
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Coordination chemistry of a calix[4]arene-based NHC ligand: dinuclear complexes and comparison to IiPr2Me2†
Ruth
Patchett
and
Adrian B.
Chaplin
 *
*
Department of Chemistry, University of Warwick, Gibbet Hill Road, Coventry CV4 7AL, UK. E-mail: a.b.chaplin@warwick.ac.uk
First published on 10th May 2016
Abstract
The preparation and coordination chemistry of 5,17-bis(3-methyl-1-imidazol-2-ylidene)-25,26,27,28-tetrapropoxycalix[4]arene (1) is described. Starting from the bis(imidazolium) pro-ligand 1·2HI, the free carbene 1 was readily generated in solution through deprotonation using K[OtBu] and its reactivity with rhodium(I) dimers [Rh(COD)Cl]2 (COD = 1,5-cyclooctadiene) and [Rh(CO)2Cl]2 investigated. Dinuclear complexes were isolated in both cases, where the calix[4]arene-based NHC ligand adopts a bridging μ2-coordination mode, and in one case characterised in the solid-state by X-ray diffraction. Using instead an isolated and well-defined (mononuclear) silver transfer agent, generated by reaction of 1·2HI with Ag2O in the presence of a halide extractor, reactions with [Rh(COD)Cl]2 and [Rh(CO)2Cl]2 produced cationic dinuclear complexes bearing μ2-1 and μ2-Cl bridging ligands. The structural formulation of the novel dinuclear adducts of 1 was aided through spectroscopic congruence with model complexes, containing monodentate 1,3-diisopropyl-4,5-dimethylimidazol-2-ylidene (IiPr2Me2).
Introduction
In addition to desirable donor properties, the structural versatility of N-heterocyclic carbenes (NHCs) has proven to be a major contributing factor in the widespread application of these ligands throughout organometallic chemistry and catalysis.1 The root of this diversity lies in the numerous and generally straightforward preparative procedures available for the construction of the respective NHC pro-ligands; synthetic methodologies that have enabled access not only a plethora of monodenate ligands, but also an extensive range of polydentate variants.2,3As polydentate ligand scaffolds, calix[4]arenes are well suited building blocks with scope for functionalisation across the upper and/or lower rims of these ‘bowl’ shaped macromolecules.4 Despite extensive investigation of heteroatom-based donor systems, the coordination chemistry of NHC-functionalized calix[4]arenes is not well developed.5 5,17-Difunctionalised examples, bearing NHC donors on opposite faces of the upper rim, are of particular interest for positioning bound metal centres across the macrocycle cavity. Well-defined complexes of these ligands are, however, limited to imidazol-2-ylidene based A–D (Chart 1). Complexes A illustrate this principle well, positioning square planar palladium centres directly over the macrocycle cavity.6 The incorporation of a methylene spacer between the calix[4]arene rim and the NHC donor group in B results instead in unfavourable twisting of the donor groups that skews the bound metal centres to one side of the ligand, while in C and D, the cavity is pinched closed as a consequence of the cis-coordination geometry of the metal centres.7 Closely related to A, phosphine-based systems E have also been described, where the ligand backbone enforces trans-coordination.8,9
 | ||
| Chart 1 Complexes of upper rim functionalised calix[4]arene NHC and phosphine ligands. | ||
In this report the preparation and coordination chemistry of an upper rim functionalised calix[4]arene pro-ligand 1·2HI are described (Chart 2). Through generation of the free NHC ligand (1) and use of transmetallation methodology, reactions with rhodium(I) dimers [Rh(COD)Cl]2 (COD = 1,5-cyclooctadiene) and [Rh(CO)2Cl]2 lead to a range of dinuclear complexes containing either discrete or chloro-bridged metal fragments depending on the reagents employed. To help understand the influence of the calix[4]arene scaffold and corroborate the structural formulations of these species, the spectroscopic characteristics of complexes of 1 are contrasted with analogues containing the monodentate NHC ligand IiPr2Me2.10
 | ||
| Chart 2 NHC systems of interest. | ||
Results and discussion
The synthesis of 5,17-(3-methyl-1-imidazole)-functionalised pro-ligand 1·2HI was achieved through straightforward adaption of literature protocols for related N-butyl and N-isopropyl analogues, involving alkylation of F with methyl iodide and isolated in 84% yield (Scheme 1).11 The known precursor F was obtained following a five step synthesis, starting from commercially available 4-tert-butylcalix[4]arene (ca. 2 weeks, 8% yield in our hands), giving an overall yield of 7% for the six step procedure.12 The formation of the new bis(imidazolium) salt was fully corroborated through a combination of NMR spectroscopy, ESI-MS, combustion analysis and X-ray crystallography. In solution the pre-carbenic centre is characterized by 1H and 13C resonances at δ 9.20 and δ 134.9 (CD2Cl2), respectively, while the mass spectrum showed a strong dication signal centred at 377.2220 m/z (calcd 377.2224) with half-integer spacing. The solid-state structure features two independent but structurally similar dications that adopt distinctive ‘pinched-cone’ conformations, reflecting intra-charge repulsion between the imidazolium groups (C2⋯C8, 13.43(2) Å/C102⋯C108, 13.09(2) Å; Fig. 1).13 In order to quantify this distortion, the angles between the least squares planes (Mpln) of opposing aryloxy groups ΘCALIX(R), where R = the aryl para substituent and the sign reflects the relative disposition of the R substituents (+ve, outwards; −ve, inwards), have been measured. Using this approach distortion from an ideal C4v symmetric ‘cone’ to C2v symmetric ‘pinched cone’ conformation of the calix[4]arene skeleton was found to be most pronounced in the independent molecule shown in Fig. 1 with ΘCALIX(NHC·H+)/ΘCALIX(H) = +80.8(2)/-19.3(2)° (ΔΘCALIX = 100.1(4)°); the other is characterised by ΘCALIX(NHC·H+)/ΘCALIX(H) = +86.4(2)/−2.8(2)° (ΔΘCALIX = 89.2(4)°). | ||
| Scheme 1 Preparation and deprotonation of pro-ligand 1·2HI. | ||
 | ||
| Fig. 1 Solid-state structure of 1·2HI. Thermal ellipsoids for selected atoms drawn at the 30% probability level; only one of the two unique molecules shown (Z′ = 2); hydrogen atoms and anions are omitted for clarity. Selected bond lengths (Å) and angles (°): C2⋯C8, 13.43(2); ∠Mpln(C13–C18,O41)–Mpln(C27–32,O49), 80.8(2); ∠Mpln(C20–C25,O45)–Mpln(C34–C39,O53), 19.3(2); C102⋯C108, 13.09(2); ∠Mpln(C113–C118,O141)–Mpln(C127–132,O149), 86.4(2); ∠Mpln(C120–C125,O145)–Mpln(C134–C139,O153), 2.8(2). | ||
Deprotonation of 1·2HI with strong hindered base K[OtBu] in C6D6 or d8-THF at 293 K resulted in quantitative formation of the free carbene 1, which was thoroughly characterised in situ using NMR spectroscopy. The deprotonation was corroborated by the absence of the 1H NCHN resonance and characteristically high frequency 13C resonance at δ 213.2 (C6D6)/216.0 (d8-THF) of the carbenic centre.14 For comparison, the equivalent signal in IiPr2Me2 is located at δ 212.7 (C6D6).10 Multiple attempts at isolating 1 from solution proved unsuccessful and instead it was generated in situ in subsequent studies.
Reactions of 1, generated as described above, with rhodium(I) dimers [Rh(COD)Cl]2 and [Rh(CO)2Cl]2 were explored in THF, using excess KI (ca. 10 equiv.) to avoid formation of mixed halide products. Using either 0.5 or 1 equiv. of rhodium precursor per ligand, the only well-defined species that could be identified were dinuclear complexes 2a and 3a, where 1 adopts a bridging μ2-coordination mode (Scheme 2). In this manner, rhodium-diene-based 2a was obtained in good isolated yield of 53% following purification over alumina. Isolation of 3a, however, proved to be very low yielding (ca. 17%) and in order to acquire a more meaningful quantity of 3a, alterative preparation from 2avia diene displacement with CO (1 atm) in CH2Cl2 solution was required (quantitative conversion by 1H NMR spectroscopy, 37% isolated yield). Mononuclear analogues of these new complexes bearing IiPr2Me2, 2b and 3b, were readily obtained through direct adaptions of the aforementioned procedures (Scheme 2).
 | ||
| Scheme 2 Reactions of 1 and IiPr2Me2 with rhodium dimers. | ||
For both NHC ligands, the rhodium-diene complexes 2 were characterised in the solid-state by X-ray crystallography (Fig. 2). The conformation of the calix[4]arene backbone in 2a is notable for a dramatic conformational inversion in comparison to the pro-ligand 1·2HI, that appears to be driven by a favourable (off-centre) π-stacking interaction between the imidazolylidene rings (Cnt-Cnt = 3.608(5) Å; ΔΘCALIX = −98.41(12)°, 2a; 100.1(4)/89.2(4)°, 1·2HI), and pseudo C2 symmetry. As expected for d8-metal complexes of this type, the rhodium centres adopt square planar coordination geometries in 2 and the associated metal–ligand bonding metrics are in line with related literature precedents.15 The similarity of the metal-NHC bonding characteristics in the new systems is apparent in both the solid-state (Rh–C = 2.021(3)/2.024(3) Å, 2a; 2.021(3) Å, 2b) and in solution by 13C NMR spectroscopy [δ 180.4 (1JRhC = 49 Hz), 2a; 178.4 (1JRhC = 49 Hz), 2b]. The structures were fully corroborated in solution by 1H and 13C NMR spectroscopy, with intact coordination of the diene ligands readily established by presence of alkene 13C signals at ca. δ 95 (1JRhC = 7 Hz) and 72 (1JRhC = 14 Hz) displaying coupling to 103Rh. Loss of Cs symmetry in the {Rh(COD)I} fragment and C2v symmetry of the ligand 1 is observed on complexation, with C2 symmetry evident on inspection of the 1H and 13C{1H} NMR spectra of 2a. Moreover, a large difference in chemical shift is observed for the now inequivalent 1H resonances of the imidazolylidene functionalised aryl ring (Δδ = 2.03), with the high frequency signal at δ 7.95 presumably a consequence of the close proximity of the halogen atom (cf. ![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) –H⋯
–H⋯![[I with combining low line]](https://www.rsc.org/images/entities/char_0049_0332.gif) = 4.139(3)/4.295(3) Å observed in the solid-state and δ 6.61 for 1·2HI), as previously noted in a related calix[4]arene-based system.5b Combined, these NMR data of 2a are consistent with retention of solid-state structure in solution.
