
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Supramolecular exo-functionalized palladium cages: fluorescent properties and biological activity†
Andrea
Schmidt
ab,
Manuela
Hollering
a,
Markus
Drees
a,
Angela
Casini
*b and
Fritz E.
Kühn
*a
aMolecular Catalysis, Catalysis Research Center and Department of Chemistry, Technische Universität München, Lichtenbergstr. 4, 85747 Garching bei München, Germany. E-mail: fritz.kuehn@ch.tum.de
bMedicinal and Bioinorganic Chemistry, School of Chemistry, Cardiff University, Park Place, CF103AT Cardiff, UK. E-mail: casinia@cardiff.ac.uk
First published on 15th April 2016
Abstract
Metallosupramolecular systems are promising new tools for pharmaceutical applications. Thus, novel self-assembled Pd(II) coordination cages were synthesized which were exo-functionalized with naphthalene or anthracene groups with the aim to image their fate in cells. The cages were also investigated for their anticancer properties in human lung and ovarian cancer cell lines in vitro. While the observed cytotoxic effects hold promise and the cages resulted to be more effective than cisplatin in both cell lines, fluorescence emission properties were scarce. Therefore, using TD-DFT calculations, fluorescence quenching observed in the naphthalene-based system could be ascribed to a lower probability of a HOMO–LUMO excitation and an emission wavelength outside the visible region. Overall, the reported Pd2L4 cages provide new insights into the chemical–physical properties of this family of supramolecular coordination complexes whose understanding is necessary to achieve their applications in various fields.
Introduction
Coordination-driven self-assembly is an ideal method for constructing discrete, two- and three-dimensional metal-based entities.1 These supramolecular coordination complexes (SCCs) with well-defined geometries and cavities have found applications not only in host–guest chemistry2 and catalytic reactions,3 but also in biochemistry and medicine.4 For medicinal purposes, SCCs have been developed as drug delivery vehicles,5–7 to recognize and interact with biomolecules8 and to function as anticancer agents.9–11Notably, Therrien et al. reported the first coordination cage, a ruthenium–arene metallocage, used as a drug delivery system for Pd and Pt acetylacetonato complexes, showing anticancer effects in cancer cell line A2780.6 In further studies, ruthenium–arene metallocages displayed increased cytotoxic effects in human lung cancer cells compared to cisplatin, thus acting as anticancer drugs per se.11 Interestingly, the first toxicity studies in vivo of SCCs as anticancer compounds were performed with rhomboidal platinum(II) assemblies showing an effect on the reduction of the tumor growth rate in mice.10
A specific and attractive area of SCCs is the self-assembly of M2L4 (M = metal, L = ligand) metallocages,12 which can enclose a variety of small molecules within their cavity, such as ions13 and neutral molecules.14 In addition, the properties of the M2L4 coordination cages can be optimized by functionalization of the ligand framework.
Up to now, only a few reports on the biological properties of Pd2L4 complexes have appeared.5,15 Recently, we investigated fluorescent palladium cages as drug delivery systems for cisplatin, which proved to be active in cancer cells, while poorly toxic ex vivo in healthy rat liver tissues.5 The obtained metallocages showed fluorescence properties due to the used ligand system, namely exo-functionalized bipyridyl ligands. However, upon cage formation, a significant quenching of the ligands’ fluorescence occurred due to the binding to Pd2+ ions. The extremely low quantum yield of this first series of cages prevented their study of cellular uptake via fluorescence microscopy in most cases. Indeed, nearly all capsules and cages composed of coordination metal–ligand bonds are non-emissive due to quenching by the heavy transition metal ions.16 Only in a few cases, highly fluorescent M2L4 cages were obtained.17,18
However, fluorescent metal-based entities would be very attractive in the medicinal inorganic chemistry field, to achieve their imaging in biological environments via fluorescence microscopy. In fact, an increasing number of reports on anticancer metal compounds bearing fluorescent moieties for both therapeutic and imaging applications have appeared.19 The reported metal-based compounds include fluorescent Ru polypyridine complexes,20 as well as several organometallic compounds among others.21
