
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thermal and near-infrared light induced spin crossover in a mononuclear iron(II) complex with a tetrathiafulvalene-fused dipyridophenazine ligand†
F.
Pointillart
*a,
X.
Liu
b,
M.
Kepenekian
a,
B.
Le Guennic
a,
S.
Golhen
a,
V.
Dorcet
a,
T.
Roisnel
a,
O.
Cador
a,
Z.
You‡
b,
J.
Hauser
b,
S.
Decurtins
b,
L.
Ouahab
a and
S.-X.
Liu
*b
aInstitut des Sciences Chimiques de Rennes, UMR 6226 CNRS - Université de Rennes 1, 35042, Rennes Cedex, France. E-mail: fabrice.pointillart@univ-rennes1.fr; Fax: +33(0)2 23 23 68 40; Tel: +33(0)2 23 23 67 52
bDepartement für Chemie und Biochemie, Universität Bern, Freiestrasse 3, CH-3012 Bern, Switzerland. E-mail: liu@dcb.unibe.ch
First published on 24th June 2016
Abstract
A mononuclear Fe(II) complex involving a tetrathiafulvalene-based ligand exhibits thermal spin-crossover (around 143 K) with pronounced hysteresis behaviour (48 K). The chromophoric and π-extended ligand allows Near-Infrared (NIR) sensitization for the light-induced excited spin-state trapping (LIESST) with T(LIESST) = 90 K.
Molecular magnetism is a very active field where both chemist and physicist communities are looking for systems with high-density memory capabilities. In this context, Single Molecule Magnets (SMM)1 and Spin Crossover (SCO) materials2 are intensively studied because of their potential applications.3 SCO behaviour has been known since the 1930s.4 In this respect, the Fe(II) ion is the most popular magnetic centre owing to its large variation of the spin value (S = 2 → S = 0), the largest possible for 3d metal centres along the high-spin (HS) to low-spin (LS) transition. A crucial parameter for device applications is the presence of magnetic bistability that is driven mainly by the cooperativity induced by intermolecular interactions in the crystal lattice. Thus, the switching of spin states can be induced by changing the size of the coordinated ligands, counter ions and/or the molecules of crystallization.5 The magnetic bistability can also be reached under kinetic conditions by light irradiation at low temperatures (light-induced excited spin state trapping, LIESST).6 All these aspects have already been studied on several derivatives of [Fe(H2Bpz2)2(X)] complexes (H2Bpz2 = dihydrobis(1-pyrazolyl)borate, X = 2,2′-bipyridine, 1,10-phenanthroline, dipyrido-[3,2-a:2′,3′-c]phenazine (dppz)).5,7 Moreover, the SCO behaviour of such derivatives has recently been observed upon deposition on a gold surface and in thin films.8 Herein, we propose to extend the π-system of the dppz ligand through fusion with a tetrathiafulvalene (TTF) derivative in order (i) to combine redox activity with SCO behaviour,9 (ii) to increase the cooperativity via enhancement of π-stacking interactions, thus the hysteretic behaviour around the transition temperature,10 and (ii) to induce strong intra-ligand charge transfer (ILCT) transitions reaching the NIR spectral region for LIESST investigation. Sensitization of the LIESST effect through ILCT excitations could present some advantages such as (i) the ILCT bands allow lower-energy excitations than the MLCT and (ii) they can be easily modulated in energy through chemical changes of the push–pull ligands and (iii) the absorption intensity is higher than forbidden MLCT transitions.
