
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnticancer activity of a chelating nitrogen mustard bearing tetrachloridoplatinum(IV) complex: better stability yet equipotent to the Pt(II) analogue†
Subhendu
Karmakar
,
Saptarshi
Chatterjee
,
Kallol
Purkait
and
Arindam
Mukherjee
*
Department of Chemical Sciences, Indian Institute of Science Education and Research Kolkata, Mohanpur-741246, India. E-mail: a.mukherjee@iiserkol.ac.in
First published on 16th May 2016
Abstract
Two Pt(IV) complexes cis,cis,trans-[PtIV(L1)Cl4] (1a) & cis,cis,trans-[PtIV(L2)Cl4] (2a) containing the nitrogen mustard moieties –N(CH2CH2Cl)2 & –NHCH2CH2Cl, were prepared in a single step from the Pt(II) complexes containing –N(CH2CH2OH)2 (1) & –NHCH2CH2OH (2) moieties respectively using only thionyl chloride. The characterization of both the Pt(IV) complexes was performed by NMR, IR, UV and elemental analysis. Complex 1a was also characterized by single crystal X-ray diffraction. 1a crystallized in the I2/a space group. 1a exhibited much higher solution stability than 2a in kinetic studies by 1H NMR. 1a shows a prodrug like activity as it converts to its Pt(II) congener, [PtII(L1)Cl2] (3) after 2 days in buffered solution. The binding experiment of 1a with model nucleobase 9-ethylguanine (9-EtG), showed that 1a converts to 3 and forms mono-adducts with 9-EtG. In the presence of reduced glutathione (GSH), the formation of 3 from 1a is quicker and upon the formation of 3 it binds almost instantaneously to GSH to form cis-[PtCl(L1)SG] (3c). Complex 3c transformed within a day to give a free aziridinium ion of L1 (3b) by dissociation. The in vitro cytotoxicity of the complexes and the clinical anticancer drug cisplatin show that 1a is potent against MCF-7, A549, HepG2 and MIA PaCa-2. The potency is highest against MIA PaCa-2 exhibiting an IC50 value of 4.4 ± 0.5 μM. The in vitro cytotoxicity data also showed that between the two complexes only 1a is active against MCF-7, A549 and MIA PaCa-2 in normoxia and hypoxia, both in the presence and absence of added GSH. Even in the presence of excess GSH in hypoxia, 1a exhibits significant cytotoxicity against MIA PaCa-2 and MCF-7 with IC50 values of 4.5 ± 0.3 and 11.2 ± 1.8 μM respectively. Platinum accumulation studies by ICP-MS display greater internalization of 1a, than 2a, 3 and cisplatin inside MCF-7 cells. 1a arrests cell cycle at the G2/M phase in MCF-7, exhibits capability to inhibit metastasis, induces apoptotic cell death and displays blood compatibility with human blood.
Introduction
The serendipitous discovery of cisplatin by Barnett Rosenberg and his co-workers was the landmark in the use of metal complexes as chemotherapeutic agents.1–3 Despite having several limitations,4 a great clinical success was achieved in using cisplatin for the treatment of various types of cancer.5–7 The limitations of the usage of cisplatin mainly lie in the area of severe toxicities leading to major side-effects8–11 and tumor resistance to drugs.12–14 In order to counter the limitations, drugs like oxaliplatin and carboplatin have emerged and are serving well in clinic for the treatment of various types of cancer.15,16 All the above drugs are square planar Pt(II) complexes. It has been found that after systematic hydrolysis at the metal centre, the drug generates labile sites which cross-link the DNA bases, followed by a cascade that triggers cell death.17,18 However, square planar Pt(II) complexes are known to start early hydrolysis i.e. before even reaching the target, a significant amount of a Pt(II) drug is hydrolyzed and reacts with various ingredients of blood plasma and several intra- and extracellular bio-molecules to generate unwanted toxicities. Since there is a loss of the drug the effective applied dosage becomes low, requiring more drug to be given for the desired effect. It is found that on oxidation of Pt(II) to Pt(IV), the resulting d6 electronic configuration stabilizes the octahedral geometry and reduces reactivity. Hence, a Pt(IV) complex may possess enough kinetic inertness to resist immature hydrolysis and other unwanted side reactions with bio-molecules.19,20The cytotoxicity of the Pt(IV) complexes is known to arise due to their reduction to Pt(II) in the cellular reducing environment, especially in the hypoxic tumor environment where bio-molecules viz. glutathione and ascorbic acid act as reducing agents (Scheme 1).19,21–23 Such a pro-drug like activity is one of the added advantages in designing Pt(IV) complexes over Pt(II) drugs.24–26 Out of the numerous Pt(IV) complexes designed so far, four Pt(IV) complexes were subjected to clinical trials (Chart 1A),26,27 but iproplatin was abandoned from clinical trials due to less reactivity compared to cisplatin (or carboplatin) and tetraplatin was abandoned due to its severe neurotoxicity.15,26,28,29 Amongst all four the orally active satraplatin showed promising clinical activity in combination therapy as well as a standalone drug against various cancers especially against prostate cancer and is still in clinical trials.15,26,28,29 LA-12 is a very similar analogue of satraplatin, whose clinical trial is ongoing (phase I & II, yet unpublished).26,29,30 Therefore, designing a suitable Pt(IV) anticancer agent still remains a challenge.
 | ||
| Scheme 1 Bioreduction of Pt(IV) complexes in the presence of reducing bio-molecules. | ||
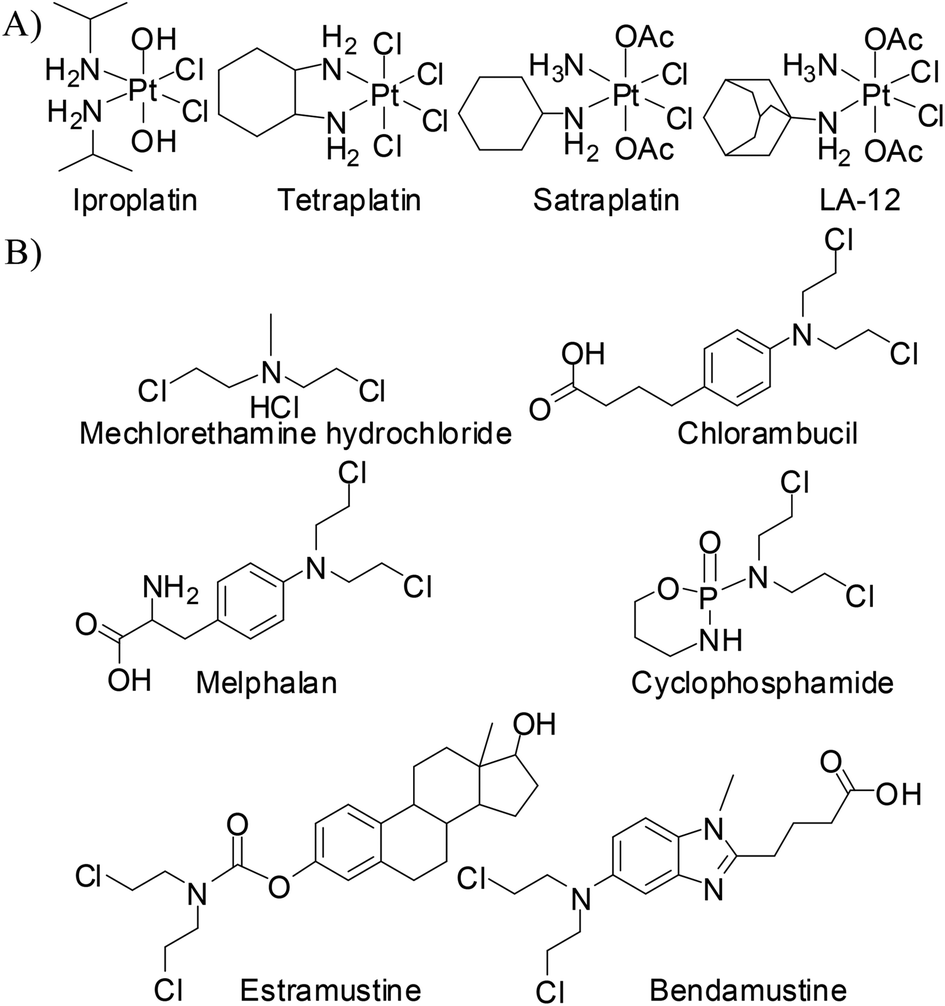 | ||
| Chart 1 (A) Pt(IV) complexes in clinical trials and (B) nitrogen mustards in clinical use. | ||
In a Pt(IV) complex the axial ligands are lost during the time of reduction to Pt(II). The presence of the axial ligands would influence the redox potentials of a Pt(IV) complex even if the equatorial ligands are the same in two different complexes. Among the numerous axial ligands used, some popular axial ligands with decreasing order of reducing ability of the Pt(IV) complex may be represented as –OCOCF3 > –Cl > –OCOCH3 > –OH.31 In this work we have chosen –Cl as the axial ligand with the thought that it would render an intermediate redox potential to the designed Pt(IV) complex. In our ligand design we have used the pyridine containing nitrogen mustard (L1) which gave us valuable information during our work on the Pt(II) complex (3, Scheme 2).32 We have synthesized the Pt(IV) mustard complex 1a in a single step from the Pt(II) complex of the ligand bis(2-hydroxyethyl)-pyridylmethylamine with SOCl2 in good yield (Scheme 2). For the sake of comparison of activity we also synthesized a Pt(IV) complex (2a) bearing a –NHCH2CH2Cl group in the ligand (L2) instead of the mustard group, –N(CH2CH2Cl)2 (Scheme 2).
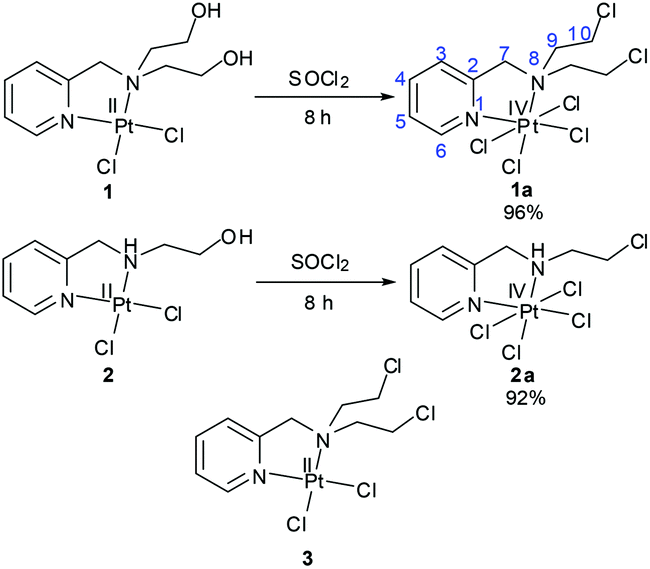 | ||
| Scheme 2 Synthetic methods for the preparation of Pt(IV) complexes (1a & 2a) and the structure of the Pt(II) counterpart of 1a (3). | ||
Complex 1a has a chelating mustard ligand at the equatorial position along with two equatorial and two axial –Cl− ions. The alkylating ability of the nitrogen mustards is well known and they have a rich history in chemotherapy (Chart 1B) against various forms of cancers.33 They form an inter-strand crosslink with N7 of guanine in DNA upon generation of the aziridinium ion as an intermediate.34,35 It is known that the reactivity of nitrogen mustards can be tuned by controlling the availability/reactivity of the lone pair of nitrogen on the mustard moiety, –N(CH2CH2Cl)2. The donation of the nitrogen lone pair to the metal centre, during the formation of complexes reduces the reactivity of the nitrogen mustard to form the aziridinium ion, thus controlling the alkylating ability. The metal bound mustard group would be reactivated upon the loss of a metal ion due to the higher availability of the lone pair on the nitrogen of the mustard group.36–43 However, in spite of this knowledge only three Pt(IV) complexes are reported so far, cis,cis,trans-[PtIVXYCl4],44cis,cis,trans-[PtIVX2Cl4]45 and trans,cis,cis-[PtIVX2Cl4]45 (where X = bis(2-chloroethyl)amine & Y = aziridine). None of them is a chelating nitrogen mustard and none of them has been probed for anticancer activity either.
