
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceC3-symmetry Mo3S4 aminophosphino clusters combining three sources of stereogenicity: stereocontrol directed by hydrogen bond interactions and ligand configuration†
Carmina
Alfonso
a,
Marta
Feliz
b,
Vicent S.
Safont
 a and
Rosa
Llusar
*a
a and
Rosa
Llusar
*a
aDepartament de Química Física i Analítica, Universitat Jaume I, Av. Sos Baynat s/n, 12071 Castelló, Spain
bInstituto de Tecnología Química, CSIC-UPV, Avda. de los Naranjos, s/n, 46022, Valencia, Spain. E-mail: rosa.llusar@uji.es; Fax: +34 964 728066; Tel: +34 964 728086
First published on 11th April 2016
Abstract
A diastereoselective synthesis of proline containing aminophosphino cubane-type Mo3S4 clusters, (P)-[Mo3S4Cl3((1S,2R)-PPro)3]Cl ((P)-[Mo-(SN,RC)]Cl) and (P)-[Mo3S4Cl3((1S,2S)-PPro)3]Cl ((P)-[Mo-(SN,SC)]Cl), has been achieved in high yields by reacting the corresponding enantiomerically pure PPro ((R)- and (S)-2-[(diphenylphosphino)methyl]pyrrolidine) ligands with the Mo3S4Cl4(PPh3)3(H2O)2 complex. Circular dichroism, nuclear magnetic resonance and X-ray techniques confirm that the (P)-[Mo-(SN,RC)]Cl and (P)-[Mo-(SN,SC)]Cl cluster cations are diastereoisomers which combine three sources of stereogenicity provided by the cluster framework, one carbon atom of the aminophosphine ligand and the nitrogen stereogenic center. The higher stability of the (P)-[Mo-(SN,SC)]+ cation is due to stabilizing vicinal Cl⋯HN interactions as well as due to the cis-fused conformation of the bicyclic system formed upon coordination of the aminophosphine ligand.
Introduction
One of the most intriguing aspects in the chemistry of transition metal clusters concerns their potential as selective catalysts beautifully exemplified in the specificity of some enzymes.1,2 In general, enantioselectivities using cluster-based chiral catalysts are low in comparison with mononuclear catalysts although evidence exists on the strong chiral induction by chiral phosphines in cluster-based hydrogenation.3 Recently we reported that diphosphino, aminophosphino and diamino Mo3S4 cuboidal clusters, represented in Fig. 1, are efficient catalysts or precatalysts for the hydrogenation of aromatic nitroderivatives.4–6 The cluster framework in these trimetallic clusters is chiral, and the terms P and M are used on the basis of the stereospecific arrangement of the halogen around the M3S4 (M = Mo or W) core, as illustrated in Fig. 1. In the case of the aminophosphino derivatives, the phosphorus atom of the ligand invariably occupies the outer position trans to the capping sulphur.7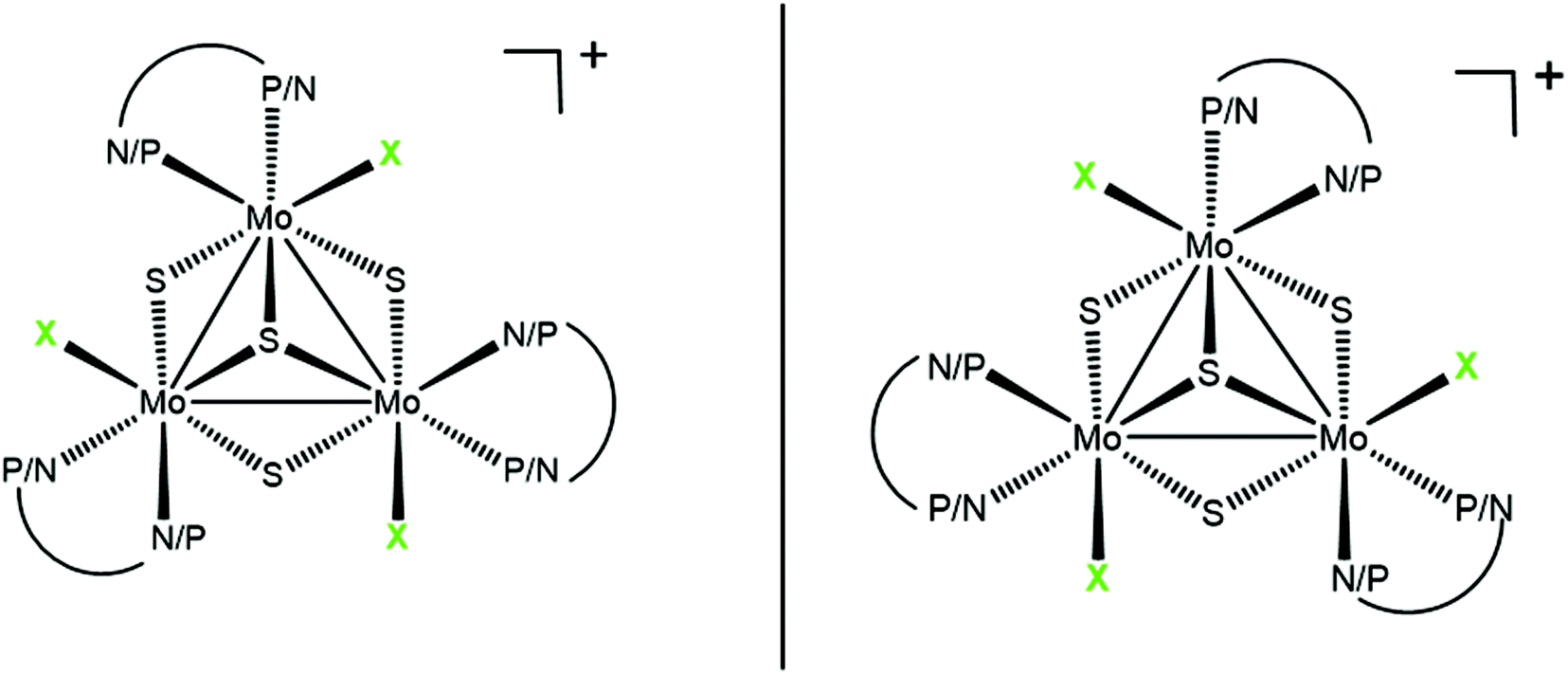 | ||
| Fig. 1 Absolute configurations of the P (left) and M (right) Mo3S4 diastereomers. The P symbol refers to a clockwise sense of rotation of the chlorine ligands around the threefold axis with the capping sulphur pointing towards the viewer. The M symbol refers to the anticlockwise rotation. | ||