In this article, we report the first TTF-based complex of Fe(II) displaying both thermal and photo-induced spin crossover. One should mention here that Oshio et al. previously proposed an iron(II) complex containing a TTF moiety that shows thermal spin-crossover but without a photomagnetism study.11 [Fe(H2Bpz2)2(L)]·(CH2Cl2)2, called 1·(CH2Cl2)2, was elaborated from the in situ formation of the Fe(H2Bpz2)2 precursor followed by coordination with the TTF-fused dppz ligand (L).12 The photophysical properties of the related [Fe(phen)2(L)]2+ complex have been reported.13
Compound 1·(CH2Cl2)2 crystallises in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2).§ The ORTEP drawing of 1 is shown in Fig. 1 (150 K). The asymmetric unit is composed of one Fe(H2Bpz2)2 moiety, one L ligand, one water molecule and 1.5 dichloromethane molecules of crystallisation. The L ligand shows a chelating N^N bonding to Fe(II). Its distorted octahedral coordination environment stems from six nitrogen atoms of one L and two H2Bpz2 ligands. The average Fe–N distance is 2.105(3) Å which is slightly shorter than that observed for a Fe(II) HS (S = 2) state in analogue compounds without the TTF moiety (Table S1†).5b,7a,c The central C9–C10 bond length (1.343(5) Å) attests the neutral charge state of L.14L is planar which allows for an optimal π-overlap between two “head-to-tail” molecules in the bulk phase (Fig. 2). The shortest intermolecular S⋯S distances are 4.002 Å (S5⋯S5) and 4.065 Å (S5⋯S6). The crystal packing reveals intermolecular CH⋯π (C16⋯H30 = 2.922 Å and C32⋯H26 = 2.869 Å) and BH⋯π interactions between H2Bpz2 and the benzyl fused to TTF (C12⋯H31 = 2.869 Å and B2⋯H16 = 2.772 Å).
(no. 2).§ The ORTEP drawing of 1 is shown in Fig. 1 (150 K). The asymmetric unit is composed of one Fe(H2Bpz2)2 moiety, one L ligand, one water molecule and 1.5 dichloromethane molecules of crystallisation. The L ligand shows a chelating N^N bonding to Fe(II). Its distorted octahedral coordination environment stems from six nitrogen atoms of one L and two H2Bpz2 ligands. The average Fe–N distance is 2.105(3) Å which is slightly shorter than that observed for a Fe(II) HS (S = 2) state in analogue compounds without the TTF moiety (Table S1†).5b,7a,c The central C9–C10 bond length (1.343(5) Å) attests the neutral charge state of L.14L is planar which allows for an optimal π-overlap between two “head-to-tail” molecules in the bulk phase (Fig. 2). The shortest intermolecular S⋯S distances are 4.002 Å (S5⋯S5) and 4.065 Å (S5⋯S6). The crystal packing reveals intermolecular CH⋯π (C16⋯H30 = 2.922 Å and C32⋯H26 = 2.869 Å) and BH⋯π interactions between H2Bpz2 and the benzyl fused to TTF (C12⋯H31 = 2.869 Å and B2⋯H16 = 2.772 Å).
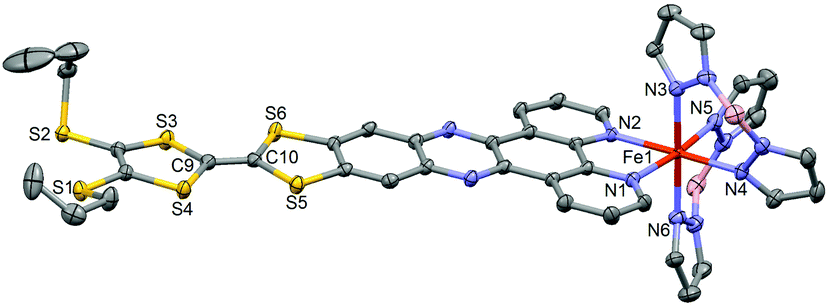 | ||
| Fig. 1 ORTEP view of the structure of 1·(CH2Cl2)2. Atomic displacement ellipsoids are drawn at the 50% probability level. Hydrogen atoms are omitted for clarity. | ||
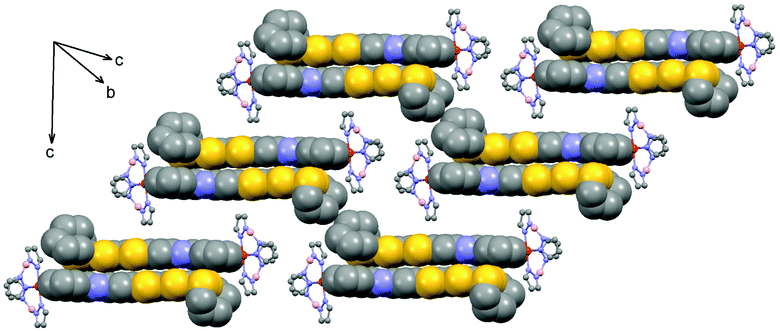 | ||
| Fig. 2 Crystal packing of 1 (HS) in 1·(CH2Cl2)2 highlighting the “head-to-tail” dimers. | ||
The cyclic voltammogram of 1 shows three single-electron redox waves at −1.11 V, 0.71 V and 1.06 V corresponding to the formation of the dppz radical anion, the radical cation and the dication of the TTF fragment, respectively (Fig. S1†). These oxidation potentials are comparable to a range of functionalized TTF donors.15 Upon coordination to Fe(II), the dppz-centred reduction wave is anodically shifted from −1.23 V (ref. 12) for the free L to −1.11 V for 1 while the TTF-centred oxidation waves remain unchanged.16 These electrochemical properties look promising for controlling magnetic properties by changing oxidation states.
