
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlteration of steric hindrance modulates glutathione resistance and cytotoxicity of three structurally related RuII-p-cymene complexes†
Kallol
Purkait‡
,
Saptarshi
Chatterjee‡
,
Subhendu
Karmakar
and
Arindam
Mukherjee
*
Department of Chemical Sciences, Indian Institute of Science Education and Research Kolkata, Mohanpur Campus, 741246, India. E-mail: a.mukherjee@iiserkol.ac.in
First published on 7th April 2016
Abstract
The effect of steric hindrance on reactivity towards biomolecules while designing RuII-η6-p-cymene based anticancer agents seems to be an important parameter in improving the activity and inducing resistance against glutathione (GSH) deactivation. Herein we present the structure, hydrolysis, anticancer activity and the effect of steric hindrance on deactivation by glutathione for three complexes, [RuII(η6-p-cym)(L1)(Cl)](PF6) (1), [RuII(η6-p-cym)(L2)(Cl)](PF6) (2) and [RuII(η6-p-cym)(L3)(Cl)](PF6) (3). The ligands L1–L3 are Schiff bases which show increasing substitution in a benzene ring, such that two ortho hydrogens are replaced by -methyl in 2 and by -isopropyl in 3. The cytotoxicity results strongly suggest that controlling the rate of hydrolysis through tuning of steric hindrance may be a feasible pathway to derive GSH resistant anticancer agents. The cellular studies show that all the three complexes show good blood compatibility (haemolysis <3%) and induce cellular death through caspase activation via the mitochondrial pathway. They have anti-angiogenic activity and prevent the healing of treated cells.
Introduction
Metallo drugs have found wide use as anticancer agents. Improving the design of a metal based anticancer agent has certain advantages viz. higher number of accessible coordination numbers, reversible mode of binding ability with different kinds of electron donor species, accessible oxidation states under normal physiological conditions1 and choice of suitable ligands. All the above factors tune the properties of the metal complexes.2,3 Platinum complexes are the predominant contributors among the metal based drugs4 despite several major drawbacks in the form of severe side effects and induced resistance to several cancer types. During the past few decades researchers have focused on non-platinum complexes among which ruthenium has emerged as a significant contributor.5–8 Ruthenium complexes are octahedral or tetrahedral unlike the case of PtII complexes which are generally square planar, and the tuning of electronic properties shows variation since they bear differences in electronic configuration and geometries. Among the promising ruthenium complexes, certain RuIII-complexes, NAMI-A, NKP1339 have moved up to clinical trial phase-II.9,10 Ru-complexes may be delivered to cells aided by plasma proteins11 and they may get reduced to RuII inside the cell in the presence of cellular reducing agents viz. glutathione, cysteine, and ascorbate.12–16 However, under hypoxic conditions of tumor17 these reducing agents can simultaneously inhibit those drugs by forming stable bonds with the metal centre18,19 although activation of the drugs due to glutathione is also reported.20 The literature data reveals that DNA is a potential site of action for Ru complexes21,22 while other possibilities like, inhibition of the active site of an enzyme8,23 are found to be alternative modes of action for RuII complexes. One reason for interest in Ru-complexes is their relatively higher stability towards aquation which is supposed to decrease the loss of effective dosage due to the formation of active species before reaching the target16,24,25 and this may minimize side effects compared to Pt-complexes. In addition Ru is also thought to be interfering with iron metabolism.11,26Several ruthenium(II) complexes are evaluated in vitro against various cancerous cell lines, especially the [RuII(η6-arene)(L)Cl] class of complexes (where L = bidentate chelating ligand) which shows activity against primary tumour cells.2,14,27,28 The library of work from Sadler et al. shows that the activity of these complexes can be tuned by changing the ligand (L) and arene.28,29 To cite an example the [RuII(η6-arene)(diamine)Cl] family of complexes show that their anticancer activity is related to the structure, hydrolytic ability,30 DNA binding31 and also deactivation by thiolate sulfur.32 A different generation but related to the above type of complex is the RAPTA series of complexes in which a phosphorus based monodentate ligand developed by Dyson et al. shows antimetastatic activity.33 Recent findings show that these RuII-anticancer agents have strong affinity for the thiolate sulfur of cysteine,34 and glutathione.32 Elevated levels of the glutathione (GSH) synthetase and other thiol based enzymes viz. γ-glutamylcysteine ligase, and γ-glutamyl-transpeptidase are observed in tumour cells35,36 which are rendered as major contributing factors for chemoresistance37 and their depletion may increase the sensitivity of the drugs and oxidative stress.38 Since Ru has an affinity for sulfur hence Ru-complexes may be deactivated by the above enzymes, GSH and other cellular thiols.32,39,40
Hence complexes that are not deactivated by GSH or show less affinity towards GSH and other cellular thiols would be of high significance provided they are cytotoxic. We recently have shown that complex 3 shows in vitro cytotoxicity in certain cancer cell lines and there is little deactivation in the presence of glutathione under hypoxic conditions.41 In addition no GSH binding was observed by NMR studies. We have synthesized two more isostructural complexes where the only difference lies in the substitution at the vicinity of the metal centre so as to correlate with the effect of steric hindrance on the hydrolysis, anticancer activity, glutathione binding and the pathways of cytotoxicity which is strongly related to the breaking and making of new bonds with the metal centre leading to the pattern of reactivity.