Within the last decade, we have developed a stereoselective synthetic route for the synthesis of complexes of the general formula [Mo3S4Cl3(diphosphino)3]+ by reacting polymeric {Mo3S7Cl4}n phases with enantiopure bis(phosphines), namely ((R,R)-Me-BPE ((+)-1,2-bis[(2R,5R)-2,5-(dimethylphospholan-1-yl)]ethane) or (S,S)-Me-BPE ((−)-1,2-bis[(2S,5S)-2,5-(dimethylphospholan-1-yl)]ethane)).8 The (R,R)-diphosphine invariably leads to (P)-[Mo3S4Cl3((R,R)-Me-BPE)3]+ while (M)-[Mo3S4Cl3((S,S)-Me-BPE)3]+ is isolated as the only product from the reaction with the (S,S)-diphosphine, both compounds being enantiomers of each other. This synthetic procedure was further extended to tungsten and selenium as well as to other optically pure diphosphines.9–11 All these complexes combine the cluster framework chirality with that of the diphosphine ligand. In addition, these trimetallic Mo3S4 compounds can act as metalloligands for a second transition metal to afford catalytically active chiral Mo3S4Cu clusters with moderate enantioselection in cyclopropanation reactions while preserving the absolute configuration of the cluster framework.8
Motivated by the important role of the NH group in catalysis, we decided to explore synthetic routes aimed to obtain optically pure aminophosphine derivatives of the Mo3S4 cluster core.12 For that, we have chosen commercially available chiral aminophosphines derived from (R) and (S)-proline, whose structures are depicted in Fig. 2. The reaction outcome invariably leads to (P)-diastereoisomers unlike the results obtained with diphosphines. To rationalize this unexpected result, theoretical calculations on the relative energies among different stereoisomers have been undertaken.
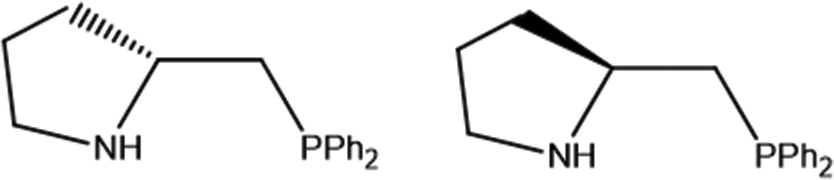 | ||
| Fig. 2 Structure of (R)-2-[(diphenylphosphino)methyl]pyrrolidine ((R)-PPro, left) and (S)-2-[(diphenylphosphino)methyl]pyrrolidine ((S)-PPro, right). | ||
Results and discussion
Synthesis and structure
The molecular Mo3S4Cl4(PPh3)3(H2O)2 precursor has been successfully employed for the preparation of [Mo3S4Cl3(diamino)3]+ and [Mo3S4Cl3(diphosphino)3]+ cluster complexes.5,13 The reaction between Mo3S4Cl4(PPh3)3(H2O)2 with stoichiometric amounts of the aminophosphine ligand (R)-PPro or its enantiomer, (S)-PPro, leads to the enantiopure cluster complexes [Mo3S4Cl3((1S,2R)-PPro)3]Cl ([Mo-(SN,RC)]Cl) and [Mo3S4Cl3((1S,2S)-PPro)3]Cl ([Mo-(SN,SC)]Cl) in ca. 85% yields, according to eqn (1), where PPro is noted as its (R)- or (S)-enantiomer, indistinctively. Structural studies show that upon N-coordination, the nitrogen atom of the pyrrolidine ring becomes stereogenic and invariably adopts an S conformation, as discussed next.| Mo3S4Cl4(PPh3)3(H2O)2 + 3PPro → [Mo3S4Cl3(PPro)3]Cl + 3PPh3 + 2H2O | (1) |