The UV-visible absorption of 1 was studied in CH2Cl2 solution (Fig. 3) and in the solid state (Fig. S2†). TD-DFT calculations were performed to rationalize the absorption spectrum (Fig. S3, S4 and Table S2†), however considering the Fe(II) ion in its LS (S = 0) state (see the ESI† for computational details). The lowest energy bands were attributed to the ILCT transitions from the TTF to the dppz moieties which in the solid state gives rise to an absorption tail that stretches out into the NIR (800 nm, 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 cm−1) region. These electronic excitations as well as those at higher energies were in good agreement with the calculated ones (Table S2†).
500 cm−1) region. These electronic excitations as well as those at higher energies were in good agreement with the calculated ones (Table S2†).
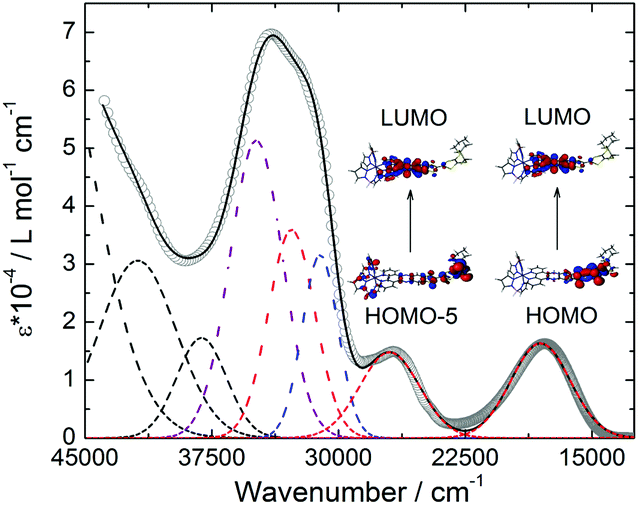 | ||
| Fig. 3 UV-visible absorption spectrum of 1 in CH2Cl2 solution (c = 2 × 10−5 mol L−1) with respective Gaussian deconvolutions (dashed lines) and best fit (full black line) (R = 0.9991). Colour code: red, ILCT; blue, LLCT (Ligand to Ligand Charge Transfer); purple IL and black IA. Representation of the molecular orbitals involved in the low-energy ILCT. | ||
Magnetic measurements clearly indicate that 1·(CH2Cl2)2 remains in the HS state (S = 2; χMT = 3.5 cm3 K mol−1) down to low temperatures (Fig. 4). Thermogravimetric analysis reveals that the sample loses the solvent of crystallization when heated up to 120 °C (Fig. S5†). After the initial loss of the wetting solvent of a fresh sample, the observed mass loss (14%) conforms to a full desolvation process (theo, 15%). After desolvation, the magnetic behaviour of 1 is radically different since it shows a HS to LS transition centred at 143 K with a wide thermal hysteresis (48 K) which can be cycled (Fig. S6†). The transition occurs at TC↓ = 119 K in the cooling mode and TC↑ = 167 K in the warming mode. Both curves have been recorded at a rate of 0.3 K min−1. The transition occurs in the same temperature range as in the parent H2Bpz2 ligand based compounds,5b,7 but with larger thermal hysteresis. It is expected that the loss of solvent from the crystal structure increases the overlap between TTF moieties (Fig. 2), leading to the extension of the interaction between the iron(II) centres. Unfortunately, the structural modifications upon the desolvation process are so severe that crystals crack and do not diffract anymore.