Our impetus for this work came from our success with Pt(II) complex 3 of the chelating nitrogen mustard ligand.32 We were eager to know the influence of the nitrogen mustard on the reactivity of Pt(IV) complexes. This encouraged us to synthesize and characterize two Pt(IV) complexes with the molecular formula cis,cis,trans-[PtIV(L1)Cl4] (1a) and cis,cis,trans-[PtIV(L2)Cl4] (2a) and probe their potential as anticancer agents.
Results & discussion
Synthesis and characterization
Preparation of the Pt(IV) complex from Pt(II) precursors needs oxidation of the Pt(II) centre. The traditional method for oxidation of the Pt(II) centre is the use of an oxidizing agent like H2O2 in aqueous medium resulting in the formation of trans-dihydroxido Pt(IV). Further reactions are performed depending on which axial ligands are needed for derivatization, these include chlorination by purging Cl2 gas, reaction with acid anhydrides, coupling reaction with any carboxylic acid derivative.46 We have used SOCl2 for the preparation of our trans-dichlorido Pt(IV) complexes. Both 1 & 2 were converted to 1a & 2a respectively within 8 h using neat SOCl2 in a single step (Scheme 2). Here two different reactions concurred| [PtIICl2] + SOCl2 → [PtIVCl4] + [SO] | (1) |
| 2[SO] → S + SO2 | (2) |
Both the synthesized complexes (1a & 2a) were characterized by multinuclear NMR techniques (1H, 13C & 195Pt NMR), FT-IR and UV–vis spectroscopy. The IR spectra clearly showed the C–Cl stretching for 1a & 2a at 768 & 770 cm−1 respectively indicating –Cl functionalization by the nucleophilic substitution of SOCl2 with the –OH group (Fig. S1†). The NMR data show that the shift of 1H peaks supports the formation of Pt(IV) which differs from its reduced counterpart (3). The aromatic region of 3 contains four types of pyridine ring hydrogen. They are numbered as H3, H4, H5 & H6 and they appear in the NMR accordingly from a lower to higher field as H6, H4, H3 & H5 respectively. Upon oxidation of the platinum from +2 to +4 except for H6 and H10 (–CH2Cl) all the other protons show a downfield shift by around 0.2–0.5 ppm. Such trends of the chemical shift are also found in the literature.55,56 Apart from changes in the chemical shift, 195Pt satellites were observed in H6 & H7 (PyCH2–) protons for 1a which are absent in 3 and the coupling constant values (3JH–Pt) were 26.3 & 17.5 Hz respectively (Fig. S2†). In the case of 2a also the 195Pt satellites were found for H6 and one of the two H7 protons where the 3JH–Pt values are 26.3 and 18.7 Hz respectively (Fig. S7†). The 195Pt chemical shift values −64.1 & −174.0 ppm for 1a (Fig. S6†) & 2a (Fig. S11†), respectively, were also in accordance with the +4 oxidation sate of platinum. The analytical purity of complexes was confirmed by CHN analysis which matches well with the calculated percentage of elements.
X-ray crystallography
A suitable single crystal of complex 1a was obtained from an acetonitrile (acidified with HCl)–diethyl ether layer. 1a crystallized in the monoclinic crystal system with the space group I2/a (Table S1†). Each unit cell contains 8 complexes. The bond lengths of the two axial chlorides are higher than the equatorial two and the bond distance for Pt–N of the pyridine moiety is shorter than Pt–N of mustard nitrogen (Table S2†). The two axial chlorides are not exactly opposite to each other with respect to the Pt centre; rather they make a Cl–Pt–Cl angle of 175.84°. The structure shows that the two –CH2CH2Cl groups are projected above and below the coordination plane containing Pt, two N and two equatorial Cl− (Fig. 1). The presence of four Cl− coordinated to the Pt centre and the absence of any other cation in the lattice supports Pt in the oxidation state +4.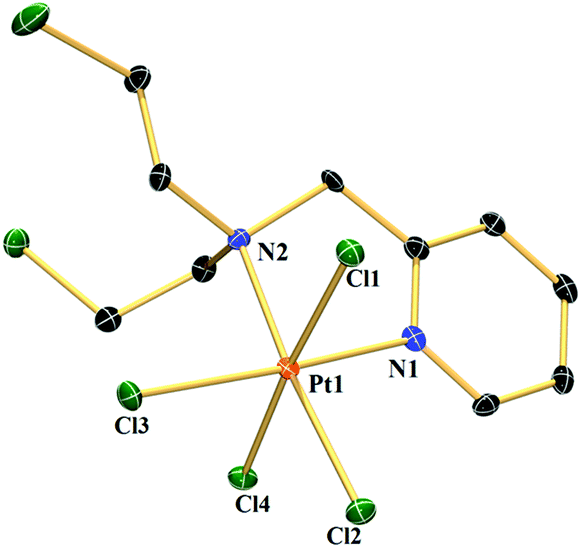 | ||
| Fig. 1 ORTEP diagram of complex 1a. Thermal ellipsoids are drawn at the 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å): Pt1–Cl1 2.3286(7), Pt1–Cl4 2.3183(7), Pt1–Cl2 2.3165(7), Pt1–Cl3 2.3178(7), Pt1–N1 2.027(2) & Pt1–N2 2.155(2); selected bond angles (°): Cl4–Pt1–Cl1 175.84(3), Cl2–Pt1–Cl3 88.66(3) & N2–Pt1–N1 80.60(10). | ||
Compound stability in solution
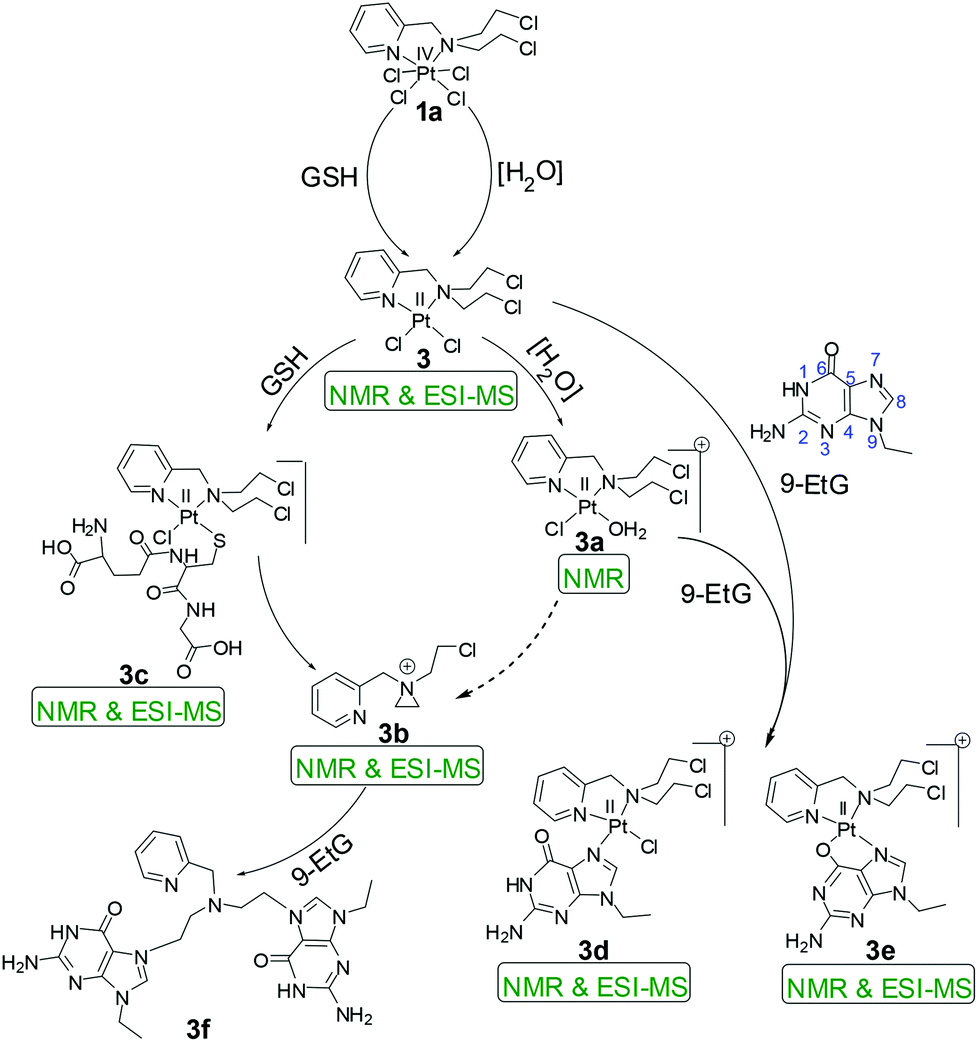
The kinetic inertness of octahedral Pt(IV) complexes is well studied.19,57 It is also known that all the complexes of Pt(IV) are not inert.19 The inert nature depends strongly on the type of the axial ligand.31 We have determined the solution stability of the Pt(IV) complex 1a in 20% PBS (pD 7.4, prepared in D2O) in DMSO-d6 by 1H NMR. The NMR data show that 1a has excellent stability in buffered medium up to 1 d and only the original complex is visible in the NMR plots (Fig. 2). Hence, the kinetic inertness of 1a towards hydrolysis is better than 3, which as per our earlier work, started to hydrolyze within 20 minutes. The additional kinetic stability should be helpful for 1a to increase its circulation lifetime and entry into the cells. 1a started to hydrolyze after 2 d and new peaks appeared at 9.08 & 9.00 (H6), 8.24 & 8.13 (H4), 7.84 & 7.65 (H3) and 7.51 (H5) ppm (see Scheme 2 for numbering; only the aromatic region of 1a is shown for clarity). Among the three pairs of new peaks, the peaks at 9.00, 8.13, 7.65 and 7.51 ppm correspond respectively to H6, H4, H3 and H5 of the reduced, square planar cis-dichlorido Pt(II) complex (3), obtained upon losing the axial Cl− ligands. The chemical shift values of 9.08, 8.24 and 7.84 ppm correspond to H6, H4 and H3, respectively, of the hydrolyzed Pt(II) complex (3a, Scheme 3) as depicted in Fig. 2. The peak of H5 of the hydrolyzed product 3a has similar chemical shift value (7.65 ppm) with the H3 of 3 as seen from the NMR plots (Fig. 2). The ESI-MS spectrum has also provided evidence for the formation of 3 and its hydrolyzed product along with the sodium or potassium ion bound to complex 1a (Fig. S12†). The m/z value at 520.9330, 538.9435, 592.8755 and 608.8354 matches well with the species of formulation [3 + Na+]+ (calcd 520.9429) (Table S3 and isotope modelling in Fig. S13†), [3 + K+]+ (calcd 538.9160) (Table S3 and isotope modelling in Fig. S14†), [1a + Na+]+ (calcd 592.8788) (Table S3 and isotope modelling in Fig. S15†) and [1a + K+]+ (calcd 608.8527) (Table S3 and isotope modelling in Fig. S16†) respectively. The m/z corresponding to hydrolyzed complex 3 appeared at 462.9764 corresponding to the speciation [3 − 2Cl− + 2OH− + H+]+ (calcd 463.0297) (Table S3 and isotope modelling in Fig. S17†). ESI-MS speciation indicates that both the Cl− might be lost upon hydrolysis. Further NMR studies presented later regarding the binding of 9-ethylguanine (9-EtG) confirm that upon monoaquation the reactivity towards nucleophilic substrates like 9-EtG may be initiated. One important point to note here is that the chemical shifts corresponding to the free aziridinium ion (of equatorial ligand L1, 3b, Scheme 3) were found to be distinctly visible during our earlier work with 3 alone as the starting complex.32 However in this case where 3 is generated in situ from 1a, we could see new peaks which may be due to aziridinium ion formation, but they did not appear distinctively and hence they cannot be assigned with certainty. | ||
Fig. 2 Stack plot of the stability kinetic study of 1a monitored by 1H NMR at different time intervals in 20% PBS (pD 7.4, prepared in D2O) – DMSO-d6 mixture, where  & &  indicate unreacted complexes 1a & 3 and indicate unreacted complexes 1a & 3 and 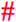 indicates hydrolyzed complex 3 (3a). indicates hydrolyzed complex 3 (3a). | ||
 | ||
| Scheme 3 Proposed reaction pathways of 1a for the hydrolysis and binding of 9-EtG/GSH. The dotted arrow in the scheme depicts that the formation of 3b from 3a shows no distinct evidence. | ||
Complex 2a showed hydrolysis right from the beginning and after 2 d most of the complex was hydrolyzed (Fig. S18†). Hence, it seems that the presence of the additional –CH2CH2Cl group on the bound nitrogen in 1a has a positive effect on the kinetic stability of the Pt(IV) complex. This additional stability might be attributed to the change in the electronic factor especially the increase in steric hindrance. To understand the system further, it was now needed that a binding experiment with 9-EtG be performed. The objective was to find out if the binding of 1a with guanine happens through the in situ formation of 3 and how many sites are hydrolyzed and accessible to 9-EtG per molecule of 1a.