The reaction products with both (R)-PPro, and (S)-PPro show one peak centered at 1330 m/z associated with the [Mo3S4Cl3(PPro)3]+ cation on the basis of the m/z value and its characteristic isotopic pattern. However, there are small differences between the chemical shifts of the unique peak observed in the 31P{1H} NMR spectra of the [Mo-(SN,RC)]+ (32.3 ppm) and [Mo-(SN,SC)]+ (32.1 ppm) complexes and those assigned to three equivalent phosphorus atoms. These differences indicate that these two cluster cations are not enantiomers, a conclusion that was supported by circular dichroism (CD) spectroscopy. Fig. 3 shows the CD spectra of sample solutions of the chlorido salts of the [Mo-(SN,RC)]+ and [Mo-(SN,SC)]+ cations in dichloromethane, in which differences in the intensities and signs of the signals are appreciated. The characteristic bands of the [Mo-(SN,RC)]+ cluster appear at 260 and 301 nm for +51.7 and +14.7 mdeg respectively, while two signals of opposite signs at 254 nm for −46 mdeg and at 313 nm for −3.2 mdeg are found in the CD spectrum of [Mo-(SN,SC)]+ so the two bands must involve proline (R or S) based ligand orbitals.8 Interestingly, the observed band around 255–260 nm shows a red shift with regard to that of the free aminophosphine ligand (220 nm) and opposite signs. The most intense bands registered for both complexes below 250 nm have the same sign and can be tentatively assigned to transitions between orbitals with a marked contribution from the S nitrogen stereogenic center.14,15 The specific rotation is approximately one order of magnitude lower than that of the analogous diphosphino complexes, which show values from −400 to +450 mdeg.8
 | ||
| Fig. 3 CD spectra of [Mo-(SN,RC)]Cl (black line) and [Mo-(SN,SC)]Cl (red line) complexes. | ||
The absolute configuration of the [Mo-(SN,RC)]+ and [Mo-(SN,SC)]+ cations was determined by X-ray crystallography from their tetrafluoroborate salts. Both structures are solved in the non-centrosymmetric P213 cubic group with Flack parameters close to zero, and the Mo3S4 cluster core located on a C3 axis passing through the capping sulphur, the absolute configuration being P for both [Mo-(SN,RC)]+ and [Mo-(SN,SC)]+ cations. An ORTEP representation of the two cations is depicted in Fig. 4. The stereochemistry of the PPro aminophosphine ligand as R for (P)-[Mo-(SN,RC)]+ and S for (P)-[Mo-(SN,SC)]+ could be established with no ambiguity. X-ray confirms that these (P)-[Mo-(SN,RC)]+ and (P)-[Mo-(SN,SC)]+ cluster cations are diastereoisomers that combine the chirality (R or S) of the PPro aminophosphine ligand with that of the cluster framework. In addition, the pyrrolidine ring can adopt two conformations upon N-coordination that allow the stereogenic nitrogen atom to be R or S (S in our case) and the concomitant introduction of a third source of stereogenicity.