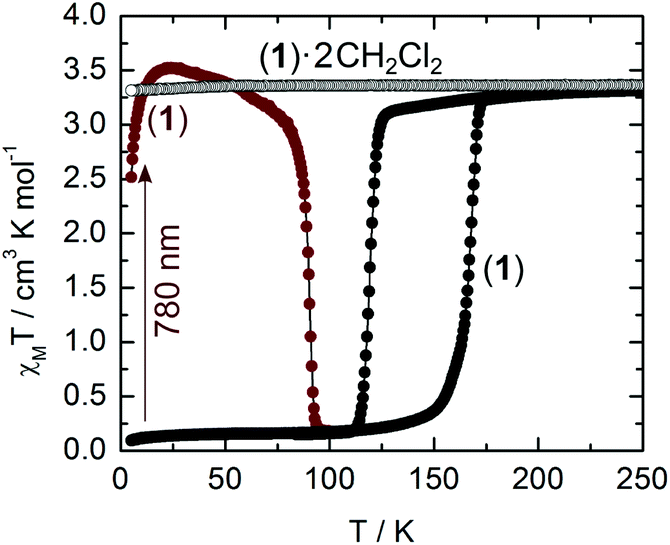 | ||
| Fig. 4 Thermal dependencies of χMT of the solvated (empty circles) and desolvated (full circles) forms of 1. Wine colour represents χMT in the low temperature range after light irradiation at 780 nm (12820 cm−1, 5 mW) for 5 min. | ||
Starting from the desolvated form (1), irradiation at 780 nm (12820 cm−1) in the tail of the ILCT band (Fig. S2†), at 10 K, induces within minutes a complete spin change from LS to HS (Fig. 4). After the light has been switched off, the population of the metastable 5T2g (Oh) state persists up to TLIESST = 90 K at a sweep rate of 0.3 K min−1. This value is much higher than the one (44 K) reported for [Fe(H2Bpz2)2phen].7b Irradiation at 530 nm (18870 cm−1) and 660 nm (15150 cm−1) produces the same quantitative LIESST effect while the irradiation at 404 nm (24750 cm−1), outside the HOMO → LUMO band (Fig. S2†), is less efficient. Excitation in the low-energy singlet ILCT band (780 nm) might induce fast relaxation to the triplet T1 level of FeII and progressive population of the quintet T2 state of the HS FeII ion. Even if we are aware that the sensitization process is controversial,17 a hypothetical proposed mechanism for the LIESST effect is given in Fig. S7.†
The spectacular difference in magnetic behaviour of 1·(CH2Cl2)2 and 1 calls for further investigation. Electronic structure calculations have been conducted on the periodic crystal structure using the density functional theory (DFT) framework as implemented in the VASP code.18 The Perdew–Buke–Ernzerhof form generalized gradient approximation was used for the exchange and correlation functional.19 Wave functions were expanded using a plane-wave basis set with a cutoff energy of 400 eV. Core electrons were treated within the projector augmented wave method.20 The simulation cell contains two molecules of 1 and, in the case of the solvated structure, three molecules of CH2Cl2 and two of H2O. The latter structure was chosen to account for (i) the presence of two CH2Cl2 molecules and (ii) the disorder created at the two half-occupied sites in the crystal structure of 1·(CH2Cl2)2, that are mimicked by two H2O molecules. Given the size of the cell and the nature of the system, Γ-point calculations turn out to give satisfactory accuracy. Geometries were optimized until the forces were smaller than 0.01 eV Å−1. Forces were corrected for the missing van der Waals interactions using the Grimme scheme.21 It is known that usual functionals fail to take into account the strongly correlated character of iron d-electrons. Therefore, a Hubbard-like term is added to the Kohn–Sham Hamiltonian,22 controlled by an effective term Ueff.23 Several propositions have been formulated regarding the appropriate value for Ueff in the case of SCO compounds ranging from 1.55 to 2.65 eV.24 As we did not intend to reach a quantitative agreement, but rather a qualitative description of the physical processes, we used a value Ueff = 2.00 eV. We checked that a value of 3.00 eV was not affecting dramatically the geometry of the systems (variations are less than 0.5%). For both solvated and non-solvated compounds, the HS (S = 2) and LS (S = 0) solutions have been considered. In the case of the HS state with solvents, the resulting optimized cell is in good agreement with the experimental structure (Table S3†). In particular, the optimized HS molecule shows deviations of less than 3% in bond lengths (Table S4†).