Results
Syntheses and characterization
The Schiff base ligands (L1–L2) were synthesized using a similar procedure as described by us earlier.41 The reaction of one mole equivalent of [RuII2(η6-p-cym)2Cl4] precursor with ca. 2.4 mole equivalents of the respective ligand in methanolic solution led to the formation of respective complexes (Scheme 1). The complexes were crystallized using 1 equivalent of ammonium hexafluorophosphate due to their residual positive charges. All of the ligands and complexes were well characterised by 1H, 13C, HMQC and DEPT 135 NMR, FT-IR, UV-Vis spectra and single crystal X-ray structure. The bulk purity was confirmed by elemental analysis.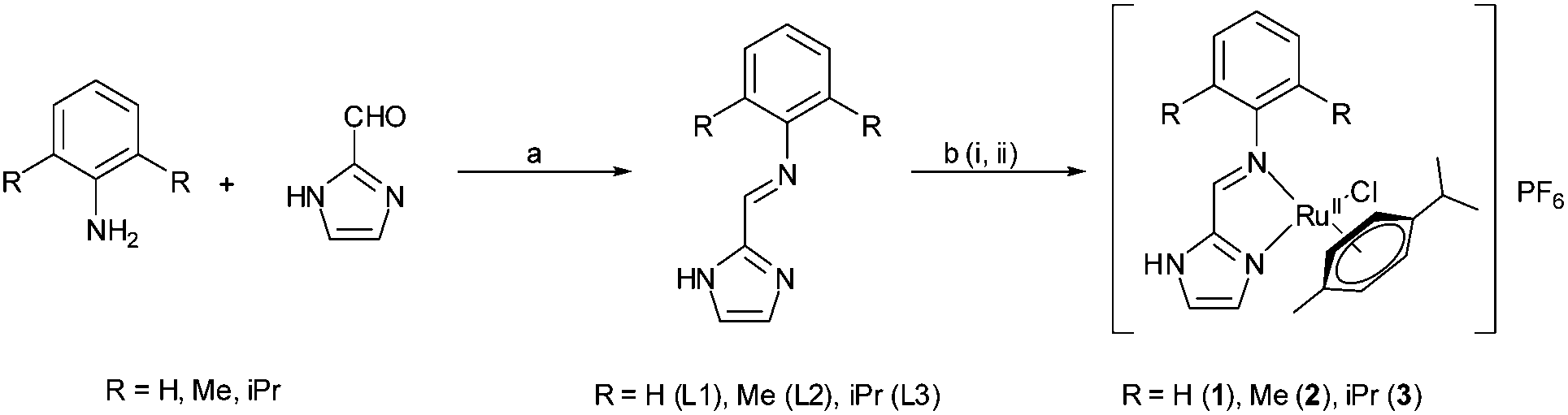 | ||
| Scheme 1 Synthetic scheme and chemical structure of ligands (L1–L3) and metal complexes (1–3). (a) Amine (1 mmol), aldehyde (1 mmol), EtOH, HCO2H (0.5%), reflux, dark, 12 h. (b) (i) Ligand (1.2 mmol), [Ru2(η6-p-cym)2Cl4] (0.5 mmol), MeOH, reflux, dark, 4 h. (ii) NH4PF6. | ||
X-ray crystallography
The single crystals of complexes 1–2 were obtained by layering petroleum benzene over a dichloromethane solution of the respective complex. They were structurally characterized by a single crystal X-ray diffraction method.Complex 1 crystallizes in the space group P21/n (ESI, Table S1†). The crystal structure shows that the RuII is bound with two nitrogens of L1, one chloride and a η6-bonded p-cymene ring. In imidazole as usual the sp2 hybridized double bonded N-atom is the donor. The structure displays that the benzene and imidazole rings are not in the same plane (Fig. 1). In 1, the methyl group of p-cymene is on the same side as that of chlorine, the isopropyl group of p-cymene seems to be moving away from the benzene ring (Fig. 1). An octahedral [PF6]− is present outside the coordination sphere and the Ru is in +2 oxidation state. The average Ru–N bond distance is 2.097 ± 0.024 Å and the Ru–Cl distance is 2.416(9) Å (ESI, Table S2†). The distances between the carbons of the p-cymene and the RuII are not uniform as described in ESI, Table S2.†