Therefore, in this work, in order to obtain cytotoxic metallocages with improved fluorescence properties, we designed a new series of self-assembled exo-functionalized Pd2L4 cages coupled to fluorescent groups. The main structural motif of the Pd2L4 cage compound 1 attached to naphthalene or anthracene moieties is shown in Fig. 1. In addition, a cage featuring a carboxy group in the exo-position was also included for comparison purposes to better characterize the effects of the bulky fluorescent tags on the biological properties of metallocages. Thus, the photophysical properties of cage compounds and ligands were investigated and DFT calculations were performed to rationalize the observed spectroscopic properties. In addition, the cages and respective ligands were studied for their antiproliferative effects in human cancer cell lines in vitro.
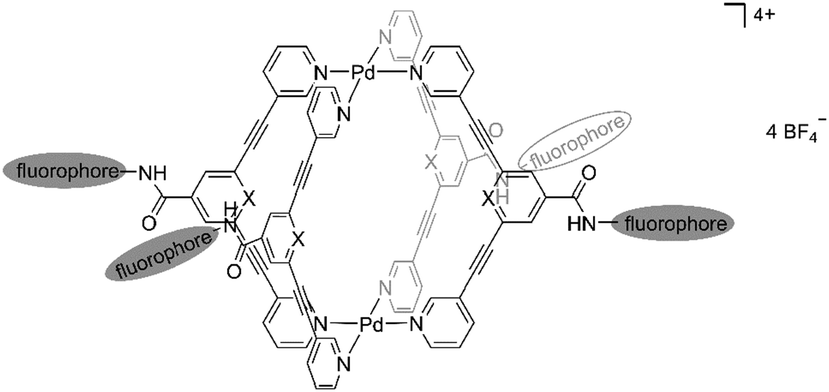 | ||
| Fig. 1 Schematic structure of Pd2L4 cage compound 1 attached to fluorophore moieties. | ||
Results and discussion
Synthesis and characterization
Rigid bidentate alkyne-based ligands 2a–2c attached to fluorophores were synthesized in a two-step synthesis relying on amide bond formation and a Sonogashira cross-coupling reaction (Scheme 1). The dibromo derivatives 3a–3c were obtained in yields ranging from 54 to 98% by treatment of the acid with naphthyl- or anthracenyl-based amines in the presence of a coupling reagent EDC and DMAP as a base. Compounds 3a–3c were coupled with 3-ethynylpyridine using Sonogashira conditions to give the ligands 2a–2c in 18 to 72% yield. In order to evaluate the optical and biological properties of fluorophore-based compounds compared to a non-fluorophore system, carboxy-functionalized ligand 2d was prepared in 17% yield by coupling 3,5-dibromobenzoic acid with 3-ethynylpyridine using a Sonogashira reaction (Scheme 1).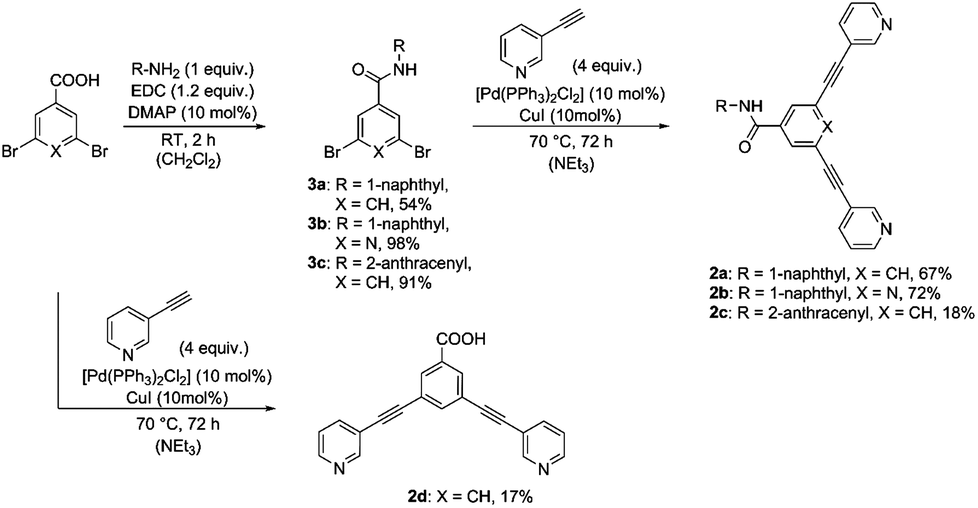 | ||
| Scheme 1 Synthesis of ligands 2a–dvia Sonogashira cross coupling. | ||
The reaction of bidentate ligands 2a–2d and a palladium precursor [Pd(NCCH3)4](BF4)2 in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ligand:metal ratio in DMSO at room temperature for 1 h yielded cage compounds [Pd2(2)4](BF4)41a–1d in 63 to 81% (Scheme 2). Successful cage formation was studied by 1H and DOSY NMR spectroscopy, ESI mass spectrometry and single crystal X-ray diffraction.
:1 ligand:metal ratio in DMSO at room temperature for 1 h yielded cage compounds [Pd2(2)4](BF4)41a–1d in 63 to 81% (Scheme 2). Successful cage formation was studied by 1H and DOSY NMR spectroscopy, ESI mass spectrometry and single crystal X-ray diffraction.
 | ||
| Scheme 2 Synthesis of palladium(II) cages 1a–dvia self-assembly using bidentate ligands 2a–d and [Pd(NCCH3)4](BF4)2 as a precursor. | ||
The 1H NMR spectra of naphthalene-based ligand 2a, carboxy-based ligand 2d and their corresponding cages 1a and 1d are exemplarily depicted in Fig. 2. Upon cage formation, the pyridyl proton signals (Ha–Hd) experience a significant downfield shift, especially the signals of Ha and Hb, which are shifted by ca. 1 ppm. However, the proton signals of naphthalene and anthracene moieties are not affected by the coordination of the pyridyl group to palladium.
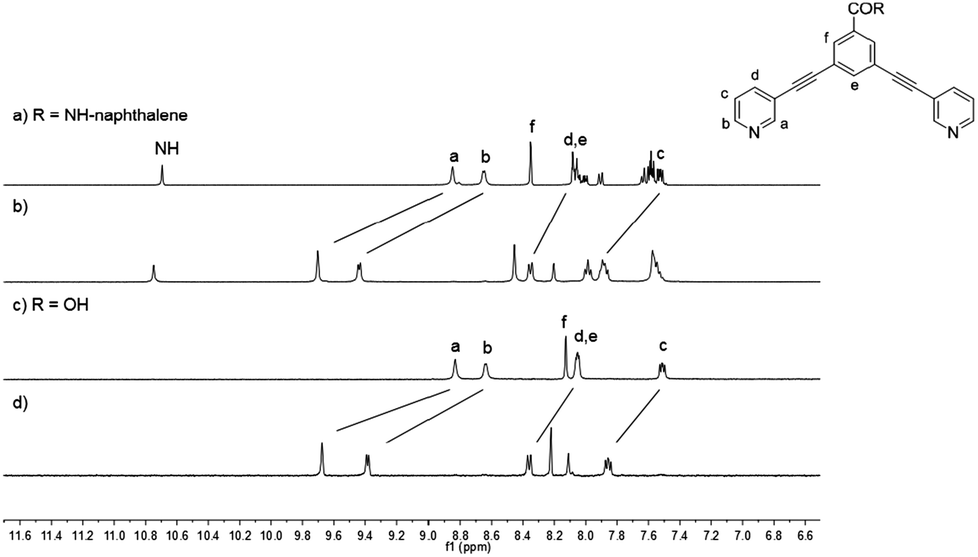 | ||
| Fig. 2 Stacked 1H NMR spectra (400 MHz, [D6]DMSO) of ligand 2a (a), cage 1a (b), ligand 2d (c) and cage 1d (d). | ||
Diffusion-ordered NMR spectroscopy (DOSY) provides additional evidence for the successful cage formation in solution (Table 1). 1H DOSY measurements of the cages 1a–d and of the ligands 2a–d show diffusion coefficients (D) in the range of 0.81 to 0.90 × 10−10 m2 s−1 and 1.76 to 2.02 × 10−10 m2 s−1, respectively, resulting in Dligand/Dcomplex ratios of about 2:1, which are in accordance with the literature values.5,17 In addition, DOSY NMR was used to estimate the size of the cage compounds applying the Stokes–Einstein equation.22 The hydrodynamic radii rs of the cage compounds were determined to be between 1.2 and 1.4 nm.
| Cage | D of cage | Ligand | D of ligand | Ratio |
|---|---|---|---|---|
| 1a | 0.90 | 2a | 2.02 | 2.25 |
| 1b | 0.88 | 2b | 1.76 | 2.00 |
| 1c | 0.81 | 2c | 1.77 | 2.19 |
| 1d | 0.87 | 2d | 2.02 | 2.32 |
The formation of Pd2L4 cages was further confirmed by ESI-MS spectrometry showing isotopically resolved peaks of [Pd2(2)4]4+ and [Pd2(2)4BF4]3+. For instance, the MS signals of cage 1d at m/z 378.0 and 532.4 were assigned to the fragments [1d – 4BF4−]4+ and [1d – 3BF4−]3+, respectively (Fig. 3).