= 4.139(3)/4.295(3) Å observed in the solid-state and δ 6.61 for 1·2HI), as previously noted in a related calix[4]arene-based system.5b Combined, these NMR data of 2a are consistent with retention of solid-state structure in solution.
 | ||
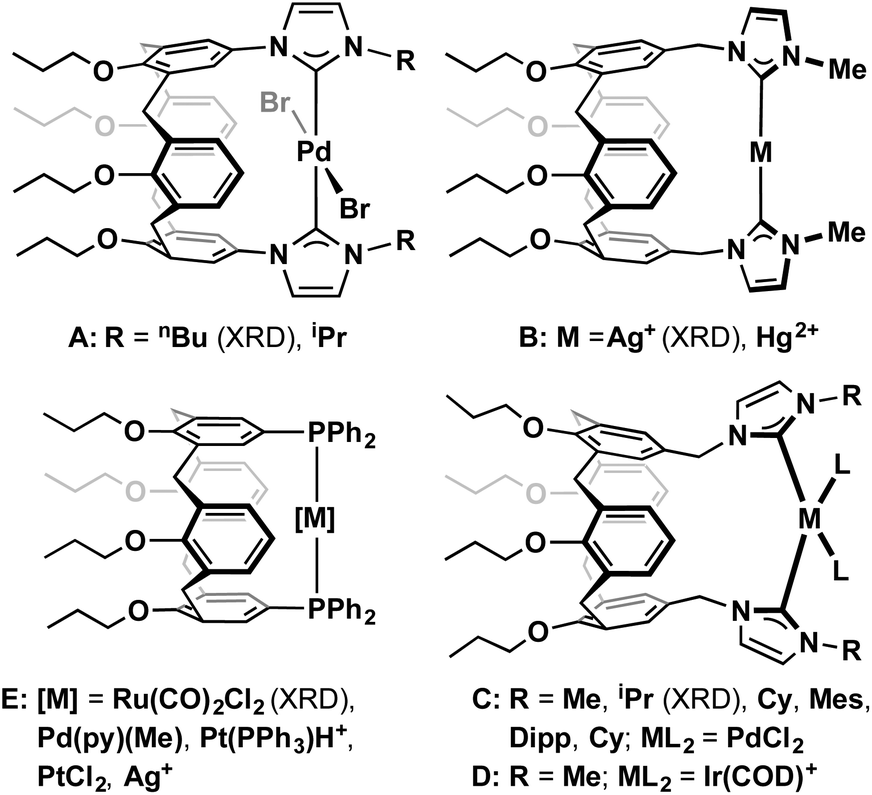
| Fig. 2 Solid-state structures of 2a and 2b. Thermal ellipsoids for selected atoms drawn at the 50% probability level; minor disordered components (two OnPr groups in 2a), and hydrogen atoms omitted for clarity. Selected bond lengths (Å) and angles (°): 2a: Rh1–I3, 2.6905(3); Rh1–Cnt(C5,C6), 2.089(3); Rh1–Cnt(C9,C10), 2.001(3); Rh1–C21, 2.021(3); Rh2–I4, 2.6934(3); Rh2–Cnt(C13,C14), 2.098(3); Rh1–Cnt(C17,C18), 1.990(4); Rh2–C27, 2.024(3); Rh1⋯Rh2, 8.5698(3); Cnt(C21–N25)–Cnt(C27–N31), 3.608(5); ∠Mpln(C33–C38,O61)–Mpln(C47–C52,O69), 22.35(6); ∠Mpln(C40–C45,O65)–Mpln(C54–C59,O73), 76.06(6); 2b: Rh1–I2, 2.6729(2); Rh1–Cnt(C3,C4), 2.109(2); Rh1–Cnt(C7,C8), 2.00(3); Rh1–C11, 2.021(3). | ||
Although, we have so far been unable to grow suitable samples of 3 for structural elucidation by X-ray crystallography, the solution data points strongly to equivalent structural formations as 2. For instance, the relative cis-configuration of the carbonyl ligands was established through observation of two bands in the respective IR spectra (υ(CO) = 2071, 2002, 3a; 2072, 1998 cm−1, 3b) and two distinct high frequency doublet resonances in the 13C{1H} NMR spectra [δ 188.4 (1JRhC = 54 Hz), 181.6 (1JRhC = 78 Hz), 3a; 188.7 (1JRhC = 53 Hz), 182.7 (1JRhC = 78 Hz), 3b]. 1H and 13C{1H} NMR spectra of 3a also exhibit C2 symmetry. In comparison to 2, the carbenic resonances are notable for a shift to lower frequency by ca. 10 ppm and there is a reduction in the magnitude of the coupling to 103Rh [δ 171.5 (1JRhC = 44 Hz), 3a; 166.8 (1JRhC = 41 Hz), 3b].
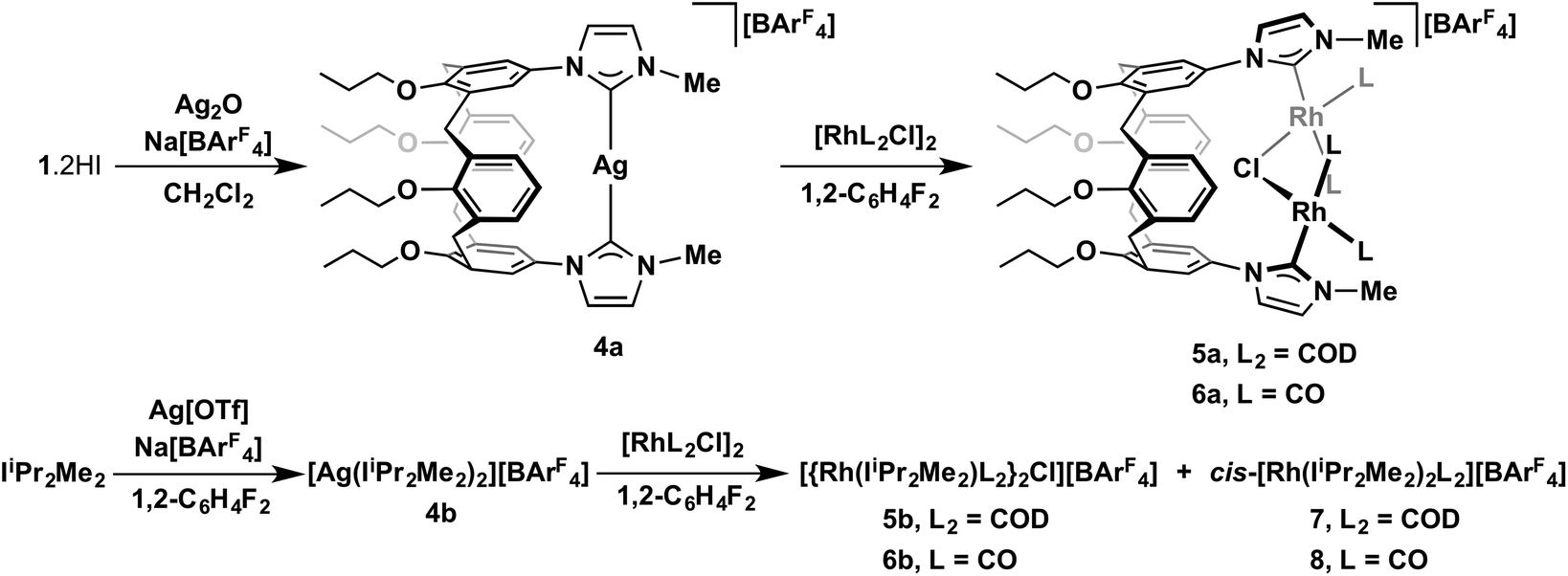
To further explore the coordination chemistry of 1, we sought to utilise widely employed transmetallation methodology based on silver transfer agents.1c,16 To this end, 1·2HI was reacted with a slight excess of Ag2O in the presence of Na[BArF4] (ArF = 3,5-C6H3(CF3)2), as a halide extractor, resulting in chelation of 1 and subsequent isolation of 4a in 73% yield (Scheme 3). The structural formulation of the new silver(I) complex was readily established in solution though a combination of 1H and 13C NMR spectroscopy and ESI-MS. The 1H NMR spectrum of isolated 4a shows C2v symmetry, with the N-methyl signal located to lower frequency than found in the pro-ligand (δ 3.80 vs. 4.17). A strong parent ion is observed by ESI-MS in positive ion mode at 859.3340 m/z (calcd 859.3347 m/z) with a correct isotope pattern. Moreover the cation![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[BArF4] ratio was verified by integration of 1H NMR data and elemental analysis. The 13C{1H} NMR spectrum is notable for the carbenic signal at δ 180.7 that shows coupling to both 109Ag (1JAgC = 211 Hz) and 107Ag (1JAgC = 183 Hz); the chemical shift and relative magnitudes of these coupling constants are in good agreement with literature values.16a Interrogation of single crystals of 4a by X-ray diffraction provides further evidence to the mononuclear formulation of 4a in solution, revealing a distorted linear geometry of the silver cation [C2–Ag1–C8 = 171.00(14)°] and essentially equivalent Ag–C bond lengths (Ag1–C2, 2.085(4); Ag1–C8, 2.083(4); Fig. 3). The biscarbene silver complex of IiPr2Me2 bearing the tetrafluoroborate counter anion [Ag(IiPr2Me2)2][BF4] G has previously been prepared,17 however, for comparison the analogue bearing the [BArF4]− counter anion 4b was generated in situ through reaction of Ag[OTf], Na[BArF4] and 2 equiv. IiPr2Me2 in 1,2-C6H4F2. The solid-state structure of 4b is notable for a significantly more orthogonal orientation of the NHC ligands in comparison to 4a and G [e.g. |N6–C2–C8–N12| = 3.7(5)°, 4a; |N3–C2–C15–N19| = 77.0(5)°, 4b; |N–C–C–N| = 20.6(6)°, G]. The near coplanar disposition of the NHC donors in 4a is fully in line with expectation, and readily attributed to the calix[4]arene scaffold [ΔΘCALIX = −96.4(2)°], while the conformational differences in 4b/G are presumably attributed to crystal packing effects, induced by disparate anion sizes, and enabled through low-energy Ag–NHC bond rotation. The Ag–C bond lengths are all within experimental error [2.085(4)/2.083(4) Å, 4a; 2.093(4)/2.092(4) Å, 4b; 2.078(11) Å, G], although we note an apparent trend of bond length contraction for 4a in comparison to 4b and that this parameter was not determined with high precision for G.