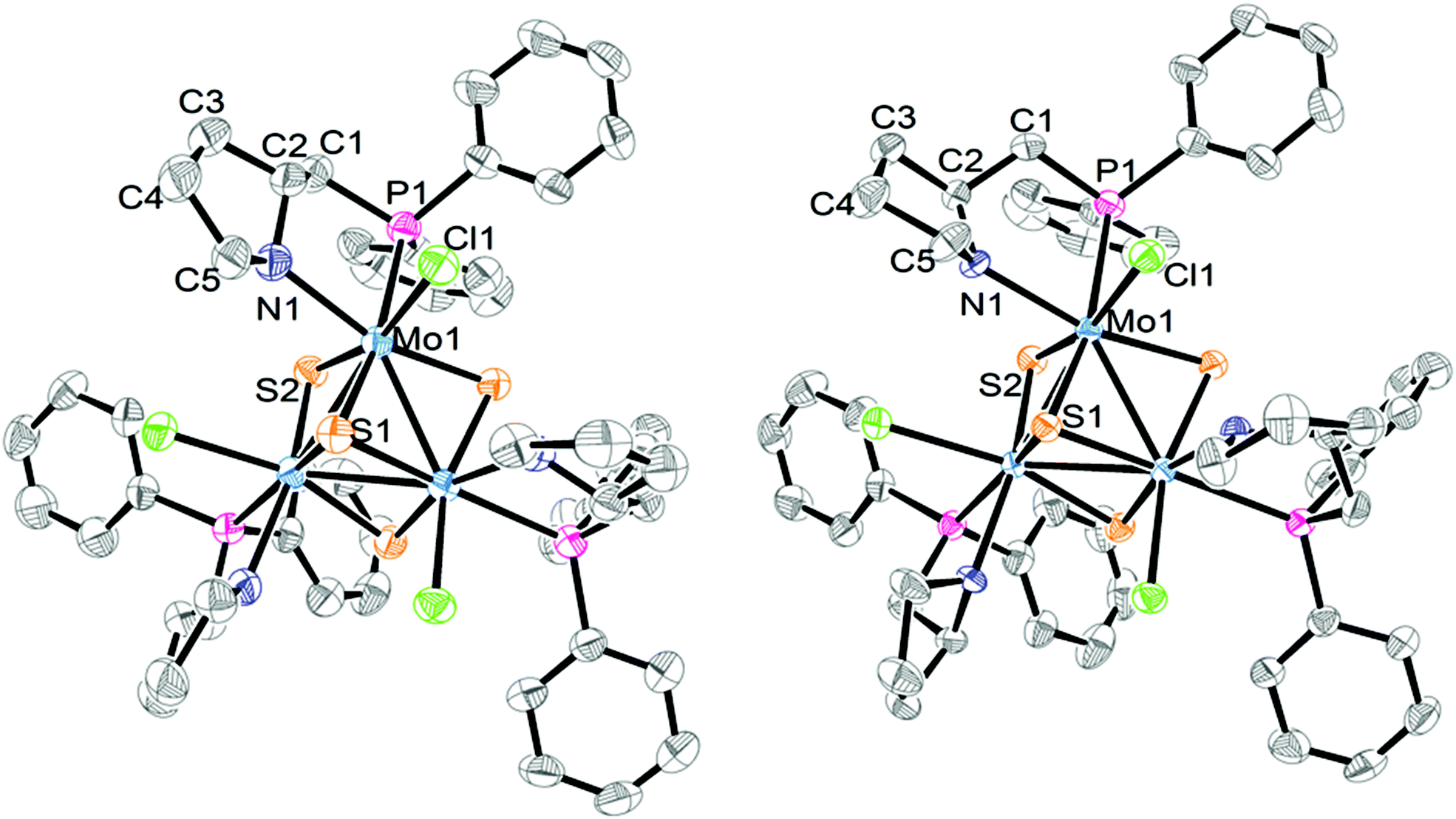 | ||
| Fig. 4 ORTEP diagrams of (P)-[Mo-(SN,RC)]+ (left) and (P)-[Mo-(SN,SC)]+ (right) with the atom numbering scheme. Hydrogen atoms are omitted for clarity. | ||
Table 1 lists the most relevant bond distances for (P)-[Mo-(SN,RC)]+ and (P)-[Mo-(SN,SC)]+ together with those of a closely related aminophosphino complex. The interatomic Mo–Mo and Mo–S distances follow the tendencies observed for other Mo3S4 cluster compounds.10,16,17 Each molybdenum atom in (P)-[Mo-(SN,RC)]+ and (P)-[Mo-(SN,SC)]+ is in a pseudooctahedral environment defined by three sulphur atoms, one chlorine atom, and the nitrogen and phosphorus atoms of the aminophosphine ligand, where the phosphorus atom is located trans to the capping sulphur. Such preferential spatial disposition of the aminophosphine ligand when coordinated to cuboidal Mo3S4 cluster units is not unprecedented, and considerably diminishes the number of potential stereoisomers, as also observed in W3S4 clusters.7 Coordination of the chiral PPro ligand to the metal results in the formation of a stereogenic nitrogen center incorporated into a [3.3.0] bicyclic system (see Fig. 4 and 5a). This bicyclic system adopts a cis-fused conformation in the (P)-[Mo-(SN,SC)]+ complex, which is known to be thermodynamically more stable than the trans-fused structure by analogy to bicyclooctanes.18 The calculated dihedral angles (C5–N–C2–C1 and C3–C2–N–Mo, see Table 1) confirm the cis conformation of the bicyclic system with values of 96.860° and 14.402° respectively. In the case of the (P)-[Mo-(SN,RC)]+ complex, both rings lie nearly in the same plane with dihedral angles of 11.269° and 179.416°. As a result, a selective formation of an S configuration at each stereogenic nitrogen center takes place for both (P)-[Mo-(SN,SC)]+ and (P)-[Mo-(SN,RC)]+ diastereoisomers. Note that once the stereochemistry of a nitrogen center is defined, that of the other two nitrogen atoms is equally fixed. The interatomic distances in both clusters are similar to those observed for [Mo3S4Cl3(edpp)3]BPh4 (edpp = (2-aminoethyl)diphenylphosphine), as can be seen in Table 1.6 The small deviations among Mo–(μ-S) bond distances in the three complexes suggest a similar trans influence of the nitrogen vs. the chlorine atoms in these compounds in contrast with the differences found in the corresponding [Mo3S4Cl3(diphosphine)3]+ cluster cations.
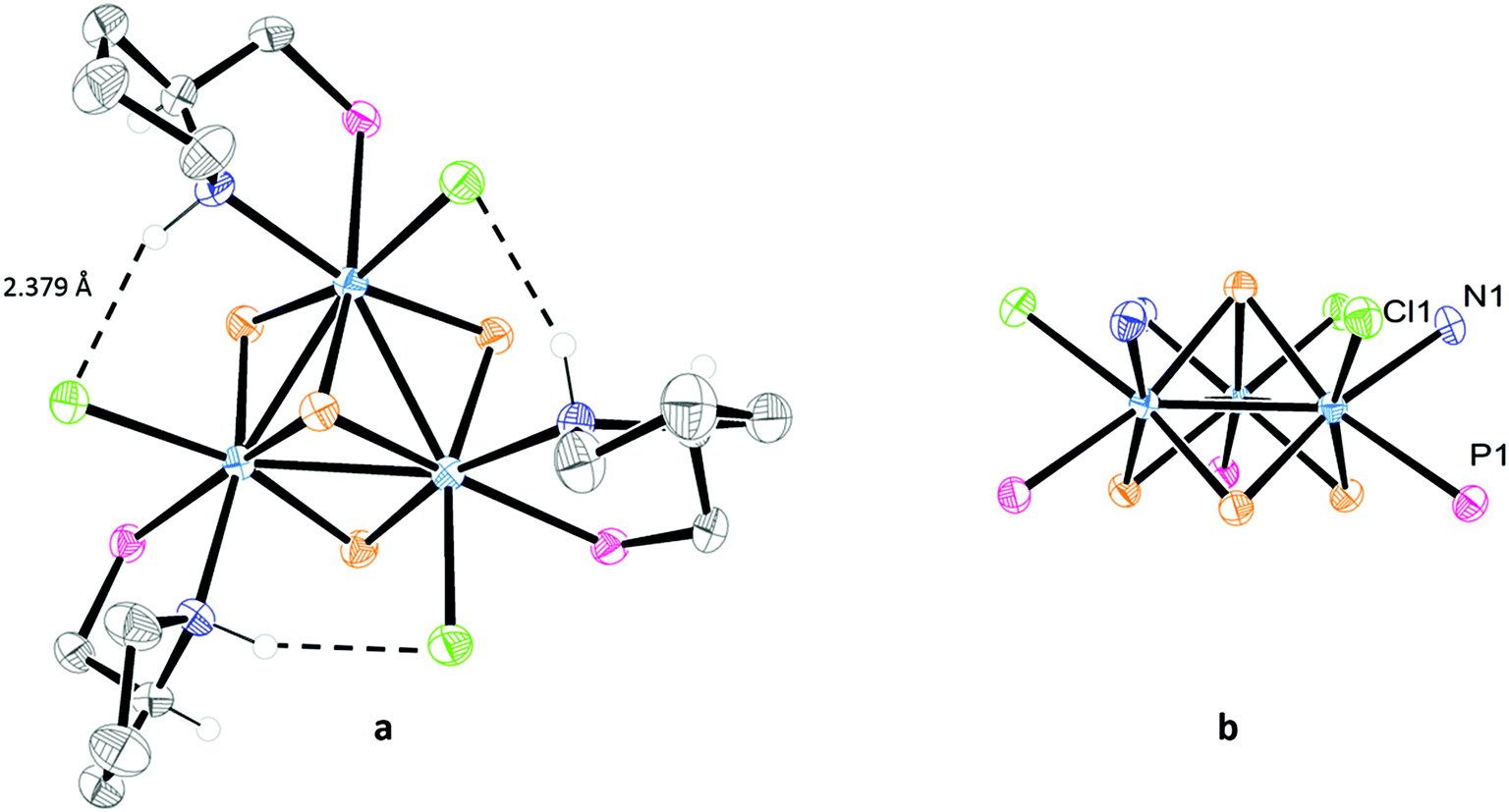 | ||
| Fig. 5 ORTEP representations of the (P)-[Mo-(SN,SC)]+ complex. Phenyl groups are omitted. (a) Hydrogen atoms have been deleted for simplicity, except for those bonded to stereogenic centers. (b) Carbon and hydrogen atoms are omitted for clarity. | ||