In order to achieve SCO, a system has to possess specific features. In particular, the enthalpy should favour the LS state since the entropic contributions (electronic and vibrational) will benefit to the HS state. This is characterized by the adiabatic gap ΔEadia = EHS − ELS, i.e. the energy difference between the HS and LS states each considered in their own geometry. As a necessary, but not sufficient condition, ΔEadia should be positive. In the case of solvated 1 (Fig. 5a), ΔEadia = −0.40 eV, which leads to an HS system whatever the temperature, in agreement with the experimental observation. The situation is different with 1. Indeed, in the absence of solvents, the LS state is stabilized and ΔEadia reaches 0.41 eV and SCO behaviour becomes possible.
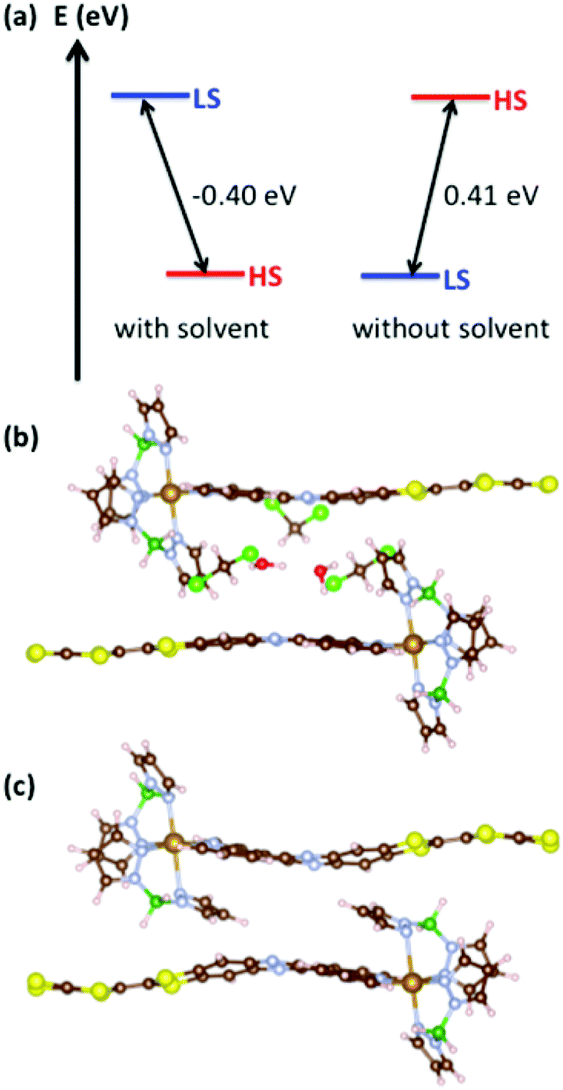 | ||
| Fig. 5 (a) Adiabatic gap ΔEadia = EHS − ELS for 1 with and without solvent molecules (CH2Cl2 and H2O), calculated at the PBE + D3 + U level (Ueff = 2.0 eV). (b), (c) Optimized geometry of the HS state with and without solvent, respectively. Fe, Cl, S, O, N, C, B and H atoms are depicted in orange, light green, yellow, red, blue, brown, dark green, and pink, respectively. Without solvent molecules, the HS geometry shows a competition between Fe-coordination and the π–π interaction between H2Bpz2 and the ligand L. | ||
When considering the optimized geometries of the HS and LS solutions with and without solvents (Fig. S8 and Table S4†), it becomes clear that the variation of the adiabatic gap can be traced back to a competition between Fe-coordination and π–π interactions. In particular, when considering the HS solution with and without solvents (Fig. 5b, c and Table S4†), one can notice that the H2Bpz2 ligand opts either for a Fe-coordination which leads to an expected Fe–N bond of 2.15 Å, or without solvent, an interaction with the π-system of L which leads to a much longer bond (2.42 Å). The distortion of the coordination sphere leads to a destabilization of the HS solution and makes SCO possible.
