
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Towards 99mTc-based imaging agents with effective doxorubicin mimetics: a molecular and cellular study†
S.
Imstepf
a,
V.
Pierroz
ab,
P.
Raposinho
c,
M.
Felber
a,
T.
Fox
a,
C.
Fernandes
c,
G.
Gasser
a,
I. R.
Santos
c and
R.
Alberto
*a
aDepartment of Chemistry, University of Zurich, Winterthurerstrasse 190, CH-8057 Zurich, Switzerland
bInstitute of Molecular Cancer Research, University of Zurich, Winterthurerstrasse 190, CH-8057 Zurich, Switzerland
cCentro de Ciências e Tecnologias Nucleares, Universidade de Lisboa, Estrada Nacional 10, PT-2695-066 Bobadela LRS, Portugal
First published on 8th April 2016
Abstract
Doxorubicin is a clinical benchmark drug, which is applied in the treatment of numerous cancers. Known for its accumulation in the nucleus and ability to intercalate into DNA, it targets quickly dividing i.e. hypermitotic cells. Through this mechanism, it could be an ideal structural motif for a new class of imaging agents, given that the new entity approximates the in vitro profile of the parent drug. Here we describe design, synthesis and biological activity of a small array of Doxorubicin-metalloconjugates (M = 99mTc, Re). We demonstrate that the conjugates preferably accumulate in the nuclear compartment, tightly bind to DNA and retain an appreciable cytotoxicity. Moreover, the Re conjugates effectively act as inhibitors of the human Topoisomerase II enzyme, which is the widely accepted mechanism of action of the parent drug. Since the conjugates effectively mimic the in vitro behavior of native Doxorubicin, the 99mTc compounds are prospective imaging agents.
Introduction
Early medical intervention is decisive for the curing of cancer or prolonging survival.1 For this reason, timely detection of malignant alterations is crucial. Since macroscopic changes in cellular structures, i.e. neoplasms, become visible only after long dwell-times in the body (sometimes years), it is important to visualize even subtle changes in cellular metabolism early on. Functional imaging techniques are indispensable to probe such metabolic changes.2 Single photon emission computed tomography (SPECT) and positron emission tomography (PET) are functional imaging techniques, employing radiotracers, which are routinely used in the clinics. Both modalities rely on radiation detector devices, which provide external, non-invasive visualization of isotope radiation in the human body. The specific accumulation, distribution, retention, perfusion and clearance of the radiotracer provides information on function and metabolism of the organism.3An effective imaging agent needs to accumulate in the target tissue and is retained therein for the time of image acquisition. Furthermore, the uptake in target versus non-target tissue should be maximal. One strategy to achieve this selectivity is the labeling of macromolecules, such as tissue-specific peptides or antibodies. While labeling of these structural motifs is attractive because of the inherent targeting function, costs, issues of in vivo stability, stability under labeling conditions and incompatibility of physical half-life and homing time to the target can be problematic.4 Functional imaging with small molecules may therefore be favorable, as prominently exemplified by the majority of small molecule imaging probes in the clinics. For small molecules, “selective” accumulation is achieved by the upregulated metabolism of cancer cells compared to normal cells. This effect enhances the uptake ratio of a radiotracer in target vs. non-target tissue and hence allows for visualization and identification of rapidly dividing cells, as found in malignant tissue alterations. However, labelling of small molecules can pose significant challenges. The incorporation of an isotope into a chemical construct or the appending of a chelated radionuclide e.g. Tc-99m to a small molecule may significantly alter the delivery, retention, binding and clearance with respect to the parent molecule.5 These effects can lead to unwarranted distribution and pharmacokinetics of the tracer and ultimately to dismissal of the probe. Nonetheless, chemotherapeutic agents are ideal structural motifs to guide radiotracers into target tissue and effect accumulation therein, given that the radiotracer closely mimics the parent drug in vivo.
Doxorubicin (ADR) is a natural, broadband chemotherapeutic agent, which is known to intercalate strongly between DNA nucleobases.6–8 ADR is clinically well-proven and shows extensive activity against a variety of cancers.9–11 Its mode of action is commonly ascribed to the inhibition of Topoisomerase II and to the resulting damage during DNA replication, inducing apoptosis, the programmed cell death pathway.12 Since DNA is the primary target of ADR, it could well serve as a lead structure for a novel class of radiotracers intended to image hypermitotic tissue. Additionally, Elsinga et al. have shown that anthracycline-based radiotracers can be employed to probe P-glycoprotein (Pgp) in vivo in view of assessing if tumors are subject to multidrug resistance (MDR).13 Overexpression of Pgp in resistant tumors leads to an enhanced drug efflux and hence reduced effective drug concentration in tumor cells. This is typically associated with a poor therapy response to anthracycline-based chemotherapy agents, such as ADR. Neoadjuvant imaging with anthracycline-based radiotracers has therefore been proposed as a prognostic factor for therapy outcome and an indication for combined treatment with Pgp-modulators (e.g. verapamil,14 cyclosporine A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 15) during chemotherapy, in order to enhance the tumor response to anthracycline drugs. If successful this strategy would follow a “theranostic” approach: neoadjuvant imaging with an anthracycline-based imaging agent and follow-up treatment with the unmodified parent drug.
15) during chemotherapy, in order to enhance the tumor response to anthracycline drugs. If successful this strategy would follow a “theranostic” approach: neoadjuvant imaging with an anthracycline-based imaging agent and follow-up treatment with the unmodified parent drug.
Inspired by ADR's clinical relevance and the wide-spread availability of 99mTc for nuclear medical applications we set out to synthesize a small array of ADR-99mTc(CO)3 bioconjugates. We have previously reported on the first ADR-metalloconjugates (1,2 see Scheme 1) and observed that these derivatives efficiently targeted the cell's nucleus or mitochondria.16,17 In this study we expand this series with structurally differing ADR metalloconjugates (3–6), which were closely examined with regard to their capabilities of mimicking the parent drug in vitro. The “cold” Re-conjugates were scrutinized for binding affinity to DNA, inhibition of human Topoisomerase II and in vitro cytotoxicity since Re organometallic complexes were found in recent years to have an interesting cytotoxic profile.18 Studies on the cellular localization of the metalloconjugates were carried out by confocal fluorescence microscopy to assess their ability to target the cell's nucleus and the in vitro distribution was corroborated by two independent quantification modalities. In addition, we present a new key intermediate for C14-derivatives of ADR, which allows for easily accessible derivatives of the parent drug, while the integrity of the daunosamine sugar moiety is conserved.
