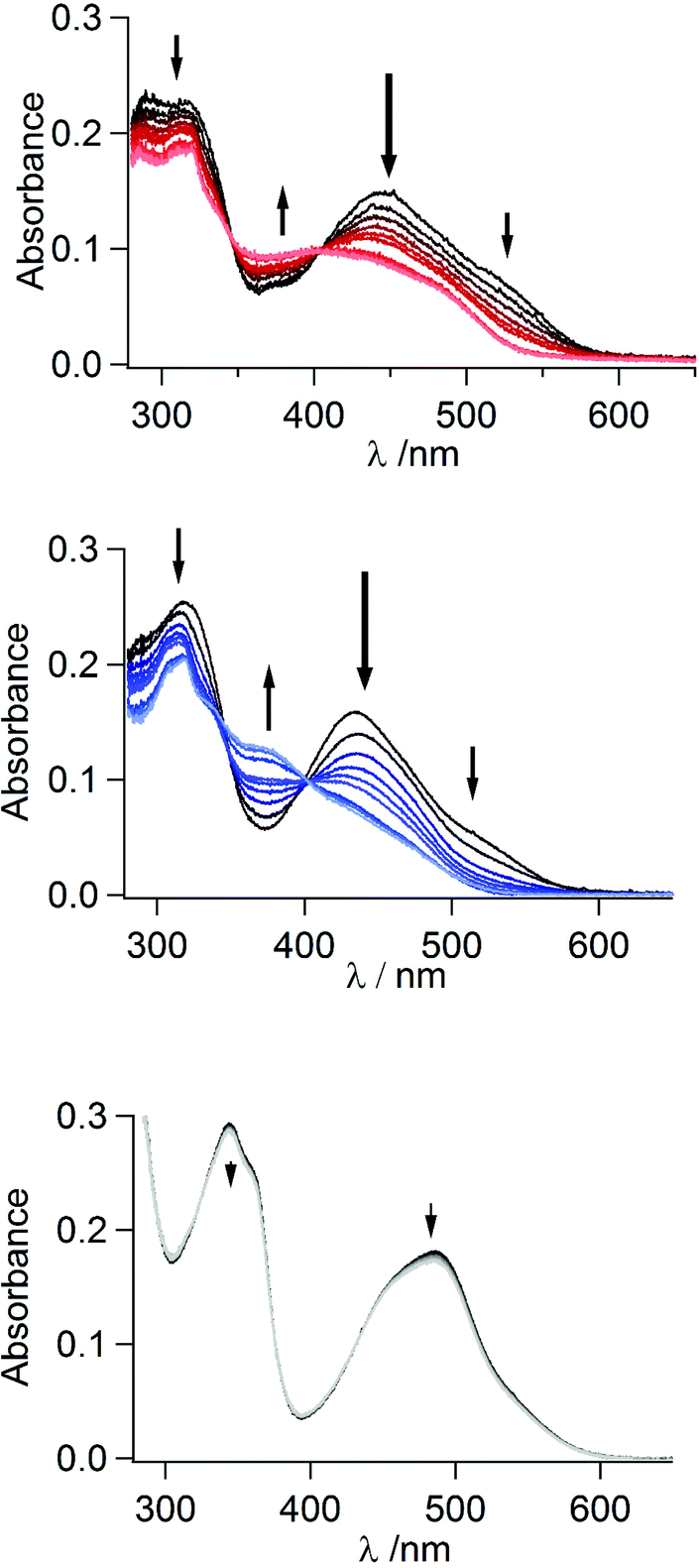 | ||
| Fig. 5 White light irradiation of mer-[Ru(Q3PzH)3]2+ (top), mer-[Ru(Q1Pz)3]2+ (middle) and mer-[Ru(DQPz)2]2+ (bottom) with 10 equiv. triflic acid in acetonitrile monitored by UV-Vis. | ||
ESI-HRMS revealed that even after irradiation in presence of 20 equiv. TfOH, the only detectable species is the parent molecular 2+ ion, suggesting no ligand dissociation or fragmentation occurs (Table S2†). 1H-NMR revealed very broad resonances following irradiation, precluding any detailed structural analysis (Fig. S10†). In contrast, both tris-bidentate complexes displayed an overall decrease in absorption under irradiation in neat acetonitrile, especially at the red end of the MLCT-band (Fig. S11†). Notably, mer-[Ru(Q1Pz)3]2+ displayed somewhat larger and more rapid changes per excitation cycles compared to mer-[Ru(Q3PzH)3]2+ (Fig. S12†). The same experiment carried out in the presence of TfOH produces absorption spectra with distinct isosbestic points, at 345 and 404 nm for mer-[Ru(Q3PzH)3]2+, and at 346 and 403 nm for mer-[Ru(Q1Pz)3]2+, indicating clean transformation of the complexes into photochemical product(s), Fig. 5. Three new resonances appeared in the 1H-NMR spectra of both complexes around 9.5 ppm, Fig. 6 (and Fig. S13†). The non-coordinated protonated ligand typically displays two quinoline resonances in this region, indicating ligand dissociation. After 2.5 h of irradiation mer-[Ru(Q3PzH)3]2+ showed less ligand dissociation compared to mer-[Ru(Q1Pz)3]2+. However, after 5 h of illumination both mer-[Ru(Q3PzH)3]2+ and mer-[Ru(Q1Pz)3]2+ displayed increased relative amounts of non-coordinated ligand and ESI-HRMS confirms that the Ru-complexes have lost one ligand, with no evidence of TfO− ligation (Table S2†). After 28 h irradiation, the new 1H-NMR resonances disappear completely, indicating that over long periods of time the ligands completely dissociate.
 | ||