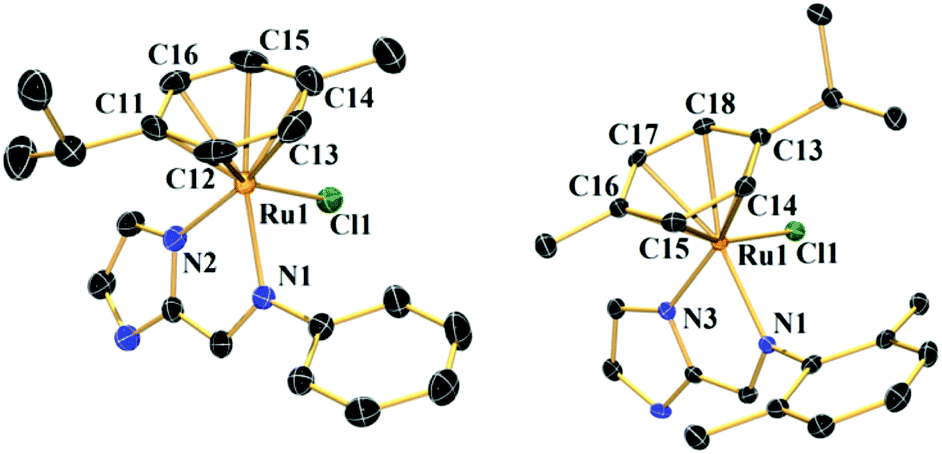 | ||
| Fig. 1 Single crystal structures of complexes 1 and 2 with 50% probability level. All hydrogen atoms, counter anions and solvent molecules are omitted for clarity. | ||
Complex 2 crystallizes with the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (ESI, Table S1†). Similar to 1 the RuII in 2 is bound to two nitrogens from L2, a chloride and the p-cymene (in a η6-fashion). However, unlike 1 the isopropyl group of the p-cymene is oriented towards the chloride rather than the methyl group. The average Ru–N distance is 2.105 ± 0.039 Å and the Ru–Cl bond shows a distance of 2.410(7) Å (ESI, Table S2†). The bond distances between the carbon atoms of the p-cymene ring and RuII are not uniform (ESI, Table S2†). The structure of complex 3 which has been described elsewhere showed similar Ru–N and Ru–Cl distances however the orientation of the chloride was like 1.41
(ESI, Table S1†). Similar to 1 the RuII in 2 is bound to two nitrogens from L2, a chloride and the p-cymene (in a η6-fashion). However, unlike 1 the isopropyl group of the p-cymene is oriented towards the chloride rather than the methyl group. The average Ru–N distance is 2.105 ± 0.039 Å and the Ru–Cl bond shows a distance of 2.410(7) Å (ESI, Table S2†). The bond distances between the carbon atoms of the p-cymene ring and RuII are not uniform (ESI, Table S2†). The structure of complex 3 which has been described elsewhere showed similar Ru–N and Ru–Cl distances however the orientation of the chloride was like 1.41
Lipophilicity
To determine the distribution coefficient we used the standard shake flask method.42 The log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Do/w values as depicted in Fig. 2, show that 3 has the highest lipophilicity (3.2 ± 0.1) whereas, complexes 1 and 2 show logDo/w of 0.42 ± 0.12 and 0.66 ± 0.1 respectively. Since among complexes 1–3, complex 1 shows quicker hydrolysis, hence we use the term distribution coefficient as a standard practice.