:[BArF4] ratio was verified by integration of 1H NMR data and elemental analysis. The 13C{1H} NMR spectrum is notable for the carbenic signal at δ 180.7 that shows coupling to both 109Ag (1JAgC = 211 Hz) and 107Ag (1JAgC = 183 Hz); the chemical shift and relative magnitudes of these coupling constants are in good agreement with literature values.16a Interrogation of single crystals of 4a by X-ray diffraction provides further evidence to the mononuclear formulation of 4a in solution, revealing a distorted linear geometry of the silver cation [C2–Ag1–C8 = 171.00(14)°] and essentially equivalent Ag–C bond lengths (Ag1–C2, 2.085(4); Ag1–C8, 2.083(4); Fig. 3). The biscarbene silver complex of IiPr2Me2 bearing the tetrafluoroborate counter anion [Ag(IiPr2Me2)2][BF4] G has previously been prepared,17 however, for comparison the analogue bearing the [BArF4]− counter anion 4b was generated in situ through reaction of Ag[OTf], Na[BArF4] and 2 equiv. IiPr2Me2 in 1,2-C6H4F2. The solid-state structure of 4b is notable for a significantly more orthogonal orientation of the NHC ligands in comparison to 4a and G [e.g. |N6–C2–C8–N12| = 3.7(5)°, 4a; |N3–C2–C15–N19| = 77.0(5)°, 4b; |N–C–C–N| = 20.6(6)°, G]. The near coplanar disposition of the NHC donors in 4a is fully in line with expectation, and readily attributed to the calix[4]arene scaffold [ΔΘCALIX = −96.4(2)°], while the conformational differences in 4b/G are presumably attributed to crystal packing effects, induced by disparate anion sizes, and enabled through low-energy Ag–NHC bond rotation. The Ag–C bond lengths are all within experimental error [2.085(4)/2.083(4) Å, 4a; 2.093(4)/2.092(4) Å, 4b; 2.078(11) Å, G], although we note an apparent trend of bond length contraction for 4a in comparison to 4b and that this parameter was not determined with high precision for G.
 | ||
| Scheme 3 Preparation and transmetallation reactions of silver NHC complexes. | ||
 | ||
| Fig. 3 Solid-state structures of 4a and 4b. Thermal ellipsoids for selected atoms drawn at the 30% probability level; minor disordered components (four OnPr groups in 4a), hydrogen atoms and anions omitted for clarity. Selected bond lengths (Å) and angles (°): 4a: Ag1–C2, 2.085(4); Ag1–C8, 2.083(4); C2–Ag1–C8, 171.00(14); ∠Mpln(C14–C19,O42)–Mpln(C28–C33,O50), 1.01(8); ∠Mpln(C21–C26,O46)–Mpln(C35–C40,O54), 97.44(10); 4b: Ag1–C2, 2.093(4); Ag1–C15, 2.092(4); C2–Ag1–C15, 176.81(15). | ||
Silver complex 4a acts as an effective carbene transfer agent, resulting in the formation of dinuclear complexes 5a and 6a bearing both μ2-1 and μ2-Cl ligands on reaction with 1 equiv. of [Rh(COD)Cl]2 and [Rh(CO)2Cl]2, respectively, in 1,2-C6H4F2 – established by NMR spectroscopy and ESI-MS. The reaction stoichiometries were maintained when using 0.5 equiv. of rhodium dimer instead, indicating selective formation 5a and 6a. Both dinuclear products were obtained in moderate isolated yields (63% and 59% respectively) and were difficult to fully purify, due to limited solution stability. Aided by comparison to 2a/3a and supported through preparation of direct IiPr2Me2 analogues (vide infra), the formulation of the structures of 5a/6a was achieved through a combination of 1H, 13C NMR and IR spectroscopy, ESI-MS and in the case of 5b a low resolution X-ray structure (Fig. 4). The ESI-MS in particular evidenced formation of these dinuclear monocationic species, with parent cation signals at 1209.3986 (calcd 1209.3973) and 1105.1680 (calcd 1105.1891) m/z, respectively for 5a and 6a, with correct isotope patterns and integer mass spacing. Moreover the cation:[BArF4] ratio was verified by integration of 1H NMR data. In both cases, single carbene resonances [δ 176.9 (1JRhC = 50 Hz), 5a; 168.1 (1JRhC = 43 Hz), 6a] with very similar chemical shifts and 1JRhC coupling constants to the respective neutral analogues [δ 180.4 (1JRhC = 49 Hz), 2a; 171.5 (1JRhC = 44 Hz), 3a], alongside 103Rh coupled alkene and carbonyl signals, were observed by 13C NMR spectroscopy. For 6a both carbonyl stretching bands are perturbed (as expected) to higher frequency relative to neutral 3a [υ(CO) = 2083, 2016, 6a; 2071, 2002 cm−1, 3a]. Bond connectivity and conformational features of 6a were established through X-ray diffraction, using a poor quality crystalline sample [R1 = 0.2744, I ≥ 2σ(I)]. The data nevertheless can affirm the structural formulation, with a reduced pinching of calix[4]arene core [ΔΘCALIX = −83.1(7)°] relative to 2a [ΔΘCALIX = −98.41(12)°] and 4a [ΔΘCALIX = −96.4(2)°].
 | ||
| Fig. 4 Solid-state structure of 6a (ball and stick, poor quality data). Minor disordered component (CO), hydrogen atoms and anion omitted for clarity. Selected bond lengths (Å) and angles(°): Rh1–Cl3, 2.353(5); Rh2–Cl3, 2.360(5); Rh1⋯Rh2, 3.861(3); Rh1–Cl3–Rh2, 110.0(3); ∠Mpln(C24–C29,O52)–Mpln(C38–C43,O60), 9.7(3); ∠Mpln(C31–C36,O56)–Mpln(C45–C50,O64), 92.8(4). | ||
Starting from the known precursors [Rh(IiPr2Me2)(COD)Cl] and [Rh(IiPr2Me2)(CO)2Cl], the new μ2-Cl bridged dinuclear complexes 5b and 6b were readily prepared by partial halide extraction using 0.5 equiv. of Na[BArF4] in CH2Cl2 (Scheme 4). These structurally simple and fully characterised species (solution and solid-state, Fig. 5) help verify the formulation of 5a and 6a, with excellent agreement of the related spectroscopic data e.g. carbene 13C signals of 5 [δ176.9 (1JRhC = 50 Hz), 5a; 175.6 (1JRhC = 50 Hz), 5b] and carbonyl stretching bands of 6 [υ(CO) = 2083, 2016, 6a; 2090, 2019 cm−1, 6b] – see Experimental section for full details. Although iridium NHC and rhodium phosphine examples have been reported, there are no proceeding crystallographically characterised examples of rhodium NHC complexes featuring a single otherwise unsupported bridging halogen atom to the best of our knowledge.18 Conformational differences, associated with the relative orientation of the NHC ligands about the Rh–Cl–Rh bridge, are evident when comparing the solid-state structures of 6a (anti) 5b (syn) 6b (anti); 1H and 13C NMR data for 5a and 6a are most consistent with C2 symmetry and anti-configurations in solution. Contrasting reactions of 4a and 4b, the calix[4]arene scaffold promotes the selective formation of dinuclear complexes. For instance, when 4b is reacted with 1 equiv. of [Rh(COD)Cl]2 or [Rh(CO)2Cl]2 in 1,2-C6H4F2 mixtures of 5b/cis-[Rh(IiPr2Me2)2(COD)][BArF4] 7 (2:1) and 6b/cis-[Rh(IiPr2Me2)2(CO)2][BArF4] 8 (5:2) are the organometallic species produced as determined by 1H NMR spectroscopy (Scheme 3) – with the structures of 7 and 8 verified by independent synthesis and full characterisation, including in the solid-state by X-ray diffraction (see experimental for full details).
