
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Spin-labelled photo-cytotoxic diazido platinum(IV) anticancer complex†
V.
Venkatesh
a,
Christopher J.
Wedge
 b,
Isolda
Romero-Canelón
a,
Abraha
Habtemariam
a and
Peter J.
Sadler
*a
b,
Isolda
Romero-Canelón
a,
Abraha
Habtemariam
a and
Peter J.
Sadler
*a
aDepartment of Chemistry, University of Warwick, Coventry CV4 7AL, UK. E-mail: P.J.Sadler@warwick.ac.uk
bDepartment of Physics, University of Warwick, Coventry CV4 7AL, UK. E-mail: c.wedge@warwick.ac.uk
First published on 18th May 2016
Abstract
We report the synthesis and characterisation of the nitroxide spin-labelled photoactivatable Pt(IV) prodrug trans,trans,trans-[Pt(N3)2(OH)(OCOCH2CH2CONH-TEMPO)(Py)2] (Pt-TEMPO, where TEMPO = 2,2,6,6-tetramethylpiperidine 1-oxyl). Irradiation with blue visible light gave rise to Pt(II) and azidyl as well as nitroxyl radicals. Pt-TEMPO exhibited low toxicity in the dark, but on photoactivation was as active towards human ovarian cancer cells as the clinical photosensitizer chlorpromazine and much more active than the anticancer drug cisplatin under the conditions used.
Platinum-based anticancer drugs are now widely used for the treatment of various types of cancer.1–3 However, their utility in the clinic is severely limited by their systemic toxicity, as well as inherent and acquired drug resistance.4–6 Photoactivated chemotherapy (PACT) has recently attracted attention because of its ability to target tumour cells selectively without causing harm to normal cells thereby minimising side effects.7 The advantages of using metal complexes in PACT stem from their rich photochemistry.8,9 This can be utilized in the design of a non-toxic prodrug that can be selectively activated in the region of a tumour-using light of specific wavelengths, with longer wavelengths penetrating more deeply into tissues.
Nitroxide spin-labelling can provide a convenient way of monitoring drug distribution and metabolic pathways using electron paramagnetic resonance (EPR).10–12 Nitroxyl radicals often exhibit low toxicity, are membrane-permeable and also possess antioxidant activity that mimics superoxide dismutase (SOD).13 The potent anticancer activity of nitroxide radicals, nitroxyl derivatives and their metal complexes towards a variety of cancer cell lines has been reported,14–17 and often involves redox-mediated signalling pathways.18 Nitroxide radicals not only have diagnostic use, but also therapeutic, and hence are potential ‘theranostics’ as well. We report here what appears to be (as far as we are aware) the first example of the photo-release of a nitroxide radical from a metal complex and its antiproliferative activity towards cancer cells.
We have synthesised the TEMPO-conjugated photoactivatable Pt(IV) prodrug trans,trans,trans-[Pt(N3)2(OH)(OCOCH2CH2CONH-TEMPO)(Py)2] (Pt-TEMPO, Fig. 1a) and studied its photocytotoxicity towards A2780 human ovarian cancer cells. The synthesis was achieved by reacting trans,trans,trans-[Pt(N3)2(OH)(succinate)(Py)2] with 4-amino TEMPO using a 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) coupling reaction (ESI, Scheme S1†). The product was purified by column chromatography and characterized by 1H NMR, elemental analysis and high resolution mass spectrometry (HRMS). The presence of the TEMPO radical in the complex was confirmed by EPR spectroscopy. The EPR spectrum of Pt-TEMPO (0.5 mM in dichloromethane) recorded at room temperature showed a typical triplet pattern with distinct 14N hyperfine splitting (Fig. 1c).