| Fig. 6 1H-NMR spectra of mer-[Ru(Q1Pz)3]2+ with 10 equiv. triflic acid after exposure to visible light for (a) 0 h after keeping the sample in the dark for 2 h, (b) 0.5 h, (c) 1 h and (d) 2.5 h, (e) 14 h, (f) 18 h, (g) 28 h, (h) 28 h with 5 μL D2O added and (i) reference spectra of ligand Q1Pz with 10 equiv. triflic acid and 5 μL D2O added. In (i) D2O were added to remove disturbing resonances from triflic acid, for comparison the same was done in (h). | ||
Excess acid makes the observed dissociation clean and irreversible as the dissociated ligands are stabilized through protonation. However, addition of triflic acid did not appreciably speed up the ligand dissociation processes, as observed from the normalized decrease in absorption at λmax as a function of absorbed photons (Fig. S12†). This decrease indicates partial ligand photo-dissociation, followed by protonation of the non-coordinated heterocycle to block re-chelation.
As seen in UV-Vis, 1H-NMR, and ESI-HRMS, photochemical ligand loss in mer-[Ru(Q1Pz)3]2+ is slightly faster than in mer-[Ru(Q3PzH)3]2+, while mer-[Ru(DQPz)2]2+ is in comparison unaffected over the experiment timeframe. It is logical that the bis-tridentate complex exhibits less ligand loss than the tris-bidentate ones in absence of acid due to expected re-chelation dynamics. However the extremely low ligand loss is either due to a hindered initial step of partial ligand dissociation, not allowing protonation of the dissociated ligand, or that the partially ejected and protonated ligand is quickly deprotonated and re-chelated. The lack of photoinduced ligand loss in mer-[Ru(DQPz)2]2+ is interesting in the context of the recently reported room temperature diastereomerization between the C-Ra and C-Sa isomers.29 The diastereomerization was proposed to proceed through a mechanism that did not include any bond-breaking steps, which is corroborated by these results as no signs of appreciable degradation is observed after extended irradiation, suggesting a dynamic yet photo-stable complex.