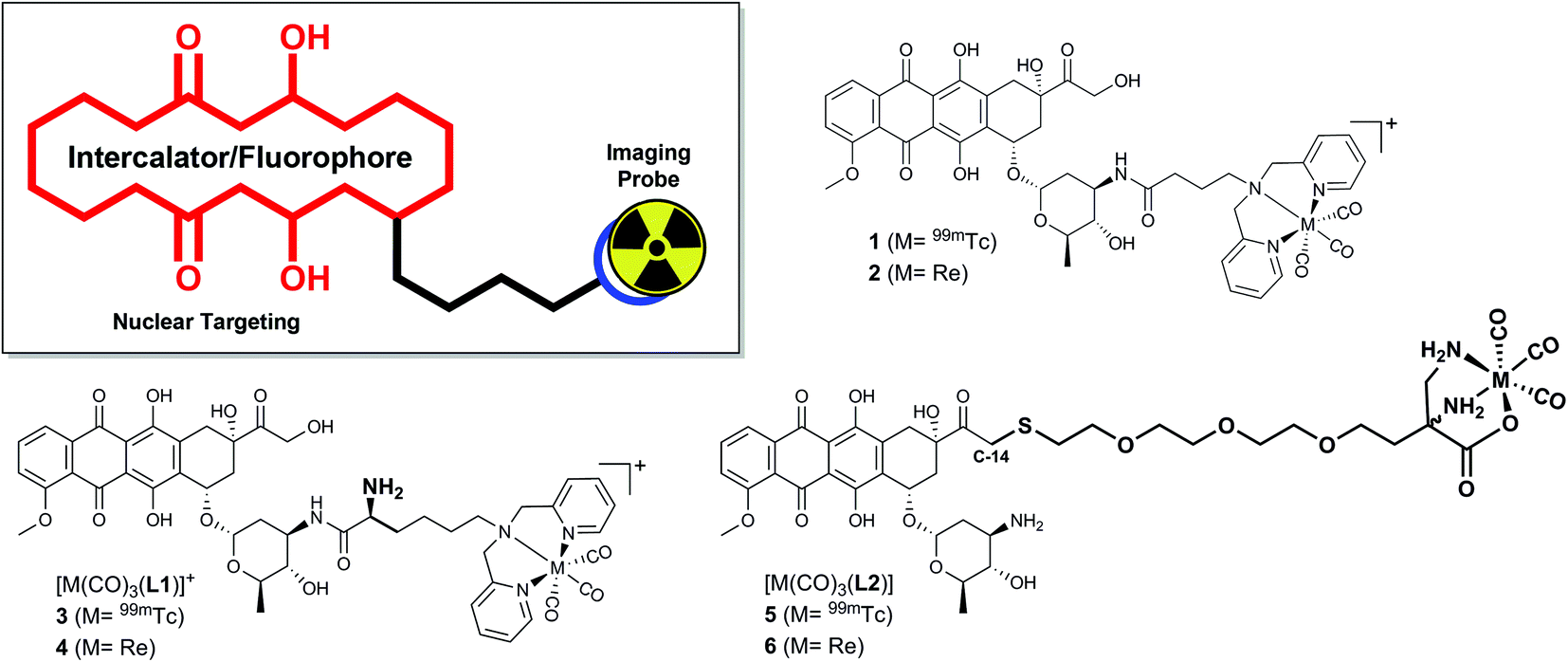 | ||
| Scheme 1 ADR-based metalloconjugates as imaging probes: previously reported complexes 1 (M = 99mTc) and 2 (M = Re) and new bioconjugates 3–6. | ||
Results & discussion
Design and synthesis of the ADR-conjugates
All synthetic data can be found in the ESI, Charts S1–S24.† Bioconjugates 3–6 (Scheme 1) were designed according to the bifunctional chelator (BFC) concept:19 The biological vector (ADR) is conjugated via a carbon linker to the chelator, which binds strongly to the fac-[M(CO)3]+ core (M = Re/99mTc). ADR is an autonomous nuclear targeting moiety for rapidly dividing cells and designated to ferry the pendant (radio-)metal into the nucleus, where intercalation into the DNA guarantees for retention. Unlike the previously reported ADR-complexes (1,2), conjugates 3 and 4 were designed with a free amine group on the α-carbon to enhance the conjugate's hydrophilicity and to mimic the underivatized daunosamine-NH2-group of the of the parent drug, which is known to be a critical point of interaction with DNA (Scheme 2).20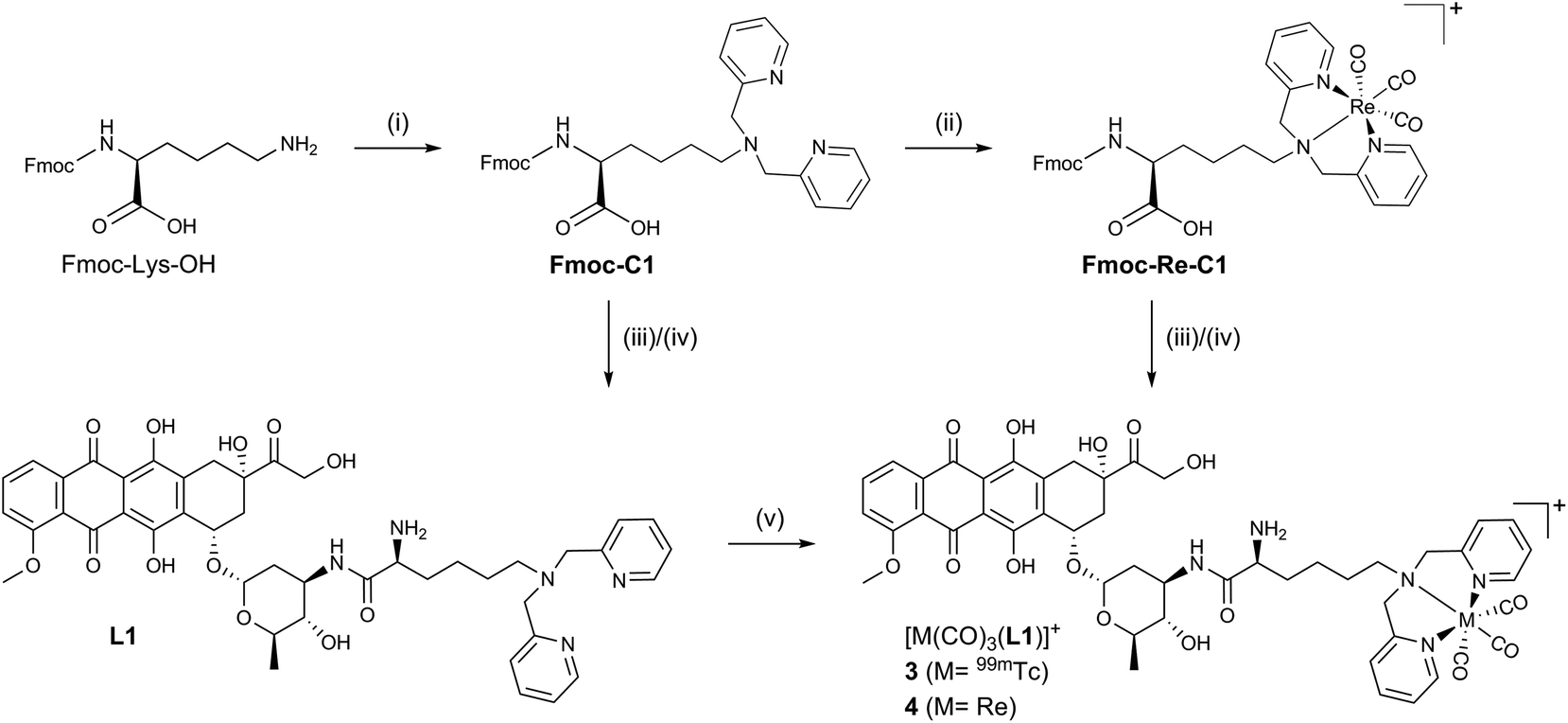 | ||
| Scheme 2 Synthesis of ADR conjugates 3 and 4. (i) 2-Pyridinecarboxaldehyde, STAB, DCE 25 °C, 62%; (ii) (NBu4)2[ReBr3(CO)3], MeOH, microwave, 110 °C, 84% (iii) ADR·HCl, HBTU, DIPEA, DMF, 25 °C, Fmoc-L1: 56%, Fmoc-4: 68% (iv) 10% piperidine in DMF, 25 °C, L1(·2TFA): 62%, 4(·2TFA): 58% (v) [99mTc(OH2)3(CO)3]+, H2O, 60 °C, 64% radiochemical yield (RCY). STAB = sodium triacetoxyborohydride, DCE = 1,2-dichloroethane, DIPEA = N,N-diisopropylethylamine, HBTU = N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate, DMF = N,N-dimethylformamide, MeOH = methanol. | ||
The Fmoc-C1 fragment was synthesized according to Zubieta and coworkers.21 The dipicolyl (DPA) chelator was introduced by direct reductive amination of commercially available Fmoc-Lys-OH with an excess of picolinaldehyde and sodium triacetoxyborohydride (STAB) as the reducing agent (Scheme 2). This chelator moiety was then reacted with the standard precursor (NEt4)2[ReBr3(CO)3] in a microwave to yield Fmoc-Re-C1. During this reaction we observed the formation of a side product, attributable to the corresponding methyl-ester.22 It likely forms due to activation of the free acid by the Lewis acidic fac-[Re(CO)3]+-fragment when methanol is the solvent. Formation of this side product, however, can be controlled by minimizing the reaction time and the excess of Re precursor. As the methyl ester is not very prone to amide formation, this species was not of concern and the crude reaction mixture was thus directly used in the following step. Reaction of commercially available ADR·HCl with either Fmoc-C1 or Fmoc-Re-C1, in a HBTU mediated amide bond formation, yielded the respective Fmoc protected ADR derivatives, which were deprotected in the last step using piperidine in DMF. After purification by preparative HPLC bifunctional chelator L1 and Re-complex 4 were obtained in yields of 21% (3 steps) and 20% (4 steps), respectively. Both species were fully characterized by detailed NMR studies (see ESI, Charts S4–S6 and S10–S12†).