Interaction with model nucleobase and plasmid DNA
The binding studies of 1a and 2a with model nucleobase 9-EtG using the 1H NMR technique provided better insight into the binding motif of the complexes with DNA. The binding studies were also performed in 20% PBS prepared in D2O pD 7.4 in DMSO-d6. N7 of 9-EtG is the most likely position58 of attack as evidenced from the chemical shift of the H8 proton of guanine (marked as H8G) downfield. In our case we do not see the binding of complex 1a directly with 9-EtG (added in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 molar ratio; 3 mM of 1a & 18 mM of 9-EtG) although certain literature reports state that nucleobases can directly bind to Pt(IV).22,59,60 The NMR data do not show any formation of free complex 3 or its aquated species rather we directly find the 9-EtG bound complex 3 after 2 d (Fig. 3). The above statement is made on the basis of the downfield shift of the H8G signal of 9-EtG from 7.69 ppm to 8.44 ppm (marked as H8′G in Fig. 3) due to the formation of a monoadduct of 9-EtG immediately upon the in situ formation of 3.
:6 molar ratio; 3 mM of 1a & 18 mM of 9-EtG) although certain literature reports state that nucleobases can directly bind to Pt(IV).22,59,60 The NMR data do not show any formation of free complex 3 or its aquated species rather we directly find the 9-EtG bound complex 3 after 2 d (Fig. 3). The above statement is made on the basis of the downfield shift of the H8G signal of 9-EtG from 7.69 ppm to 8.44 ppm (marked as H8′G in Fig. 3) due to the formation of a monoadduct of 9-EtG immediately upon the in situ formation of 3.
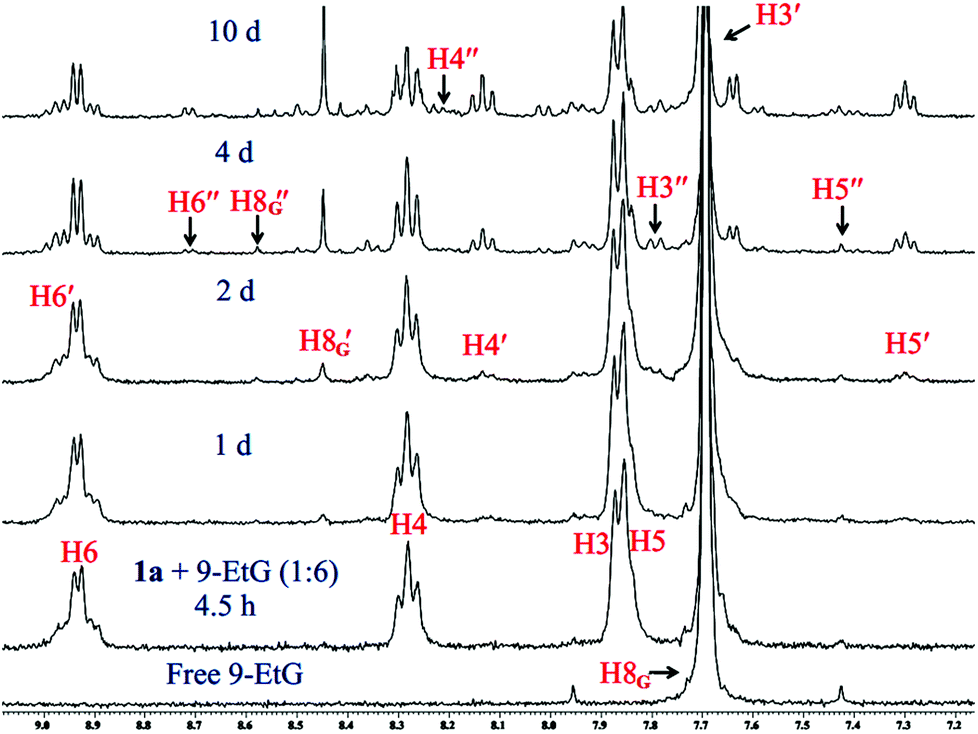 | ||
| Fig. 3 Stack plot of 1H NMR data obtained by the reaction of 1a with 9-EtG (1:6) in 20% PBS (pD 7.4, prepared in D2O) – DMSO-d6 mixture. The chemical shifts depict the presence of excess reactants (1a & 9-EtG) and two types of 9-EtG bound adducts (3d & 3e) after 10 d. H8G of free 9-EtG is shifted downfield to H8′G & H8′′G of 9-EtG bound complexes 3d & 3e respectively. | ||
The earlier depicted hydrolysis study with 1a shows that the formation of the reduced Pt(II) complex 3 happens to have a very similar time scale as found for the 9-EtG bound speciation. Hence, the presence of 9-EtG during the formation of 3 might be immediately leading to the 9-EtG conjugation. The NMR data do not show any evidence of the formation of 3 or its aquated species in this case. However, this was not the case for our earlier studies with 3 as a starting compound,32 where we could find evidence of both the 9-EtG adduct and hydrolyzed Pt(II) complex formation. This suggests that the kinetic pathway for DNA binding may not be the same i.e. the nucleophilic attack by 9-EtG may be taking place during the formation of the Pt(II) form leading to an activated intermediate which forms the 9-EtG bound Pt(II) adduct, without an intermediate hydrolysed Pt(II) species. It is also possible that the formation of the 9-EtG adduct is very quick after the aquation step hence the aquated species could not be observed. It should be borne in mind however, that the chemistry of the Pt(II) complex (3) presented earlier32 does not support such a fast 9-EtG binding upon aquation hence the above prediction about the difference in the reaction pathway is made.
The binding of 9-EtG is well supported by the ESI-MS results from the same experiment, under ESI-MS conditions we also find the Pt(II) diaquated species as per the m/z peak of 462.9832 (calcd 463.0297) corresponding to [3 − 2Cl− + 2OH− + H+]+ (Fig. S19, Table S3†). Since the intensity is low for the diaquated species and we do not find it in NMR hence we feel that the small amount of diaquated species is a result of speciation arising under the ESI-MS conditions. We could find the most intense m/z at 642.0576 which matches well with the mono-guanine adduct of formulation [(1a − 3Cl−) + 9-EtG]+ (calcd 642.0657) (Fig. S19, Table S3 and isotope modelling at Fig. S20†). The H8′G singlet is in good resemblance with the singlet found in the binding experiment of 3 with 9-EtG (1:3 molar ratio) maintaining the same experimental conditions.32 The singlet at 8.57 ppm (marked as H8′′G in Fig. 3) corresponding to the scarcely found N7, O6 chelation of 9-EtG with 3 (3e, Scheme 3) was also observed but in a low amount.32,61,62 However, it must be noted that such a chelated adduct may be difficult to form under in vivo conditions when the guanine is part of a DNA strand. The small amount of the chelated adduct formation is also found in ESI-MS at a m/z of 606.0793 (calcd 606.0896) corresponding to formulation [(3 − 2Cl−) + (9-EtG − H+)]+ (Table S3 and isotope modelling in Fig. S21†). The peaks corresponding to the pyridyl protons (H6, H4 H3, H5) for both the 9-EtG adducts, 3d and 3e appeared at (8.96, 8.15, 7.71, 7.31) and (8.73, 8.23, 7.78, 7.41) ppm respectively. The aliphatic region of the binding kinetics also contains the signals corresponding to 3d & 3e, but there is a merging of signals leading to poor resolution although we could identify most of them (Fig. S22†).
The 2D NMR (HMQC) of the reaction mixture was performed at two different time points (10 min and 10 d) in order to obtain more clarity on the speciation and comment on the certainty of the signals from the bound species 3d & 3e respectively. The plots of NMR data are summarized in Fig. S23–S37 and Table S4A, S4B.† The experimental results support the predicted adducts 3d & 3e along with some other species that cannot be assigned properly. Since we used higher concentrations of 1a (15 mM) and 9-EtG (90 mM) it was possible to obtain information which we missed earlier. Herein we could find a signal of H8′G just after the mixing of 9-EtG with 1a suggesting that a small amount of the adduct is formed right after mixing (Fig. S23†). The fact that the speciation is in a very small amount is more evident since we could not obtain any 13C signal (Fig. S24†) or a correlation spectrum (Fig. S25 and S26†) for the same. We observed a downfield shift of the protons (Fig. S27 and S28†) and carbons (Fig. S29–S33†) of unreacted 9-EtG after 10 d which is not completely understood but may be due to the insufficiency of the buffer capacity of the reaction mixture.
The 195Pt NMR studies of the 9-EtG binding with 1a also provide support for the generation of predicted adducts 3d & 3e. Since the 9-EtG adduct formation is slow to generate the minimum signal to noise ratio, which is needed for the adducts to be visible, we probed the adduct formation after 10 days and found a tiny signal for the 195Pt chemical shift at ca. −2210 ppm which does not provide enough resolution to distinguish between 3d & 3e. The major species remains to be the parent complex 1a (195Pt at −74.7 ppm, Fig. S38†) displaying a higher stability for 1a as compared to 3.