Comparing the series: effect of pyrazole orientation
Altering the orientation of the pyrazole yields complexes with different electrochemical and photochemical properties. While all complexes displayed similar electronic structure, with largely quinoline localized LUMO's and Ru localized HOMO's, they display different absorption spectra and, especially, oxidation and reduction potentials. Depending on the pyrazole orientation, the bidentate ligands can be proton donors or protolytically inactive, dramatically influencing the sensitivities of the complexes to their environment, such as pH or possibility for hydrogen bonding. In addition, both tris-bidentate mer-[Ru(Q3PzH)3]2+ and mer-[Ru(Q1Pz)3]2+ displayed white light photo-induced ligand dissociation, with slightly different rates apparently also related to the orientation of the pyrazole unit, while the bis-tridentate mer-[Ru(DQPz)2]2+ did not show any appreciable net photo-dissociation. With triflic acid present, the partially dissociated ligands were protonated, precluding re-chelation and resulting in irreversible, and eventually complete, ligand dissociation; determining the partial ligand dissociation to be the initial step.Conclusions
Three homoleptic ruthenium complexes with unsymmetric quinoline–pyrazole ligands, Q3PzH, Q1Pz and DQPz, have been evaluated experimentally and theoretically. The two bidentate ligands differ in their orientation of the pyrazole subunit in relation to the quinoline, while the tridentate ligand is a superposition of the two. It is evident that the orientation of the pyrazole unit in relation to the quinoline results in significantly different properties in the homoleptic complexes, affecting both the geometric and electronic structure, influencing the electro- and photochemistry. This difference highlights the importance of understanding the delicate structure-function relationship in ligand and complex design, important in several major fields such as catalysis, sensing, and supramolecular assemblies and for photochemical applications. Furthermore, the non-degenerate protolyzable forms of mer-[Ru(Q3PzH)3]2+ can be envisioned as useful in stereoselective processes related to catalysis or host–guest chemistry. Finally, mer-[Ru(DQPz)2]2+ display a very high photostability, despite exhibiting a dynamic ground state surface.Experimental section
Synthesis
All chemicals were purchased from Sigma-Aldrich and used as received. Anhydrous solvents were used fresh or dried further over 4 Å molecular sieves activated at 250 °C. Solvents were, if necessary, degassed by bubbling N2-gas through them for >30 min. The silica used for flash chromatography was 230–400 mesh (Sigma-Aldrich). The NMR spectra were recorded on a Varian Inova 500 MHz spectrometer, solvents were >99.9% deuterated grade; all 1H-NMR resonances were referenced to the solvent residual signals. Combustion analysis was performed by Eurofins BioPharma Product Testing (Sweden) on a FlashEA 1112 (Thermo Fisher Scientific) CHN-analyzer using helium as carrier gas and a copper catalyst for reduction of nitrous gases. Sulfur and halogens were removed by silver cobalt oxide. The complexes mer-[Ru(Q3PzH)3]2+ and mer-[Ru(DQPz)2]2+ were available from previous studies.27,281-(8-Quinolinyl)-pyrazole (Q1Pz)
To a dry round-bottom flask was added 401 mg (1.93 mmol) 8-bromo-quinoline (Alfa Aesar), 137 mg (2.01 mmol) pyrazole, 225 mg (1.95 mmol) L-proline and 550 mg (3.98 mmol) anhydrous K2CO3; which was purged N2 gas. 2.3 mL of degassed anhydrous dimethyl formamide (DMF) was added, followed by 184 mg (0.966 mmol) copper(I) iodide. Some gas bubbles and a yellow solid formed. The solution was stirred at 120 °C and gradually turned blue and then blue-green over 15 min. The solution was heated overnight to completed conversion. Phase separation was carried out subsequent to addition of 25 mL ethyl acetate and 20 mL de-ionized water. The aqueous phase was extracted using 3 × 25 mL ethyl acetate and the combined organic phases were dried over Na2SO4, filtered and evaporated to yield a yellowish crude product, which was chromatographed on silica using ethyl acetate/heptane 1:2 yielding 205 mg (54%) of a yellowish oil after evaporating the solvent. Elemental analysis C12H9N3·0.1H2O·0.05C4H8O2Calc. % C, 72.75; H, 4.80 N, 20.86; Observ. C, 72.37; H, 4.41; N, 20.55; 1H-NMR (500 MHz, CD3CN)δ 8.96 (dd, 1H, J = 1.7 Hz, J = 4.1 Hz) 8.75 (d, 1H, J = 2.4 Hz) 8.39 (dd, 1H, J = 1.7 Hz, J = 8.4 Hz) 8.15 (dd, 1H, J = 1.2 Hz, J = 7.6 Hz) 7.93 (dd, 1H, J = 1.2 Hz, J = 8.2 Hz) 7.75 (d, 1H, J = 1.1 Hz) 7.70 (dd, 1H, J = 7.9 Hz, J = 7.9 Hz) 7.58 (dd, 1H, J = 4.1 Hz, J = 8.4 Hz) 6.55 (m, 1H).