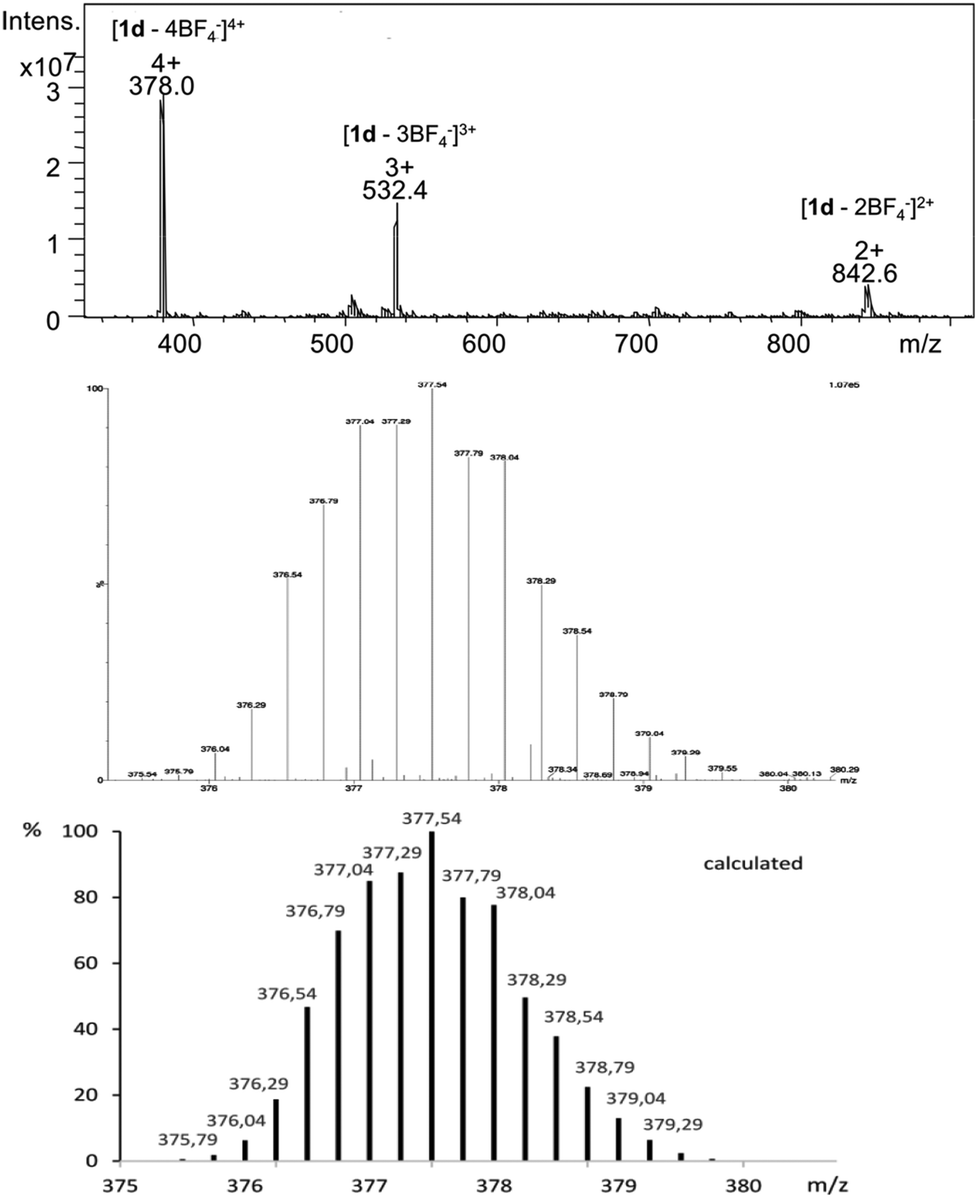 | ||
| Fig. 3 ESI mass spectrum of cage 1d (top), measured isotopic pattern of the molecule signal [1d – 4BF4−]4+ (middle) and calculated isotopic distribution (bottom). | ||
Single-crystal X-ray diffraction provided structural verification of cage 1b (Fig. 4). Suitable single crystals of 1b were grown by vapor diffusion of diethyl ether into a DMF solution of the cage. The crystal structure revealed the Pd2L4 cage configuration in which the four bidentate ligands 2b are coordinated to two Pd(II) ions in a square-planar fashion. The cavity size is defined by the Pd⋯Pd distance of 11.9 Å and by the distance between the two opposing pyridyl moieties of 10.5 Å. Based on spectroscopic evidence it can be assumed that the solid-state structures of cages 1a, 1b, 1c and 1d are similar.
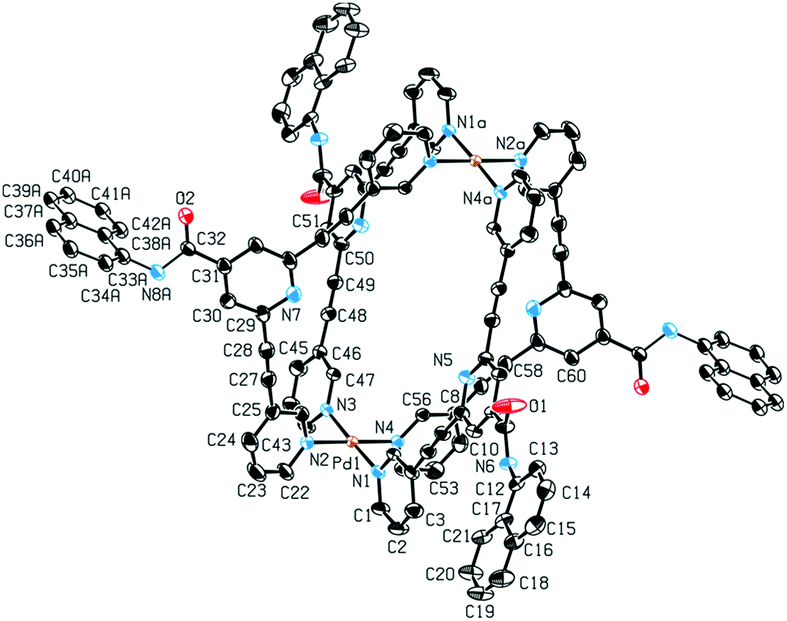 | ||
| Fig. 4 Solid state molecular structure of 1b. Ellipsoids are shown at the 50% probability level. Hydrogen atoms, counterions and solvent molecules are omitted for clarity. Selected bond lengths (Å) and angles (°): Pd1–N1 2.013(3), Pd1–N2 2.031(3), Pd1⋯Pd1a 11.9308(6), N7⋯N7a 10.5029(6), N1–Pd1–N2 88.54(12), N2–Pd1–N3 92.45(12). | ||
The palladium cages 1a–d are stable for months under air and light in both the solution and solid state. The cage compounds are soluble in DMSO and DMF, as well as in mixtures of DMF/acetone, DMSO/acetonitrile and DMSO/water, and not soluble in less polar solvents. The cages are soluble in water up to a concentration of 100 μM.