To our surprise complex 2a showed faster hydrolysis but no trace of 9-EtG binding up to 2 days in 20% PBS (Fig. S39†). After 2 days most of the complex was found to be hydrolyzed, yet no new peak for H8 of 9-EtG corresponding to the binding was observed and hence, no more data were recorded. However, in the presence of plasmid DNA (pUC19) 2a showed more interaction than 1a (Fig. S40A†), suggesting that the affinity for 9-EtG may not be strong enough but it may interact with a plasmid DNA having multiple targets. It would appear that 2a should be more toxic however, the faster reactivity of 2a may prohibit its cellular uptake due to binding with other potential nucleophiles in the assay solution. Cisplatin showed more retardation of DNA at similar concentrations (Fig. S40A†). We could see that there is some amount of cleavage of DNA as per the gel diagram which may be during the reduction of the Pt(IV) to Pt(II) species in the solution. The amount of the cleavage product observed is in the order 1a > 2a. For cisplatin the cleavage product is similar to the control. The cleavage of DNA seems ROS based since when GSH is used there is no more cleavage to be found (Fig. S40B†). In addition the retardation has also decreased suggesting the interference of GSH with the DNA binding.63 However, the cytotoxicity assay performed in the presence of at least 30 equivalents of GSH showed that the decrease in cytotoxicity is not much (details in cell viability studies).
Interaction with glutathione and ascorbate
Glutathione (GSH), the cellular thiol containing tripeptide, is responsible for the drug resistance of platinum drugs64,65 and also contributes in the bio-reduction of various prodrugs including Pt66–68 and Ru69,70 drugs. Pt(IV) drugs are reduced to Pt(II) by GSH after the removal of their axially coordinated ligands. Our results also show that complex 1a loses its axial chloro group and gets transformed into the Pt(II) analogue (3) almost instantaneously upon mixing with GSH (1:6 molar ratio, monitored by 1H NMR) in 20% PBS (pD 7.4, prepared in D2O) in DMSO-d6 (Fig. 4). Careful observation suggests that the transformation could be visualized by the naked eye through fading of the yellow colour of the Pt(IV) solution rendering a faint yellow solution corresponding to the Pt(II) species (3). Each GSH molecule would contribute one electron to the reduction process. The final product of the reduction would be a Pt(II) species (3), two molecules of HCl and one molecule of oxidized glutathione (GSSG). After the reduction, the in situ generated Pt(II) complex (3) starts to form a GSH adduct at a faster pace. It is also evident from the binding experiments that the complex only forms a monoadduct of GSH (3c, Scheme 3). The chemical shifts for the GSH bound adduct are 9.22, 8.10, 7.62 and 7.46 ppm for H6, H4, H3 and H5 type protons, respectively (Fig. 4). Mass spectral evidence provides additional support to the NMR data suggesting the formation of a mono-adduct of GSH with the Pt(II) species (Fig. S41†). The corresponding m/z value of 770.0723 matches well with the species [(3 − Cl−) + (GSH − H+) + H+]+ (calcd m/z 770.0688) (Table S3 and isotope modelling at Fig. S42†). The sodium ion containing m/z corresponds to the proposed adduct where 792.0598 shows a good match with the formulations [(3 − Cl−) + (GSH − 2H+ + Na+) + H+]+ (calcd 792.0508) (Table S3 and isotope modelling at Fig. S43†). However, the ESI-MS data provide an additional peak at a m/z of 734.0967 (calcd 734.0927) which corresponds to the S, N– chelated GSH bound Pt(II) after the replacement of both the chlorido ligand of Pt(II) species, [(3 − 2Cl−) + (GSH − H+)]+ as shown in Table S3 and isotope modelling in Fig. S44.† This species is not observed in NMR and although ESI-MS conditions are softer, yet the formation of this chelated GSH species may be under ESI-MS conditions. The oxidation of GSH is also supported by the ESI-MS through the observation of a m/z of 613.1584 (calcd 613.1598) and 635.1370 (calcd 635.1417) signifying the formulation to be [GSSG + H+]+ and [GSSG + Na+]+ respectively (Table S3 and isotope modelling at Fig. S45, S46†). The GSH binding was completed at around 5 h, then the peaks corresponding to the adduct started to diminish and new peaks corresponding to the aziridinium ion of L1 emerged in the NMR plot at 8.67, 8.37 and 7.86 ppm (Fig. 4, for H6, H4 and H3 (merged with H5) protons respectively). After a day all the GSH adduct peaks disappeared as observed in our earlier work.32 Since the GSH bound species formed in situ is the same hence we attribute it to the same cause as earlier. The release of the L1 ligand is due to the high trans effect of the S-donor of coordinated GSH in the GSH adduct which labilizes the coordinated nitrogen mustard moiety L1 resulting in an aziridinium ion. Hence, complex 1a reduced to Pt(II) complex 3, then was bound to GSH and finally dissociated to form the free aziridinium ion (3b, Scheme 3). The aliphatic region of the GSH binding is too complicated as most of the peaks corresponding to 3, 3b or 3c are merged within the peaks of GSH or oxidized GSH (Fig. S47†) and hence not mentioned in the discussion. In addition the released Pt(II) might form a S bridged polymeric Pt species71 which corroborates with our earlier evidence.32 Hence, only the peaks corresponding to this aziridinium ion were visible. We did not see any peaks corresponding to the Pt species as commented on in the literature and we also do not observe any precipitate in the solution used.71 The ESI-MS data (Fig. S48†) also support the formation of the aziridinium ion exhibiting a m/z of 197.0805 (calcd 197.0845) (Table S3 and isotope modelling at Fig. S49†). In addition we observe the GSH m/z to be 308.0923 (calcd 308.0916) (isotope modelling at Fig. S50†) and its oxidized species with a m/z of 613.1584 and 635.1491 corresponding to [GSSG + H+]+ (calcd 613.1598) (isotope modelling at Fig. S51†) and [GSSG + Na+]+ (calcd 635.1417) (isotope modelling at Fig. S52†) respectively. Hence, the ESI-MS data corroborate well with the NMR observations signifying that the Pt(II) from the decomposed 3 might have formed a S bridged polymeric Pt species.
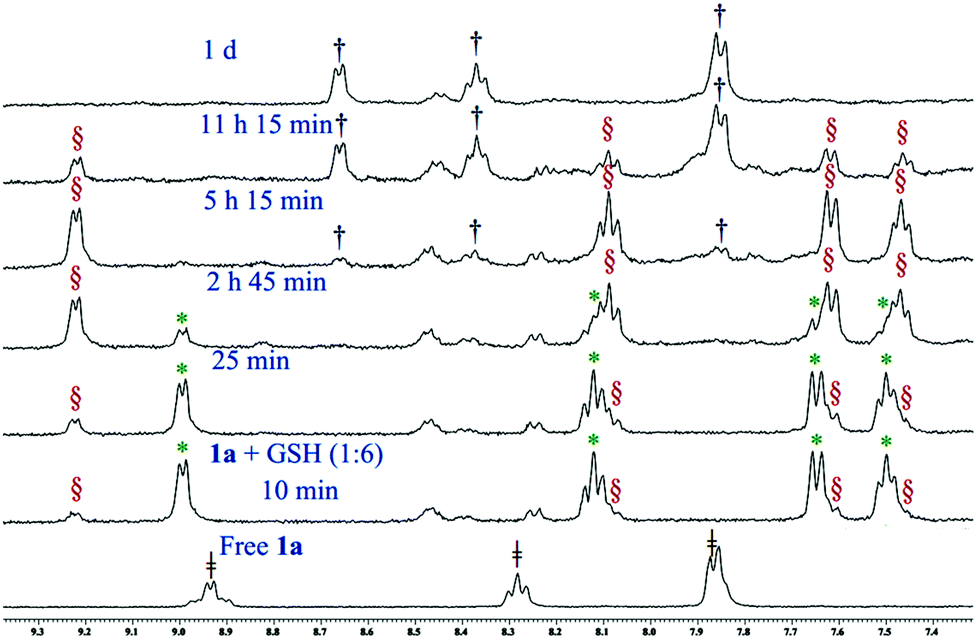 | ||
Fig. 4 Stack plot of 1H NMR data obtained by the reaction of 1a with GSH (1:6) in 20% PBS (pD 7.4, prepared in D2O) – DMSO-d6 mixture, where  , ,  , ,  and and  indicate the signals of intact complex 1a, 3, GSH bound complex 3c and aziridinium ion 3b respectively. indicate the signals of intact complex 1a, 3, GSH bound complex 3c and aziridinium ion 3b respectively. | ||
195Pt NMR spectra were also in accordance with the reduction of 1a by GSH to form 3 followed by the formation of 3c and vanishing of the NMR signal (Fig. S53†) due to the formation of polymeric species.
We have also checked the reduction reaction of complex 1a with ascorbic acid (1:6 molar ratio) by proton NMR under the same conditions as followed for GSH. We found that here too the reduction of 1a to 3 was very fast as found for GSH. We only obtained the peaks corresponding to the Pt(II) complex (3) within 10 minutes of mixing with ascorbic acid which degrades to the aziridinium ion within 10 h (Fig. S54 and S55†).
Cell viability studies
Complexes 1a & 2a were probed for in vitro cytotoxicity against various cancerous cell lines. The studies of 1a against MCF-7, A549, HepG2 & MIA PaCa-2, non-tumor (HEK293) and non-tumorigenic mouse fibroblast (NIH 3T3) cell lines under normoxic conditions showed 1a to be most toxic to MIA PaCa-2 (IC50 = 4.4 ± 0.5 μM, p < 0.05), although it is also potent with IC50 in the range of ca. 8–18 μM for MCF-7, A549, HepG2, HEK293 and NIH 3T3. The dose response of 1a in MIA PaCa-2 was seven fold better than cisplatin (IC50 = 31.8 ± 4.8 μM, p < 0.05) under our experimental conditions (Table 1).| IC50 ± SDa (μM) | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Normoxiab | Hypoxiac | |||||||||||||
| MCF-7 | MCF-7 + GSH | A549 | A549 + GSH | HepG2 | MIA PaCa-2 | HEK293 | NIH 3T3 | MCF-7 | MCF-7 + GSH | A549 | A549 + GSH | MIA PaCa-2 | MIA PaCa-2 + GSH | |
| a SD means standard deviation, IC50 values were calculated by nonlinear curve fitting in dose response inhibition – variable slope model using Graph pad prism, the data presented have significance (p value) less than 0.05 or better; Cell lines: MCF-7 (human breast adenocarcinoma), A549 (human lung carcinoma), Hep G2 (human hepatocellular carcinoma), MIA PaCa-2 (human pancreatic carcinoma), HEK293 (human embryonic kidney), NIH 3T3 (mouse embryonic fibroblast). b IC50 was determined under normoxic conditions. c IC50 was determined under hypoxic conditions (1.5% O2). d The amount of GSH used in all cases was 400 μM. e ND means not determined. f Data are taken from ref. 32. | ||||||||||||||
| 1a | 11.4 ± 2.0 | 12.9 ± 1.2d | 18.2 ± 1.0 | 24.1 ± 1.5d | 8.6 ± 0.6 | 4.4 ± 0.5 | 8.6 ± 2.3 | 8.8 ± 1.2 | 8.6 ± 0.8 | 11.2 ± 1.8d | 16.1 ± 1.1 | 26.3 ± 2.5d | 3.3 ± 0.2 | 4.5 ± 0.3d |
| 2a | >100 | >100 | >100 | >100 | NDe | NDe | NDe | NDe | >100 | >100 | >100 | >100 | NDe | NDe |
| 3 | 12.6 ± 0.8f | 16.4 ± 1.2f | 18.2 ± 1.8f | 31.2 ± 4.2f | 8.8 ± 0.3 | 4.2 ± 1.0f | 14.9 ± 0.8f | 6.8 ± 0.5 | 9.4 ± 2.2f | 12.5 ± 1.1f | 20.8 ± 1.5f | 26.6 ± 3.5f | 3.4 ± 0.2f | 4.4 ± 0.4f |
| Cisplatin | 14.1 ± 1.2f | 27.8 ± 2.1f | 22.8 ± 1.2f | 41.3 ± 1.5f | 14.3 ± 0.8 | 31.8 ± 4.8f | >50f | 7.0 ± 1.0 | 18.7 ± 1.4f | 29.0 ± 1.1f | 24.4 ± 1.1f | 39.3 ± 1.3f | 18.1 ± 2.9f | 29.7 ± 4.1f |
The in vitro cytotoxicity studies of 2a against MCF-7 and A549 showed that it is not toxic up to 100 μM. The kinetic instability may be one of the reasons for the poor cytotoxic profile of 2a, it may be decomposing fast as it encounters the culture media components and hence fails to reach the cellular target. In addition as per the NMR studies, upon hydrolysis 2a does not show any binding to the model nucleobase 9-EtG. Hence no more cytotoxicity experiments were carried out with 2a.