| Distancea (Å) | (P)-[Mo-(SN,RC)]BF4 | (P)-[Mo-(SN,SC)]BF4 | [Mo3S4Cl3(edpp)3]BPh4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
|---|---|---|---|
| a Standard deviations for averaged values are given in brackets. b Data taken from ref. 6. c Distance trans to the Mo–N bond. d Distance trans to the Mo–Cl bond. | |||
| Mo–Mo | 2.752(1) | 2.757(1) | 2.7463[7] |
| Mo–(μ3-S) | 2.364(3) | 2.353(3) | 2.3611[13] |
| Mo–(μ-S)c | 2.277(2) | 2.293(2) | 2.2863[12] |
| Mo–(μ-S)d | 2.297(2) | 2.294(2) | 2.2960[12] |
| Mo–P | 2.547(2) | 2.550(2) | 2.5414[14] |
| Mo–N | 2.300(7) | 2.289(7) | 2.2733[4] |
| Mo–Cl | 2.461(2) | 2.484(2) | 2.4625[15] |
| Dihedral C5–N–C2–C1 | 11.269° | 96.860° | — |
| Dihedral C3–C2–N–Mo | 179.416° | 14.402° | — |
The chlorine and nitrogen atoms in (P)-[Mo-(SN,RC)]+ and (P)-[Mo-(SN,SC)]+ are found on the same side of a trimetallic plane, as emphasized in Fig. 5b, allowing multiple Cl⋯HN interactions. Short Cl⋯HN interactions are observed in (P)-[Mo-(SN,SC)]+ between the chlorine atom coordinated to one metal and the hydrogen atom of the amino group coordinated to the adjacent metal, resulting in a vicinal Cl⋯HN bond length of 2.379 Å (see Fig. 5a) vs. the 2.941 Å found in (P)-[Mo-(SN,RC)]+. Similar values, ranging from 2.436 to 2.995 Å, are reported for the aminophosphine [Mo3S4Cl3(edpp)3]+ cluster analogue.6 These vicinal Cl⋯HN interactions are expected to confer to the (P)-[Mo-(SN,SC)]+ cation an additional stability. On the other hand, the Cl⋯N distances associated with the geminal Cl⋯HN interactions, in which the NH and the Cl belong to the coordination sphere of the same metal centre of the cluster unit, (3.055 Å for (P)-[Mo-(SN,RC)]+, and 3.170 Å for (P)-[Mo-(SN,SC)]+) are slightly longer than the corresponding distance reported for the [Mo3S4Cl3(edpp)3]+ cation (3.021 Å).6 This confirms the similar chelating mode for both edpp and (R)- or (S)-PPro aminophosphines.
To summarize, the reaction between Mo3S4Cl4(PPh)3(H2O)2 and (R)-PPro or (S)-PPro is diastereoselective towards the (P)-Mo3S4 configuration with the preservation of the stereochemistry of the aminophosphine and formation of an S stereogenic nitrogen center. This result is unexpected based on our previous observations on diphosphino Mo3S4 clusters that showed the stereoselective formation of the (P)-M3Q4 (M = Mo, W; Q = S, Se) configuration upon coordination of the enantiomerically pure (R,R)-diphosphine, and the (M)-M3Q4 configuration when the (S,S)-ligand was reacted instead. In both cases, the chirality of the diphosphine was preserved.8–11 In order to explain the preferential diastereoselective formation of [Mo3S4Cl3(PPro)3]+ complexes, DFT calculations were performed to study the differences between relative energies among the four possible isomers.