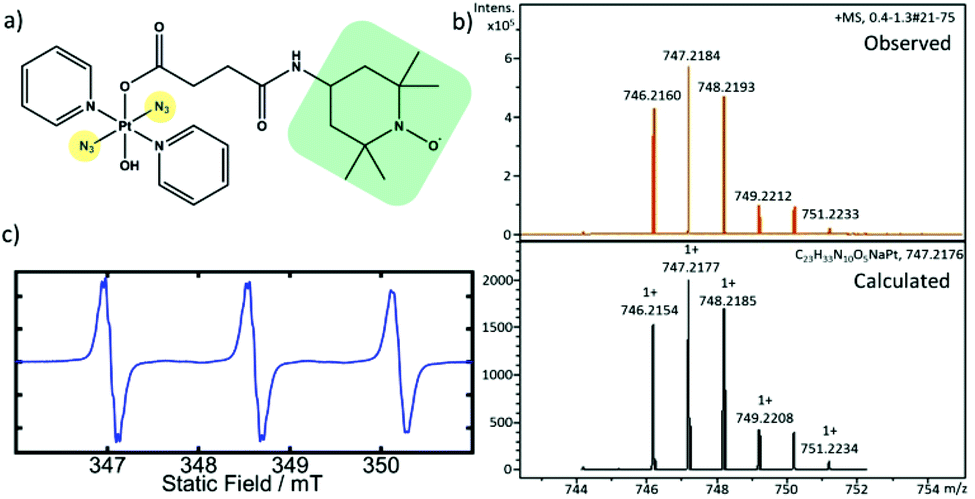 | ||
| Fig. 1 (a) Molecular structure of Pt-TEMPO complex; (b) calculated and observed HR-ESI-MS spectra of Pt-TEMPO with isotopic distribution; (c) EPR spectrum of Pt-TEMPO (0.5 mM) in degassed dichloromethane. | ||
The 1H NMR spectrum of Pt-TEMPO in CDCl3 shows broadened peaks (Fig. S1 ESI†) indicating the presence of the TEMPO radical attached to the diazido Pt(IV) complex. The peaks in the aromatic region of the spectrum indicate the presence of pyridine ligands and the signals at (2.3–2.8 ppm) correspond to the methylene protons in the succinate chain. The signals at (0.9–1.3 ppm) confirmed the successful conjugation of TEMPO unit in Pt-TEMPO. The observed HRMS is in good agreement with the spectrum calculated for [M + Na]+, Fig. 1b.
Pt-TEMPO was irradiated with 420 nm light in aqueous solution. The progress of the photoreaction was monitored at different time intervals by UV-Vis spectroscopy and the decomposition products were subsequently analysed by ESI-MS. The change in ligand-to-metal charge-transfer (LMCT) band for Pt-TEMPO was monitored after every 30 min of photoactivation at 420 nm up to 2 h Fig. 2c. The reduction in LMCT band indicates that there is loss of an azido ligand and/or electron transfer from azido ligands to Pt(IV) resulting in the formation of azidyl radicals and Pt(IV/II) species. ESI-MS shows the formation of a mixture of products 1–4 in solution after photoactivation Fig. 2a (ESI, Fig. S3†). When the photoactivation was carried out using green light (517 nm), the reduction in intensity of the LMCT band was very slow (ESI, Fig. S5†) due to the weaker absorbance of Pt-TEMPO at 517 nm.