All the IC50 values determined for 1a were compared with the IC50 value of 3 (Table 1) in an urge to know the dependence of cytotoxicity (IC50 values) on the alteration of the oxidation state from 3 to 1a. Among the tested cell lines the highest toxicity exhibited by 1a was against MIA PaCa-2 (IC50 = 4.4 ± 0.5 μM, p < 0.05) which is comparable with the IC50 (4.2 ± 1.0 μM, p < 0.05) of 3. In HepG2 also both 1a and 3 showed similar IC50 of 8.6 ± 0.6 μM (p < 0.05) and 8.8 ± 0.3 μM (p < 0.05) respectively. Similarly in the case of MCF-7 and A549 no alterations of IC50 were found between 1a and 3. Hence it can be stated that derivatization from the kinetically labile Pt(II) to apparently kinetically inert Pt(IV) showed almost no effect in terms of the cytotoxicity profile but there was improvement in the solution stability profile under similar conditions.
The cytotoxicity pattern of 1a in HEK293 is even better than 3. It is known that HEK293 overexpresses two human copper transporting ATPases (ATP7A & ATP7B),72 which are responsible for the efflux of cisplatin.73–77 The ligand environment around 1a or its reduced form (3) might have prohibited the ligation through the cys rich region of ATP7A & ATP7B due to the enhanced steric factor rendering better activity against that particular cell line. Earlier also we have observed a higher cytotoxicity of the Pt(II) analogue (3, IC50 = 14.9 ± 0.8 μM, p < 0.01) but the activity of 1a is better (IC50 = 8.6 ± 2.3 μM, p < 0.05). This might be because the higher solution stability of 1a is due to its relatively kinetically inert nature which made the deactivation further difficult.
The cytotoxicity profile of 1a when compared in non-tumorigenic mouse fibroblast (NIH 3T3) vs. MIA PaCa-2 showed that the IC50 in NIH 3T3 needs double the dosage than that against MIA PaCa-2. This is encouraging, although it should be still better, for a good anticancer agent. In addition it must be noted that the in vitro cytotoxicity of the clinical drug cisplatin in NIH 3T3 is also similar to that of 1a (Table 1) although 1a is more efficient in MIA PaCa-2.
Although the above data are encouraging the indifference of the effect of the kinetic inertness found in the in vitro activity led us to probe the effect of the cytotoxicity of 1a in the presence of GSH. NMR studies showed that 1a gets almost instantly converted to its Pt(II) analogue (3) in the presence of GSH and also binds with GSH. Hence it seemed that detoxification of 1a by GSH is inevitable. To evaluate this we have determined the IC50 of 1a in the presence of an excess amount of GSH (400 μM) against MCF-7 and A549 under normoxic conditions. In both the cell lines (MCF-7 and A549) the IC50 of 1a showed an increase in the dosage amount but the activity was still better than that observed for 3 or cisplatin (Table 1). The above results suggest that 1a may be converted to 3 but the reactivity towards GSH may not follow the same trend as found in the NMR experiment.
We have also studied the cytotoxicity of 1a under hypoxic conditions against MCF-7, A549 and MIA PaCa-2 in the presence and absence of excess GSH. The IC50 value in the absence of excess added GSH showed that the cytotoxic activity of 1a stays similar or is better than normoxic conditions (Table 1) signifying that the activity of 1a inside a tumor may not be hindered due to hypoxia. On addition of excess GSH (400 μM) compared to the respective complexes we find that the activity of 1a worsens but the dosage is still similar to the normoxic IC50 values without any added GSH especially for the MIA PaCa-2 and MCF-7 cell lines (Table 1). The activity is comparatively poorer for the A549 cell line which shows that the compound is not active against all cancer cell lines. Cisplatin shows much worsening of the activity against A549, compared to 1a (Table 1) under similar conditions. In MIA PaCa-2 although the activity of cisplatin in hypoxia is better yet a ca. 6 times higher dosage than 1a is required.
Cellular internalization and mechanistic studies
The accumulation study of the metal in the cancer cells is known to provide insight about the internalization of a given complex.78 We used the ICP-MS technique to quantify the amount of Pt in the cancer cells after 24 h of exposure with equal concentrations (9 μM) of each complex. 1a shows a higher uptake based on the Pt content inside MCF-7 cells, compared to 2a (Fig. 5). The platinum accumulation of 2a is significantly less than even cisplatin or the reduced Pt(II) complex 3. This is in good agreement with the lower IC50 values of 1a. The slow hydrolysis of 1a helps to keep it neutral and hence renders a greater lipophilic character which might be acting in favour of greater internalization.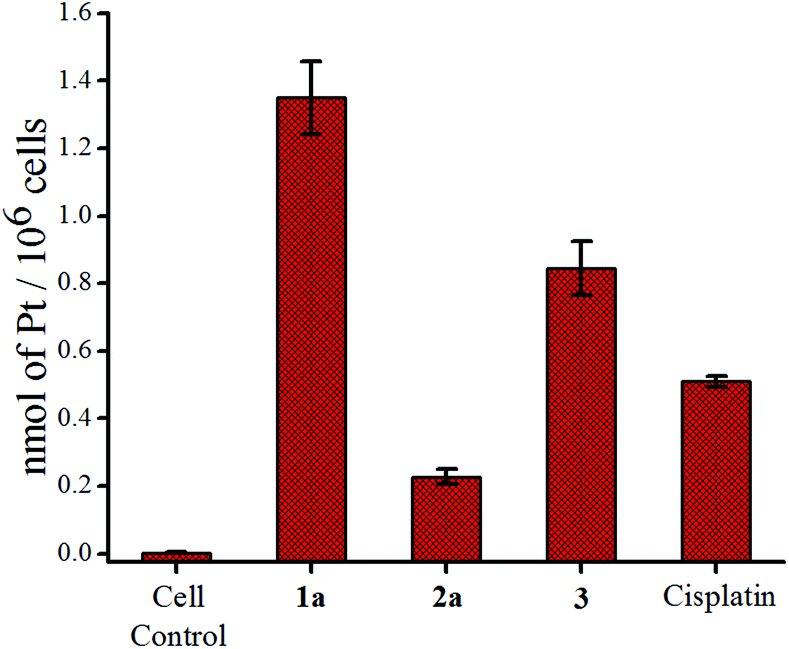 | ||
| Fig. 5 Accumulation of platinum in MCF-7 cells after treatment with 9 μM of 1a, 2a, 3 and cisplatin respectively. | ||
Complex 1a was probed to gain insight about its effect on the cell cycle. Flow cytometry data show that 1a arrests the MCF-7 cell cycle most prominently in the G2/M phase after 24 h exposure of 1a, which is observed by an increase of the population in this phase with an increasing concentration of 1a w.r.t. untreated control cells (Fig. 6 & S60†). The G2/M phase arrest was also found for MIA PaCa-2 but the effect was less pronounced in MIA PaCa-2 compared to MCF-7 (Fig. 6 & S61†).
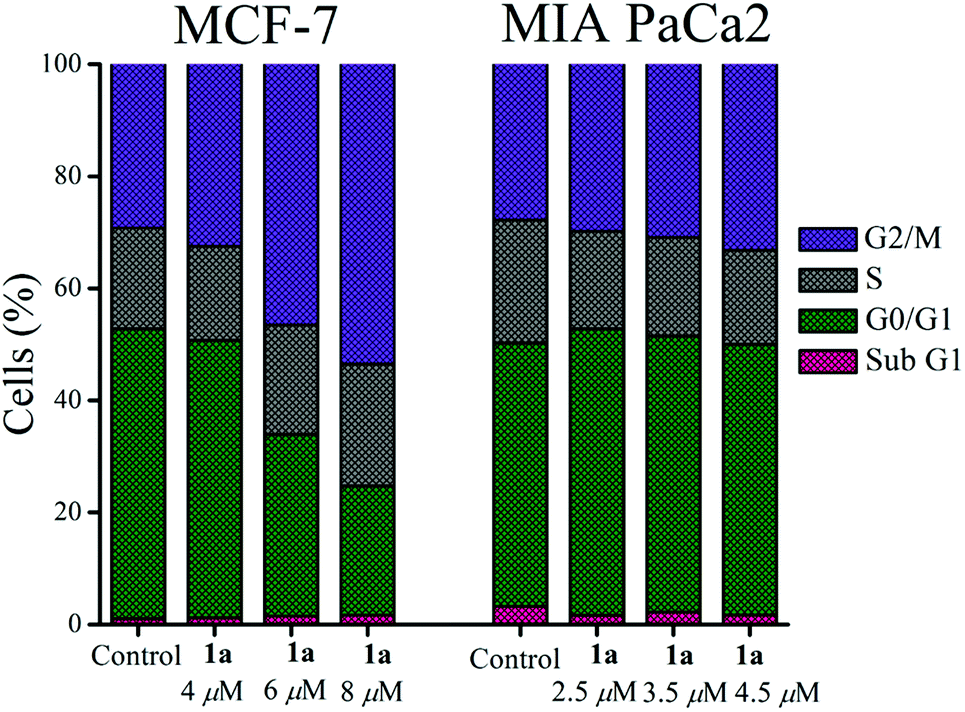 | ||
| Fig. 6 Cell cycle arrest by 1a showing G2/M phase arrest both in MCF-7 and MIA PaCa-2. | ||
The efficiency of 1a led us to probe if the killing pathway is apoptotic in nature. DNA extracted from MCF-7 cells treated with sub IC50 concentrations of 1a (6 μM, 8 μM and 10 μM respectively) for 24 h showed a ladder pattern in agarose gel electrophoresis, while intact DNA was observed for the untreated cells (Fig. 7). The above results suggest that 1a kills through apoptosis since inter-nucleosomal DNA fragmentation79,80 with sections of around 180 bp of endonucleases is a hallmark of apoptosis.81–83
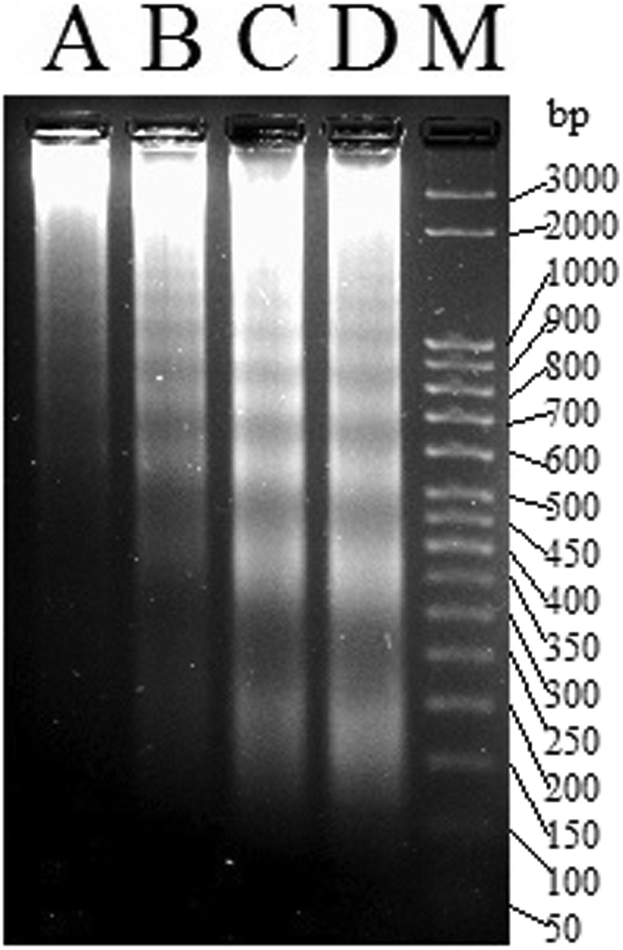 | ||
| Fig. 7 Gel image of the DNA ladder formation of MCF-7 treated with (A) DMSO, (B) 6 μM of 1a, (C) 8 μM of 1a and (D) 10 μM of 1a respectively over 24 h. M denotes the known bp ladder. | ||
The apoptotic pathway was further strengthened by probing the change in mitochondrial transmembrane potential and caspase activation as described below. Among the two major pathways of apoptosis, the intrinsic mechanism is regulated via mitochondria.84,85 In this event the mitochondrial membrane potential (ΔΨm) disintegrates.86 We could observe a significant change in the mitochondrial membrane potential by 1a, using the mitochondria specific fluorescent dye JC-1. The JC-1 dye emits red fluorescence (λex = 575 nm, λem = 590 nm) when the mitochondrial membrane is intact with the usual ΔΨm (since JC-1 remains in aggregates) and when the membrane potential is no longer intact it emits green fluorescence (λex = 510 nm, λem = 550 nm) due to the monomeric form.87 Therefore, a decrease in the ratio of the red/green fluorescence intensity of JC-1 reflects depolarization of the mitochondrial membrane potential.88 The strong depolarization of the mitochondrial membrane potential by 1a shows concentration dependence (Fig. 8) with respect to untreated MCF-7 cells determined by flow cytometry. This may be taken as an indication that 1a possibly goes through an intrinsic pathway which involves depolarization of the mitochondrial membrane potential.