Theoretical calculations
Four diastereoisomers ((P)-[Mo-(SN,RC)]+, (M)-[Mo-(SN,RC)]+, (P)-[Mo-(SN,SC)]+ and (M)-[Mo-(SN,SC)]+) have been taken as cluster models for the B3LYP calculations. The initial structural parameters of (P)-[Mo-(SN,RC)]+ and (P)-[Mo-(SN,SC)]+ cations have been extracted from crystallographic data, whereas the (M)-isomers have been built by changing the chlorine and aminophosphine orientation, but preserving the configuration of the stereogenic centers. The optimized bond distances and dihedral angles as well as the calculated relative energies are listed in Table 2. The experimentally isolated (P)-[Mo-(SN,RC)]+ and (P)-[Mo-(SN,SC)]+ stereoisomers are the most stable compounds and the highest stability calculated for (P)-[Mo-(SN,SC)]+ agrees with the expected stabilization attributed to the vicinal Cl⋯HN interaction observed in the crystal structure and also to the ligand configuration. Whereas (P)-[Mo-(SN,RC)]+ turned out to be 15.4 kcal mol−1 more stable than its corresponding M diastereoisomer, the (P)-[Mo-(SN,SC)]+ isomer is 20.0 kcal mol−1 less energetic than the (M)-[Mo-(SN,SC)]+ cation. These differences suggest a thermodynamic preference for the formation of P isomers.| Distances (Å) | (P)-[Mo-(SN,RC)] + | (P)-[Mo-(SN,SC)] + | (M)-[Mo-(SN,RC)] + | (M)-[Mo-(SN,SC)] + |
|---|---|---|---|---|
| a Distance trans to the Mo–N bond. b Distance trans to the Mo–Cl bond. c Relative to (P)-[Mo-(SN,SC)]+. | ||||
| Mo–Mo | 2.807 | 2.805 | 2.834 | 2.845 |
| Mo–(μ3-S) | 2.438 | 2.443 | 2.461 | 2.449 |
| Mo–(μ-S)a | 2.380 | 2.387 | 2.376 | 2.376 |
| Mo–(μ-S)b | 2.379 | 2.367 | 2.356 | 2.371 |
| Mo–P | 2.661 | 2.631 | 2.673 | 2.633 |
| Mo–N | 2.289 | 2.291 | 2.297 | 2.362 |
| Mo–Cl | 2.550 | 2.570 | 2.600 | 2.544 |
| Dihedral C5–N–C2–C1 | 8.770° | 95.723° | 24.866° | 81.800° |
| Dihedral C3–C2–N–Mo | 179.573° | 14.052° | 6.480° | 4.885° |
| Energies (kcal mol−1)c | 4.8 | 0.0 | 20.2 | 20.0 |
The calculated bond distances listed in Table 2 show a good agreement with the experimental data for the (P)-clusters (see Table 1), whereas the longer Mo–Mo and Mo–(μ3-S) distances for (M)-[Mo-(SN,RC)]+ and (M)-[Mo-(SN,SC)]+ suggest a slight distortion of the Mo3S4 cluster core for these theoretically calculated species. The calculated values for the dihedral angles that describe the conformation of the bicyclic system in the ligand are also in good agreement with the experimental data, with differences ranging less than three degrees. Therefore, both the experimental as well as the theoretical studies establish that the (P)-[Mo-(SN,SC)]+ complex exhibits a cis-fused conformation of the bicyclic system that confers additional stabilization to the system. In addition, the stabilizing effect of the vicinal Cl⋯HN interaction can also be observed in the (P)-[Mo-(SN,SC)]+ calculated structure, that shows an interatomic Cl⋯H distance of only ca. 2.32 Å.
A similar Cl⋯HN interaction can also be noticed in the calculated (M)-[Mo-(SN,RC)]+ species, with a Cl⋯H distance of 2.255 Å, although in this case the interaction takes place between the NH and the Cl belonging to the coordination sphere of the same metal centre of the cluster unit. In order to gain a deeper insight into the nature of such Cl⋯HN interactions, we have performed Natural Bond Order (NBO) and topological Electron Localization Function (ELF) analyses. The bonding character of the shortest Cl⋯HN interactions for (M)-[Mo-(SN,RC)]+ and (P)-[Mo-(SN,SC)]+ has been confirmed by their higher bond orders (0.043 and 0.052, respectively) in comparison with those calculated for geminal and vicinal Cl⋯HN interactions found for the respective (P)-[Mo-(SN,RC)]+ and (M)-[Mo-(SN,SC)]+ isomers (bond orders of ca. 0.003). As can be seen, the Cl–H bond order calculated for the (M)-[Mo-(SN,RC)]+ species is ca. 17% lower than the bond order calculated for (P)-[Mo-(SN,SC)]+, which qualitatively agrees with the calculated stabilities: the (P)-[Mo-(SN,SC)]+ species has a higher bond order and is more stable than the (M)-[Mo-(SN,RC)]+ cation.
On the other hand, the ELF analysis of the Cl⋯HN molecule fragment shows the disynaptic V(N–H) basin accounting for the covalent N–H bond, and two monosynaptic V(Cl) basins, one roughly oriented to the hydrogen atom and the other to the molybdenum atom. There is no disynaptic V(Cl–H) basin, as expected due to the ionic character of this Cl⋯H interaction. The population of the V(Cl) basin pointing to the H atom and therefore involved in the Cl⋯HN interaction is higher for the (P)-[Mo-(SN,SC)]+ structure (3.91e−) than the one found in the (M)-[Mo-(SN,RC)]+ case (3.77e−). This indicates a more electronic contribution of the V(Cl) to the vicinal Cl⋯HN interaction in (P)-[Mo-(SN,SC)]+ as compared with the geminal Cl⋯HN in (M)-[Mo-(SN,RC)]+, which supports from a topological point of view the higher stabilizing effect of such an interaction found for the (P)-[Mo-(SN,SC)]+ structure.
Therefore, in spite of the similar Cl⋯HN distance calculated either for the vicinal interaction in (P)-[Mo-(SN,SC)]+ or for the geminal interaction in (M)-[Mo-(SN,RC)]+, both the NBO as well as the ELF analysis suggest a stronger interaction in the vicinal case, thus contributing to its higher stability.