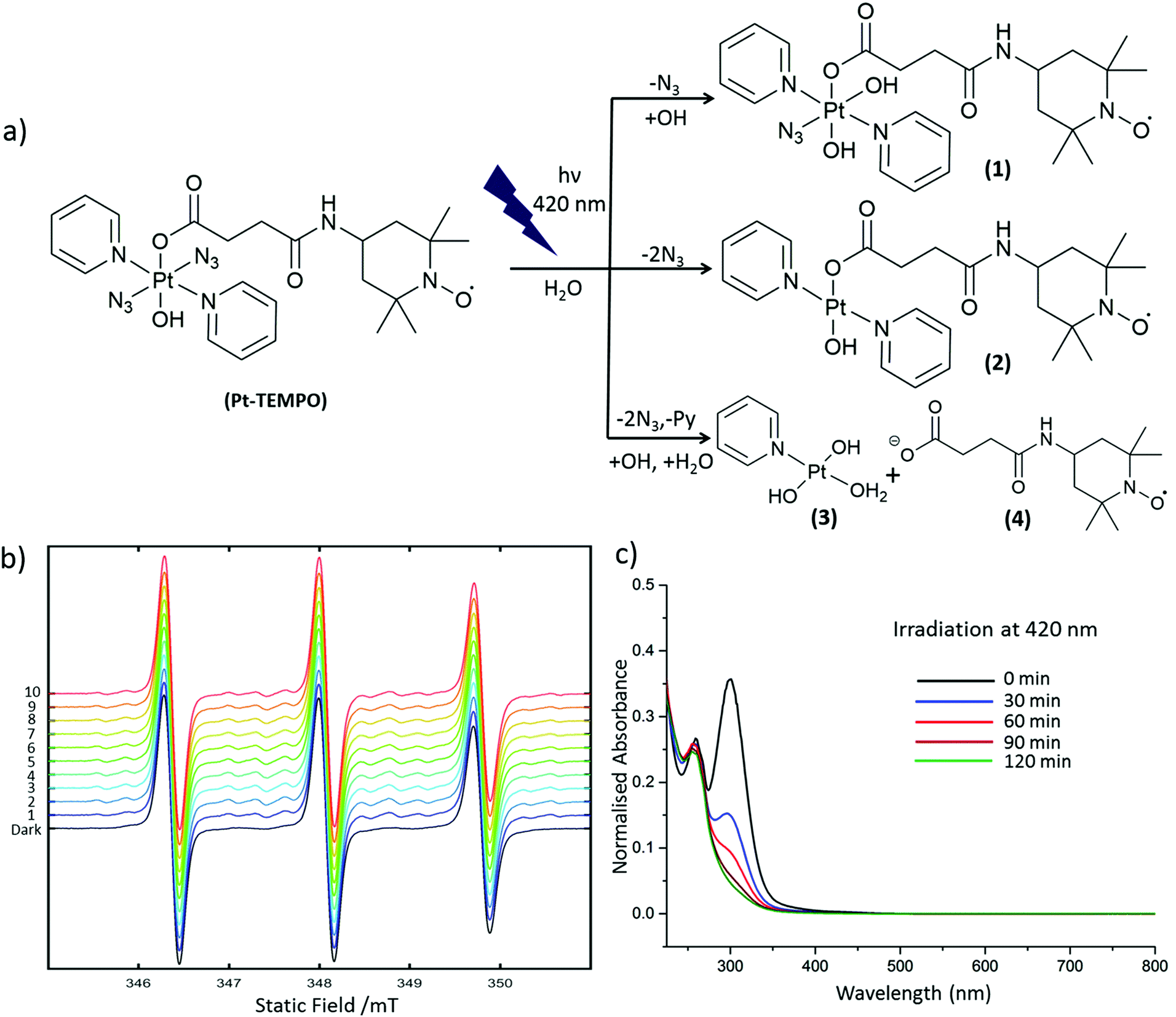 | ||
| Fig. 2 (a) Photoreaction pathways for Pt-TEMPO (0.1 mM in H2O) on irradiation with blue visible light showing formation of products 1–4; (b) EPR spectrum of Pt-TEMPO with DMPO in the dark as well as after irradiation with λirr = 405 nm laser (each consecutive scan took 21 s to record); (c) UV-Vis spectrum of Pt-TEMPO after every 30 min of irradiation (λirr = 420 nm). | ||
To capture the reactive azidyl radicals, spin-trapping experiments were performed. An aqueous solution containing 1 mM of Pt-TEMPO and 25 mM 5,5-dimethyl-1-pyrroline-N-oxide (DMPO), giving a 12.5 fold molar excess of spin-trap over azide, was used for the experiment. The EPR spectrum was recorded in the dark and during irradiation with a 405 nm laser (42 mW). In the absence of irradiation the EPR spectrum showed a triplet signal characteristic of the 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) radical present in Pt-TEMPO (Fig. S4†). Upon irradiation with the blue laser the azidyl adduct of DMPO was detected. This gave a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2:1 quartet of triplets signal along with the triplet signal of the stable TEMPO radical. During irradiation, spectra were recorded continuously with each successive field-sweep taking 21 s (Fig. 2b).
:2:2:1 quartet of triplets signal along with the triplet signal of the stable TEMPO radical. During irradiation, spectra were recorded continuously with each successive field-sweep taking 21 s (Fig. 2b).