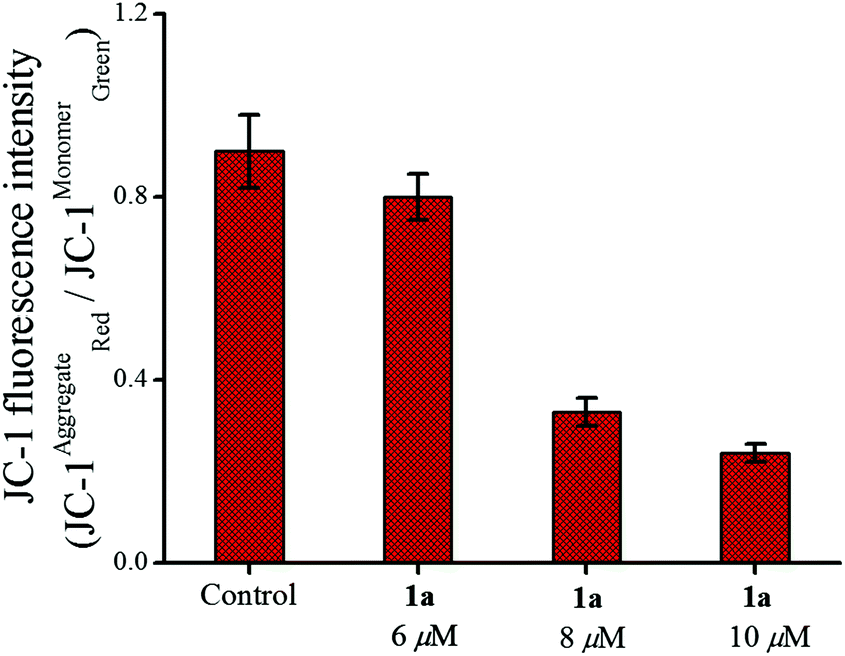 | ||
| Fig. 8 Change in the mitochondrial membrane potential of MCF-7 cells after treatment with 1a over 24 h showing the effect of increasing concentration. | ||
The release of cytochrome c into the cytoplasm is triggered by the depolarization of the mitochondrial membrane potential which in turn activates initiator (e.g. caspase-9) and effector (e.g. caspase-7) caspases leading to cell death after proteolytic cleavage of a wide range of cellular targets.89,90 Our initial studies in this area show that caspase-7 (cleaves Ac-DEVD-pNA substrate)91 was activated in MCF-7 by 24 h of treatment with 1a (Fig. 9) and this activation also showed concentration dependence as found from the plot of the pNA released (nmol min−1 ml−1) vs. the concentration used.
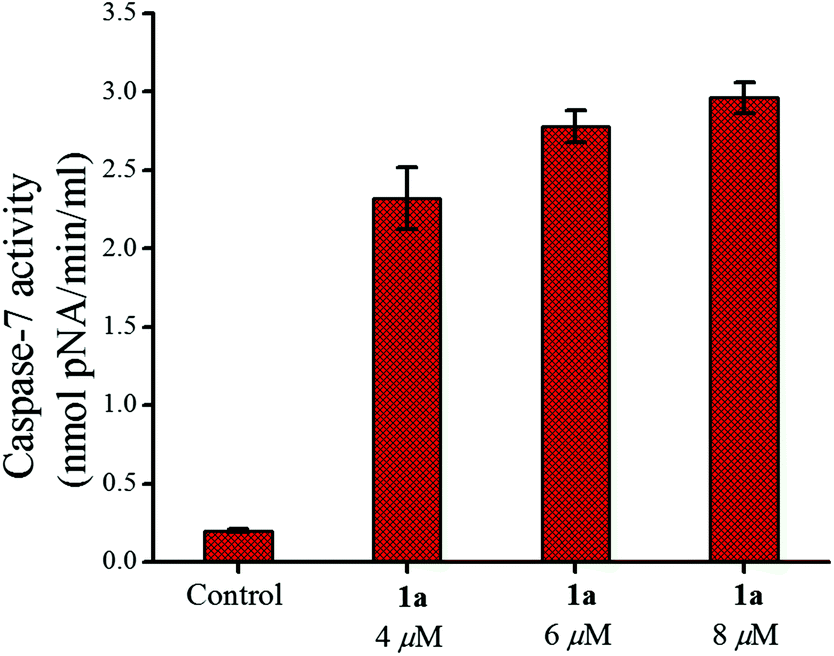 | ||
| Fig. 9 Caspase-7 activity in MCF-7 cells after treatment with 1a over 24 h showing the enhancement of activity with an increase in concentration. | ||
One more characteristic of apoptosis is causing cellular morphological changes that include cell shrinkage, plasma membrane blebbing and chromatin condensation.92 The above signatures of apoptosis were also observed for the 1a treated MCF-7 cells (Fig. S62†) using DAPI staining while the cells from the control experiment remained intact.
The above studies including DNA ladder formation, depolarization of the mitochondrial membrane potential, caspase activation and nuclear morphological changes strongly suggest that the cells are killed through an apoptotic pathway by 1a.
Growth kinetics of MCF-7 under the influence of 1a
We had a quest as chemists that what is the fate of the cells after removal of the anticancer agents. We were curious and relevant data appeared to be scarce.93 We incubated the MCF-7 cells with 1a for 48 h. The results showed that the cells ceased to grow, even after removal of 1a and the effect was observed until 48 h (Fig. 10). The clinical drug cisplatin also showed similar behaviour as 1a in this experiment. Hence, the decrease in viability is encouraging and indicative that compound 1a imposes a permanent damage in the cancer cells ceasing its growth even after the drug-removal.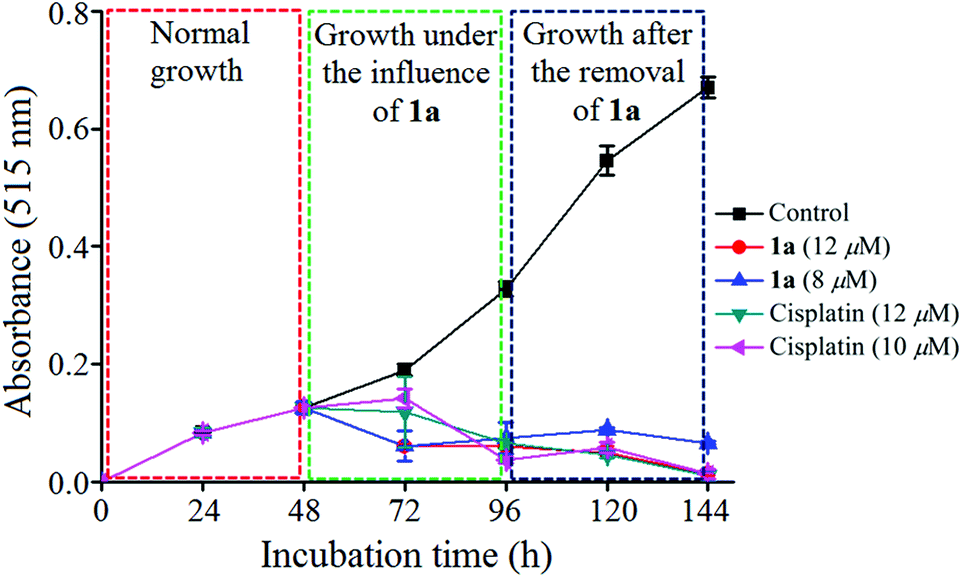 | ||
| Fig. 10 Plot of growth kinetics of MCF-7 under the influence of 1a determined by MTT assay. | ||
Wound assay (migration)
Migration is an essential property exhibited by cells in order to safeguard tissue functions, wound healing and related processes. It also plays a predominant role in several diseases, especially in cancer, leading to invasion and metastasis.94 Therefore the effect of 1a on cancer cell migration in the MCF-7 cell line was assessed using the scratch wound healing assay. From Fig. 11 it can be seen that the untreated control cells rapidly migrate over time due to the metastatic property resulting in almost full healing in 36 h. Whereas in the case of 1a added cells the wide denuded area remained mostly unhealed, indicating the anti-proliferative nature of the cells, attained upon treatment with 1a.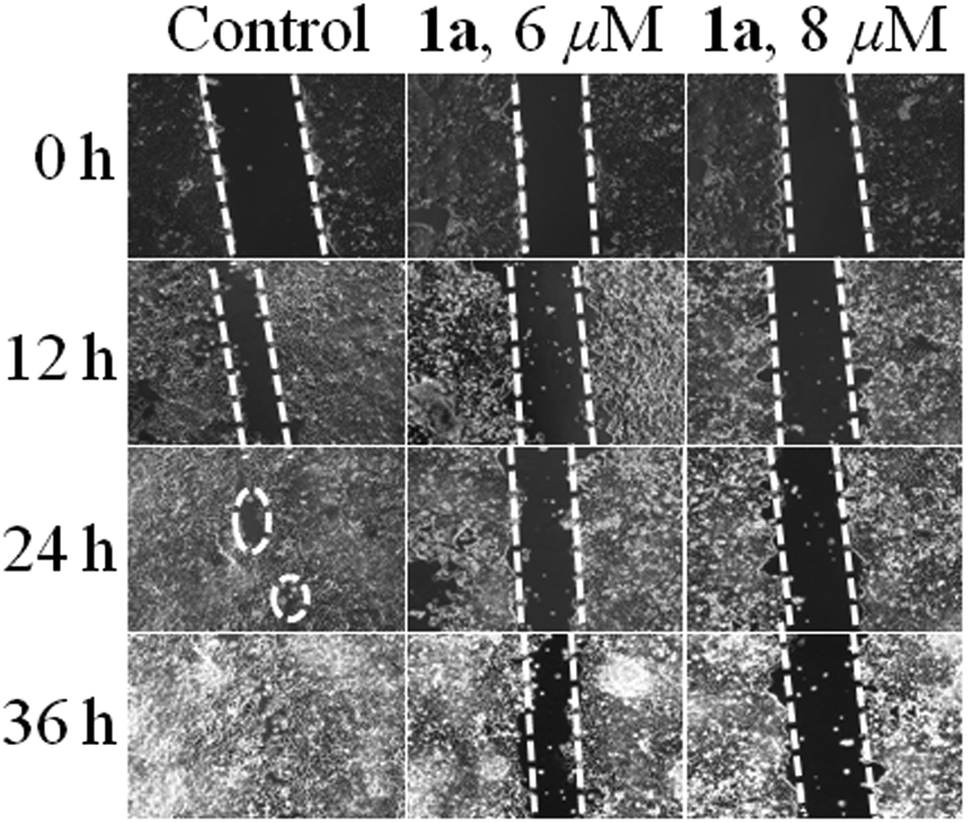 | ||
| Fig. 11 Wound healing assay in MCF-7 cells where untreated cells move rapidly towards the scratched area with an increase in time as compared to cells treated with 6 & 8 μM of 1a respectively. | ||
In addition to the above 1a was found to have a very much tolerant level for hemolysis95–97 as found from its interaction with blood components particularly RBCs (using 15 μM of 1a, hemolysis was <2.1%, Fig. S63 & Table S5†). Hence, complex 1a appears to be a blood compatible, cytotoxic and anti-metastatic anticancer agent.