 | ||
| Scheme 4 Preparation of dinuclear complexes 5b and 6b. | ||
 | ||
| Fig. 5 Solid-state structures of 5b and 6b. Thermal ellipsoids drawn at the 50% probability level; minor disordered components (two iPr groups in 6b), hydrogen atoms, solvent (6a), and anions omitted for clarity. Selected bond lengths (Å) and angles (°): 5b: Rh1–Cl3, 2.3988(9); Rh1–Cnt(C4,C5), 2.092(5); Rh1–Cnt(C8,C9), 1.974(4); Rh1–C20, 2.025(3); Rh2–Cl3, 2.4067(9); Rh2–Cnt(C12,C13), 2.101(4); Rh2–Cnt(C16,C17), 1.972(4); Rh2–C33, 2.022(3); Rh1–Cl3–Rh2, 144.13(4); 6b: Rh1–Cl3, 2.4045(9); Rh1–C4, 1.915(4); Rh1–C6, 1.833(4); Rh1–C22, 2.069(3); Rh2–Cl3, 2.3987(9); Rh2–C8, 1.920(4); Rh2–C10, 1.817(4); Rh2–C25, 2.069(3); Rh1–Cl3–Rh2, 122.26(4). | ||
Summary
Using both free carbene and transmetallation methodologies the coordination chemistry of bis(imidazolium) pro-ligand 1·2HI with rhodium(I) dimers [Rh(COD)Cl]2 and [Rh(CO)2Cl]2 has been investigated, culminating in the isolation of neutral and cationic dinuclear complexes where the calix[4]arene based-NHC is bound in a μ2-coordination mode. These dinuclear complexes have been well characterised in solution using NMR spectroscopy, IR spectroscopy (CO derivatives) and ESI-MS (cationic complexes), and their structural formulation corroborated through excellent agreement of these data with that associated with simpler model complexes containing instead IiPr2Me2. Structures determined though X-ray diffraction highlight the flexibility of the calix[4]arene ligand scaffold, which undergoes significant conformational change driven by charge repulsion (bis-cationic 1·2HI), π-stacking of the imidazolylidene rings (2a), trans-coordination to silver (4a), or presence of a bridging μ2-Cl ligand (6a).Experimental
General considerations
All manipulations were performed under an atmosphere of argon, using Schlenk and glove box techniques unless otherwise stated. Glassware was oven dried at 150 °C overnight and flamed under vacuum prior to use. Anhydrous CH2Cl2, Et2O, and pentane (<0.005% H2O) were purchased from ACROS or Aldrich and freeze–pump–thaw degassed three times before being placed under argon. THF was dried over sodium/benzophenone, vacuum distilled, and freeze–pump–thaw degassed three times before being placed under argon. 1,2-C6H4F2 was stirred over neutral alumina, filtered, dried over CaH2, vacuum distilled, and freeze–pump–thaw degassed three times before being placed under argon over 3 Å molecular sieves. CD2Cl2 was dried over CaH2, vacuum distilled, and freeze–pump–thaw degassed three times before being placed under argon. C6D6 was dried over Na, vacuum distilled, and freeze–pump–thaw degassed three times before being placed under argon. d8-THF was dried over 3 Å molecular sieves and freeze–pump–thaw degassed three times before being placed under argon. 5,17-bis(imidazolium)-25,26,27,28-tetrapropoxycalix[4]arene (F),12 IiPr2Me2,10 [Rh(COD)Cl]2,19 [Rh(CO)2Cl]2,20 Na[BArF4],21 [Rh(IiPr2Me2)(COD)Cl],22 and cis-[Rh(IiPr2Me2)(CO)2Cl]22 were synthesised using literature protocols. All other solvents and reagents are commercial products and were used as received. NMR spectra were recorded on Bruker AV spectrometers at 298 K unless otherwise stated. 1H NMR spectra recorded in 1,2-C6H4F2 were referenced using the highest intensity peak of the highest (δ 6.865) frequency fluoroarene multiplet. 13C{1H} NMR spectra recorded in 1,2-C6H4F2 were referenced using an internal sealed capillary of C6D6. Chemical shifts are quoted in ppm and coupling constants in Hz. IR spectra were recorded on a PerkinElmer Spectrum One FT-IR spectrometer at 293 K. ESI-MS analyses were recorded on Bruker Maxis Impact instrument. Microanalyses were performed by Stephen Boyer at London Metropolitan University.Preparation of 1·2HI
A solution of F (1.00 g, 1.38 mmol) was stirred with iodomethane (0.84 mL, 13.8 mmol) in THF (30 mL) at 70 °C for 14 hours. The product was isolated by filtration following addition of Et2O (ca. 20 mL), washed with Et2O (2 × 20 mL) and dried in vacuo. Yield = 1.17 g (84%, fine off-white powder).
1
H NMR (CD2Cl2, 400 MHz): δ 9.20 (br, 2H, NCHN), 8.04 (t, 3JHH = 1.8, 2H, imid.), 7.23 (d, 3JHH = 7.5, 4H, Ar), 7.05 (t, 3JHH = 7.5, 2H, Ar), 6.72 (t, 3JHH = 1.8, 2H, imid.), 6.61 (s, 4H, Ar), 4.54 (d, 2JHH = 13.5, 4H, ArC![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2Ar), 4.17 (s, 6H, NCH3), 4.03–4.09 (m, 4H, OCH2), 3.74 (t, 3JHH = 6.8, 4H, OCH2), 3.28 (d, 2JHH = 13.5, 4H, ArC2Ar), 1.87–2.06 (m, 8H, C2CH3), 1.12 (t, 3JHH = 7.4, 6H, CH2C3), 0.93 (t, 3JHH = 7.5, 6H, CH2C3). 13C{1H} NMR (CD2Cl2, 101 MHz): δ 157.5 (s, OCH2), 157.3 (s, OCH2), 137.7 (s, Ar{CH2}), 135.7 (s, Ar{CH2}), 134.9 (s, NCHN), 130.2 (s, Ar), 129.2 (s, Ar{CN}), 125.6 (s, imid.), 124.4 (s, Ar), 120.8 (s, Ar), 119.9 (s, imid.), 78.1 (s, OCH2), 77.4 (s, OCH2), 37.8 (s, NCH3), 31.4 (s, ArH2Ar), 23.9 (s, H2CH3), 23.5 (s, H2CH3), 10.9 (s, CH2H3), 10.2 (s, CH2H3). ESI-MS (CH3CN, 180 °C, 3 kV) positive ion: 377.2220 m/z, [M]2+ (calcd 377.2224 m/z). Anal. Calcd For C48H58I2N4O4 (1008.81 g mol−1): C, 57.13; H, 5.80; N, 5.55. Found: C, 56.88; H, 6.00; N, 5.55.
2Ar), 4.17 (s, 6H, NCH3), 4.03–4.09 (m, 4H, OCH2), 3.74 (t, 3JHH = 6.8, 4H, OCH2), 3.28 (d, 2JHH = 13.5, 4H, ArC2Ar), 1.87–2.06 (m, 8H, C2CH3), 1.12 (t, 3JHH = 7.4, 6H, CH2C3), 0.93 (t, 3JHH = 7.5, 6H, CH2C3). 13C{1H} NMR (CD2Cl2, 101 MHz): δ 157.5 (s, OCH2), 157.3 (s, OCH2), 137.7 (s, Ar{CH2}), 135.7 (s, Ar{CH2}), 134.9 (s, NCHN), 130.2 (s, Ar), 129.2 (s, Ar{CN}), 125.6 (s, imid.), 124.4 (s, Ar), 120.8 (s, Ar), 119.9 (s, imid.), 78.1 (s, OCH2), 77.4 (s, OCH2), 37.8 (s, NCH3), 31.4 (s, ArH2Ar), 23.9 (s, H2CH3), 23.5 (s, H2CH3), 10.9 (s, CH2H3), 10.2 (s, CH2H3). ESI-MS (CH3CN, 180 °C, 3 kV) positive ion: 377.2220 m/z, [M]2+ (calcd 377.2224 m/z). Anal. Calcd For C48H58I2N4O4 (1008.81 g mol−1): C, 57.13; H, 5.80; N, 5.55. Found: C, 56.88; H, 6.00; N, 5.55.
Deprotonation of 1·2HI
A suspension of 1·2HI (10.2 mg, 0.0101 mmol) and K[OtBu] (3.1 mg, 0.0276 mmol) in d8-THF (0.5 mL) was agitated and sonicated for several minutes inside a sealed J. Young's NMR tube. Analysis by NMR spectroscopy indicated quantitative formation of 1 with the concomitant formation of tBuOH (δH 5.33, OH; 1.15, CH3; δC 68.1,CH3; 31.6, CH3). The reaction was repeated in C6D6 with the same outcome. The 1H NMR spectra remained unchanged on standing at 293 K for 24 h in both cases.