Do/w values as depicted in Fig. 2, show that 3 has the highest lipophilicity (3.2 ± 0.1) whereas, complexes 1 and 2 show logDo/w of 0.42 ± 0.12 and 0.66 ± 0.1 respectively. Since among complexes 1–3, complex 1 shows quicker hydrolysis, hence we use the term distribution coefficient as a standard practice.
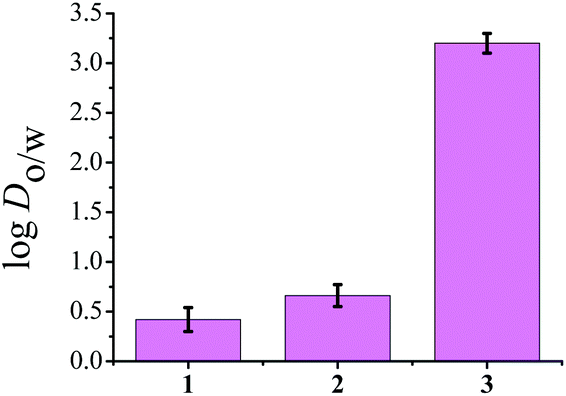 | ||
| Fig. 2 Representative diagram of lipophilicity of 1–3 represented by a bar diagram showing comparative logDo/w values of complexes in the octanol–aqueous phosphate buffer system, where error bars in the graph indicate the standard deviation in measurement. For 3 data was obtained from ref. 41. | ||
Hydrolysis study
To gain better insight into the properties of 1–3 we performed hydrolysis studies under various conditions depicted in Table 1 (a more elaborative table is present in the ESI, Table S3†). The results show that under acidic conditions the rate of hydrolysis increases, leading to decrease in solution half-life and in the presence of higher chloride concentration (40 and 110 mM) the half-life increases. At pH 6.7 the complexes show a higher rate of hydrolysis in 4 mM chloride containing buffer compared to that observed for water (Table 1, ESI, Table S3 and Fig. S13–S15†). The time dependent 1H NMR study in the 7:3 v/v D2O–DMSO-d6 mixture shows evidence of formation of hydrolyzed species after 1 to 3 h (Fig. 3) for 1 and 2. The hydrolysis study using 1H NMR (7:3 v/v D2O–DMSO-d6, 110 mM chloride) shows excellent stabilities for up to 10 days for all the complexes (ESI, Fig. S16†). In the NMR studies 1–3 do not show any peak corresponding to the dissociation of p-cymene from the metal complexes up to 23 h (beyond which we did not check anymore) even when the 7:3 v/v D2O–DMSO-d6 mixture with no chloride was used.