Conclusions
In a single step the Pt(II) to Pt(IV) conversion was achieved along with the generation of nitrogen mustards from the coordinated ethanolamine/diethanolamine moiety. The direct coordination of the nitrogen mustard to kinetically inert Pt(IV), is scarce, and very much increases the solution stability compared to the Pt(II) analogue. The bis(2-chloroethyl)amine moiety in 1a ensures the provision of the required electronic factors which allow binding to 9-EtG, higher cytotoxicity and solution stability compared to 2a, which has the 2-chloroethylamine moiety. The dose effectiveness of 1a was detected even in the presence of excess GSH, unlike cisplatin. A prominent G2/M phase arrest by complex 1a is noticed in two different cell lines, MCF-7 and MIA PaCa-2. The in vitro data against MCF-7 also suggest that 1a induces the loss of the mitochondrial membrane potential and caspase-7 activation leading to apoptosis. In addition an initial study on the anti-metastatic properties of 1a shows a positive effect, which confirms it to be a potential candidate as an anticancer agent. We have gained a better understanding of the chelating nitrogen mustard bearing Pt(II/IV) complexes, revealing appreciable in vitro cytotoxicity profiles, which are getting fine tuned with our progress.Experimental section
Materials and methods
The solvents and chemicals used were purchased from various authentic commercial sources. 9-EtG and GSH were purchased from Carbosynth and used as received. Analytical grade solvents were purchased from Merck India and distilled prior to use. Spectroscopy grade solvents (Spectrochem) were used in spectroscopic studies. 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT), 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) and penicillin–streptomycin were purchased from USB and Hyclone respectively. Dulbecco's Modified Eagle's Medium (DMEM), Minimum Essential Medium (MEM) and Phosphate-Buffered Saline (PBS) were purchased from Invitrogen. 5,5′,6,6′-Tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide (JC-1) and propidium iodide (PI) were purchased from Sigma-Aldrich. UV–visible data were recorded using a Perkin Elmer lambda 35 spectrophotometer. FT-IR spectra were recorded using a Perkin-Elmer SPECTRUM RX I spectrometer in KBr pellets. 1H & 13C (proton decoupled) and 195Pt (proton coupled) NMR spectra of the complexes were recorded using a Bruker Avance III 500 MHz spectrometer at room temperature. All chemical shifts are reported in parts per million (ppm). 195Pt NMR chemical shifts are described against the standard K2[PtCl4] for which the chemical shift in D2O is at −1628.0 ppm. Elemental analyses reported were performed using a Perkin-Elmer 2400 series II CHNS/O analyzer. Electrospray ionization mass spectra (ESI-MS) were performed using a Micromass Q-Tof micro™ (Waters) by +ve mode electrospray ionization. The synthetic yields reported are of isolated analytically pure compounds.The full details of the experimental procedures followed are given in the ESI.†
Statistical analysis
All data were given as the mean of independent experiments. The statistical analyses were analyzed using Graph pad prism® software 5.0 with Student's t-test.Acknowledgements
This research work was financially supported by the Department of Science and Technology, vide project no. SB/S1/IC-02/2014. We also sincerely acknowledge IISER Kolkata for financial and infrastructural support including NMR, single crystal XRD, ESI-MS, ICP-MS, FACS and fluorescence microscope. SK & KP thank UGC-India and SC thanks IISER Kolkata for research fellowships.Notes and references
- B. Rosenberg, L. Van Camp and T. Krigas, Nature, 1965, 205, 698–699 CrossRef CAS PubMed.
- B. Rosenberg, L. VanCamp, J. E. Trosko and V. H. Mansour, Nature, 1969, 222, 385–386 CrossRef CAS PubMed.
- B. Rosenberg and L. VanCamp, Cancer Res., 1970, 30, 1799–1802 CAS.
- C. A. Rabik and M. E. Dolan, Cancer Treat. Rev., 2007, 33, 9–23 CrossRef CAS PubMed.
- P. J. O'Dwyer, J. P. Stevenson and S. W. Johnson, Cisplatin, 1999, 31–69 Search PubMed.
- M. A. Jakupec, M. Galanski and B. K. Keppler, Rev. Physiol., Biochem. Pharmacol., 2003, 146, 1–53 CAS.
- L. Kelland, Nat. Rev. Cancer, 2007, 7, 573–584 CrossRef CAS PubMed.
- N. Pabla and Z. Dong, Kidney Int., 2008, 73, 994–1007 CrossRef CAS PubMed.
- P. J. O'Dwyer, J. P. Stevenson and S. W. Johnson, Drugs, 2000, 59, 19–27 CrossRef.
- S. R. McWhinney, R. M. Goldberg and H. L. McLeod, Mol. Cancer Ther., 2009, 8, 10–16 CrossRef CAS PubMed.
- J. H. Van Den Berg, J. H. Beijnen, A. J. M. Balm and J. H. M. Schellens, Cancer Treat. Rev., 2006, 32, 390–397 CrossRef CAS PubMed.
- L. R. Kelland, Drugs, 2000, 59, 1–8 CrossRef CAS PubMed.
- P. Heffeter, U. Jungwirth, M. Jakupec, C. Hartinger, M. Galanski, L. Elbling, M. Micksche, B. Keppler and W. Berger, Drug Resist. Updates, 2008, 11, 1–16 CrossRef CAS PubMed.
- B. Koeberle, M. T. Tomicic, S. Usanova and B. Kaina, Biochim. Biophys. Acta, Rev. Cancer, 2010, 1806, 172–182 CrossRef CAS PubMed.
- N. J. Wheate, S. Walker, G. E. Craig and R. Oun, Dalton Trans., 2010, 39, 8113–8127 RSC.
- B. W. Harper, A. M. Krause-Heuer, M. P. Grant, M. Manohar, K. B. Garbutcheon-Singh and J. R. Aldrich-Wright, Chem. – Eur. J., 2010, 16, 7064–7077 CrossRef CAS PubMed.
- A. Eastman, Cancer Cells, 1990, 2, 275–280 CAS.
- Z. H. Siddik, Oncogene, 2003, 22, 7265–7279 CrossRef CAS PubMed.
- M. D. Hall and T. W. Hambley, Coord. Chem. Rev., 2002, 232, 49–67 CrossRef CAS.
- N. Graf and S. J. Lippard, Adv. Drug Delivery Rev., 2012, 64, 993–1004 CrossRef CAS PubMed.
- E. E. Blatter, J. F. Vollano, B. S. Krishnan and J. C. Dabrowiak, Biochemistry, 1984, 23, 4817–4820 CrossRef CAS PubMed.
- O. Novakova, O. Vrana, V. I. Kiseleva and V. Brabec, Eur. J. Biochem., 1995, 228, 616–624 CrossRef CAS PubMed.
- M. Galanski and B. K. Keppler, Anti-Cancer Agents Med. Chem., 2007, 7, 55–73 CrossRef CAS PubMed.
- W. H. Ang, S. Pilet, R. Scopelliti, F. Bussy, L. Juillerat-Jeanneret and P. J. Dyson, J. Med. Chem., 2005, 48, 8060–8069 CrossRef CAS PubMed.
- M. D. Hall, H. R. Mellor, R. Callaghan and T. W. Hambley, J. Med. Chem., 2007, 50, 3403–3411 CrossRef CAS PubMed.
- T. C. Johnstone, K. Suntharalingam and S. J. Lippard, Chem. Rev., 2016, 116, 3436–3486 CrossRef CAS PubMed.
- V. Pichler, P. Heffeter, S. M. Valiahdi, C. R. Kowol, A. Egger, W. Berger, M. A. Jakupec, M. Galanski and B. K. Keppler, J. Med. Chem., 2012, 55, 11052–11061 CrossRef CAS PubMed.
- H. S. Oberoi, N. V. Nukolova, A. V. Kabanov and T. K. Bronich, Adv. Drug Delivery Rev., 2013, 65, 1667–1685 CrossRef CAS PubMed.
- X. Han, J. Sun, Y. Wang and Z. He, Med. Res. Rev., 2015, 35, 1268–1299 CrossRef CAS PubMed.
- G. Apps Michael, H. Y. Choi Eugene and J. Wheate Nial, Endocr.-Relat. Cancer, 2015, 22, R219–R233 CAS.
- S. Choi, C. Filotto, M. Bisanzo, S. Delaney, D. Lagasee, J. L. Whitworth, A. Jusko, C. Li, N. A. Wood, J. Willingham, A. Schwenker and K. Spaulding, Inorg. Chem., 1998, 37, 2500–2504 CrossRef CAS.
- S. Karmakar, K. Purkait, S. Chatterjee and A. Mukherjee, Dalton Trans., 2016, 45, 3599–3615 RSC.
- B. A. Chabner and T. G. Roberts, Nat. Rev. Cancer, 2005, 5, 65–72 CrossRef CAS PubMed.
- S. R. Rajski and R. M. Williams, Chem. Rev., 1998, 98, 2723–2795 CrossRef CAS PubMed.
- D. M. Noll, A. M. Noronha, C. J. Wilds and P. S. Miller, Front. Biosci., 2004, 9, 421–437 CrossRef CAS.
- D. C. Ware, P. J. Brothers, G. R. Clark, W. A. Denny, B. D. Palmer and W. R. Wilson, Dalton Trans., 2000, 925–932 RSC.
- D. C. Ware, W. A. Denny and G. R. Clark, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1997, C53, 1058–1059 CAS.