Optical properties and DFT calculations
As previously mentioned, the photophysical properties of metallocages, particularly the emissive ones, are of special interest to follow the uptake and distribution of cages in cells by fluorescence microscopy. The absorption and emission properties of our series are shown in Table 2 while the corresponding spectra are depicted in Fig. 5 and 6. The excitation spectra are shown in the ESI.†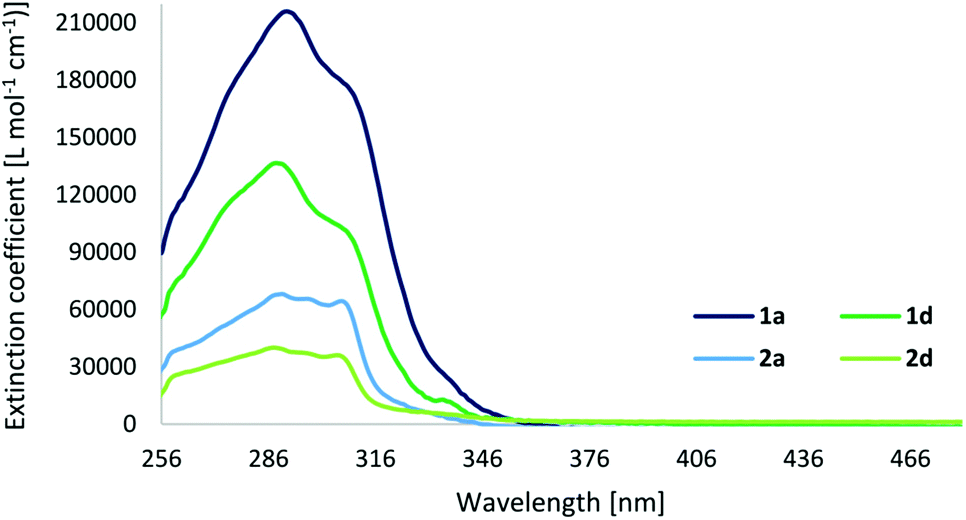 | ||
| Fig. 5 Absorption spectra of cage compounds 1a and 1d, and ligands 2a and 2d in DMSO (c = 5 × 10−6 M and 10−5 M). | ||
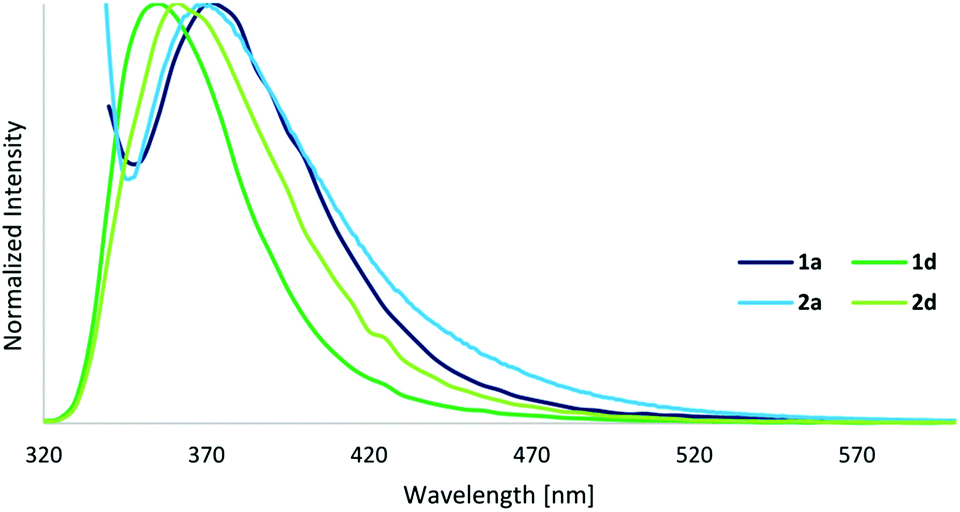 | ||
| Fig. 6 Emission spectra of cage compounds 1a and 1d (λ(ex) = 330/280 nm), and ligands 2a and 2d (λ(ex) = 325/280 nm) in DMSO (c = 10−5 M). | ||
| Compound | ε max [L mol−1 cm−1] | λ max(abs) [nm] | λ(ex) [nm] | λ max(em) [nm] | Φ [%] |
|---|---|---|---|---|---|
| a Mean ± probable error of three measurements. | |||||
| 1a | 216000 |
291, 311 | 330 | 370 | 0.3 ± 0.02 |
| 2a | 68100 |
290, 306 | 325 | 370 | 0.4 ± 0.03 |
| 1b | 125800 |
285, 325 | 350 | 405 | 0.1 ± 0.01 |
| 2b | 36700 |
287, 329 | 305 | 375, 450 | 0.2 ± 0.02 |
| 1c | 256800 |
291, 309, 381 | 330 | — | — |
| 2c | 73700 |
290, 310 | 280 | 430 | 0.2 ± 0.01 |
| 1d | 136600 |
288, 310 | 280 | 355 | 1.2 ± 0.07 |
| 2d | 40000 |
288, 305 | 280 | 360 | 7.6 ± 0.31 |
The UV-Vis spectra of cages display strong ligand-based absorption bands in the range of 260–350 nm deriving from π–π* transitions of the highly conjugated ligands. The cages exhibit a three- to four-times higher extinction coefficient compared to their respective ligands. As normally observed for this type of cage compound, the carboxy-based ligand 2d (with a quantum yield of 8%) presents a significant quenching of the fluorescence upon cage formation, resulting in quantum yields of 1% for 1d. However, in contrast to our expectations, the fluorophore-based ligands and cages display fluorescence quantum yields even lower, below 1%, and the anthracene-based cage 1c exhibits no fluorescence at all.
In order to investigate the unexpected photophysical properties of fluorophore-based systems, a time-dependent density functional theory (TD-DFT) approach was used to calculate the probability of the HOMO–LUMO excitation and the emission wavelength. Also, the absorption and fluorescence properties of the naphthalene-based ligand 2a (Φ < 1%) were compared with those of the carboxy- and amine-based ligands 2d and 2e (Φ = 8 and 52%,5 respectively) (Fig. 7).