As a final remark, let us note that in the absence of solvent, the Fe–Fe distance goes from 13.48 Å to 13.42 Å when going from HS to LS. It is known that a limited contraction in the Fe–Fe distance along the HS to LS transition favours the electrostatic contribution to cooperativity hence to the opening of a hysteresis loop.25 In addition to the existing π-network, this strongly supports the potential of TTF-based compounds as candidates for SCO systems with large hysteresis.
To summarize, compound 1 was obtained from the association of the TTF-fused dipyrido-[3,2-a:2′,3′-c]phenazine ligand (L) and the metallo-precursor Fe(H2Bpz2)2. This neutral heteroleptic complex presents both thermal and light-induced spin transitions. The π-extended chromophoric ligand L exhibits an intense and energetically low-lying ILCT absorption band, which enables sensitization of LIESST in the NIR spectral region. The analogous NIR antenna effect of this ligand has recently been described for the NIR emission of Yb(III) doped silica nanoparticles.26 The large π-system of L increases as well the interaction between the magnetic centres which leads to one of the largest thermal hysteresis for a mononuclear spin transition complex of Fe(II). The use of a TTF-based ligand provides a promising possibility to have high temperature LIESST effects and large thermal hysteresis loops as well as to use the redox activity of the TTF ligand to switch the magnetic properties.
This work was supported by the CNRS, Rennes Métropole, Université de Rennes 1, Région Bretagne, FEDER, Agence Nationale de la Recherche (no. ANR-13-BS07-0022-01) and the Swiss National Science Foundation (no. 200021-147143).
Notes and references
-
(a) R. Sessoli, D. Gatteschi, A. Caneschi and M. A. Novak, Nature, 1993, 365, 141–143 CrossRef CAS
; (b) G. Aromi and E. K. Brechin, in Single-Molecule Magnets and Related Phenomena, ed. R. Winpenny, 2006, p. 1–67 Search PubMed
-
(a) P. Gütlich, Y. Garcia and H. A. Goodwin, Chem. Soc. Rev., 2000, 29, 419–427 RSC
-
(a) L. Bogani and W. Wernsdorfer, Nat. Mater., 2008, 7, 179–186 CrossRef CAS PubMed
- L. Cambi and A. Gagnasso, Atti. Accad. Naz. Lincei, 1931, 13, 809 CAS
-
(a) D. T. Roberts, A. M. Little, F. Tuna, C. A. Kilnera and M. A. Halcrow, Chem. Commun., 2013, 49, 6280–6282 RSC
-
(a) S. Decurtins, P. Gütlich, C. P. Köhler, H. Spiering and A. Hauser, Chem. Phys. Lett., 1984, 105, 1–4 CrossRef CAS
-
(a) J. A. Real, M. C. Muñoz, J. Faus and X. Solans, Inorg. Chem., 1997, 36, 3008–3013 CrossRef CAS PubMed
-
(a) T. G. Gopakumar, F. Matino, H. Naggert, A. Bannwarth, F. Tuczek and R. Berndt, Angew. Chem., Int. Ed., 2012, 51, 6262–6266 CrossRef CAS PubMed
- H. S. Scott, C. J. Gartshore, S.-X. Guo, B. Moubaraki, A. M. Bond, S. R. Batten and K. S. Murray, Dalton Trans., 2014, 43, 15212–15220 RSC
- A. Santoro, L. J. K. Cook, R. Kulmaczewski, S. A. Barret, O. Cespedes and M. A. Halcrow, Inorg. Chem., 2015, 54, 682–693 CrossRef CAS PubMed
- M. Nihei, N. Takahashi, H. Nishikawa and H. Oshio, Dalton Trans., 2011, 40, 2154–2156 RSC
- C. Jia, S.-X. Liu, C. Tanner, C. Leiggener, A. Neels, L. Sanguinet, E. Levillain, S. Leutwyler, A. Hauser and S. Decurtins, Chem. – Eur. J., 2007, 13, 3804–3812 CrossRef CAS PubMed