Conjugates 5 and 6 (Scheme 3) were designed according to a different concept. As evident from crystal structures, the daunosamine glycoside binds to the minor groove of DNA.23 While the artificially introduced, free NH2-group in 3 and 4 may enhance the integrity of the ADR–DNA interaction, blocking of the glycosidic amine still likely disrupts the natural binding mode of ADR to DNA. In contrast, the hydroxyl group, located on C14, clearly points away from the double helix during intercalation. L2 was designed to maximize the binding capability of the conjugates to DNA and was hence derivatized with a chelator on C14. HS-DAP contains the 2,3-diaminopropionic acid (DAP) moiety – an excellent chelator for the fac-[M(CO)3]+-core (M = Re, 99mTc) – and its synthesis was described by Felber et al.24 The polyethylene glycol linker was chosen for solubility reasons and its well tolerated biocompatibility.
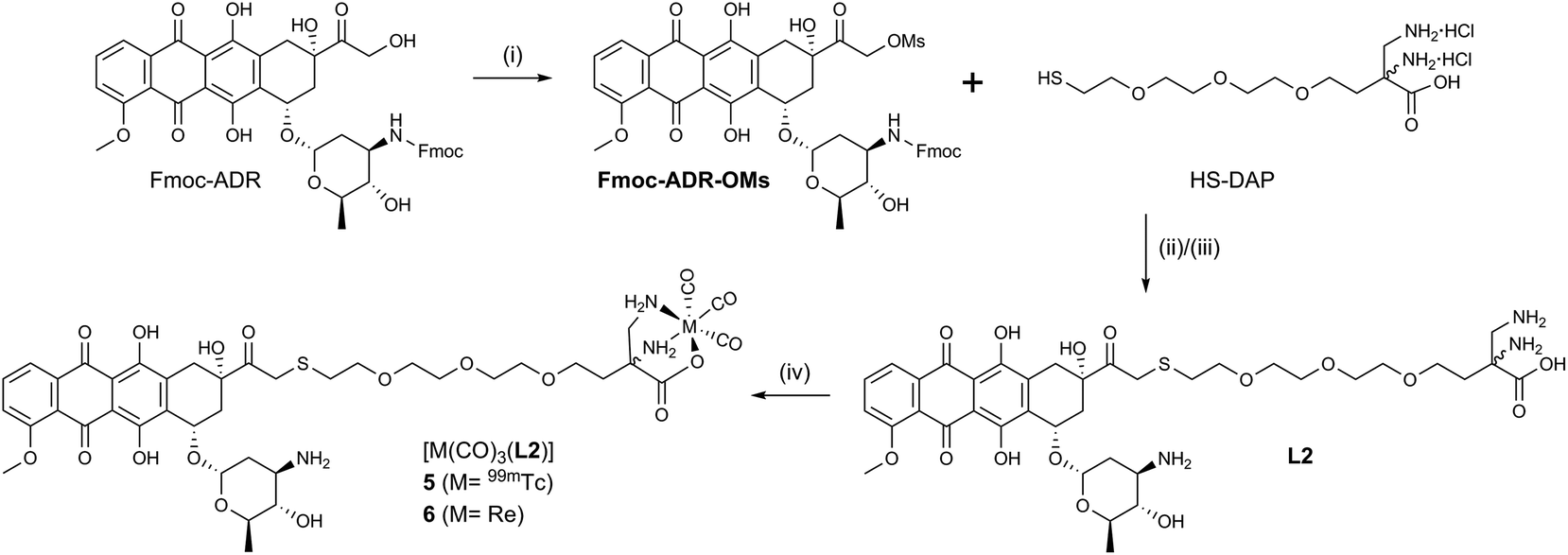 | ||
| Scheme 3 Synthesis of ADR-conjugates 5 and 6. (i) MsCl, TEA, CH2Cl2, 0–25 °C,72%, (ii) K2CO3, DMF, 25 °C, 62%, (iii) 10% Piperidine in DMF, 25 °C, 54%, (iv) 5: [99mTc(OH2)3(CO)3]+, H2O, 60 °C, 78% RCY, 6: (NBu4)2[ReBr3(CO)3], DIPEA, H2O, 25 °C, 55%. MsCl = methanesulfonyl chloride, TEA = triethylamine, DMF = N,N-dimethylformamide, DIPEA = N,N-diisopropylethylamine. | ||
Reaction of N-protected ADR with an excess of mesylchloride (MsCl) in dry CH2Cl2 afforded the C14-mesylate Fmoc-ADR-OMs which, to the best of our knowledge, is a new key intermediate for ADR derivatives. Under carefully controlled conditions, the C14–OH reacts rather selectively with MsCl, as it is the only primary hydroxyl and the most nucleophilic alcohol of ADR. Fmoc-ADR-OMs can be obtained in good to excellent yields, ready for the following step or for storage after preparative HPLC purification. Even though a C14 bromide of Daunorubicin has been published by Israel et al.,25 literature is void of a method to convert ADR directly into a strong electrophile. The mesylate obtained in this new way is easily displaced by nucleophiles, under mild conditions, and may alleviate laborious synthetic procedures to access C14 derivatives of ADR, such as the clinically applied Valrubicin (Valstar™), the C14-valerate ester of ADR.26,27
The soft thiol nucleophile in HS-DAP efficiently displaced the mesylate in a SN2 reaction from Fmoc-ADR-OMs under conditions adapted from Seshadri et al.28 After cleavage of the Fmoc group with piperidine in DMF and HPLC purification L2 was obtained in 24% overall yield (3 steps). Reacting L2 with (NEt4)2[ReBr3(CO)3] and HPLC purification afforded ADR-conjugate 6 in 55% yield. Chelator L2 and Re-complex 6 have been fully characterized by NMR (see ESI, Charts S16–S18 and S22–S24†). We note that L2 and 6 form a mixture of diastereomers by coupling racemic HS-DAP to enantiomerically pure ADR. No attempts were undertaken to separate the diastereomers, as neither the passive uptake of ADR nor intercalation into DNA requires stereochemically pure substances. Table 1 summarizes the synthesized ADR-conjugates and the relative hydrophilicities, reflected in the partition coefficients between n-octanol and PBS buffer (logDO/PBS).