1
H NMR (d8-THF, 500 MHz): δ 7.41 (s, 4H, Ar), 7.24 (d, 3JHH = 1.6, 2H, imid.), 6.94 (d, 3JHH = 1.6, 2H, imid.), 6.40 (d, 3JHH = 7.5, 4H, Ar), 6.31 (app t, 2H, J = 8, Ar), 4.51 (d, 2JHH = 13.1, 4H, ArC2Ar), 4.00–4.04 (m, 4H, OCH2), 3.76–3.79 (m, 4H, OCH2), 3.76 (s, 6H, NCH3), 3.20 (d, 2JHH = 13.1, 4H, ArC2Ar), 1.99–2.07 (m, 4H, C2CH3), 1.92–1.99 (m, 4H, C2CH3), 1.10 (t, 3JHH = 7.4, 6H, CH2C3), 0.98 (t, 3JHH = 7.5, 6H, CH2C3). 13C{1H} NMR (d8-THF, 126 MHz): δ 216.0 (s, NCN), 156.7 (s, OCH2), 156.3 (s, OCH2), 137.9 (s, Ar{CN}), 137.6 (s, Ar{CH2}), 134.5 (s, Ar{CH2}),128.9 (s, Ar), 123.1 (s, Ar), 121.6 (s, Ar), 121.1 (s, imid.), 118.1 (s, imid.), 77.9 (s, OCH2), 77.7 (s, OCH2), 38.3 (s, NCH3), 32.0 (s, ArH2Ar), 24.5 (s, H2CH3), 24.2 (s, H2CH3), 11.2 (s, CH2H3), 10.7 (s, CH2H3). 1H NMR (C6D6, 500 MHz): δ 7.40 (s, 4H, Ar), 6.74 (d, 3JHH = 7.5, 4H, Ar), 6.71 (br, 2H, imid.), 6.61 (t, 3JHH = 7.5, 2H, Ar), 6.18 (br, 2H, imid.), 4.53 (d, 2JHH = 13.2, 4H, ArC2Ar), 3.87 (t, 3JHH = 7.6, 4H, OCH2), 3.76 (t, 3JHH = 7.4, 4H, OCH2), 3.42 (s, 6H, NCH3), 3.17 (d, 2JHH = 13.3, 4H, ArC2Ar), 1.84–1.95 (m, 8H, C2CH3), 0.93 (app. t, J = 7, 12H, CH2C3). 13C{1H} NMR (C6D6, 126 MHz): δ 213.2 (s, NCN), 156.7 (s, OCH2), 155.5 (s, OCH2), 137.1 (s, Ar{CN}), 136.4 (s, Ar{CH2}), 134.7 (s, Ar{CH2}), 128.8 (s, Ar), 123.0 (s, Ar), 121.5 (s, Ar), 120.1 (s, imid.), 117.8 (s, imid.), 77.1 (s, OCH2), 77.0 (s, OCH2), 37.8 (s, NCH3), 31.6 (s, ArH2Ar), 23.7 (s, H2CH3), 23.6 (s, H2CH3), 10.6 (s, CH2H3), 10.5 (s, CH2H3).
Preparation of 2a
A solution of 1·2HI (99.7 mg, 0.0987 mmol) and K[OtBu] (27.8 mg, 0.248 mmol) was stirred in THF (10 mL) for 1 hour then added to a flask charged with [Rh(COD)Cl]2 (51.6 mg, 0.105 mmol) and KI (165.1 mg, 1.42 mmol). After a further hour the volatiles were removed in vacuo. The product was extracted with CH2Cl2 (2 × 10 mL) and purified over alumina (1:1 EtOAc/CH2Cl2, Air). Yield = 75.5 mg (53%, yellow powder).
1
H NMR (CD2Cl2, 400 MHz): δ 7.95 (d, 4JHH = 2.8, 2H, Ar), 7.29–7.39 (m, 2H, Ar), 7.04–7.14 (m, 2H, Ar), 6.97 (t, 3JHH = 7.4, 2H, Ar), 6.44 (d, 3JHH = 2.0, 2H, imid.), 6.19 (d, 3JHH = 2.0, 2H, imid.), 5.92 (d, 4JHH = 2.8, 2H, Ar), 5.24–5.29 (m, 2H, COD{CH}), 4.92–5.09 (m, 2H, COD{CH}), 4.62 (d, 2JHH = 13.3, 2H, ArC2Ar), 4.50 (d, 2JHH = 13.5, 2H, ArC2Ar), 3.99–4.18 (m, 4H, OCH2), 3.85 (s, 6H, NCH3), 3.65–3.82 (m, 4H, OCH2), 3.38 (d, 2JHH = 13.4, 2H, ArC2Ar), 3.21–3.26 (m, 2H, COD{CH}), 3.19 (d, 2JHH = 13.7, 2H, ArC2Ar), 2.29–2.39 (m, 2H, COD{CH}), 2.09–2.29 (m, 6H, COD{CH2}), 1.97–2.09 (m, 4H, C2CH3), 1.83–1.97 (m, 6H, C2CH3 + COD{CH2}), 1.58–1.79 (m, 4H, COD{CH2}), 1.27–1.46 (m, 4H, COD{CH2}), 1.15 (t, 3JHH = 7.4, 6H, CH2C3), 0.95 (t, 3JHH = 7.5, 6H, CH2C3). 13C{1H} NMR (CD2Cl2, 101 MHz): δ 180.4 (d, 1JRhC = 49, NCN), 158.2 (s, OCH2), 155.0 (s, OCH2), 137.0 (s, Ar{CH2}), 136.9 (s, Ar{CH2}), 134.9 (s, Ar{C}), 134.7 (s, Ar{C}), 134.3 (s, Ar{C}), 131.5 (s, Ar), 129.6 (s, Ar), 123.6 (s, Ar), 123.4 (s, imid.), 122.9 (s, Ar), 122.5 (s, imid.), 122.5 (s, Ar), 95.0 (d, 1JRhC = 7, COD{CH}), 95.0 (d, 1JRhC = 7, COD{CH}), 77.7 (s, OCH2), 77.3 (s, OCH2), 71.4 (d, 1JRhC = 14, 2 × COD{CH}), 39.1 (s, NH3), 33.9 (s, COD{CH2}), 31.7 (s, CH2), 31.4 (s, CH2), 31.0 (s, CH2), 30.7 (s, CH2), 29.2 (s, COD{CH2}), 23.8 (s, H2CH3), 23.4 (s, H2CH3), 11.3 (s, CH2H3), 10.3 (s, CH2H3). Anal. Calcd For C64H80I2N4O4 (1428.24 g mol−1): C, 53.79; H, 5.64; N, 3.92. Found: C, 53.71; H, 5.75; N, 4.04.
Preparation of 2b
A solution of IiPr2Me2 (61.2 mg, 0.336 mmol), [Rh(COD)Cl]2 (166.1 mg, 0.337 mmol) and KI (1072.2 mg, 6.46 mmol) in THF (10 mL) was stirred for 1 hour. The volatiles were removed in vacuo and the product obtained after purification over silica (CH2Cl2, Air) and subsequent recrystallisation from CH2Cl2/pentane. Yield = 55.0 mg (31%, yellow crystals).
1
H NMR (CD2Cl2, 500 MHz): δ 5.97 (sept, 3JHH = 7.1, 2H, NCH), 5.03–5.09 (m, 2H, COD{CH}), 3.50–3.57 (m, 2H, COD{CH}), 2.26–2.36 (m, 4H, COD{CH2}), 2.16 (s, 6H, CC3), 1.87–1.98 (m, 2H, COD{CH2}), 1.73–1.83 (m, 2H, COD{CH2}), 1.56 (d, 3JHH = 7.1, 6H, CHC3), 1.50 (d, 3JHH = 7.1, 6H, CHC3). 13C{1H} NMR (CD2Cl2, 126 MHz): δ 178.4 (d, 1JRhC = 49, NCN), 126.0 (s, CH3), 95.2 (d, 1JRhC = 7, COD{CH}), 71.5 (d, 1JRhC = 14, COD{CH}), 53.7 (s, NCH), 32.8 (s, COD{CH2}), 30.0 (s, COD{CH2}), 22.4 (s, CHH3), 21.4 (s, CHH3), 10.7 (s, CH3). Anal. Calcd for C19H32I2N2Rh (518.29 g mol−1): C, 44.03; H, 6.22; N, 5.41. Found: C, 44.11; H, 6.14; N, 5.47.
Preparation of 3a
Method A: A solution of 1·2HI (102.1 mg, 0.101 mmol) and K[OtBu] (28.9 mg, 0.258 mmol) was stirred in THF (10 mL) for 1 hour then added to a flask charged with [Rh(CO)2Cl]2 (40.7 mg, 0.105 mmol) and KI (165.4 mg, 0.996 mmol). After a further hour the volatiles were removed in vacuo. The product was extracted with CH2Cl2 (2 × 10 mL) and purified over alumina (1:1 Et2O/CH2Cl2, Air). Yield = 22.4 mg (17%, yellow powder). Method B: A solution of 2a (121.2 mg, 0.0848 mmol) in CH2Cl2 (10 mL) was stirred under CO (1 atm) for 12 hours, resulting in a colour change from bright to pale yellow. The mixture was dried in vacuo and washed with pentane (3 × 5 mL). Yield = 42.1 mg (37%, yellow powder).
1
H NMR (CD2Cl2, 500 MHz): δ7.26 (s, 2H, Ar), 7.02 (br, 4H, Ar), 6.82 (t, 3JHH = 7.4, 2H, Ar), 6.77 (s, 2H, imid.), 6.46 (s, 2H, imid.), 6.41 (s, 2H, Ar), 4.51 (d, 2JHH = 13.3, 2H, ArC2Ar), 4.48 (d, 2JHH = 13.6, 2H, ArC2Ar), 3.80–4.09 (m, 8H, OCH2), 3.77 (s, 6H, NCH3), 3.24 (d, 2JHH = 14.1, 2H, ArC2Ar), 3.22 (d, 2JHH = 14.1, 2H, ArC2Ar), 1.79–2.16 (m, 8H, C2CH3), 1.03–1.13 (m, 6H, CH2C3), 0.92–1.03 (m, 6H, CH2C3). 13C{1H} NMR (CD2Cl2, 126 MHz): δ 188.4 (d, 1JRhC = 54, CO), 181.6 (d, 1JRhC = 78, CO), 171.5 (d, 1JRhC = 44, NCN), 157.5 (s, OCH2), 156.3 (s, OCH2), 136.2, 136.1, 135.9, 135.6, 130.6, 129.4, 123.8, 123.4, 123.2, 122.8, 77.8 (OCH2), 77.4(OCH2), 40.0 (s, NCH3), 31.5 (br, ArH2Ar), 24.0 (H2CH3), 23.7 (H2CH3), 11.0 (s, CH2H3), 10.4 (s, CH2H3). Not all signals were unambiguously identified. IR (CH2Cl2, cm−1): ν(CO) 2071, 2002. Anal. Calcd for C52H56I2N4O8Rh2 (1324.02 g mol−1): C, 47.15; H, 4.26; N, 4.23. Found: C, 46.91; H, 4.12; N, 4.25.