- N. Dupont, Y.-F. Ran, S.-X. Liu, J. Grilj, E. Vauthey, S. Decurtins and A. Hauser, Inorg. Chem., 2013, 52, 306–312 CrossRef CAS PubMed
- P. Guionneau, C. J. Kepert, G. Bravic, D. Chasseau, M. R. Truter, M. Kurmoo and P. Day, Synth. Met., 1997, 86, 1973 CrossRef CAS
-
(a) D. Lorcy, N. Bellec, M. Fourmigué and N. Avarvari, Coord. Chem. Rev., 2009, 253, 1398–1438 CrossRef CAS
- F. Pointillart, J. Jung, R. Berraud-Pache, B. Le Guennic, V. Dorcet, S. Golhen, O. Cador, O. Maury, Y. Guyot, S. Decurtins, S.-X. Liu and L. Ouahab, Inorg. Chem., 2015, 54, 5384–5397 CrossRef CAS PubMed
- G. Auböck and M. Chergui, Nat. Chem., 2015, 7, 629–633 CrossRef PubMed
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed
-
(a) P. E. Blöchl, Phys. Rev. B: Condens. Matter, 1994, 50, 17953–17979 CrossRef
- S. Grimme, J. Antony, S. Ehrlich and S. Krieg, J. Chem. Phys., 2010, 132, 154104–154124 CrossRef PubMed
-
(a) V. I. Anisimov, I. V. Solovyev, M. A. Korotin, M. T. Czyzyk and G. A. Sawatzky, Phys. Rev. B: Condens. Matter, 1993, 48, 16929–16934 CrossRef CAS
- S. L. Dudarev, G. A. Botton and S. Y. Savrasov, Phys. Rev. B: Condens. Matter, 1998, 57, 1505–1509 CrossRef CAS
-
(a) S. Lebègue, S. Pillet and J. G. Ángyán, Phys. Rev. B: Condens. Matter, 2008, 78, 024433 CrossRef
-
(a) M. Kepenekian, B. Le Guennic and V. Robert, Phys. Rev. B: Condens. Matter, 2009, 79, 094428 CrossRef
- G. Lapadula, D. Trummer, M. P. Conley, M. Steinmann, Y. F. Ran, S. Brasselet, Y. Guyot, O. Maury, S. Decurtins, S.-X. Liu and C. Copéret, Chem. Mater., 2015, 27, 2033–2039 CrossRef CAS
- S. Trofimenko, J. Am. Chem. Soc., 1967, 89, 3170–3177 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: Details of crystallography and physical measurements; Fig. S1–S6. CCDC 1448431. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt00920d |
| ‡ Present address: Department of Chemistry and Chemical Engineering, Liaoning Normal University, Dalian 116029, P.R. China. |
| § [Fe(H2Bpz2)2(L)]·(CH2Cl2)2 ((1)·(CH2Cl2)2). 16 mg of FeCl2·4H2O (0.04 mmol) were dissolved in 5 mL of degassed H2O under argon and then 14.8 mg of solid KH2Bpz2 (0.08 mmol)27 were added in one portion under stirring. The light yellow solid formed was dissolved by addition of 10 mL of CH2Cl2. The aqueous phase was removed and the organic phase was dried over MgSO4. A solution of 5 mL of CH2Cl2 containing 12.1 mg of L (0.02 mmol)12 was added to the organic phase. After 15 min of stirring, n-hexane was layered for a few days leading to the formation of dark blue single crystals of (1)·(CH2Cl2)2 which are suitable for X-ray diffraction studies. Yield (dried in air for two hours, based on L), 16.2 mg (71%). Anal. Calcd (%) for C42H40N12B2Cl4FeS6 (1)·(CH2Cl2)2: C 44.86, H 3.56, N 14.95; found: C 45.63, H 3.84, N 15.48. Crystal data for (1)·(CH2Cl2)2: C42H40N12B2Cl4FeS6, M = 1123.45 g mol−1, T = 150(2) K, triclinic space group P [Fe(H2Bpz2)2(L)] (1). The desolvation is followed by TGA measurement. 1 is obtained after heating the sample up to 120 °C. Anal. Calcd (%) for C40H36N12B2FeS6 (1): C 50.34, H 3.78, N 17.62; found: C 50.23, H 3.74, N 17.48. |
| This journal is © The Royal Society of Chemistry 2016 |