99mTc radiolabeling
For the labeling of chelators L1 and L2, [99mTc(OH2)3(CO)3]+ was prepared according to a modified procedure.30,31 The three labile water ligands in this 99mTc precursor are displaced by the strong chelators DPA (L1) and DAP (L2) to yield excellent radiolabeling efficiencies of up to 95%. RP-HPLC purification of the crude solutions afforded ADR-99mTc conjugates 3 and 5 in good radiochemical yields (RCY) of 64% and 78% respectively. However, labeling conditions needed to be mild and carefully controlled. We observed a general trade-off between labeling at lower pHs (4–5), which kept the protonated chelators L1 and L2 in solution, but reduced the radiolabeling efficiency due to unavailability of free electron pairs on protonated nitrogens. Additionally, lower pHs promoted α-glycosidic cleavage. Vice versa, at pH values exceeding 6–7 the labeling efficiency was markedly enhanced but now, the neutral, hydrophobic chelators precipitated from the aqueous solution. Labeling experiments carried out in methanol were in support of the observed pH dependencies, as in this solvent byproduct formation was almost completely suppressed. The optimal conditions for fully aqueous labeling of ADR-chelates were T = 55–60 °C for 30 min, after careful quenching of the kit-contained boranocarbonate and readjusting the pH to 6–7. Since co-injection with the “cold” rhenium homologue is the accepted procedure for authentication of new 99mTc radioproducts at the tracer level, Fig. 1 shows HPLC chromatograms of the purified ADR-conjugates 3 and 5 and the respective co-injections with Re-complexes 4 and 6. For the in vitro experiments, 3 and 5 were purified by HPLC to radiochemical purities (RCP) ≥95% (see ESI, Chart S25†).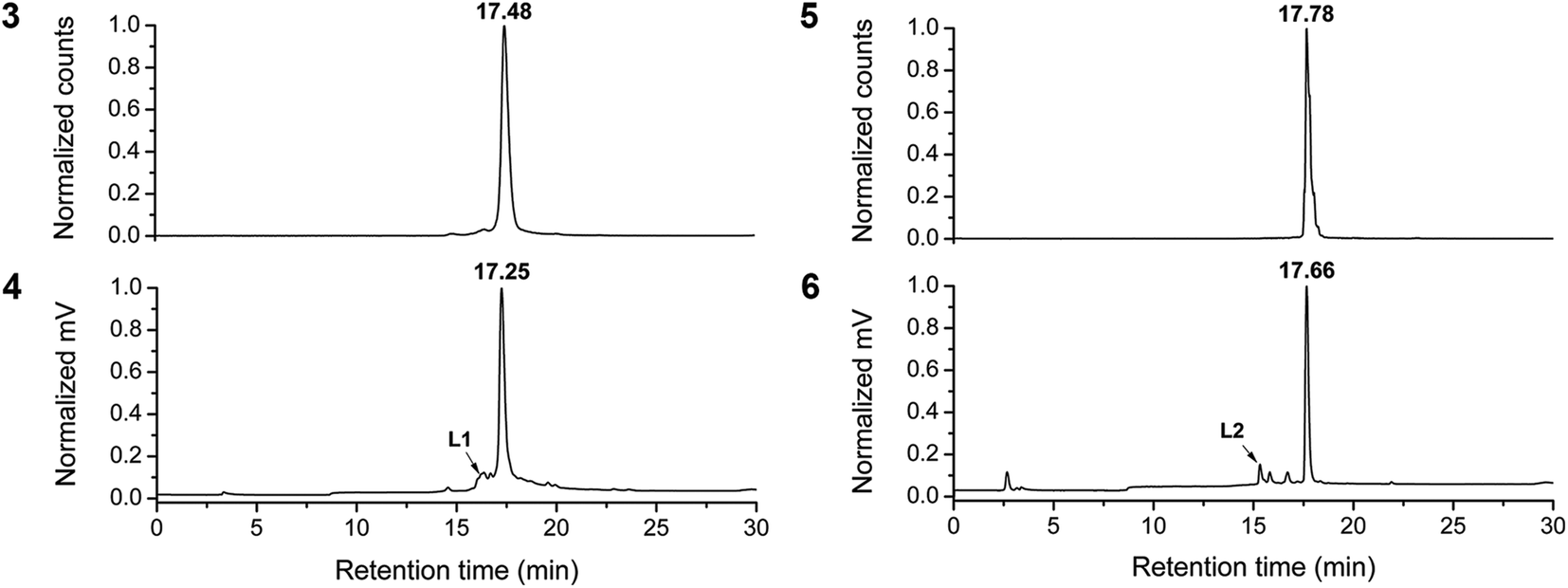 | ||
| Fig. 1 Normalized HPLC co-injections. Top left: γ-trace of 3 (17.48 min, RCP >95%), bottom left: UV-trace of rhenium complex 4 (17.25 min); top right: γ-trace of 5 (17.78 min, RCP >98%), bottom right: UV-trace of rhenium complex 6 (17.66 min). Differences in retention time result from a serial detector setup. RCP = radiochemical purity. | ||
Cytotoxicity studies
ADR is a strongly cytostatic agent with excellent inhibitory potencies against various cancer cell lines. It was hence of interest to assess if the new ADR-bioconjugates retained this cytotoxicity. The activity of conjugates 4 and 6 against the human cervix carcinoma (HeLa) cell line was studied by the fluorometric Resazurin cell viability assay.32 For comparison purposes, cisplatin was used in the same assay as internal reference. As shown in Table 2, bioconjugates 4 and 6 exhibited appreciable toxicities after 48 h of incubation. Like conjugate 2, these derivatives exhibit IC50 values which approximate the one of cisplatin, yet, the activities are markedly reduced with respect to native ADR. Still, conjugates 4 and 6 showed somewhat lower IC50 values than 2, likely owed to the more rationale design of the new bioconjugates: the artificially introduced amine group in 4 and the underivatized daunosamine in 6 afforded ADR derivatives which draw closer to the parent ADR, both in terms of hydrophilicity (see Table 1) and the possibility to bind DNA through hydrogen bonds rather than an electrostatic interaction via the charged metal center. It is evident that metalloconjugates of ADR could exhibit potentially useful cytotoxicity profiles after careful optimization of the pendant complexes. Such bioconjugates could yield new metallo-drugs that follow a different mechanism of action and possibly overcome the acquired MDR, observed for the parent drug.| Compound | Cell line |
|---|---|
| HeLa (μM) | |
| a Cisplatin was used as internal reference. Values indicate means ± standard deviations of three independent experiments. | |