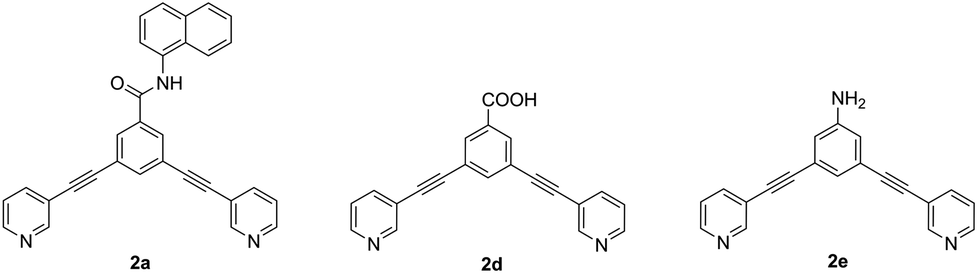 | ||
| Fig. 7 Structure of ligands 2a, 2d and 2e used for computational studies. | ||
TD-single point calculations were performed to determine the excitation probabilities of 2a, 2d and 2e (Table 3). Of interest is the relevant excitation from HOMO to LUMO. Further calculated transitions and the calculated UV-Vis spectra are shown in the ESI.† Notably, the probability of a HOMO–LUMO transition with 2% for ligand 2a is significantly lower than for 2d and 2e with 24 and 31%, respectively. The calculated probability is also in correlation with the quantum yield, as 2a is the least and 2e the most fluorescent ligand.
| Compound | λ(abs) [nm] | Probability | Origin (MO) | Destination (MO) |
|---|---|---|---|---|
| 2a | 360.27 | 0.0184 | 117 | 118 |
| 2d | 346.06 | 0.2358 | 84 | 85 |
| 2e | 359.87 | 0.3099 | 77 | 78 |
To determine the wavelength of a possible fluorescence, the energy difference between the energy after excitation and the energy after relaxation (applying excited state geometry) was calculated and transformed to a wavelength (Table 4). The calculated emission wavelengths of 2d and 2e are 420 and 489 nm, respectively. In contrast the emission wavelength of the naphthalene-based ligand 2a is calculated to be in the IR region at 2000 nm.
| Compound | State A (a.u.) | State B (a.u.) | ΔE (eV) | λ (em) [nm] |
|---|---|---|---|---|
| 2a | −1432.137122 | −1432.159841 | 0.6181 | 2000 |
| 2d | −1067.250746 | −1067.358863 | 2.9420 | 420 |
| 2e | −934.038722 | −934.131580 | 2.5268 | 489 |
According to the obtained results, the low fluorescence of 2a compared to 2d and 2e is attributable to two factors. First, a HOMO–LUMO excitation is less likely for 2a than for the other two compounds. Second, the energy differences between the highest excited state and the non-relaxed ground state reveal no emission of 2a in the visible region if emission arises between these states. In contrast, 2d and 2e emit light in the visible part, with the calculated wavelengths being slightly longer than those of the absorbed photons.
Taking into consideration the geometry of the excited state compared to the pure ground state, an explanation for the emission properties of 2a can be proposed (Fig. 8). In the ground state, the amide bond is almost planar with a dihedral angle of 177° from the centered benzene ring to the naphthalene substituent. However, in the excited state, this angle is reduced to 99°, thus the amide bond is nearly orthogonal. Therefore, the chromophoric system is disrupted leading to a higher energy difference between the lowest point that can be reached after fluorescence and the relaxed ground state. Accordingly, a smaller amount of energy is converted into light by emission resulting in a higher wavelength outside the visible region.
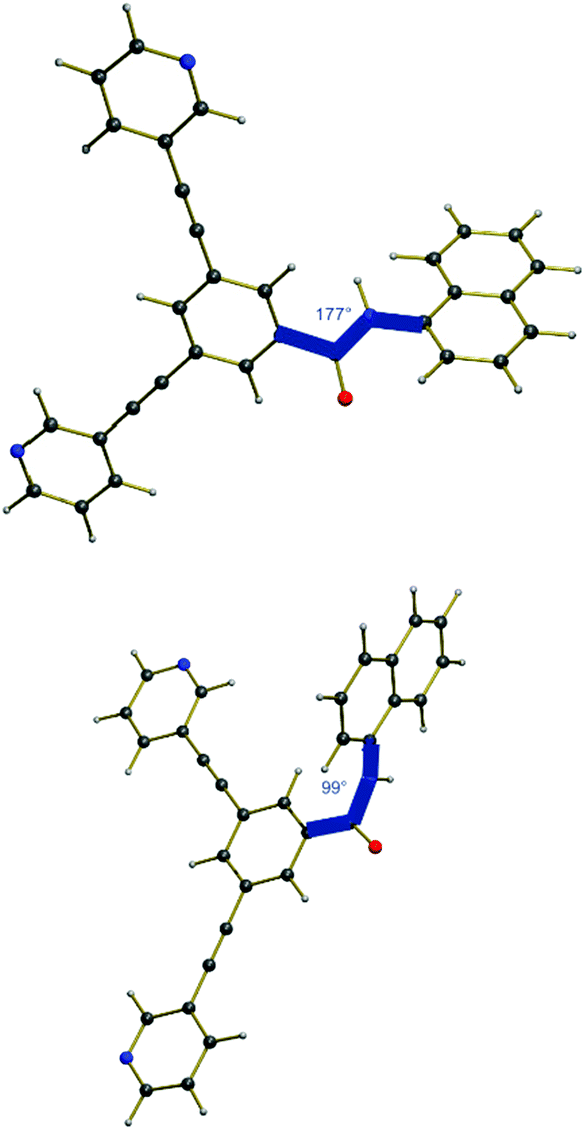 | ||
| Fig. 8 Ground state of 2a (top) and the optimized first excited state after light absorption (bottom). | ||
Anticancer activity
The antiproliferative effects of the coordination cages 1a–d, the ligands 2a–d and the palladium precursor were evaluated against the human cancer cell lines A549 (lung carcinoma) and SKOV-3 (ovarian carcinoma) using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. The IC50 (inhibitory concentration to reduce viability to 50%) values of the compounds are presented in Table 5 in comparison with cisplatin.| Compound | IC50a [μM] | |
|---|---|---|
| A549 | SKOV-3 | |
| a Mean ± SD of at least three independent measurements. | ||
| Cage 1a | 5.9 ± 1.4 | 8.0 ± 1.4 |
| Ligand 2a | 35.7 ± 5.9 | 46.6 ± 11.0 |
| Cage 1b | 1.4 ± 0.5 | 1.2 ± 0.7 |
| Ligand 2b | 3.8 ± 0.4 | 5.4 ± 2.1 |
| Cage 1c | 1.1 ± 0.3 | 1.1 ± 0.6 |
| Ligand 2c | 11.3 ± 0.8 | 23.6 ± 6.2 |
| Cage 1d | 82.6 ± 15.1 | 94.4 ± 7.9 |
| Ligand 2d | >100 | >100 |
| [(Cisplatin)2 ⊂ 1d] | — | 12.8 ± 1.2 |
| [Pd(NCCH3)4](BF4)2 | >100 | >100 |
| Cisplatin | 8.9 ± 4.2 | 15.4 ± 2.2 |
Overall, the fluorophore-based cages 1a–c are more toxic for all tested cancer cells than cisplatin with the anthracenyl-tagged 1c being the most cytotoxic, and the naphthyl-tagged 1a the least. The corresponding ligands alone elicit low to moderate antiproliferative activities against the tested cancer cells, with the exception of ligand 2b (IC50ca. 3.8 μM). The latter is the only ligand bearing a pyridyl moiety as the central aromatic scaffold, which appears to induce a pronounced cytotoxic effect compared to the benzyl moiety of ligand 2a. The carboxy-based complex 1d, however, is less effective in cell lines A549 and SKOV-3 compared to cisplatin. Notably, the palladium precursor [Pd(NCCH3)4](BF4)2 and the bidentate ligand 2d are inactive (IC50 > 100 μM) for the selected cell lines. Based on previous investigations,5 cage compound 1d can possibly function as a drug delivery system for cisplatin due to its encapsulation properties and its low cytotoxicity. The encapsulation of cisplatin within cage 1d has been studied by 1H NMR spectroscopy. The obtained spectrum (see Fig. S15†) shows that the inward directed proton signals of the benzyl proton He undergo significant downfield shifts upon encapsulation of two equivalents of cisplatin, while broadening of the proton signals takes place, in accordance with previous observations.5 Cage 1d encapsulating cisplatin (IC50 = 12.8 ± 1.2 μM) has slightly more potent anticancer activity than cisplatin alone in SKOV-3. These preliminary results hold promise for the development of new Pd2L4 cages as drug delivery systems.