Due to the overlap of peaks for Pt-TEMPO and the DMPO-N3 adduct, and the significant intensity difference between them, it is difficult to obtain accurate spin Hamiltonian parameters of the weaker component through a multi-component fit. For simulations of the DMPO azidyl adduct the hyperfine couplings were therefore fixed to literature values (aNNO = 1.45 mT, aHβ = 1.49 mT, aNα = 0.316 mT).19 By subtracting the experimental data recorded before illumination from those recorded after illumination, the Pt-TEMPO signal intensity was significantly reduced allowing the twelve-line azidyl spin-adduct signal to be observed clearly (Fig. 3a). As aNNO ≈ aHβ the usual DMPO doublet of triplets overlaps to form a 1:2:2:1 quartet, with each line further split into a triplet by the small coupling to the azidyl nitrogen. This spectral assignment has been confirmed previously in photoactivation studies of a closely related Pt-azidyl anti-cancer compound by use of 15N labelled azide,19 hence it was not considered necessary to perform isotopic substitution experiments.
 | ||
| Fig. 3 (a) Overlap of experimental and simulated DMPO-N3 adduct spectra. The experimental trace shows the subtraction of spectra recorded immediately before and after switch on of the light source. Stars indicate the location of the three main lines in the TEMPO spectrum which are not fully subtracted; (b) comparison between simulated and experimental EPR spectra of Pt-TEMPO and DMPO-N3; (c) azide spin-adduct concentration as a function of illumination time (42 mW at 405 nm) determined by least-squares fitting of the sum of simulated Pt-TEMPO and DMPO-N3 spectra to the experimental EPR traces. | ||
Once individual spectral simulations for Pt-TEMPO and the azidyl radical adduct had been completed using EasySpin20 it was possible to reproduce the overlapping experimental spectra as linear combinations of the two (Fig. 3b). The scaling parameter between simulated and experimental spectra was determined from a least-squares fit of the simulated Pt-TEMPO spectra to the experimental spectra prior to illumination. Treating this value as a constant and using the known initial Pt-TEMPO concentration of 1 mM as a reference, it was possible to determine the azidyl adduct concentration from the weighting factor of the combined fit. Running the spectrometer in incremental scan mode, a series of field-swept spectra was recorded consecutively, which by individual fitting with the simulated spectra allowed measurement of the azidyl-adduct concentration as a function of time since illumination began (Fig. 3c).
The photocytotoxicity of Pt-TEMPO was determined by using the sulforhodamine B (SRB) colorimetric assay which quantifies protein content of treated samples against untreated controls. A2780 human ovarian carcinoma cells were treated with Pt-TEMPO for 1 h in the dark and also treated with Pt-TEMPO followed by irradiation with blue light (50 mW at 465 nm) for 1 h. The complex was not toxic in the dark but potently cytotoxic when irradiated in the cells with blue visible light, giving a phototoxicity index (PI) of >8. A2780 cells are >14× more sensitive to Pt-TEMPO than to cisplatin when treated under the same conditions. The activity of cisplatin was not affected by the irradiation. The photocytotoxicity of Pt-TEMPO is similar to that21 of the parent trans-dihydroxido complex [Pt(N3)2(OH)2(Py)2] which shows that replacement of one axial hydroxide ligand by a TEMPO-conjugated succinate ligand does not diminish the activity. Interestingly, the activity of the new complex Pt-TEMPO is as high as the organic photosensitizer chlorpromazine (CPZ) (Table 1).
| Compound | IC50(μM) | |
|---|---|---|
| +465 nm | Dark | |
| Pt-TEMPO | 5.9 ± 0.2 | >50 |
| [Pt(N3)2(OH)2(Py)2] | 7.1 ± 0.4 | >60 |
| CPZ | 6.2 ± 0.3 | >100 |
| CDDP | >80 | >80 |
In summary, a diazido Pt(IV) pro-drug trans,trans,trans-[Pt(N3)2(OH)(OCOCH2CH2CONH-TEMPO)(Py)2] (Pt-TEMPO) spin-labelled with a nitroxyl radical has been synthesised. It is non-toxic in the dark but potently cytotoxic towards cancer cells when irradiated with visible light. The photocytotoxicity is as high as the clinical photosensitizer chlorpromazine and much more cytotoxic than anticancer drug cisplatin under the same conditions. EPR experiments showed that photoactivation of Pt-TEMPO with blue light leads to the formation of azidyl and TEMPO radicals in solution. The reactive azidyl radicals were captured by the spin trap DMPO. The anticancer activity of Pt-TEMPO may be the result of attack on DNA by the Pt(II) photoproducts as well as the activity of the reactive azidyl and appended TEMPO radicals. The complex should be suitable for the treatment of surface cancers such as bladder and oesophagus, and if deeper light penetration is required then either two-photon activation or coupling to an upconversion nanoparticle system may be possible.22,23
Acknowledgements
We thank the Newton International Fellowship program (VV), EPSRC (grant no EP/J500045/1, CJW), and ERC (grant no. 247450, IRC, PJS) for support, and Mr James Coverdale for his excellent assistance with ICP experiments.Notes and references
- F. M. Muggia, A. Bonetti, J. D. Hoeschele, M. Rozencweig and S. B. Howell, J. Clin. Oncol., 2015, 33, 4219 CrossRef CAS PubMed.