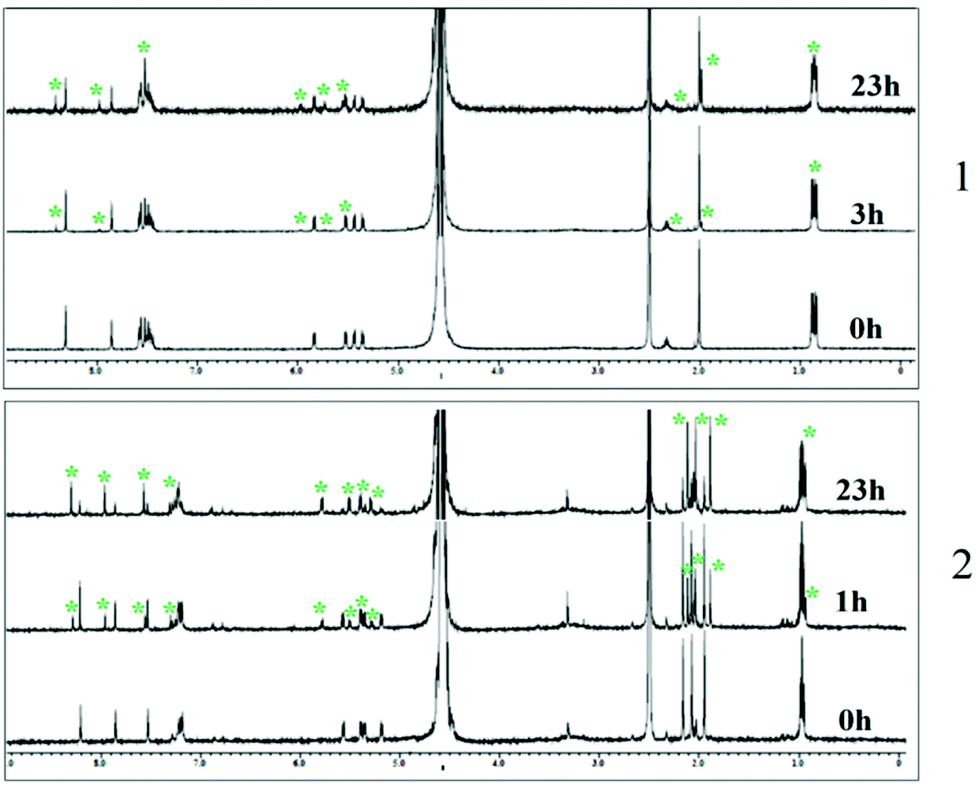 | ||
| Fig. 3 Hydrolysis study of complexes 1–2 in 3:7 v/v DMSO-d6 and D2O mixtures measured by 1H NMR at 25 °C with time. * indicates the peak corresponding to the hydrolysis product. | ||
| pH | Chloride conc. (mM) | t 1/2 (h) | ||
|---|---|---|---|---|
| 1 | 2 | 3 | ||
| a Detected by 1H NMR. | ||||
| 7.4 | 4 | 2.79 ± 0.66 | 5.67 ± 0.91 | 17.75 ± 0.02 |
| 40 | 3.91 ± 0.91 | 22.22 ± 2.13 | 49.40 ± 1.90 | |
| 6.7 | 4 | 1.18 ± 0.36 | 4.37 ± 0.31 | 8.07 ± 1.1 |
| 40 | 2.55 ± 0.18 | 9.98 ± 2.90 | 23.96 ± 2.98 | |
| Water | — | 2.01 ± 0.24 | 7.56 ± 1.04 | 11.70 ± 2.48 |
| — | 110a | Stable for 10 days | Stable for 10 days | Stable for 10 days |
Binding studies
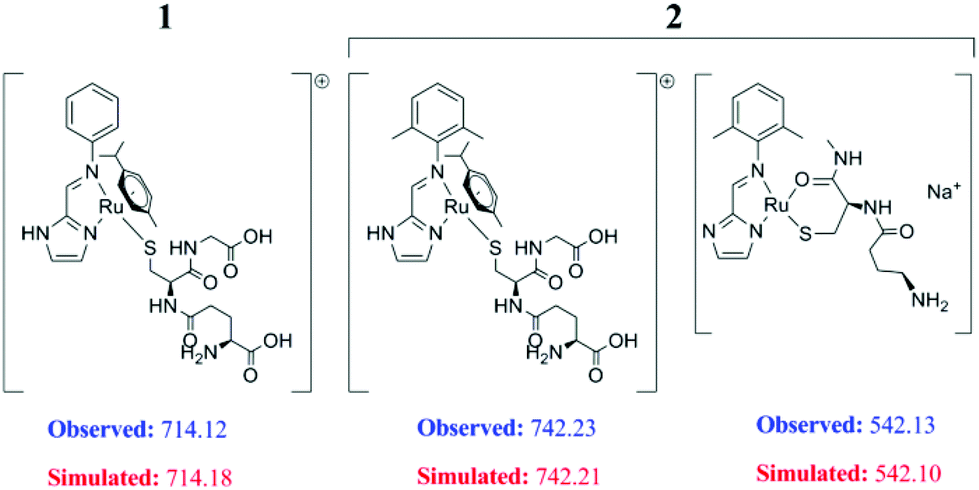 | ||
| Fig. 4 ESI MS speciations of complexes 1 and 2 upon treatment with excess glutathione showing the proposed speciation as per the m/z peaks obtained. Complex 3 did not provide any GSH bound species, hence not shown. | ||
But when we probed the concentration dependent binding with excess GSH using ESI-MS, we found the mono-GSH adduct of the complexes at a higher concentration of GSH (at least 25 equivalents) for both 1 and 2. It appears that –SH groups of GSH bind to the Ru-centre in the complexes rendering m/z of 714.12 and 742.23 for 1 and 2 respectively (Fig. 4, ESI, Fig. S21†). Another GSH bound fragmented peak with loss of p-cymene from 2, at m/z 542.13, was identified. The isotopic distribution ratio confirms the speciation (ESI, Fig. S21 and S22†). At this concentration it was not possible to obtain any significant NMR data. The intensity of the mono-GSH adduct peak was more for 1 compared to 2. In contrast even at the 100 equivalent concentration of GSH, no GSH bound peak was obtained for 3 even with ESI-MS.