Preparation of 3b
A solution of 2b (40.1 mg, 0.0774 mmol) in CH2Cl2 (10 mL) was placed under an atmosphere of CO (1 atm) for 2 hours. The volatiles were removed in vacuo to afford the product, which was washed with pentane (3 × 10 mL) and dried thoroughly under vacuum. Yield = 21.4 mg (59%, fine yellow powder).
1
H NMR (CD2Cl2, 500 MHz): δ 5.26 (sept, 3JHH = 7.1, 2H, NCH), 2.22 (s, 6H, CCH3), 1.501 (d, 3JHH = 7.1, 6H, CHC3), 1.496 (d, 3JHH = 2.7, 6H, CHC3). 13C{1H} NMR (CD2Cl2, 126 MHz): δ 188.7 (d, 1JRhC = 53, CO), 182.7 (d, 1JRhC = 78, CO), 166.8 (d, 1JRhC = 41, NCN), 127.2 (s, CH3), 54.3 (s, HCH3), 22.2 (s, CHH3), 21.1 (s, CHH3), 10.7 (s, CH3). IR (CH2Cl2, cm−1): ν(CO) 2072, 1998. Anal. Calcd for C13H20I2N2O2Rh (466.12 g mol−1): C, 33.50; H, 4.33; N, 6.01. Found: C, 33.41; H, 4.25; N, 6.03.
Preparation of 4a
A suspension of 1·2HI (79.5 mg, 0.0788 mmol), Ag2O (20.0 mg, 0.0862 mmol) and Na[BArF4] (69.0 mg, 0.0778 mmol) in CH2Cl2 (20 mL) was sonicated periodically over two hours. The solution was filtered and the solvent was removed in vacuo to afford the product. Yield = 100.0 mg (73%, white crystals).
1
H NMR (400 MHz, CD2Cl2): δ 7.72–7.76 (m, 8H, ArF), 7.57 (br, 4H, ArF), 7.10 (d, 3JHH = 7.4, 4H, Ar), 6.92 (d, 3JHH = 1.7, 2H, imid.), 6.85 (t, 3JHH = 7.5, 2H, Ar), 6.81 (d, 3JHH = 1.7, 2H, imid.), 6.40 (s, 4H, Ar), 4.57 (d, 2JHH = 12.8, 4H, ArC2Ar), 4.10–4.22 (m, 4H, OCH2), 3.80 (s, 6H, NCH3), 3.74 (t, 3JHH = 7.1, 4H, OCH2), 3.24 (d, 2JHH = 12.8, 4H, ArC2Ar), 2.10–2.21 (m, 4H, C2CH3), 1.99 (app. sex., J = 7, 4H, C2CH3), 1.12 (t, 3JHH = 7.4, 6H, CH2C3), 0.98 (t, 3JHH = 7.5, 6H, CH2C3). 13C{1H} NMR (CD2Cl2, 101 MHz): δ 180.7 (two coincident d, 1J109AgC = 211, 1J107AgC = 183, NCN), 162.3 (q, 1JBC = 50.5, ArF), 157.8 (OCH2), 156.7 (s, OCH2), 136.5 (s, 2 × Ar{C}), 135.4 (s, ArF), 134.3 (s, Ar{C}), 129.4 (qq, 2JFC = 32, 3JBC = 3, ArF), 129.3 (s, Ar), 118.0 (sept, 3JFC = 4, ArF), 126.4 (s, Ar), 125.2 (q, 1JFC = 272, ArF), 124.7 (d, 3JAgC = 6, imid.), 123.4 (s, Ar), 121.9 (d, 3JAgC = 6, imid.), 118.0 (sept, 3JFC = 4, ArF), 78.7 (s, OCH2), 77.2 (s, OCH2), 38.8 (d, 3JAgC = 3, NCH3), 31.4 (s, ArH2Ar), 24.0 (s, H2CH3), 23.5 (s, H2CH3), 11.0 (s, CH2H3), 10.1 (s, CH2H3). ESI-MS (CH3CN, 180 °C, 3 kV) positive ion: 859.3340 m/z, [M]+ (calcd 859.3347 m/z). Anal. Calcd For C80H68AgBF24N4O4 (1724.09 g mol−1): C, 55.73; H, 3.98; N, 3.25. Found: C, 55.85; H, 4.08; N, 3.33.
In situ reactions of 4b
A suspension of IiPr2Me2 (9.9 mg, 0.0549 mmol), Ag[OTf] (7.2 mg, 0.0277 mmol) and Na[BArF4] (24.0 mg, 0.0271 mmol) in difluorobenzene (0.5 mL) was agitated for several minutes. Analysis in situ by NMR spectroscopy indicated quantitative formation of 4b (data below). Solutions of 4b, prepared in this way, where then filtered onto either [Rh(COD)Cl]2 (13.6 mg, 0.0276 mmol) or [Rh(CO)2Cl]2 (10.8 mg, 0.0278 mmol), resulting in the formation of 5b:7 (2:1) and 6b/8 (5:2).
1
H NMR (1,2-C6H4F2, 500 MHz): δ 8.11–8.13 (m, 8H, ArF), 7.49 (br, 4H, ArF), 4.20 (sept, 3JHH = 6.8, 4H, NCH), 1.87 (s, 12H, CCH3), 1.31 (d, 3JHH = 6.8, 24H, CHC3). 13C{1H} NMR (1,2-C6H4F2, 126 MHz): δ162.5 (q, 1JBC = 50, ArF), 135.1 (s, ArF), 129.7 (qq, 2JFC = 32, 3JBC = 3, ArF), 124.9 (q, 1JFC = 272, ArF), 124 (obscured, CH3), 117.6 (sept, 3JFC = 4, ArF), 50.1 (s, NCH), 23.3 (s, CHC3), 8.1 (s, CH3). The carbene resonance was not located. 19F{1H} NMR (1,2-C6H4F2, 282 MHz): δ-62.25 (ArF). No signal for [OTf]− detected. ESI-MS (CH3CN, 180 °C, 3 kV) positive ion: 467.2297 m/z, [M]+ (calcd 467.2298 m/z).
Preparation of 5a
A solution of 4a (29.9 mg, 0.0173 mmol) and [Rh(COD)Cl]2 (9.4 mg, 0.0191 mmol) in 1,2-C6H4F2 (0.5 mL) was stirred for 1 hour with the exclusion of light. The solvent was removed in vacuo and the resulting crude material washed with pentane and dried in vacuo. Yield = 22.3 mg (63%, yellow powder). The product was characterised in situ, although has limited stability in solution.
1
H NMR (1,2-C6H4F2/C6D6, 500 MHz): δ 8.09–8.15 (m, 8H, ArF), 7.49 (br, 4H, ArF) 5.44 (br, 2H, COD{CH}), 5.38 (br, 2H, COD{CH}), 4.58 (d, 2JHH = 13.6, 2H, ArC2Ar), 4.49 (d, 2JHH = 13.6, 2H, ArC2Ar), 4.20 (s, 6H, NCH3), 3.96–4.24 (m), 3.67–3.76 (m), 3.55–3.63 (m), 3.49–3.55 (m), 3.46 (d, 2JHH = 13.6, 2H, ArC2Ar), 3.23–3.33 (m), 3.09 (d, 2JHH = 13.6, 2H, ArC2Ar), 1.35–2.50 (m, CH2), 1.03 (t, 3JHH = 6.9, 6H, CH2C3), 0.85 (t, 3JHH = 6.9, 6H, CH2C3), 0.7–1.1 (m, COD{CH2}). Not all signals unambiguously assigned and some signals obscured by solvent peak. 13C{1H} NMR (1,2-C6H4F2/C6D6, 126 MHz, selected signals only): δ 176.9 (d, 1JRhC = 50, NCN), 100.3 (br, COD{CH}), 96.4 (br, COD{CH}), 78.5 (d, 1JRhC = 14, COD{CH}), 71.8 (d, 1JRhC = 13, COD{CH}), 97.7 (d, 1JRhC = 7, COD{CH}), 70.2 (d, 1JRhC = 16, COD{CH}), 37.8 (s, NCH3). ESI-MS (CH3CN, 180 °C, 3 kV): positive ion: 1209.3986 m/z, [M]+ (calcd 1209.3973 m/z).
Preparation of 5b
A solution of [Rh(IiPr2Me2)(COD)Cl] (23.6 mg, 0.0553 mmol) and Na[BArF4] (24.5 mg, 0.0275 mmol) in CH2Cl2 (10 mL) was stirred for 1 hour. The solution was then filtered and layered with pentane to afford the product on diffusion. Yield = 32.0 mg (69%, yellow crystals).