- T. C. Johnstone, K. Suntharalingam and S. J. Lippard, Chem. Rev., 2016, 116, 3436 CrossRef CAS PubMed.
- M. S. Robillard and J. Reedijk, Encyclopedia of Inorganic and Bioinorganic Chemistry, 2011, DOI:10.1002/9781119951438.eibc0178.
- J. T. Hartmann and H. P. Lipp, Expert Opin. Pharmacother., 2003, 4, 889 CrossRef CAS PubMed.
- J. T. Hartmann, C. Kollmannsberger, L. Kanz and C. Bokemeyer, Int. J. Cancer, 1999, 83, 866 CrossRef CAS PubMed.
- A. Florea and D. Büsselberg, Cancer, 2011, 3, 1351 CrossRef CAS PubMed.
- N. J. Farrer, L. Salassa and P. J. Sadler, Dalton Trans., 2009, 10690 RSC.
- C. Daniel, Coord. Chem. Rev., 2015, 282–283, 19 CrossRef CAS.
- N. A. Smith and P. J. Sadler, Philos. Trans. R. Soc. London, Ser. A, 2013, 371, 1 CrossRef PubMed.
- S. Hirosawa, S. Araib and S. Takeoka, Chem. Commun., 2012, 48, 4845 RSC.
- D. Tatlidil, M. Ucuncu and Y. Akdogan, Phys. Chem. Chem. Phys., 2015, 17, 22678 RSC.
- G. Martini and L. Ciani, Phys. Chem. Chem. Phys., 2009, 11, 211 RSC.
- T. Offer, A. Russo and A. Samuni, FASEB J., 2000, 14, 1215 CAS.
- S. Suy, J. B. Mitchell, D. Ehleiter, A. H. Friedman and U. Kasid, J. Biol. Chem., 1998, 273, 17871 CrossRef CAS PubMed.
- V. D. Sen’, Russ. Chem. Bull., 1993, 42, 507 CrossRef.
- V. D. Sen’, V. A. Golubev, L. M. Volkova and N. P. Konovalova, J. Inorg. Biochem., 1996, 64, 69 CrossRef.
- V. B. Arion, A. Dobrov, S. Goschl, M. A. Jakupec, B. K. Keppler and P. Rapta, Chem. Commun., 2012, 48, 8559 RSC.
- G. T. Wondrak, Antioxid. Redox Signaling, 2009, 11, 3013 CrossRef CAS PubMed.
- J. S. Butler, J. A. Woods, N. J. Farrer, M. E. Newton and P. J. Sadler, J. Am. Chem. Soc., 2012, 134, 16508 CrossRef CAS PubMed.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42 CrossRef CAS PubMed.
- N. J. Farrer, J. A. Woods, L. Salassa, Y. Zhao, K. S. Robinson, G. Clarkson, F. S. Mackay and P. J. Sadler, Angew. Chem., Int. Ed., 2010, 49, 8905 CrossRef CAS PubMed.
- Y. Zhao, G. M. Roberts, S. E. Greenough, N. J. Farrer, M. J. Paterson, W. H. Powell, V. G. Stavros and P. J. Sadler, Angew. Chem., Int. Ed., 2012, 51, 11263 CrossRef CAS PubMed.
- Y. Min, J. Li, F. Liu, E. K. L. Yeow and B. Xing, Angew. Chem., Int. Ed., 2014, 53, 1012 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6dt01382a. Data created during this research are openly available from the University of Warwick Research Archive Portal at http://wrap.warwick.ac.uk/79055/ |
| This journal is © The Royal Society of Chemistry 2016 |