mer-[Ru(Q1Pz)3](PF6)2via mer/fac mixture
In a 10 mL borosilicate glass vial 47.3 mg (0.242 mmol) Q1Pz and 38.9 mg (0.0803 mmol) Ru(DMSO)4Cl2 were suspended in 1.0 mL of ethylene glycol. The vial was wrapped in foil to protect from light and was heated at 195 °C in an oil bath, with regular shaking. After 7 min of heating the vial was cooled in a water bath, and 1.0 mL of methanol was added followed by 0.35 mL 1.0 M NaPF6 (aq). A red solid formed which was filtered and washed quickly with 3 × 0.5 mL cold methanol. The solid was dried under vacuum in the dark overnight yielding 56 mg of a red powder.To extract the pure mer-isomer from the mer![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) :fac mixture, 44.0 mg of the crude product was dissolved in 1.0 mL acetonitrile and was irradiated for 3 h using a cold light source (Leica, model CLS 150 XE) at power 4/6 at a distance of about 5 cm. Methanol was added dropwise until a precipitate started forming, until no further precipitate formed, at which time the solution was filtered. The solid was washed with 3 × 0.5 mL of a mixture of methanol/water 1:2 and then dried under vacuum to yield 22.5 mg (51%) of a red powder. Elemental analysis C36H27F12N9P2Ru·1CH3OH Calc. % C, 44.06; H, 3.10; N, 12.50; Observ. C, 44.1; H, 2.8; N, 12.3; 1H-NMR (500 MHz, CD3CN)δ 8.80 (d, 1H, J = 2.9 Hz) 8.79 (dd, 1H, J = 1.0 Hz, J = 5.3 Hz) 8.77 (dd, 1H, J = 1.1 Hz, J = 5.4 Hz) 8.57 (dd, 1H, J = 0.9 Hz, J = 8.2 Hz) 8.49 (dd, 1H, J = 0.8 Hz, J = 7.8 Hz) 8.45 (dd, 1H, J = 1.0 Hz, J = 5.2 Hz) 8.29 (m, 3H) 8.10–8.03 (m, 3H) 7.99 (d, 1H, J = 1.9 Hz) 7.74 (dd, 1H, J = 0.7 Hz, J = 7.6 Hz) 7.71 (dd, 1H, J = 0.8 Hz, J = 7.7 Hz) 7.67 (t, 2H, J = 7.8 Hz) 7.53 (dt, 2H, J = 8.2 Hz) 7.35 (d, 1H, J = 1.9 Hz) 7.29 (d, 1H, J = 1.9 Hz) 7.23 (m, 2H) 7.15 (dd, 1H, J = 5.3 Hz, J = 8.2 Hz) 6.80 (t, 1H, J = 2.5 Hz) 6.58 (t, 2H, J = 2.5 Hz); High-resolution Mass Spectrometry (HRMS) (ESI+, CH3CN)m/z {rel. intensity} 343.57110 [Ru(Q1Pz)3]2+ {100} (calc. C36H27N9Ru2+ 343.57110), 832.10831 [M2+ + PF6−]+ {10} (calc. C36H27F6N9PRu2+ 832.10692), 605.04636 [M2+ − Q1Pz + DMSO + Cl−]+ {5} (calc. C26H24ClN6ORuS+ 605.04588).