Interestingly, the fluorophore-based ligands, especially 2b, and the respective cages can act as anticancer drugs alone, although their imaging in cancer cells via fluorescence microscopy may not be possible in pharmacologically relevant concentrations.
Conclusions
A series of exo-functionalized Pd2L4 coordination cages coupled to fluorophore moieties has been prepared and characterized including their photophysical properties. The fluorophore-based compounds were compared with a carboxy-functionalized system in terms of their emission properties and anticancer activity in cancer cell lines. Interestingly, the carboxy-based ligand exhibits a higher fluorescence quantum yield than the ligands attached to fluorescent tags. The quenching of the fluorescence in these systems can be explained by TD-DFT calculations, showing a lower probability for HOMO–LUMO transitions and emission in the IR region (2000 nm). The cage compounds attached to fluorophore groups display a higher cytotoxicity in all tested cancer cells than cisplatin, making them suitable as anticancer agents. In contrast, cage 1d has a very low anticancer activity with respect to the cell lines A549 and SKOV-3, which may still make it suitable as a non-toxic drug delivery system for cisplatin or other drugs, and ongoing studies in our group are exploring this possibility. As a matter of fact preliminary cytotoxicity data on the [(cisplatin)2 ⊂ 1d] system reinforce the relevance of our strategy. Other fluorophore-based ligands and corresponding metallocages are currently under investigation in our laboratories, where the length of the linker between the bipyridyl ligands and the fluorescent tags is increased to avoid the predicted torsion of the amide bond angle in the excited state. Finally, it should be pointed out that still the mechanisms of anticancer action of metallocages have not yet been clarified in most cases, although a few studies suggest different modes of action with respect to cisplatin, for instance palladium helicates induce the disruption of the cell membrane.15aExperimental
General remarks
All reagents were purchased from commercial sources and used without further purification. Chromatographic separations were performed using silica gel (60–200 μm). NMR spectra were recorded with a Bruker Avance DPX 400 or a Bruker Avance III 400 or a Bruker Avance I 500 spectrometer at a temperature of 298 K. The spectra were referenced to the residual 1H and 13C{1H} signals of the solvents in parts per million (ppm). Abbreviations for NMR multiplicities are: singlet (s), doublet (d), triplet (t), multiplet (m). Coupling constants J are given in Hz. The IR spectra were collected on a Varian ATR-FTIR instrument. Electrospray ionization (ESI) and fast atomic bombardment (FAB) mass spectra were obtained on a Thermo Scientific LCQ/Fleet spectrometer and a Finnigan MAT spectrometer, respectively. UV-Vis absorption spectra were acquired with a Jasco V-550 UV-Vis spectrometer.General procedure for the synthesis of dibromo derivatives
Acid (1.0 equiv.), amine (1.0 equiv.), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (1.2 equiv.) and 4-dimethylaminopyridine (10 mol%) were dissolved in dry dichloromethane and stirred under an argon atmosphere at room temperature for 2 h. To the reaction mixture, 2 M HCl (40 mL) was added and the aqueous phase was extracted three times with dichloromethane (40 mL). The organic phase was dried over MgSO4 and concentrated under reduced pressure. The crude product was recrystallized in dichloromethane to yield dibromide as a solid.General procedure for the synthesis of ligands
Dibromide (1.0 equiv.), 3-ethynylpyridine (4.0 equiv.), [Pd(PPh3)2Cl2] (10 mol%) and CuI (10 mol%) were added to degassed triethylamine and heated under an argon atmosphere at 70 °C. After 72 hours, the reaction mixture was diluted with ethyl acetate (50 mL) and filtered over celite. The solvent was removed under reduced pressure and the crude residue was further purified by column chromatography (CH2Cl2:MeOH = 100:5) to give the ligand as a solid.
General procedure for the synthesis of cages
A solution of [Pd(NCCH3)4](BF4)2 (2.0 equiv.) and ligand (4.0 equiv.) in DMSO was stirred at RT for one hour. After precipitation by addition of acetone and diethyl ether, the solid was filtered and washed with acetone and diethyl ether to yield the cage compound as a solid.Crystallographic details
For detailed information see the ESI.† Crystallographic Data (excluding structure factors) for the structure reported in this paper has been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC 1454096 (1b).Fluorescence spectroscopy and determination of quantum yield
The emission spectra were recorded on a Varian Cary Eclipse fluorescence spectrophotometer. For each compound, dilutions in DMSO at a concentration of 10 μM were prepared. First, UV-Vis spectra of compounds were recorded in DMSO, to determine the wavelength of the absorbance maximum. The absorbance maximum should be between 0.3 and 1. The measured absorbance wavelength was used as the excitation wavelength for fluorescence microscopy.For the determination of quantum yields,23,24 the choice of the right quantum standard is important. Depending on the excitation and emission wavelengths of the compounds, quinine sulfate or naphthalene was used as a standard. Different dilutions of the compounds and standards were prepared in DMSO and sulfuric acid or cyclohexane, respectively. Absorbance spectra were recorded using three different concentrated solutions, not exceeding an absorbance of 0.1 to minimize reabsorption effects. The same solutions were used for recording the fluorescence spectra. The excitation wavelength should be the same for the compound and standard. In addition, all fluorescence spectra must be recorded with constant excitation and emission slits. The quantum yield of a compound was calculated using the Stokes Einstein equation.24
Computational details
All calculations have been carried out by using the GAUSSIAN 09 D.01 package.25 The hybrid functional B3LYP26,27 has been chosen together with the basis set 6-31+G*.28–30 For the excited states optimization, a time-dependent DFT formalism31,32 has been applied using the B3LYP functional. All calculations have been performed in solution with PCM as an implicit solvation model and DMSO as a solvent.Cell viability assay (MTT assay)
Human lung cancer cell line A549 and human ovarian cancer cell line SKOV-3 were obtained from the European Centre of Cell Cultures (ECACC, Salisbury, UK) and maintained in culture as described by the provider. The cells were cultured in DMEM (Dulbecco's Modified Eagle Medium) containing 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin at 37 °C and 5% CO2. For evaluation of growth inhibition, cells were seeded in 96-well plates at a concentration of 11000 cells per well and grown for 24 h in complete medium. Solutions of the compounds with the required concentration (0.5 to 150 μM) were prepared by diluting a freshly prepared stock solution (10−2 M in DMSO) of the corresponding compound in aqueous DMEM medium. 200 μL of the dilutions were added to each well and the cells were incubated for 72 h. Following drug exposure, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was added to the cells at a final concentration of 0.5 mg mL−1 and incubated for 2 h. Afterwards the culture medium was removed and violet formazan was dissolved in DMSO. The optical density was quantified in tetraplicates at 550 nm using a multi-well plate reader. The percentage of surviving cells was calculated from the ratio of absorbance of treated to untreated cells. The IC50 values were calculated as inhibitory concentration of cell growth at 50% by plotting the percentage of surviving cells against the drug concentration and presented as mean ± SE of at least three independent experiments.