| Cisplatin | 9.6 ± 1.1 |
| ADR | 0.093 ± 0.02 |
| 2 | 19.7 ± 2.1 |
| 4 | 6.2 ± 2.3 |
| 6 | 12.2 ± 2.4 |
DNA binding affinity
Nuclear DNA is the primary target of ADR and anthracene glycosides are known to strongly intercalate into DNA. This durable interaction with DNA is the cause of manifold toxic actions such as inhibition of DNA biosynthesis,33–35 DNA crosslinking,36,37 interference with DNA unwinding,38 strand separation and helicase activity.39,40 Ideally, an effective ADR mimic should thus target nuclear DNA and the affinity of conjugates 4 and 6 for DNA was evaluated in this respect. The binding affinity of ADR derivatives is frequently studied by means of their auto-fluorescence, which is quenched in the presence of increasing amounts of double stranded calf-thymus DNA (ctDNA). Fitting this titration data to the non-cooperative model for DNA binding28,29 allows for determination of the binding constant (Kb) and the size of the interaction site (s), which are reported in Table 3. As observed for native ADR, the initial fluorescence of the bioconjugates is maximal in the absence of ctDNA (see ESI, Chart 26†). In all instances it was strongly quenched at increasing DNA concentrations but no shift in the emission maximum (593 nm) was observed. Fig. 2 exemplarily shows the emission data and binding curves for ADR in the presence of ctDNA. All ADR derivatives showed a reduced affinity to ctDNA compared to the parent drug; we previously attributed this phenomenon to derivatization at the glycoside's amine.16 Conjugate 4, which comprises an additional amino group, exhibited a comparable binding affinity as native ADR. Apparently, the introduction of this free NH2-group strengthens the binding to DNA – possibly via the formation of hydrogen bonds to DNA – as this introduced amine is the only structural difference compared to derivative 2. On the contrary, the Kb of conjugate 6 is markedly lower than those of ADR and 4, even though this construct comprises an unaltered daunosamine moiety. We hypothesize that the lengthy linker and the bulky Re-complex impede an effective intercalation. Nevertheless, the binding constants obtained for the examined ADR bioconjugates indicate strong affinities for DNA. Even at the tracer level (99mTc concentrations: <μM), the great majority of all molecules are bound to DNA, theoretically enabling an effective retention of the radiotracers in the cell's nuclei.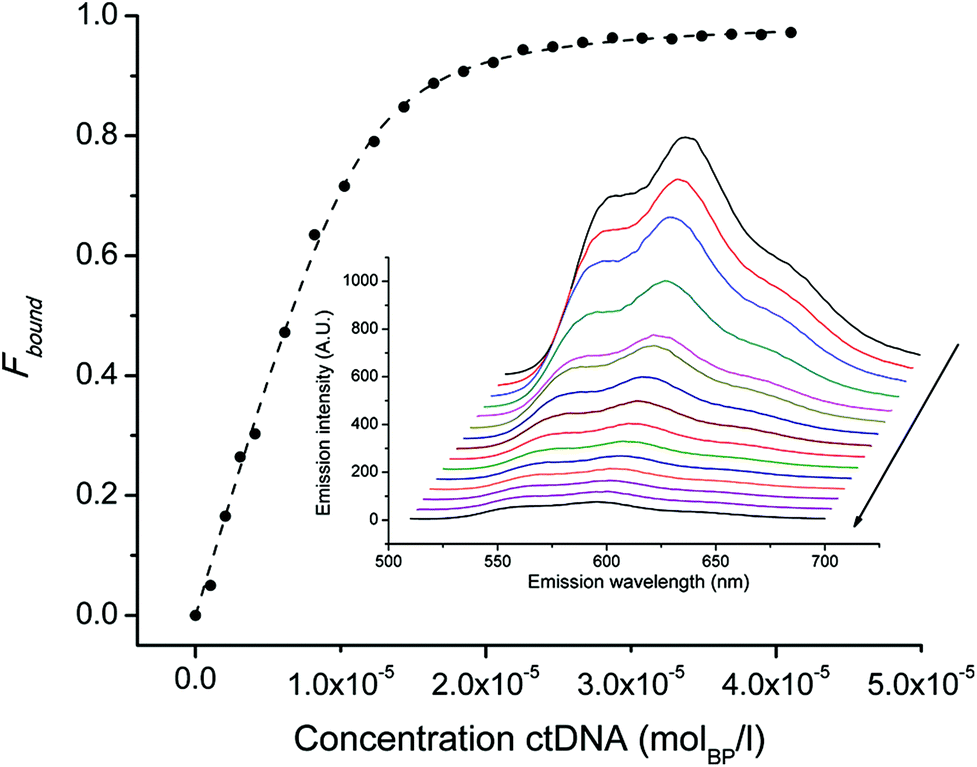 | ||
| Fig. 2 Binding curve of ADR (2.8 μM) to DNA. Symbols indicate Fbound calculated by equation (2, ESI†) and the dashed line represents the non linear squares fit to the non-cooperative binding model. Inset: emission spectra (510–700 nm) of ADR in PBS buffer. Note that the initial fluorescence is quenched upon increasing the concentration of ctDNA (arrow). | ||
| Compound (μM) | K b (M−1 per nucl) × 106 | s (bp) |
|---|---|---|
| a K b: affinity constant, s: binding site size. | ||
| ADR (2.8) | 4.98 ± 0.45 | 2.00 ± 0.04 |
| 2 (6.5) | 0.23 ± 0.03 | 2.20 ± 0.15 |
| 4 (0.68) | 4.11 ± 0.29 | 0.29 ± 0.01 |
| 6 (3.8) | 0.63 ± 0.11 | 1.26± 0.12 |
Inhibition of human Topoisomerase II