:fac mixture, 44.0 mg of the crude product was dissolved in 1.0 mL acetonitrile and was irradiated for 3 h using a cold light source (Leica, model CLS 150 XE) at power 4/6 at a distance of about 5 cm. Methanol was added dropwise until a precipitate started forming, until no further precipitate formed, at which time the solution was filtered. The solid was washed with 3 × 0.5 mL of a mixture of methanol/water 1:2 and then dried under vacuum to yield 22.5 mg (51%) of a red powder. Elemental analysis C36H27F12N9P2Ru·1CH3OH Calc. % C, 44.06; H, 3.10; N, 12.50; Observ. C, 44.1; H, 2.8; N, 12.3; 1H-NMR (500 MHz, CD3CN)δ 8.80 (d, 1H, J = 2.9 Hz) 8.79 (dd, 1H, J = 1.0 Hz, J = 5.3 Hz) 8.77 (dd, 1H, J = 1.1 Hz, J = 5.4 Hz) 8.57 (dd, 1H, J = 0.9 Hz, J = 8.2 Hz) 8.49 (dd, 1H, J = 0.8 Hz, J = 7.8 Hz) 8.45 (dd, 1H, J = 1.0 Hz, J = 5.2 Hz) 8.29 (m, 3H) 8.10–8.03 (m, 3H) 7.99 (d, 1H, J = 1.9 Hz) 7.74 (dd, 1H, J = 0.7 Hz, J = 7.6 Hz) 7.71 (dd, 1H, J = 0.8 Hz, J = 7.7 Hz) 7.67 (t, 2H, J = 7.8 Hz) 7.53 (dt, 2H, J = 8.2 Hz) 7.35 (d, 1H, J = 1.9 Hz) 7.29 (d, 1H, J = 1.9 Hz) 7.23 (m, 2H) 7.15 (dd, 1H, J = 5.3 Hz, J = 8.2 Hz) 6.80 (t, 1H, J = 2.5 Hz) 6.58 (t, 2H, J = 2.5 Hz); High-resolution Mass Spectrometry (HRMS) (ESI+, CH3CN)m/z {rel. intensity} 343.57110 [Ru(Q1Pz)3]2+ {100} (calc. C36H27N9Ru2+ 343.57110), 832.10831 [M2+ + PF6−]+ {10} (calc. C36H27F6N9PRu2+ 832.10692), 605.04636 [M2+ − Q1Pz + DMSO + Cl−]+ {5} (calc. C26H24ClN6ORuS+ 605.04588).
Mass-spectrometry
Electrospray ionization in positive mode was performed on a Thermo Scientific LTQ Velos Pro Orbitrap instrument. The mass spectrometer was operated in FT-mode at a resolution of 30000 and Leucine Enkephalin was used for lockmass correction. Solvent was acetonitrile unless noted otherwise.
Theoretical
The relaxed ground state electronic structure of the mer- and fac- isomers of [Ru(Q3PzH)3]2+ and [Ru(Q1Pz)3]2+, as well as two mer-, two trans-fac- and four cis-fac- isomers of [Ru(DQPz)2]2+ have been investigated using density functional theory (DFT) calculations. In addition, all unique once, twice and three times deprotonated mer-[Ru(Q3PzH)3−x(QPz)x](2−x)+ were optimized. All quantum chemical calculations have been performed using the Gaussian09 program31 using the PBE032–34 functional in conjunction with standard Gaussian type orbital (GTO) basis sets of double-ζ quality, 6-31G(d,p), and the SDD Stuttgart/Dresden effective core potential (ECP) was used to provide an effective core potential for Ru35,36 with a complete polarizable continuum model (PCM) solvent description for acetonitrile and the spin-restricted singlet formalism. All calculations were run with a total charge on the complex based on the protonation state of the ligands, all fully protonated ligands are neutral, and no symmetry constraints were applied, allowing for possible Jahn–Teller effects. All fully relaxed minima were verified using vibrations calculated at the same level of theory following geometry optimizations. TD-DFT calculations were performed at the same level of theory from the ground state optimized structure to obtain the calculated absorption spectra with a half-width/half-height of 0.25 eV.The free energies of organometallic species were calculated using G298K = Eelec + Gsolv. + ZPE + Hvib + nKT/2 + T(Selec + Svib) where Gsolv is the free energy of solvation, ZPE is the zero point energy correction, Svib and Selec are the vibrational and electronic entropies, and n = 12 accounts for the potential and kinetic energies of the translational and rotational modes. Frequency calculations were conducted using the same functional as the optimizations with a triple-ζ standard Gaussian type orbital (GTO) basis set, 6-311G*, and the SDD Stuttgart/Dresden ECP for Ru.