Acknowledgements
A. C. acknowledges support from Cardiff University and the August-Wilhelm Scheer Visiting Professorship at the Technical University of Munich. Authors acknowledge the support of the Technische Universität München – Institute for Advanced Study, funded by the German Excellence Initiative and the European Union Seventh Framework Programme under grant agreement no. 291763. A. S. is grateful to Boehringer Ingelheim Fonds for financial support. EU COST action CM1105 is gratefully acknowledged for funding and fruitful discussion. A. S. and M. H. are grateful for the financial support of the TUM Graduate School of Chemistry. The authors thank Leibnitz Rechenzentrum of the Bavarian Academy of Science for provision of computing time. Dr Alexander Pöthig's support with crystallographic data is greatly appreciated.Notes and references
- T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001–7045 CrossRef CAS PubMed.
- M. Han, D. M. Engelhard and G. H. Clever, Chem. Soc. Rev., 2014, 43, 1848–1860 RSC.
- (a) M. D. Pluth, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2009, 42, 1650–1659 CrossRef CAS PubMed; (b) D. M. Vriezema, M. Comellas Aragonès, J. A. A. W. Elemans, J. J. L. M. Cornelissen, A. E. Rowan and R. J. M. Nolte, Chem. Rev., 2005, 105, 1445–1490 CrossRef CAS PubMed; (c) M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418–3438 CrossRef CAS PubMed; (d) S. H. A. M. Leenders, R. Gramage-Doria, B. de Bruin and J. N. H. Reek, Chem. Soc. Rev., 2015, 44, 433–448 RSC.
- (a) R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS PubMed; (b) T. R. Cook, V. Vajpayee, M. H. Lee, P. J. Stang and K.-W. Chi, Acc. Chem. Res., 2013, 46, 2464–2474 CrossRef CAS PubMed.
- A. Schmidt, V. Molano, M. Hollering, A. Pöthig, A. Casini and F. E. Kühn, Chem. – Eur. J., 2016, 22, 2253–2256 CrossRef CAS PubMed.
- B. Therrien, G. Süss-Fink, P. Govindaswamy, A. K. Renfrew and P. J. Dyson, Angew. Chem., Int. Ed., 2008, 47, 3773–3776 CrossRef CAS PubMed.
- (a) Y.-R. Zheng, K. Suntharalingam, T. C. Johnstone and S. J. Lippard, Chem. Sci., 2015, 6, 1189–1193 RSC; (b) O. Zava, J. Mattsson, B. Therrien and P. J. Dyson, Chem. – Eur. J., 2010, 16, 1428–1431 CrossRef CAS PubMed; (c) A. Pitto-Barry, N. P. E. Barry, O. Zava, R. Deschenaux, P. J. Dyson and B. Therrien, Chem. – Eur. J., 2011, 17, 1966–1971 CrossRef CAS PubMed; (d) F. Schmitt, J. Freudenreich, N. P. E. Barry, L. Juillerat-Jeanneret, G. Süss-Fink and B. Therrien, J. Am. Chem. Soc., 2012, 134, 754–757 CrossRef CAS PubMed; (e) J. Mattsson, O. Zava, A. K. Renfrew, Y. Sei, K. Yamaguchi, P. J. Dyson and B. Therrien, Dalton Trans., 2010, 39, 8248–8255 RSC; (f) N. P. E. Barry, O. Zava, P. J. Dyson and B. Therrien, Chem. – Eur. J., 2011, 17, 9669–9677 CrossRef CAS PubMed.
- (a) H. Ahmad, D. Ghosh and J. A. Thomas, Chem. Commun., 2014, 50, 3859–3861 RSC; (b) R. Kieltyka, P. Englebienne, J. Fakhoury, C. Autexier, N. Moitessier and H. F. Sleiman, J. Am. Chem. Soc., 2008, 130, 10040–10041 CrossRef CAS PubMed; (c) A. Mishra, S. Ravikumar, S. H. Hong, H. Kim, V. Vajpayee, H. Lee, B. Ahn, M. Wang, P. J. Stang and K.-W. Chi, Organometallics, 2011, 30, 6343–6346 CrossRef CAS PubMed; (d) N. P. E. Barry, N. H. Abd Karim, R. Vilar and B. Therrien, Dalton Trans., 2009, 10717–10719 RSC.
- (a) A. Mishra, S. Chang Lee, N. Kaushik, T. R. Cook, E. H. Choi, N. Kumar Kaushik, P. J. Stang and K.-W. Chi, Chem. – Eur. J., 2014, 20, 14410–14420 CrossRef CAS PubMed; (b) A. Dubey, J. W. Min, H. J. Koo, H. Kim, T. R. Cook, S. C. Kang, P. J. Stang and K.-W. Chi, Chem. – Eur. J., 2013, 19, 11622–11628 CrossRef CAS PubMed; (c) A. Garci, A. A. Dobrov, T. Riedel, E. Orhan, P. J. Dyson, V. B. Arion and B. Therrien, Organometallics, 2014, 33, 3813–3822 CrossRef CAS; (d) G. Gupta, J. Kumar, A. Garci, N. Nagesh and B. Therrien, Molecules, 2014, 19, 6031–6046 CrossRef PubMed; (e) G. Gupta, B. Murray, P. Dyson and B. Therrien, Materials, 2013, 6, 5352–5366 CrossRef CAS; (f) A. Mishra, Y. J. Jeong, J.-H. Jo, S. C. Kang, M. S. Lah and K.-W. Chi, ChemBioChem, 2014, 15, 695–700 CrossRef CAS PubMed.
- V. Grishagin, J. B. Pollock, S. Kushal, T. R. Cook, P. J. Stang and B. Z. Olenyuk, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 18448–18453 CrossRef PubMed.
- V. Vajpayee, Y. J. Yang, S. C. Kang, H. Kim, I. S. Kim, M. Wang, P. J. Stang and K.-W. Chi, Chem. Commun., 2011, 47, 5184–5186 RSC.