- D. C. Ware, B. D. Palmer, W. R. Wilson and W. A. Denny, J. Med. Chem., 1993, 36, 1839–1846 CrossRef CAS PubMed.
- D. C. Ware, H. R. Palmer, F. B. Bruijn, R. F. Anderson, P. J. Brothers, W. A. Denny and W. R. Wilson, Anti-Cancer Drug Des., 1998, 13, 81–103 CAS.
- D. C. Ware, B. G. Siim, K. G. Robinson, W. A. Denny, P. J. Brothers and G. R. Clark, Inorg. Chem., 1991, 30, 3750–3757 CrossRef CAS.
- D. C. Ware, W. R. Wilson, W. A. Denny and C. E. F. Richard, J. Chem. Soc., Chem. Commun., 1991, 1171–1173, 10.1039/c39910001171.
- P. R. Craig, P. J. Brothers, G. R. Clark, W. R. Wilson, W. A. Denny and D. C. Ware, Dalton Trans., 2004, 611–618, 10.1039/b311091e.
- B. A. Teicher, M. J. Abrams, K. W. Rosbe and T. S. Herman, Cancer Res., 1990, 50, 6971–6975 CAS.
- S. V. Yakovlev, O. M. Nozdrina and V. B. Ukraintsev, Zh. Obshch. Khim., 1992, 62, 967–971 CAS.
- S. V. Yakovlev, V. B. Ukraintsev, O. M. Nozdrina and Y. N. Kukushkin, Zh. Obshch. Khim., 1990, 60, 1321–1325 CAS.
- J. J. Wilson and S. J. Lippard, Chem. Rev., 2014, 114, 4470–4495 CrossRef CAS PubMed.
- V. Y. Kukushkin, S. V. Yakovlev and V. B. Ukraintsev, Koord. Khim., 1988, 14, 969–971 CAS.
- G. Schmid and G. Ritter, Z. Anorg. Allg. Chem., 1975, 415, 97–103 CrossRef CAS.
- J. Barry, J. G. H. Du Preez, E. Els, H. E. Rohwer and P. J. Wright, Inorg. Chim. Acta, 1981, 53, L17–L18 CrossRef CAS.
- H. Schmidbaur and P. Jandik, Inorg. Chim. Acta, 1983, 74, 97–99 CrossRef CAS.
- W. H. Beattie, W. B. Maier II and R. F. Holland, J. Inorg. Nucl. Chem., 1978, 40, 1885–1887 CrossRef CAS.
- F. Bottomley, J. Darkwa, L. Sutin and P. S. White, Organometallics, 1986, 5, 2165–2171 CrossRef CAS.
- H. G. Alt, T. Frister, E. E. Trapl and H. E. Engelhardt, J. Organomet. Chem., 1989, 362, 125–134 CrossRef CAS.
- J. A. Broomhead, J. Am. Chem. Soc., 1968, 90, 4480–4482 CrossRef CAS.
- F. S. Mackay, J. A. Woods, P. Heringova, J. Kasparkova, A. M. Pizarro, S. A. Moggach, S. Parsons, V. Brabec and P. J. Sadler, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20743–20748 CrossRef CAS PubMed.
- F. S. Mackay, S. A. Moggach, A. Collins, S. Parsons and P. J. Sadler, Inorg. Chim. Acta, 2009, 362, 811–819 CrossRef CAS.
- L. Cubo, T. W. Hambley, P. J. Sanz Miguel, A. Carnero, C. Navarro-Ranninger and A. G. Quiroga, Dalton Trans., 2011, 40, 344–347 RSC.
- J. Reedijk, Chem. Commun., 1996, 801–806, 10.1039/cc9960000801.
- R. M. Roat and J. Reedijk, J. Inorg. Biochem., 1993, 52, 263–274 CrossRef CAS.
- E. G. Talman, Y. Kidani, L. Mohrmann and J. Reedijk, Inorg. Chim. Acta, 1998, 283, 251–255 CrossRef CAS.
- J. Lorberth, M. El-Essawi, W. Massa and L. Labib, Angew. Chem., Int. Ed. Engl., 1988, 100, 1194–1195 CAS.
- U. Warnke, C. Rappel, H. Meier, C. Kloft, M. Galanski, C. G. Hartinger, B. K. Keppler and U. Jaehde, ChemBioChem, 2004, 5, 1543–1549 CrossRef CAS PubMed.
- M. A. Medrano, A. Alvarez-Valdes, J. Perles, J. Lloret-Fillol, S. Munoz-Galvan, A. Carnero, C. Navarro-Ranninger and A. G. Quiroga, Chem. Commun., 2013, 49, 4806–4808 RSC.
- J. Reedijk, Chem. Rev., 1999, 99, 2499–2510 CrossRef CAS PubMed.
- M. A. Fuertes, C. Alonso and J. M. Perez, Chem. Rev., 2003, 103, 645–662 CrossRef CAS PubMed.
- G. R. Gibbons, S. Wyrick and S. G. Chaney, Cancer Res., 1989, 49, 1402–1407 CAS.
- A. Eastman, Biochem. Pharmacol., 1987, 36, 4177–4178 CrossRef CAS PubMed.
- Y. Kido, A. R. Khokhar and Z. H. Siddik, Biochem. Pharmacol., 1994, 47, 1635–1642 CrossRef CAS PubMed.
- A. Levina, A. Mitra and P. A. Lay, Metallomics, 2009, 1, 458–470 RSC.
- R. Trondl, P. Heffeter, C. R. Kowol, M. A. Jakupec, W. Berger and B. K. Keppler, Chem. Sci., 2014, 5, 2925–2932 RSC.
- S. J. Berners-Price and P. W. Kuchel, J. Inorg. Biochem., 1990, 38, 305–326 CrossRef CAS.
- N. Barnes, M. Y. Bartee, L. Braiterman, A. Gupta, V. Ustiyan, V. Zuzel, J. H. Kaplan, A. L. Hubbard and S. Lutsenko, Traffic, 2009, 10, 767–779 CrossRef CAS PubMed.
- M. Komatsu, T. Sumizawa, M. Mutoh, Z.-S. Chen, K. Terada, T. Furukawa, X.-L. Yang, H. Gao, N. Miura, T. Sugiyama and S.-I. Akiyama, Cancer Res., 2000, 60, 1312–1316 CAS.
- G. Samimi, R. Safaei, K. Katano, A. K. Holzer, M. Rochdi, M. Tomioka, M. Goodman and S. B. Howell, Clin. Cancer Res., 2004, 10, 4661–4669 CrossRef CAS PubMed.
- N. V. Dolgova, D. Olson, S. Lutsenko and O. Y. Dmitriev, Biochem. J., 2009, 419, 51–56 CrossRef CAS PubMed.
- O. Y. Dmitriev, Biochem. Cell Biol., 2011, 89, 138–147 CrossRef CAS PubMed.
- V. Calandrini, F. Arnesano, A. Galliani, T. H. Nguyen, E. Ippoliti, P. Carloni and G. Natile, Dalton Trans., 2014, 43, 12085–12094 RSC.
- Z. Liu, A. Habtemariam, A. M. Pizarro, S. A. Fletcher, A. Kisova, O. Vrana, L. Salassa, P. C. A. Bruijnincx, G. J. Clarkson, V. Brabec and P. J. Sadler, J. Med. Chem., 2011, 54, 3011–3026 CrossRef CAS PubMed.
- A. H. Wyllie, J. F. Kerr and A. R. Currie, Int. Rev. Cytol., 1980, 68, 251–306 CAS.
- M. J. Arends, R. G. Morris and A. H. Wyllie, Am. J. Pathol., 1990, 136, 593–608 CAS.
- A. H. Wyllie, Nature, 1980, 284, 555–556 CrossRef CAS PubMed.
- M. Enari, H. Sakahira, H. Yokoyama, K. Okawa, A. Iwamatsu and S. Nagata, Nature, 1998, 391, 43–50 CrossRef CAS PubMed.
- P. T. Daniel, I. Sturm, S. Ritschel, K. Friedrich, B. Dorken, P. Bendzko and T. Hillebrand, Anal. Biochem., 1999, 266, 110–115 CrossRef CAS PubMed.
- S. Nagata, Cell, 1997, 88, 355–365 CrossRef CAS PubMed.
- D. R. Green and J. C. Reed, Science, 1998, 281, 1309–1312 CrossRef CAS PubMed.
- D. R. Green and G. Kroemer, Science, 2004, 305, 626–629 CrossRef CAS PubMed.
- G. Kroemer and J. C. Reed, Nat. Med., 2000, 6, 513–519 CrossRef CAS PubMed.
- S. T. Smiley, M. Reers, C. Mottola-Hartshorn, M. Lin, A. Chen, T. W. Smith, G. D. Steele Jr. and L. B. Chen, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 3671–3675 CrossRef CAS.
- E. A. Slee, M. T. Harte, R. M. Kluck, B. B. Wolf, C. A. Casiano, D. D. Newmeyer, H.-G. Wang, J. C. Reed, D. W. Nicholson, E. S. Alnemri, D. R. Green and S. J. Martin, J. Cell Biol., 1999, 144, 281–292 CrossRef CAS PubMed.
- M. O. Hengartner, Nature, 2000, 407, 770–776 CrossRef CAS PubMed.
- N. A. Thornberry, T. A. Rano, E. P. Peterson, D. M. Rasper, T. Timkey, M. Garcia-Calvo, V. M. Houtzager, P. A. Nordstrom, S. Roy, J. P. Vaillancourt, K. T. Chapman and D. W. Nicholson, J. Biol. Chem., 1997, 272, 17907–17911 CrossRef CAS PubMed.
- J. F. Kerr, A. H. Wyllie and A. R. Currie, Br. J. Cancer, 1972, 26, 239–257 CrossRef CAS PubMed.
- C. M. Seynaeve, M. Stetler-Stevenson, S. Sebers, G. Kaur, E. A. Sausville and P. J. Worland, Cancer Res., 1993, 53, 2081–2086 CAS.
- P. Friedl and K. Wolf, Nat. Rev. Cancer, 2003, 3, 362–374 CrossRef CAS PubMed.
- B. Naeye, H. Deschout, M. Roding, M. Rudemo, J. Delanghe, K. Devreese, J. Demeester, K. Braeckmans, S. C. De Smedt and K. Raemdonck, Biomaterials, 2011, 32, 9120–9127 CrossRef CAS PubMed.
- H.-W. An, S.-L. Qiao, C.-Y. Hou, Y.-X. Lin, L.-L. Li, H.-Y. Xie, Y. Wang, L. Wang and H. Wang, Chem. Commun., 2015, 51, 13488–13491 RSC.
- T.-Y. Liu, W. M. Hussein, A. K. Giddam, Z. Jia, J. M. Reiman, M. Zaman, N. A. J. McMillan, M. F. Good, M. J. Monteiro, I. Toth and M. Skwarczynski, J. Med. Chem., 2015, 58, 888–896 CrossRef CAS PubMed.
- A. Sarkar, S. Bhattacharyya, S. K. Dey, S. Karmakar and A. Mukherjee, New J. Chem., 2014, 38, 817–826 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional crystallographic data, NMR data of synthesized compounds, NMR & ESI-MS data of stability and binding kinetics, tables containing ESI-MS speciation, isotope modelling of ESI-MS speciation, plots of MTT assay, cell cycle arrest histograms, fluorescence microscopy image, data of hemolysis assay, detailed experimental procedures. CCDC 1449897. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt00831c |
| This journal is © The Royal Society of Chemistry 2016 |