Absorption spectroscopy
UV-visible molar absorptivities were determined by linear regression on a concentration series of 10 dilute samples (5.0–50 μM) in neat spectrophotometric grade acetonitrile (Sigma-Aldrich, >99.5%) on a Varian Cary 5000 UV-vis-NIR spectrophotometer between 800–200 nm. The first and second pKa of mer-[Ru(Q3PzH)3]2+ were estimated by measuring UV-Vis spectra coupled to pH-monitoring during base-titrations. Spectra were recorded on a Varian Cary Bio 50 spectrophotometer, and pH was initially set by addition of hydrochloric acid to mer-[Ru(Q3PzH)3]2+ dissolved in milli-Q filtered water followed by NaOH titration. The relative concentrations of the different protonation states were extracted via Lambert-Beer's law by localizing isosbestic points between the different protonation states (at 468 nm and 529 nm for the first and second deprotonation respectively). Molar absorptivities was taken at pH 3.5 and pH 11.45 for the fully protonated and twice deprotonated forms respectively.Electrochemistry
Cyclic voltammetry was carried out in argon-purged (>10 min) acetonitrile with 0.10 M TBAPF6 (Sigma-Aldrich) in a conventional three electrode setup, with a platinum disk working electrode, a rod-shaped glassy carbon auxiliary electrode and a salt-bridged (acetonitrile/0.1 M TBAPF6) SCE reference electrode. A scan speed of 0.1 V s−1 was used. Irreversible reductions were evaluated by differential pulse voltammetry. Subsequent to measurement, ferrocene was added as an internal standard, and the Fc0/+ couple appeared at +417.5 mV with a peak-to-peak separation of 114 mV. The differential pulse voltammetry for the reductions were collected using pulse width 60 ms, pulse period 200 ms, pulse amplitude 50 mV and sampling width 20 ms.Photolysis experiments
The stability of the complexes under light exposure was studied by UV/vis and 1H-NMR. UV-Vis light stability measurements were performed in neat acetonitrile with and without 10 equivalents of triflic acid, and were carried out on a Varian Cary Bio 50 spectrophotometer. Broad band visible illumination was achieved by employing two IR-filters in front of a 150 W Xe arc lamp, yielding an irradiation spectrum between 390–750 nm, with a minor contribution from 320–350 nm. The intensity of the resulting spectrum was measured to 400 mW cm−2 with a thermocoupled power meter (407A, Spectra Physics), and the number of absorbed photons at any given time was integrated from the fractional absorption spectra times the irradiance spectrum with respect to time. Note that the number of absorbed photons is calculated for all species in the sample, apart from the solvent, not only the parental complexes.For the NMR experiments a 2.5 mM solution of the complex was prepared in CD3CN. An excess of triflic acid was added, 10–20 equiv., where the larger excess was to make sure the large resonance of the triflic acid was shifted outside the region of the proton resonances of the complex. After collecting reference spectra the sample(s) were illuminated by a 250 W white light lamp (Luma) at a distance of 60 cm. Temperature was kept low by fixing the samples in a test tube on top of ice in a transparent container, both made of borosilicate glass. Temperature remained 10–30 °C during the experiment. Spectra were collected after 30, 60, 90 and 150 min; and sometimes for longer times.
Acknowledgements
The authors wish to thank the Swedish Research council (VR), the Craaford foundation, Ångpanneföreningens forskningsstiftelse (Åforsk), the Knut and Alice Wallenberg (KAW) foundation, and the Swedish Energy Agency (Energimyndigheten) for financial support. In addition, M. A. and J. H. W. thank the Area of Advance Nano at Chalmers for financial support. P. P. acknowledges support from the National Supercomputer Centre (NSC) and The Center for Scientific and Technical Computing at Lund University (LUNARC).Notes and references
- A. E. Friedman, J. C. Chambron, J. P. Sauvage, N. J. Turro and J. K. Barton, J. Am. Chem. Soc., 1990, 112, 4960–4962 CrossRef CAS.
- M. R. Gill and J. A. Thomas, Chem. Soc. Rev., 2012, 41, 3179–3192 RSC.
- K. K.-W. Lo and S. P.-Y. Li, RSC Adv., 2014, 4, 10560–10585 RSC.
- S. Ardo and G. J. Meyer, Chem. Soc. Rev., 2009, 38, 115–164 RSC.
- M. K. Nazeeruddin, C. Klein, P. Liska and M. Grätzel, Coord. Chem. Rev., 2005, 249, 1460–1467 CrossRef CAS.
- J. W. Tucker and C. R. J. Stephenson, J. Org. Chem., 2012, 77, 1617–1622 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879–5918 CrossRef CAS PubMed.