- A. Schmidt, A. Casini and F. E. Kühn, Coord. Chem. Rev., 2014, 275, 19–36 CrossRef CAS.
- (a) R. Sekiya, M. Fukuda and R. Kuroda, J. Am. Chem. Soc., 2012, 134, 10987–10997 CrossRef CAS PubMed; (b) M. Han, R. Michel, B. He, Y.-S. Chen, D. Stalke, M. John and G. H. Clever, Angew. Chem., Int. Ed., 2013, 52, 1319–1323 CrossRef CAS PubMed; (c) G. H. Clever, W. Kawamura, S. Tashiro, M. Shiro and M. Shionoya, Angew. Chem., Int. Ed., 2012, 51, 2606–2609 CrossRef CAS PubMed; (d) S. Freye, J. Hey, A. Torras-Galán, D. Stalke, R. Herbst-Irmer, M. John and G. H. Clever, Angew. Chem., Int. Ed., 2012, 51, 2191–2194 CrossRef CAS PubMed; (e) S. Freye, R. Michel, D. Stalke, M. Pawliczek, H. Frauendorf and G. H. Clever, J. Am. Chem. Soc., 2013, 135, 8476–8479 CrossRef CAS PubMed.
- (a) N. Kishi, M. Akita and M. Yoshizawa, Angew. Chem., Int. Ed., 2014, 53, 3604–3607 CrossRef CAS PubMed; (b) N. Kishi, Z. Li, K. Yoza, M. Akita and M. Yoshizawa, J. Am. Chem. Soc., 2011, 133, 11438–11441 CrossRef CAS PubMed; (c) J. E. M. Lewis, E. L. Gavey, S. A. Cameron and J. D. Crowley, Chem. Sci., 2012, 3, 778–784 RSC; (d) J. E. M. Lewis, C. John McAdam, M. G. Gardiner and J. D. Crowley, Chem. Commun., 2013, 49, 3398–3400 RSC; (e) P. Liao, B. W. Langloss, A. M. Johnson, E. R. Knudsen, F. S. Tham, R. R. Julian and R. J. Hooley, Chem. Commun., 2010, 46, 4932–4934 RSC; (f) S. Löffler, J. Lübben, L. Krause, D. Stalke, B. Dittrich and G. H. Clever, J. Am. Chem. Soc., 2015, 137, 1060–1063 CrossRef PubMed; (g) M. Yamashina, M. M. Sartin, Y. Sei, M. Akita, S. Takeuchi, T. Tahara and M. Yoshizawa, J. Am. Chem. Soc., 2015, 137, 9266–9269 CrossRef CAS PubMed.
- (a) S. M. McNeill, D. Preston, J. E. M. Lewis, A. Robert, K. Knerr-Rupp, D. O. Graham, J. R. Wright, G. I. Giles and J. D. Crowley, Dalton Trans., 2015, 44, 11129–11136 RSC; (b) A. Ahmedova, D. Momekova, M. Yamashina, P. Shestakova, G. Momekov, M. Akita and M. Yoshizawa, Chem. - Asian J., 2016, 11, 474–477 CrossRef CAS PubMed.
- (a) J. K. Klosterman, Y. Yamauchi and M. Fujita, Chem. Soc. Rev., 2009, 38, 1714–1725 RSC; (b) B. J. Holliday and C. A. Mirkin, Angew. Chem., Int. Ed., 2001, 40, 2022–2043 CrossRef CAS; (c) M. Fujita, M. Tominaga, A. Hori and B. Therrien, Acc. Chem. Res., 2005, 38, 369–378 CrossRef CAS PubMed.
- A. M. Johnson, O. Moshe, A. S. Gamboa, B. W. Langloss, J. F. K. Limtiaco, C. K. Larive and R. J. Hooley, Inorg. Chem., 2011, 50, 9430–9442 CrossRef CAS PubMed.
- (a) J. E. M. Lewis, A. B. S. Elliott, C. J. McAdam, K. C. Gordon and J. D. Crowley, Chem. Sci., 2014, 5, 1833–1843 RSC; (b) Z. Li, N. Kishi, K. Hasegawa, M. Akita and M. Yoshizawa, Chem. Commun., 2011, 47, 8605–8607 RSC.
- K. Y. Zhang and K. K.-W. Lo, Metal Complexes for Cell and Organism Imaging, In Inorganic Chemical Biology, John Wiley & Sons Ltd, 2014 Search PubMed.
- M. Wenzel, A. de Almeida, E. Bigaeva, P. Kavanagh, M. Picquet, P. Le Gendre, E. Bodio and A. Casini, Inorg. Chem., 2016, 55, 2544–2557 CrossRef CAS PubMed.
- (a) A. Citta, E. Schuh, F. Mohr, A. Folda, M. L. Massimino, A. Bindoli, A. Casini and M. P. Rigobello, Metallomics, 2013, 5, 1006 RSC; (b) D. C. Crans, E. Nordlander, B. Bertrand, A. de Almeida, E. P. M. van der Burgt, M. Picquet, A. Citta, A. Folda, M. P. Rigobello, P. Le Gendre, E. Bodio and A. Casini, Eur. J. Inorg. Chem., 2014, 2014, 4532–4536 CrossRef; (c) M. Patra and G. Gasser, ChemBioChem, 2012, 13, 1232–1252 CrossRef CAS PubMed.
- (a) Y. Cohen, L. Avram and L. Frish, Angew. Chem., Int. Ed., 2005, 44, 520–554 CrossRef CAS PubMed; (b) D. Li, G. Kagan, R. Hopson and P. G. Williard, J. Am. Chem. Soc., 2009, 131, 5627–5634 CrossRef CAS PubMed.
- (a) D. F. Eaton, J. Photochem. Photobiol., B, 1988, 2, 523–531 CrossRef CAS; (b) S. Fery-Forgues and D. Lavabre, J. Chem. Educ., 1999, 76, 1260–1264 CrossRef CAS.
- A. T. Rhys Williams and S. A. Winfield, Analyst, 1983, 108, 1067–1071 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785–789 CrossRef CAS.
- R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724–728 CrossRef CAS.
- T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. v. R. Schleyer, J. Comput. Chem., 1983, 4, 294–301 CrossRef CAS.
- M. J. Frisch, J. A. Pople and J. S. Binkley, J. Chem. Phys., 1984, 80, 3265–3269 CrossRef CAS.
- G. Scalmani, M. J. Frisch, B. Mennucci, J. Tomasi, R. Cammi and V. Barone, J. Chem. Phys., 2006, 124, 1–15 CrossRef PubMed.
- M. E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, J. Chem. Phys., 1998, 108, 4439–4449 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR, ESI-MS, excitation spectra, crystallographic details and DFT calculations. CCDC 1454096. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt00654j |
| This journal is © The Royal Society of Chemistry 2016 |