Despite the ubiquitous clinical use of ADR, its cytotoxic action is still under debate. Apart from interference with DNA biosynthesis (vide supra) different mechanisms have been proposed in literature. Yet it is accepted that ADR is a strong inhibitor of human topoisomerase II (hTopoII).41 We showed previously that ADR conjugate 2 effectively inhibited human hTopoII α and β, which was an incentive to look for a possible correlation between the strong binding to DNA, the toxicity of ADR conjugates 4 and 6 and inhibitory activity towards hTopoII α and β. We employed a method of Shapiro et al.42,43 which exploits the preferred binding of the fluorescently labeled (TTC)3T oligonucleotide to double-stranded plasmid DNA, containing a triplex forming sequence, after relaxation by hTopoII α or β versus the supercoiled plasmid.39,40 Changes in fluorescence anisotropy were monitored at increasing inhibitor concentrations to calculate the %-inhibition and the IC50 was determined by nonlinear least-squares fit of the data points to an adaptation of the Hill equation. The concentration dependent inhibition of hTopoII α and β by complexes 4 and 6 and fitting of the data to the Hill equation can be found in the ESI, Chart 28.†Table 4 indicates that Re complexes 4 as well as 6 effectively inhibited the activity of hTopoII α and β at similar concentrations as reported for native ADR. The similar structures of conjugates 2 and 4 can be directly compared and it is evident that the enhanced ability of 4 to inhibit hTopoII α and β is consistent with the above-shown superior IC50 and stronger binding to DNA. No correlation between toxicity in HeLa cells and hTopoII inhibition can be found for 6: it outperforms all presented inhibitors in this assay, but is less toxic than 4 and ADR. This absence of a one-to-one relationship between Topoisomerase inhibition and cytotoxicity is reminiscent of the above-mentioned multiple modes of action of ADR. Nonetheless, it is remarkable that the structural changes of ADR, by derivatizations at different positions with metal complexes, do not impede the inhibition of hTopoII α and β. We note that in addition to retaining toxicity and DNA binding, the novel ADR bioconjugates mimic also this feature of the parent drug.Cellular localization and distribution
The red autofluorescence of the anthraquinone chromophore (λabs = 488 nm, λem = 592 nm) allows for cellular localization of ADR derivatives by confocal microscopy. Fig. 3 displays the fluorescence distribution of conjugates 4 (20 μM) and 6 (20 μM) compared to native ADR (1 μM), after 2 h of incubation in HeLa cells. Both derivatives showed an efficient cellular uptake, as cytosolic fluorescence was detected already at 10 min post incubation. After the full 2 h period, complex 4 exhibited a predominantly cytoplasmic and enhanced perinuclear staining. Although we observed some fluorescence in the nucleus, it was diminishingly weak, yet somewhat stronger in nucleolar regions. We note that by closer examination the fluorescence distribution of 4 resembles a negative image of the parent ADR and that comparable fluorescent staining observations were made for 2.16 In contrast, conjugate 6 exhibited a different picture: while a fluorescence signal was observed both in the cytosol and nucleus, the nuclear luminescence was now clearly enhanced and the nucleoli stained to a reduced extent, much like the parent drug.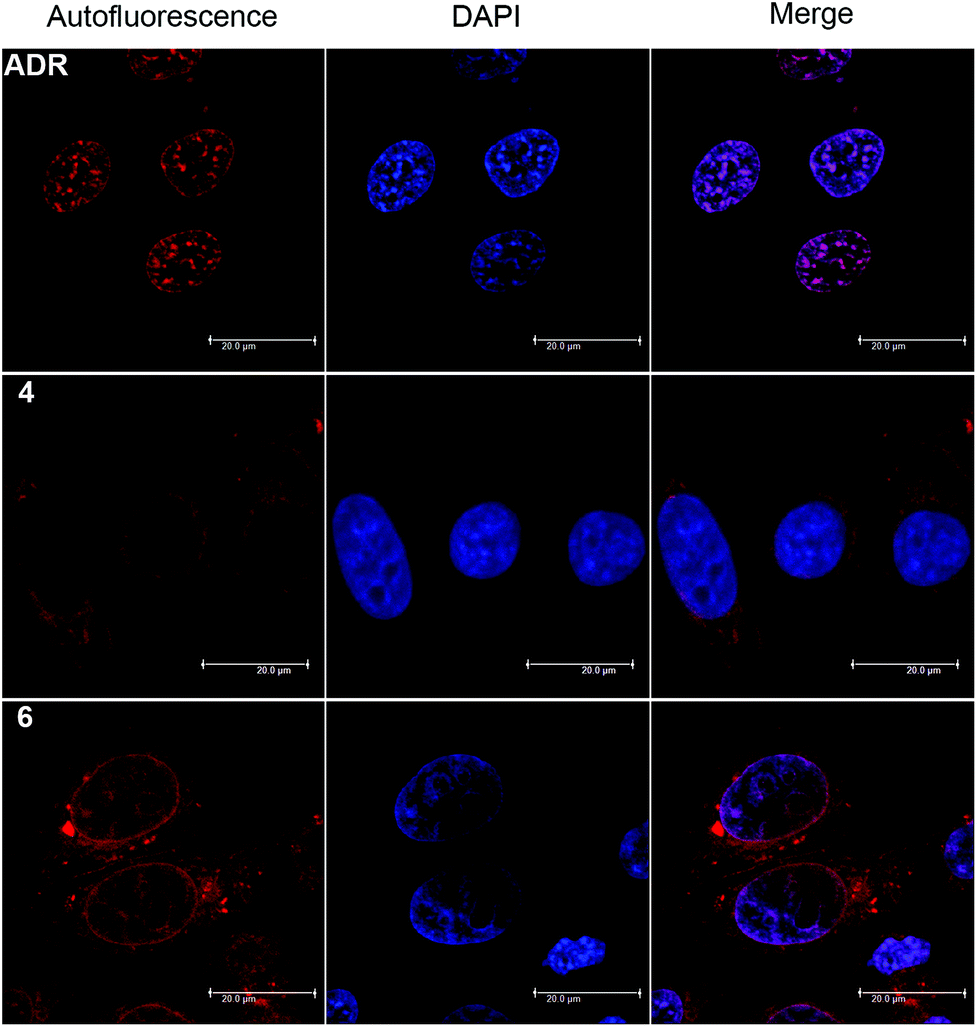 | ||
| Fig. 3 Confocal fluorescence microscopy of native ADR (1 μM) and Re conjugates 4 (20 μM) and 6 (20 μM) in HeLa cells after 2 h incubation. Left: red autofluorescence (excitation at 488 nm, emission above 600 nm), center: DAPI nuclear stain, right: merge of autofluorescence and DAPI staining. DAPI = 4′,6-diamidin-2-phenylindol. | ||
When examining the fluorescence distribution of ADR derivatives, caution is advised due to altering fluorescence features which strongly depend on their immediate environment.44 For example, complex 2 showed a nucleus void of fluorescence, but revealed a predominant accumulation in the nuclear compartment after quantifications of the metal content.16 Thus, we directly proceeded with quantifying the sub-cellular distribution of conjugates 3–6 in order to make a firm assessment.