Conclusions
Two cluster complexes [Mo3S4Cl3(PPro)3]Cl have been prepared in high yields by reacting stoichiometric amounts of the (R)- and (S)-PPro ligands with the Mo3S4Cl4(PPh3)3(H2O)2 complex. 31P{1H} NMR and CD analyses suggest that both cluster compounds are diastereoisomers. Single-crystal X-ray confirms the preferential disposition of the chiral PPro aminophosphine ligand once coordinated to the trinuclear backbone to allow two (P)-stereoisomers, namely (P)-[Mo-(SN,RC)]Cl and (P)-[Mo-(SN,SC)]Cl, with the preservation of the stereochemistry of the aminophosphine and the selective formation of a stereogenic nitrogen center with S configuration. These results confirm the presence of three sources of stereogenicity integrated in each cluster compound. A short stabilizing Cl⋯HN distance has been found in the (P)-[Mo-(SN,SC)]Cl complex from experimental and B3LYP methods. The calculated relative energies of the four possible (P)- and (M)-[Mo3S4Cl3(PPro)3]+ stereoisomers support the thermodynamic preference of the formation of (P)-isomers. The cis-fused conformation of the molybdenum–PPro bicyclic system observed in (P)-[Mo-(SN,SC)]+ confers some additional stabilization to the system, and the stabilizing effect of the Cl⋯HN interaction has been supported by electronic population (NBO) and topological (ELF) analyses. These new aminophosphine trimetallic complexes which combine three sources of stereogenicity constitute an ideal “proof of concept” system to evidence metal cluster catalysis.Experimental section
General remarks
Precursor Mo3S4Cl4(PPh3)3(H2O)2 was prepared according to literature methods.19,20 ESI-MS spectra were recorded with a Quattro LC (quadrupole–hexapole–quadrupole) mass spectrometer with an orthogonal Z-spray electrospray interface (Micromass, Manchester, UK). The cone voltage was set at 20 V unless otherwise stated using CH3CN as the mobile phase solvent. Nitrogen was employed as a drying and nebulising gas. 31P NMR spectra were recorded on a Varian Innova 300 MHz using CD2Cl2 as a solvent. Circular dichroism measurements were recorded on a JASCO J-810 spectropolarimeter.Preparation of (P)-[Mo3S4Cl3((1S,2R)-PPro)3]Cl, (P)-[Mo-(SN,RC)]Cl
To a green suspension of [Mo3S4Cl4(PPh)3(solv)2] (57.0 mg, 0.0423 mmol) in ethanol (5 mL) were added 3.2 equivalents of the enantiomerically pure ligand (R)-2-[(diphenylphosphino)methyl]pyrrolidine, (R)-PPro, (36.5 mg, 0.1355 mmol) under a nitrogen atmosphere. After 3 hours, the green solution was concentrated under reduced pressure and the desired product was precipitated by adding hexane. Finally, the green solid was separated by centrifugation and washed with hexane to yield 46.7 mg (81%).31P {1H} NMR (121 MHz, CD2Cl2) δ (ppm): 32.3 (s, 3P). ESI-MS (20 V, CH3CN) m/z: 1330 (M+).
Preparation of (P)-[Mo3S4Cl3((1S,2S)-PPro)3]Cl, (P)-[Mo-(SN,SC)]Cl
This compound was prepared and purified following the procedure described for (P)-[Mo-(SN,RC)]Cl, but using the enantiomerically pure aminophosphine (S)-2-[(diphenylphosphino)methyl]pyrrolidine, (S)-PPro. A green solid was obtained (89%).31P {1H} NMR (121 MHz, CD2Cl2) δ (ppm): 32.1 (s, 3P). ESI-MS (20 V, CH3CN) m/z: 1330 (M+).
X-ray data collection and structure refinement
Diffraction data for enantiomerically pure compounds (P)-[Mo-(SN,RC)]BF4 and (P)-[Mo-(SN,SC)]BF4 were collected on an Agilent Supernova diffractometer equipped with an Atlas CCD detector using Mo Kα radiation (λ = 0.71073 Å). Absorption corrections based on the multiscan method were applied.21,22 Structures were solved using direct methods in SHELXS-13 and refined by the full matrix method based on F2 using the OLEX software package.23,24 Suitable crystals for X-ray studies of (P)-[Mo-(SN,RC)]BF4 and (P)-[Mo-(SN,SC)]BF4 were grown by slow diffusion of toluene into a sample solution in CH2Cl2. Anion exchange, Cl− by BF4−, was carried out by elution with a NaBF4 solution in acetone after absorption of a CH2Cl2 solution of (P)-[Mo-(SN,RC)]Cl or (P)-[Mo-(SN,SC)]Cl in a silica gel column. Crystal data for (P)-[Mo-(SN,RC)]BF4: C51H60BCl3F4Mo3N3P3S4, M = 1417.15, cubic, space group P213 (no. 198), a = b = c = 19.0728 (2) Å, α = β = γ = 90.00°, V = 6938.1 (2) Å3, T = 200.00 (10) K, Z = 4, μ = 0.881 mm−1. Reflections (collected/unique): 14383/3964 (Rint = 0.0309). The final refinement converged with R1 = 0.0484 and wR2 = 0.1535 for all reflections, GOF = 1.158, max/min residual electron density 0.91/−0.45 e Å−3. Flack parameter: −0.07 (2). Anisotropic displacement parameters were refined for all non-H atoms. After reaching convergence large solvent voids appear in the structure. However, the absence of electron density in the voids makes any model meaningless.
Crystal data for (P)-[Mo-(SN,SC)]BF4·(C7H8): C51H60BCl3F4Mo3N3P3S4·(C7H8), M = 1509.27, cubic, space group P213 (no. 198), a = b = c = 19.5506 (3) Å, α = β = γ = 90.00°, V = 7472.7 (19) Å3, T = 200.00 (14) K, Z = 4, μ = 0.832 mm−1. Reflections (collected/unique): 28631/6384 (Rint = 0.0471). The final refinement converged with R1 = 0.0694 and wR2 = 0.1184 for all reflections, GOF = 1.164, max/min residual electron density 0.80/−0.53 e Å−3. Flack parameter: −0.03 (2). Anisotropic displacement parameters were refined for all non-H atoms.