1
H NMR (CD2Cl2, 500 MHz): δ 7.70–7.74 (m, 8H, ArF), 7.56 (br, 4H, ArF), 5.91 (sept, 3JHH = 7.1, 4H, NCH), 4.75 (s, 4H, COD{CH}), 3.51 (s, 4H, COD{CH}), 2.22–2.36 (m, 8H, COD{CH2}), 2.11 (s, 12H, CCH3), 1.81–1.93 (m, 8H, COD{CH2}), 1.57 (d, 3JHH = 7.0, 12H, CHC3), 1.36 (d, 3JHH = 7.0, 12H, CHC3). 13C{1H} NMR (CD2Cl2, 126 MHz): δ 175.6 (d, 1JRhC = 50, NCN), 162.3 (q, 1JBC = 51, ArF), 135.4 (s, ArF), 129.4 (qq, 2JFC = 31, 3JBC = 3, ArF), 126.3 (s, CH3), 125.2 (q, 1JFC = 272, ArF), 118.0 (sept, 3JFC = 4, ArF), 97.7 (d, 1JRhC = 7, COD{CH}), 70.2 (d, 1JRhC = 16, COD{CH}), 54.3 (s, NCH), 33.1 (s, COD{CH2}), 29.1 (s, COD{CH2}), 22.5 (s, CHH3), 22.2 (s, CHH3), 10.5 (s, CH3). ESI-MS (CH3CN, 180 °C, 3 kV) positive ion: 817.2872 m/z, [M]+ (calcd 817.2924 m/z). Anal. Calcd For C70H76BF24N4Rh2Cl (1434.39 g mol−1): C, 49.97; H, 4.56; N, 3.33. Found: C, 50.18; H, 4.52; N, 3.23.
Preparation of 6a
A solution of 4a (32.0 mg, 0.0186 mmol) and [Rh(CO)2Cl]2 (7.8 mg, 0.0201 mmol) in 1,2-C6H4F2 (10 mL) was stirred for one hour with the exclusion of light. The cloudy yellow solution was filtered, reduced to dryness and the resulting residue washed with pentane. Yield = 21.4 mg (31%, yellow powder). The product was characterised in situ, although has limited stability in solution.
1
H NMR (1,2-C6H4F2/C6D6, 500 MHz): δ 8.11–8.15 (m, 8H, ArF), 7.49 (br, 4H, ArF), 4.52 (d, 2JHH = 12.6, 2H, ArC2Ar), 4.51 (d, 2JHH = 12.6, 2H, ArC2Ar), 4.05–4.19 (m, 2H, OCH2), 3.88–3.99 (m, 2H, OCH2), 3.84 (s, 6H, NCH3), 3.51–3.65 (m, 4H, OCH2), 3.18 (d, 2JHH = 12.6, 2H, ArC2Ar), 3.15 (d, 2JHH = 12.6, 2H, ArC2Ar), 2.11–2.23 (m, 4H, C2CH3), 1.79–1.92 (m, 4H, C2CH3), 0.94 (t, 3JHH = 7.5, 6H, CH2C3), 0.93 (t, 3JHH = 7.5, 6H, CH2C3). Some signals obscured by solvent peak. 13C{1H} NMR (1,2-C6H4F2/C6D6, 126 MHz, selected signals only): δ 182.7 (d, 1JRhC = 53, CO), 181.0 (d, 1JRhC = 80, CO), 168.1 (d, 1JRhC = 43, NCN), 36.3 (s, NCH3). ESI-MS (CH3CN, 180 °C, 3 kV) positive ion: 1105.1680 m/z, [M]+ (calcd 1105.1891 m/z). IR (CH2Cl2, cm−1): ν(CO) 2083, 2016.
Preparation of 6b
A solution of [Rh(IiPr2Me2)(CO)2Cl] (26.5 mg, 0.709 mmol) and Na[BArF4] (32.6 mg, 0.368 mmol) in CH2Cl2 (5 mL) was stirred for one hour. The solution was then filtered and layered with pentane to afford the product on diffusion. Yield = 37.1 mg (66%, yellow crystals).
1
H NMR (CD2Cl2, 400 MHz): δ 7.67–7.77 (m, 8H, ArF), 7.56 (br, 4H, ArF), 5.20 (sept, 3JHH = 7.1, 4H, NCH), 2.18 (s, 12H CC3), 1.55 (d, 3JHH = 7.1, 12H, CHC3), 1.50 (d, 3JHH = 7.0, 12H, CHC3). 13C{1H} NMR (CD2Cl2, 101 MHz): δ 185.4 (d, 1JRhC = 53, CO), 181.0 (d, 1JRhC = 84, CO), 162.3 (q, 1JBC = 50, ArF), 135.4 (s, ArF), 129.4 (qq, 2JFC = 32, 3JBC = 3, ArF), 128.0 (s, CCH3), 125.2 (q, 1JFC = 272, ArF), 54.8 (s, NCH), 22.8 (s, CHH3)2, 22.4 (s, CHH3)2, (s, 10.6 CH3). The carbene resonance was not located. ESI-MS (CH3CN, 180 °C, 3 kV) positive ion: 713.0829 m/z, [M]+ (calcd 713.0843 m/z). IR (CH2Cl2, cm−1): ν(CO) 2090, 2019. Anal. Calcd For C58H52BClF24N4O4Rh2Cl (1577.11 g mol−1): C, 44.17; H, 3.32; N, 3.55. Found: C, 44.51; H, 3.57; N, 3.49.
Preparation of 7
A solution of IiPr2Me2 (50.8 mg, 0.282 mmol), [Rh(COD)Cl]2 (34.3 mg, 0.0696 mmol) and Na[BArF4] (123.4 mg, 0.139 mmol) was stirred in 1,2-C6H4F2 (5 mL) for 2.5 h. The solution was then filtered and layered with pentane to afford the product on diffusion. Yield = 114.0 mg (56%, yellow crystals).
1
H NMR (CD2Cl2, 500 MHz): δ 7.71–7.75 (m, 8H, ArF), 7.56 (br, 4H, ArF), 5.56 (sept, 3JHH = 7.1, 4H, NCH), 4.27 (s, 4H, COD{CH}), 2.31–2.40 (m, 4H, COD{CH2}), 2.16 (s, 12H, CCH3), 2.01–2.11 (m, 4H, COD{CH2}), 1.55 (d, 3JHH = 7.1, 12H, CHC3), 1.22 (d, 3JHH = 7.1, 12H, CHC3). 13C{1H} NMR (CD2Cl2, 126 MHz): δ 178.1 (d, 1JRhC = 55.8, NCN), 162.3 (q, 1JBC = 50, ArF), 135.4 (s, ArF), 129.4 (qq, 2JFC = 32, 3JBC = 3, ArF), 127.1 (s, CH3), 125.2 (q, 1JFC = 272, ArF), 118.0 (sept, 3JFC = 4, ArF), 87.2 (d, 1JRhC = 8, COD{CH}), 54.5 (s, NCH), 31.8 (s, COD{CH}), 22.9 (s, CHH3), 21.6 (s, CHH3), 10.9 (s, CH3). ESI-MS (CH3CN, 180 °C, 3 kV) positive ion: 571.3215 m/z [M]+ (calcd 571.3242 m/z). Anal. Calcd For C62H64BF24N4Rh (1434.39 g mol−1): C, 51.96; H, 4.49; N, 3.90. Found: C, 52.05; H, 4.59; N, 3.93.
Preparation of 8
A solution of 7 (113.9 mg, 0.0794 mmol) in 1,2-C6H4F2 (5 mL) was stirred under an atmosphere of CO (1 atm) for 24 hours. The solvent was removed in vacuo and the resulting crude material washed with pentane and recrystallised from CH2Cl2/pentane. Yield = 72.4 mg (66%, orange powder).
1
H NMR (CD2Cl2, 500 MHz): δ 7.71–7.75 (m, 8H, ArF), 7.56 (br, 4H, ArF), 5.05 (sept, 3JHH = 7.1, 4H, NCH), 2.20 (s, 12H, CCH3), 1.46 (d, 3JHH = 7.1, 12H, CHC3), 1.19 (d, 3JHH = 7.1, 12H, CHC3). 13C{1H} NMR (CD2Cl2, 126 MHz): δ 186.9 (d, 1JRhC = 57, CO), 168.2 (d, 1JRhC = 47, NCN), 162.3 (q, 1JBC = 50, ArF), 135.3 (s, ArF), 129.4 (qq, 2JFC = 32, 3JBC = 3, ArF), 128.2 (s, CH3), 125.2 (q, 1JFC = 272, ArF), 118.0 (sept, 3JFC = 4, ArF), 55.3 (s, NCH), 22.0 (s, CHH3), 21.1 (s, CHH3), 10.9 (s, CH3). ESI-MS (CH3CN, 180 °C, 3 kV) positive ion: 519.2249 m/z [M]+ (calcd 520.2201 m/z). IR (CH2Cl2, cm−1): ν(CO) 2077, 2018. Anal. Calcd For C56H52BF24N4Rh (1382.29 g mol−1): C, 48.64; H, 3.79; N, 4.05. Measured: C, 48.51; H, 3.63; N, 4.15.
Crystallography
Full details about the collection, solution and refinement are documented in the CIF, which have been deposited with the Cambridge Crystallographic Data Centre under CCDC 1448987–1448996.Acknowledgements
We thank the Royal Society (A. B. C.), EPSRC (R. P.) and University of Warwick for financial support. Crystallographic and high-resolution mass-spectrometry data were collected using instruments purchased through support from Advantage West Midlands and the European Regional Development Fund.References
- (a) S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612–3676 CrossRef PubMed; (b) C. Samojłowicz, M. Bieniek and K. Grela, Chem. Rev., 2009, 109, 3708–3742 CrossRef PubMed; (c) F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122–3172 CrossRef CAS PubMed; (d) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed.
- L. Benhamou, E. Chardon, G. Lavigne, S. Bellemin-Laponnaz and V. Cesar, Chem. Rev., 2011, 111, 2705–2733 CrossRef CAS PubMed.