- C. Gunanathan and D. Milstein, Chem. Rev., 2014, 114, 12024–12087 CrossRef CAS PubMed.
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2009, 42, 1890–1898 CrossRef CAS PubMed.
- H. A. Younus, W. Su, N. Ahmad, S. Chen and F. Verpoort, Adv. Synth. Catal., 2015, 357, 283–330 CrossRef CAS.
- Y. Sun, M. El Ojaimi, R. Hammitt, R. P. Thummel and C. Turro, J. Phys. Chem. B, 2010, 114, 14664–14670 CrossRef CAS PubMed.
- S. I. Gorelsky and A. B. P. Lever, J. Organomet. Chem., 2001, 635, 187–196 CrossRef CAS.
- C. Daniel, Coord. Chem. Rev., 2015, 282, 19–32 CrossRef.
- A. Vlček and S. Záliš, Coord. Chem. Rev., 2007, 251, 258–287 CrossRef.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. Von Zelewsky, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef CAS.
- M. Abrahamsson, H. Wolpher, O. Johansson, J. Larsson, M. Kritikos, L. Eriksson, P.-O. Norrby, J. Bergquist, L. Sun, B. Åkermark and L. Hammarström, Inorg. Chem., 2005, 44, 3215–3225 CrossRef CAS PubMed.
- P. A. Mabrouk and M. S. Wrighton, Inorg. Chem., 1986, 25, 526–531 CrossRef CAS.
- M. Maestri, N. Armaroli, V. Balzani, E. C. Constable and A. M. C. Thompson, Inorg. Chem., 1995, 34, 2759–2767 CrossRef CAS.
- M. Abrahamsson, M. Jäger, T. Österman, L. Eriksson, P. Persson, H.-C. Becker, O. Johansson and L. Hammarström, J. Am. Chem. Soc., 2006, 128, 12616–12617 CrossRef CAS PubMed.
- O. Schuster, L. Yang, H. G. Raubenheimer and M. Albrecht, Chem. Rev., 2009, 109, 3445–3478 CrossRef CAS PubMed.
- M. Abrahamsson, H.-C. Becker, L. Hammarström, C. Bonnefous, C. Chamchoumis and R. P. Thummel, Inorg. Chem., 2007, 46, 10354–10364 CrossRef CAS PubMed.
- D. P. Rillema, D. S. Jones, C. Woods and H. A. Levy, Inorg. Chem., 1992, 31, 2935–2938 CrossRef CAS.
- Q. Zeng, F. W. Lewis, L. M. Harwood and F. Hartl, Coord. Chem. Rev., 2015, 304, 88–101 CrossRef.
- A. J. Metherell, W. Cullen, A. Stephenson, C. A. Hunter and M. D. Ward, Dalton Trans., 2014, 43, 71–84 RSC.
- A. B. Tamayo, B. D. Alleyne, P. I. Djurovich, S. Lamansky, I. Tsyba, N. N. Ho, R. Bau and M. E. Thompson, J. Am. Chem. Soc., 2003, 125, 7377–7387 CrossRef CAS PubMed.
- S. L. Dabb and N. C. Fletcher, Dalton Trans., 2015, 44, 4406–4422 RSC.
- M. Jarenmark, L. A. Fredin, J. H. Hedberg, I. Doverbratt, P. Persson and M. Abrahamsson, Inorg. Chem., 2014, 53, 12778–12790 CrossRef CAS PubMed.
- M. Jarenmark, G. Carlström, L. A. Fredin, J. Hedberg Wallenstein, I. Doverbratt, M. Abrahamsson and P. Persson, Inorg. Chem., 2016, 55, 3015–3022 CrossRef CAS PubMed.
- A. B. P. Lever, Inorg. Chem., 1990, 29, 1271–1285 CrossRef CAS.
- V. V. Pavlishchuk and A. W. Addison, Inorg. Chim. Acta, 2000, 298, 97–102 CrossRef CAS.
- M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. Petersson, Theor. Chem. Acc., 2008, 120, 215 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 1396 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- M. Dolg, U. Wedig, H. Stoll and H. Preuss, J. Chem. Phys., 1987, 86, 866–872 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S13 and Tables S1–S20. See DOI: 10.1039/c6dt01070a |
| This journal is © The Royal Society of Chemistry 2016 |