Computational details
All geometry optimizations and NBO calculations were performed with the Gaussian09 program suite.25 Density functional theory was applied with the Becke hybrid B3LYP functional.26–28 The double-ξ pseudo-orbital basis set LanL2DZ, in which all atoms are represented by the relativistic core LanL2 potential of Los Alamos, was used. The geometry optimizations have been performed in the gas phase without any symmetry constraint followed by analytical frequency calculations to confirm that a minimum has been reached. The ELF analysis has been performed by means of the TopMod package,29 considering a cubical grid a step size smaller than 0.05 Bohr, on the wavefunction obtained at the B3LYP/3-21G//B3LYP/LanL2DZ theoretical level.Acknowledgements
The financial support of the Spanish Ministerio de Economía y Competitividad (Grant CTQ2011-23157 and CTQ2015-65207-P), Universitat Jaume I (Research Project P1·1B2013-19) and Generalitat Valenciana (PROMETEOII/2014/022) is gratefully acknowledged. The authors also thank the Servei Central d'Instrumentació Científica (SCIC) of the University Jaume I for providing us with the mass spectrometry, NMR and X-ray facilities. C. A. thanks the Spanish Ministerio de Economía y Competitividad for a predoctoral fellowship (FPI).References
- A. Okrut, R. C. Runnebaum, X. Y. Ouyang, J. Lu, C. Aydin, S. J. Hwang, S. J. Zhang, O. A. Olatunji-Ojo, K. A. Durkin, D. A. Dixon, B. C. Gates and A. Katz, Nat. Nanotechnol., 2014, 9, 459 CrossRef CAS PubMed.
- L. Quintanar, J. J. Yoon, C. P. Aznar, A. E. Palmer, K. K. Andersson, R. D. Britt and E. I. Solomon, J. Am. Chem. Soc., 2005, 127, 13832 CrossRef CAS PubMed.
- V. Moberg, R. Duquesne, S. Contaldi, O. Rohrs, J. Nachtigall, L. Damoense, A. T. Hutton, M. Green, M. Monari, D. Santelia, M. Haukka and E. Nordlander, Chem. – Eur. J., 2012, 18, 12458 CrossRef CAS PubMed.
- I. Sorribes, G. Wienhofer, C. Vicent, K. Junge, R. Llusar and M. Beller, Angew. Chem., Int. Ed., 2012, 51, 7794 CrossRef CAS PubMed.
- E. Pedrajas, I. Sorribes, K. Junge, M. Beller and R. Llusar, ChemCatChem, 2015, 7, 2675 CrossRef CAS.
- T. F. Beltrán, PhD Thesis, Universitat Jaume I, 2013 Search PubMed.
- T. F. Beltran, J. A. Pino-Chamorro, M. J. Fernandez-Trujillo, V. S. Safont, M. G. Basallote and R. Llusar, Inorg. Chem., 2015, 54, 607 CrossRef CAS PubMed.
- M. Feliz, E. Guillamon, R. Llusar, C. Vicent, S. E. Stiriba, J. Perez-Prieto and M. Barberis, Chem. – Eur. J., 2006, 12, 1486 CrossRef CAS PubMed.
- E. Guillamón, R. Llusar, J. Pérez-Prieto and S. E. Stiriba, J. Organomet. Chem., 2008, 693, 1723 CrossRef.
- A. G. Algarra, M. G. Basallote, M. J. Fernandez-Trujillo, M. Feliz, E. Guillamon, R. Llusar, I. Sorribes and C. Vicent, Inorg. Chem., 2010, 49, 5935 CrossRef CAS PubMed.
- E. M. Guillamon, M. Blasco and R. Llusar, Inorg. Chim. Acta, 2015, 424, 248 CrossRef CAS.
- B. G. Zhao, Z. B. Han and K. L. Ding, Angew. Chem., Int. Ed., 2013, 52, 4744 CrossRef CAS PubMed.
- T. F. Beltran, R. Llusar, M. Sokolov, M. G. Basallote, M. J. Fernandez-Trujillo and J. A. Pino-Chamorro, Inorg. Chem., 2013, 52, 8713 CrossRef CAS PubMed.
- F. Egidi, R. Russo, I. Carnimeo, A. D'Urso, G. Mancini and C. Cappelli, J. Phys. Chem. A, 2015, 119, 5396 CrossRef CAS PubMed.
- B. C. Ringdahl and J. C. Craig, Acta Chem. Scand. Ser. B, 1980, 34, 731 CrossRef.
- R. Llusar and S. Uriel, Eur. J. Inorg. Chem., 2003, 1271 CrossRef CAS.
- C. Alfonso, T. F. Beltran, M. Feliz and R. Llusar, J. Cluster Sci., 2015, 26, 199 CrossRef CAS.
- V. F. Kuznetsov, G. P. A. Yap and H. Alper, Organometallics, 2001, 20, 1300 CrossRef CAS.
- F. A. Cotton, P. A. Kibala, M. Matusz, C. S. McCaleb and R. B. W. Sandor, Inorg. Chem., 1989, 28, 2623 CrossRef CAS.
- G. S. M. Sasaki, T. Ouchi and T. Shibahara, J. Cluster Sci., 1998, 9, 25 CrossRef.
- CrysAlisPro, Agilent Technologies, Santa Clara CA, 2012 Search PubMed.
- R. C. Clark and J. S. Reid, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1995, 51, 887 CrossRef.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- A. D. Becke, Phys. Rev. A, 1988, 3098 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 5648 CrossRef CAS.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 785 CrossRef CAS.
- X. Krokidis, S. Noury, F. Fuster and B. Silvi, Comput. Chem., 1999, 597 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Optimized electronic energies and Cartesian coordinates. CCDC 1455624 ((P)-[Mo3S4Cl3((1S,2R)-PPro)3]BF4) and 1455733 ((P)-[Mo3S4Cl3((1S,2S)-PPro)3]BF4). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt00755d |
| This journal is © The Royal Society of Chemistry 2016 |