- (a) R. E. Andrew, L. González-Sebastián and A. B. Chaplin, Dalton Trans., 2016, 45, 1299–1305 RSC; (b) X. Hu, I. Castro-Rodriguez and K. Meyer, J. Am. Chem. Soc., 2003, 125, 12237–12245 CrossRef CAS PubMed; (c) E. Peris and R. H. Crabtree, Coord. Chem. Rev., 2004, 248, 2239–2246 CrossRef CAS; (d) J. A. Mata, M. Poyatos and E. Peris, Coord. Chem. Rev., 2007, 251, 841–859 CrossRef CAS; (e) D. Pugh and A. A. Danopoulos, Coord. Chem. Rev., 2007, 251, 610–641 CrossRef CAS; (f) M. Poyatos, J. A. Mata and E. Peris, Chem. Rev., 2009, 109, 3677–3707 CrossRef CAS PubMed; (g) C. Radloff, H.-Y. Gong, C. Schulte to Brinke, T. Pape, V. M. Lynch, J. L. Sessler and F. E. Hahn, Chem. – Eur. J., 2010, 16, 13077–13081 CrossRef CAS PubMed; (h) R. E. Andrew and A. B. Chaplin, Dalton Trans., 2014, 43, 1413–1423 RSC.
- (a) V. Böhmer, Angew. Chem., Int. Ed. Engl., 1995, 34, 713–745 CrossRef; (b) B. R. Cameron, S. J. Loeb and G. P. A. Yap, Inorg. Chem., 1997, 36, 5498–5504 CrossRef CAS; (c) C. Wieser, C. Dieleman and D. Matt, Coord. Chem. Rev., 1997, 165, 93–161 CrossRef CAS; (d) P. Molenveld, J. Engbersen and D. Reinhoudt, Chem. Soc. Rev., 2000, 29, 75–86 RSC; (e) P. D. Harvey, Coord. Chem. Rev., 2002, 233–234, 289–309 CrossRef CAS; (f) C. Jeunesse, D. Armspach and D. Matt, Chem. Commun., 2005, 2005, 5603–5614 RSC; (g) D. M. Homden and C. Redshaw, Chem. Rev., 2008, 108, 5086–5130 CrossRef CAS PubMed; (h) B. S. Creaven, D. F. Donlon and J. McGinley, Coord. Chem. Rev., 2009, 253, 893–962 CrossRef CAS.
- Selected examples relevant to this work. (a) T. Brendgen, M. Frank and J. Schatz, Eur. J. Org. Chem., 2006, 2378–2383 CrossRef CAS; (b) E. Brenner, D. Matt, M. Henrion, M. Teci and L. Toupet, Dalton Trans., 2011, 40, 9889–9898 RSC; (c) H. Ren, Y. Xu, E. Jeanneau, I. Bonnamour, T. Tu and U. Darbost, Tetrahedron, 2014, 70, 2829–2837 CrossRef CAS.
- I. Dinarès, C. Garcia de Miguel, M. Font-Bardia, X. Solans and E. Alcalde, Organometallics, 2007, 26, 5125–5128 CrossRef.
- (a) M. Frank, G. Maas and J. Schatz, Eur. J. Org. Chem., 2004, 607–613 CrossRef CAS; (b) T. Fahlbusch, M. Frank, G. Maas and J. Schatz, Organometallics, 2009, 28, 6183–6193 CrossRef CAS.
- (a) C. Wieser-Jeunesse, D. Matt and A. De Cian, Angew. Chem., Int. Ed., 1998, 37, 2861–2864 CrossRef CAS; (b) M. Lejeune, C. Jeunesse, D. Matt, N. Kyritsakas, R. Welter and J.-P. Kintzinger, J. Chem. Soc., Dalton Trans., 2002, 1642–1650 RSC.
- Dinuclear complexes, polymeric complexes, and mononuclear complexes with cis-coordination geometries can also be obtained using this and related diphosphine ligands: (a) I. A. Bagatin, D. Matt, H. Thönnessen and P. G. Jones, Inorg. Chem., 1999, 38, 1585–1591 CrossRef CAS; (b) X. Fang, B. L. Scott, J. G. Watkin, C. A. G. Carter and G. J. Kubas, Inorg. Chim. Acta, 2001, 317, 276–281 CrossRef CAS; (c) K. Takenaka, Y. Obora, L. H. Jiang and Y. Tsuji, Organometallics, 2002, 21, 1158–1166 CrossRef CAS; ref. 8b; (d) F. Plourde, K. Gilbert, J. Gagnon and P. D. Harvey, Organometallics, 2003, 22, 2862–2875 CrossRef CAS; (e) M. Lejeune, D. Sémeril, C. Jeunesse, D. Matt, F. Peruch, P. J. Lutz and L. Ricard, Chem. – Eur. J., 2004, 10, 5354–5360 CrossRef CAS PubMed; (f) L. Monnereau, D. Sémeril, D. Matt, L. Toupet and A. J. Mota, Adv. Synth. Catal., 2009, 351, 1383–1389 CrossRef CAS; (g) S. Sameni, C. Jeunesse, M. Awada, D. Matt and R. Welter, Eur. J. Inorg. Chem., 2010, 2010, 4917–4923 CrossRef; (h) L. Monnereau, D. Sémeril, D. Matt and L. Toupet, Polyhedron, 2013, 51, 70–74 CrossRef CAS.
- N. Kuhn and T. Kratz, Synthesis, 1993, 561–562 CrossRef CAS.
- I. Dinarès, C. Garcia de Miguel, N. Mesquida and E. Alcalde, J. Org. Chem., 2009, 74, 482–485 CrossRef PubMed.
- Preparation from 4-tert-butylcalix[4]arene: (a) C. D. Gutsche and L.-G. Lin, Tetrahedron, 1986, 42, 1633–1640 CrossRef CAS (alkyl group transfer; 1 day, 51% yield); (b) A. Dondoni, C. Ghiglione, A. Marra and M. Scoponi, Macromol. Chem. Phys., 1999, 200, 77–86 CrossRef CAS (alkylation; overnight reaction, 66% yield); (c) M. Larsen and M. Jørgensen, J. Org. Chem., 1996, 61, 6651–6655 CrossRef CAS PubMed (bromination; overnight reaction, 80% yield; lithium halogen exchange; 1 day, 49% yield); (d) Ref. 11 (Cu-catalyzed Ullmann reaction; ca. 7 day reaction, 62% yield).
- For two representative examples see: (a) J. Scheerder, R. H. Vreekamp, J. F. J. Engbersen, W. Verboom, J. P. M. van Duynhoven and D. N. Reinhoudt, J. Org. Chem., 1996, 61, 3476–3481 CrossRef CAS; (b) M. Conner, V. Janout and S. L. Regen, J. Am. Chem. Soc., 1991, 113, 9670–9671 CrossRef CAS.
- (a) D. Tapu, D. A. Dixon and C. Roe, Chem. Rev., 2009, 109, 3385–3407 CrossRef CAS PubMed; (b) T. Dröge and F. Glorius, Angew. Chem., Int. Ed., 2010, 49, 6940–6952 CrossRef PubMed.
- (a) S.-I. Fuku-en, J. Yamamoto, M. Minoura, S. Kojima and Y. Yamamoto, Inorg. Chem., 2013, 52, 11700–11702 CrossRef CAS PubMed; (b) G. T. S. Andavan, E. B. Bauer, C. S. Letko, T. K. Hollis and F. S. Tham, J. Organomet. Chem., 2005, 690, 5938–5947 CrossRef CAS; (c) M. V. Baker, S. K. Brayshaw, B. W. Skelton and A. H. White, Inorg. Chim. Acta, 2004, 357, 2841–2849 CrossRef CAS; (d) M. Poyatos, M. Sanau and E. Peris, Inorg. Chem., 2003, 42, 2572–2576 CrossRef CAS PubMed.
- (a) I. J. B. Lin and C. S. Vasam, Coord. Chem. Rev., 2007, 251, 642–670 CrossRef CAS; (b) J. C. Garrison and W. J. Youngs, Chem. Rev., 2005, 105, 3978–4008 CrossRef CAS PubMed.
- E. Oehlke, T. Kückmann and U. Abram, Z. Anorg. Allg. Chem., 2007, 633, 830–834 CrossRef CAS.
- (a) S. A. Hauser, R. Tonner and A. B. Chaplin, Organometallics, 2015, 34, 4419–4427 CrossRef CAS; (b) L. J. Sewell, M. A. Huertos, M. E. Dickinson, A. S. Weller and G. C. Lloyd-Jones, Inorg. Chem., 2013, 52, 4509–4516 CrossRef CAS PubMed; (c) C. Y. Tang, J. Lednik, D. Vidovic, A. L. Thompson and S. Aldridge, Chem. Commun., 2011, 47, 2523–2525 RSC; (d) F. Ekkehardt Hahn, B. Heidrich, T. Pape, A. Hepp, M. Martín, E. Sola and L. A. Oro, Inorg. Chim. Acta, 2006, 359, 4840–4846 CrossRef.
- G. Giordano, R. H. Crabtree, R. M. Heintz, D. Forster and D. E. Morris, Inorg. Synth., 1990, 28, 88–90 CrossRef CAS.
- J. A. McCleverty, G. Wilkinson, L. G. Lipson, M. L. Maddox and H. D. Kaesz, Inorg. Synth., 1990, 28, 84–86 CrossRef CAS.
- W. E. Buschmann, J. S. Miller, K. Bowman-James and C. N. Miller, Inorg. Synth., 2002, 33, 83–91 CAS.
- A. Neveling, G. R. Julius, S. Cronje, C. Esterhuysen and H. G. Raubenheimer, Dalton Trans., 2005, 181–192 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR, IR and ESI-MS spectra of new complexes. CCDC 1448987–1448996. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt01001f |
| This journal is © The Royal Society of Chemistry 2016 |