The presence of 99mTc (conjugate 3 and 5) and Re (conjugates 4 and 6) in homologous compounds allows for very sensitive quantification of metal contents in cellular compartments. 99mTc can be quantified by direct measurement of the decay-corrected activity and the Re content by inductively-coupled mass spectrometry (ICP-MS). Consequently, HeLa cells were incubated with either purified radioconjugates (3/5) or Re conjugates (4/6) for 2 h. Post incubation cells were washed with buffer to remove unbound metal and nuclei and mitochondria were isolated from whole cells by a modified procedure for mitochondria extraction from cultured cells. At this point the metal content in the nucleus and mitochondria pellets was determined by a dose calibrator or ICP-MS, after chemical digestion in aqua regia, and was expressed relative (%) to the content in the whole cell pellet. Fig. 4 and Table 5 summarize the results. Evidently, all ADR conjugates 3–6 target the nucleus as the great majority (70–80%) of metal content was recovered from this organelle, whereas 2–3% of the compounds accumulate in mitochondria. Furthermore, the compiled data showed no significant differences in relative uptake values between the examined compounds and quantification modalities. Since the quantification results of ADR conjugate 4 and 6 are in excellent agreement with those previously reported for complex 2, it must be concluded that a lacking nuclear fluorescence is observed due to a reduced overall uptake of the metal conjugates compared to native ADR and nearly complete quenching upon intercalation into nuclear DNA. The appearance of in vitro fluorescence from ADR derivatives is highly concentration dependent and thus ill-suited as a standalone tool for an unambiguous determination of the cellular localization.
 | ||
| Fig. 4 Relative Re (ICP-MS) and 99mTc (dose calibrator) uptake in the different cellular compartments of HeLa cells treated for 2 h with 20 μM of 4 and 6 or ∼12 MBq of 3 and 5. | ||
| Conjugate 4 | 6 | |
|---|---|---|
| 185Re | [ng/106 cells] | [ng/106 cells] |
| Whole cell | 13.38 ± 1.41 | 24.37 ± 0.75 |
| Nucleus | 10.28 ± 0.71 (76.8%) | 21.99 ± 2.05 (80.34%) |
| Mitochondria | 0.29 ± 0.13 (2.1%) | 1.27 ± 0.03 (5.2%) |
| Conjugate 3 | 5 | |
|---|---|---|
| 99mTc | [kBq/106 cells] | [kBq/106 cells] |
| a Rhenium was quantified by ICP-MS (20 μM, n = 3). 99mTc was quantified in a dose calibrator (∼12 MBq, n = 3). Incubation time was 2 h. Values represent means ± standard deviations per 106 cells. Values in brackets are relative to the whole cell fraction. | ||
| Whole cell | 19.92 ± 0.88 | 45.49 ± 0.64 |
| Nucleus | 16.01 ± 0.57 (80.4%) | 36.25 ± 2.02 (79.7%) |
| Mitochondria | 0.41 ± 0.04 (2.06%) | 1.73 ± 0.12 (3.80%) |
In terms of absolute uptake values (see Table 5), the cellular accumulation of conjugates 5 and 6 is greater compared to bioconjugates 3 and 4. We attribute these differences to the higher lipophilicity of 5 and 6, as lipophilic ADR derivatives are known to enhance the cellular uptake.45 Interestingly, we found that conjugate 5 and 6 show a twofold greater absolute uptake compared to 3 and 4. According to ICP-MS, the calculated concentration of 6 in the cell nucleus is roughly 140 μM, a value which markedly exceeds the one of 4 (65 μM) and previously 2 (33 μM). Moreover, it draws much closer to the nuclear concentration of native ADR, which can reach up to 340 μM at saturation.46 The enhanced nuclear concentration of 6 is well in line with the observed excess luminescence signal, as seen in confocal microscopy (vide supra).
Summary and conclusion
Doxorubicin (ADR) is a widely applied chemotherapeutic agent. Because of its ability to target hypermitotic tissue, it could serve as a structural motif for new imaging agents and/or a molecular imaging probe for multidrug resistance.In this work we studied four new ADR bioconjugates (3–6) with regard to their ability to mimic native ADR. The cold rhenium surrogates 4 and 6 were shown to be cytotoxic and to strongly bind to DNA. Moreover, the scrutinized compounds are capable of effectively inhibiting the nuclear human Topoismorease II enzyme at concentrations well comparable to native ADR. Cellular localization and uptake were studied by confocal microscopy and corroborated with two independent quantification modalities. Compared to the parent drug, the rhenium derivatives 4 and 6 are taken up to a reduced extent but still effectively target the nucleus. Especially conjugate 6 seems to be an attractive candidate for further evaluation, as its nuclear uptake is clearly articulated in comparison to 2 and 4. Overall, the presented data evidences that Re-conjugates 4 and 6, as well as 99mTc-conjugates 3 and 5 are excellent mimics of native ADR in vitro, despite substantial structural deviations from the natural molecule. Indeed, they have a prospect as imaging agents. In vivo evaluations are currently ongoing to study their biodistribution. Furthermore, the 99mTc bioconjugates are subject of experiments in ADR resistant and sensitive cells to determine their potential as in vivo probes for P-glycoprotein.
Acknowledgements
We would like to express our sincere gratitude to A. B. Shapiro for the hTopoII inhibition assays, F. Wild for the ICP-MS measurements and C. Richmond for proofreading of the manuscript. We thank the Graduate School of Chemical and Molecular Sciences Zurich (CMSZH) for financial support. This work was financially supported by the Swiss National Science Foundation (Professorships no. PP00P2_133568 and PP00P2_157545 to G. G.), the University of Zurich (G. G. and R. A.), the Stiftung für wissenschaftliche Forschung of the University of Zurich (G. G.), the Portuguese Science Foundation (C2TN strategic project ID/Multi/04349/2013) and COST Action CM1105 (G. G. and R. A.).References
- D. Chandy, G. Maguire and W. S. Aronow, Compr. Ther., 2009, 35, 18–23 Search PubMed.
- D. A. Torigian, S. S. Huang, M. Houseni and A. Alavi, CA-Cancer J. Clin., 2007, 57, 206–224 CrossRef PubMed.
- M. N. Wernick and J. N. Aarsvold, Emission tomography: the fundamentals of PET and SPECT, Academic Press, 2004 Search PubMed.
- D. A. Goodwin, J. Nucl. Med., 1987, 28, 1358–1362 CAS.
- E. D. Agdeppa and M. E. Spilker, AAPS J., 2009, 11, 286–299 CrossRef CAS PubMed.
- F. Arcamone, G. Francesco, S. Penco and A. Selva, Tetrahedron Lett., 1969, 1007–1010 CrossRef CAS.
- F. Arcamone, G. Cassinelli, G. Fantini, A. Grein, P. Orezzi, C. Pol and C. Spalla, Biotechnol. Bioeng., 2000, 67, 704–713 CrossRef CAS PubMed.
- M. J. Waring, Annu. Rev. Biochem., 1981, 50, 159–192 CrossRef CAS PubMed.
- J. W. Lown, Chem. Soc. Rev., 1993, 22, 165–176 RSC.
- C. Carvalho, R. X. Santos, S. Cardoso, S. Correia, P. J. Oliveira, M. S. Santos and P. I. Moreira, Curr. Med. Chem., 2009, 16, 3267–3285 CrossRef CAS PubMed.
- G. Minotti, P. Menna, E. Salvatorelli, G. Cairo and L. Gianni, Pharmacol. Rev., 2004, 56, 185–229 CrossRef CAS PubMed.
- D. A. Gewirtz, Biochem. Pharmacol., 1999, 57, 727–741 CrossRef CAS PubMed.
- P. H. Elsinga, E. J. F. Franssen, N. H. Hendrikse, L. Fluks, A. M. A. Weemaes, W. T. A. vanderGraaf, G. E. deVries, G. M. Visser and W. Vaalburg, J. Nucl. Med., 1996, 37, 1571–1575 CAS.
- T. Tsuruo, H. Iida, S. Tsukagoshi and Y. Sakurai, Cancer Res., 1981, 41, 1967–1972 CAS.
- P. R. Twentyman, J. G. Reeve, G. Koch and K. A. Wright, Br. J. Cancer, 1990, 62, 89–95 CrossRef CAS PubMed.
- S. Imstepf, V. Pierroz, P. Raposinho, M. Bauwens, M. Felber, T. Fox, A. B. Shapiro, R. Freudenberg, C. Fernandes, S. Gama, G. Gasser, F. Motthagy, I. R. Santos and R. Alberto, Bioconjugate Chem., 2015, 26, 2397–2407 CrossRef CAS PubMed.
- S. Imstepf, V. Pierroz, R. Rubbiani, M. Felber, T. Fox, G. Gasser and R. Alberto, Angew. Chem., Int. Ed., 2016, 55, 2792–2795 CrossRef CAS PubMed.
- A. Leonidova and G. Gasser, ACS Chem. Biol., 2014, 9, 2180–2193 CrossRef CAS PubMed.
- C. F. Ramogida and C. Orvig, Chem. Commun., 2013, 49, 4720–4739 RSC.
- A. Dimarco and F. Arcamone, Arzneim. Forsch., 1975, 25, 368–375 CAS.
- M. K. Levadala, S. R. Banerjee, K. P. Maresca, J. W. Babich and J. Zubieta, Synthesis, 2004, 1759–1766 CAS.
- A. F. Armstrong, N. Oakley, S. Parker, P. W. Causey, J. Lemon, A. Capretta, C. Zimmerman, J. Joyal, F. Appoh, J. Zubieta, J. W. Babich, G. Singh and J. F. Valliant, Chem. Commun., 2008, 5532–5534 RSC.
- C. A. Frederick, L. D. Williams, G. Ughetto, G. A. Van der Marel, J. H. Van Boom, A. Rich and A. H. J. Wang, Biochemistry, 1990, 29, 2538–2549 CrossRef CAS PubMed.
- M. Felber, M. Bauwens, J. M. Mateos, S. Imstepf, F. M. Mottaghy and R. Alberto, Chem. – Eur. J., 2015, 21, 6090–6099 CrossRef CAS PubMed.
- M. Israel, P. G. Potti and R. Seshadri, J. Med. Chem., 1985, 28, 1223–1228 CrossRef CAS PubMed.
- M. Israel, E. J. Modest and E. Frei, Cancer Res., 1975, 35, 1365–1368 CAS.
- I. C. Cotterill and J. O. Rich, Org. Process. Res. Dev., 2005, 9, 818–821 CrossRef CAS.
- R. Seshadri, M. Israel and W. J. Pegg, J. Med. Chem., 1983, 26, 11–15 CrossRef CAS PubMed.
- C. Rousselle, P. Clair, J.-M. Lefauconnier, M. Kaczorek, J.-M. Scherrmann and J. Temsamani, Mol. Pharmacol., 2000, 57, 679–686 CAS.
- R. Alberto, R. Schibli, A. Egli, A. P. Schubiger, U. Abram and T. A. Kaden, J. Am. Chem. Soc., 1998, 120, 7987–7988 CrossRef CAS.
- R. Alberto, K. Ortner, N. Wheatley, R. Schibli and A. P. Schubiger, J. Am. Chem. Soc., 2001, 123, 3135–3136 CrossRef CAS PubMed.
- S. A. Ahmed, R. M. Gogal and J. E. Walsh, J. Immunol. Methods, 1994, 170, 211–224 CrossRef CAS PubMed.
- M. F. Goodman, M. J. Bessman and N. R. Bachur, Proc. Natl. Acad. Sci. U. S. A., 1974, 71, 1193–1196 CrossRef CAS.
- K. Wassermann, L. A. Zwelling, T. D. Mullins, L. E. Silberman, B. S. Andersson, M. Bakic, E. M. Acton and R. A. Newman, Cancer Res., 1986, 46, 4041–4046 CAS.
- B. Schott and J. Robert, Biochem. Pharmacol., 1989, 38, 167–172 CrossRef CAS PubMed.
- A. Skladanowski and J. Konopa, Biochem. Pharmacol., 1994, 47, 2269–2278 CrossRef CAS PubMed.
- A. Skladanowski and J. Konopa, Biochem. Pharmacol., 1994, 47, 2279–2287 CrossRef CAS PubMed.
- F. A. Fornari, J. K. Randolph, J. C. Yalowich, M. K. Ritke and D. A. Gewirtz, Mol. Pharmacol., 1994, 45, 649–656 CAS.
- A. Montecucco, G. Pedralinoy, S. Spadari, E. Zanolin and G. Ciarrocchi, Nucleic Acids Res., 1988, 16, 3907–3918 CrossRef CAS PubMed.
- N. Tuteja, T. N. Phan, R. Tuteja, A. Ochem and A. Falaschi, Biochem. Biophys. Res. Commun., 1997, 236, 636–640 CrossRef CAS PubMed.
- Y. Pommier, E. Leo, H. Zhang and C. Marchand, Chem. Biol., 2010, 17, 421–433 CrossRef CAS PubMed.
- A. B. Shapiro and C. A. Austin, Anal. Biochem., 2014, 448, 23–29 CrossRef CAS PubMed.
- A. B. Shapiro, Biochem. Pharmacol., 2013, 85, 1269–1277 CrossRef CAS PubMed.
- P. Mohan and N. Rapoport, Mol. Pharm., 2010, 7, 1959–1973 CrossRef CAS PubMed.
- B. S. Chhikara, D. Mandal and K. Parang, J. Med. Chem., 2012, 55, 1500–1510 CrossRef CAS PubMed.
- M. Gigli, T. W. D. Rasoanaivo, J. M. Millot, P. Jeannesson, V. Rizzo, J. C. Jardillier, F. Arcamone and M. Manfait, Cancer Res., 1989, 49, 560–564 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6dt00871b |
| This journal is © The Royal Society of Chemistry 2016 |