Cellular studies
| IC50 (μM) ± S.D.a | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Normoxia | Hypoxiab | Hypoxia + glutathioned | ||||||||
| MCF-7 | A549 | HFF-1 | MIA PaCa-2 | Hep G2 | NIH 3T3 | MCF-7 | A549 | MCF-7 | A549 | |
| a IC50 values were calculated by non-linear curve fitting in dose response inhibition–variable slope model using Graph pad prism. S.D. = standard deviation. The data presented are mean of three independent experiments, in a single experiment each concentration was assayed in triplicate. The statistical significance (P) of the data is >0.001 to <0.05. b Hypoxia (1.5% O2). c Not determined. d With 1 mM of reduced L-glutathione. e 20 molar equivalents of reduced L-glutathione used with respect to IC50 dosage of the respective cell line in hypoxia. f Data was obtained from ref. 41. | ||||||||||
| 1 | 9 ± 1 | 21 ± 1 | 10.2 ± 1 | 7 ± 1 | 6 ± 1 | 10 ± 1 | 9 ± 1 | 18 ± 1 | 16 ± 1 | 37 ± 1 |
| 2 | 7 ± 1 | 19 ± 1 | 10.5 ± 1 | 12 ± 1 | 10 ± 1 | 11 ± 1 | 11 ± 1 | 17 ± 1 | 15 ± 1 | 27 ± 1 |
| 3 | 13.8 ± 1f | 23.2 ± 1f | 6 ± 1 | 10 ± 1 | 8 ± 1 | 9.2 ± 1 | 9.1 ± 1f | 15.6 ± 1f | 10.8 ± 1f | 16.7 ± 1f |
| C1 | 43.9 ± 3.6 | 36.7 ± 2.3 | N.D.c | N.D.c | N.D.c | N.D.c | 31.7 ± 1.7 | 31.2 ± 1.5 | 49.3 ± 1.8 | 31.7 ± 2.6 |
| Cisplatin | 15 ± 1 | 24 ± 1 | N.D.c | 13.8 ± 5 | 14.3 ± 1 | 7 ± 1 | 19 ± 2 | 27 ± 1 | 29 ± 2e | 40 ± 2e |
| Sub G1 | G0/G1 | S | G2/M | |
|---|---|---|---|---|
| a Cells were treated for 24 h with 1 and 2. Cells were treated with propidium iodide and analyzed by FACS. Cell populations were analyzed and expressed as the percentage of cells in each phase. The data presented is an average of two independent experiments. | ||||
| DMSO control | 5.19 | 57.8 | 21.62 | 15.96 |
| 1, 4 μM | 4.07 | 50.6 | 26.33 | 19.69 |
| 1, 6 μM | 1.66 | 50.09 | 24.96 | 24.02 |
| 2, 4 μM | 2.42 | 46.71 | 25.41 | 26.03 |
| 2, 6 μM | 3.62 | 43.39 | 28.88 | 24.91 |
 | ||
| Fig. 5 Agarose gel image of DNA ladder formation due to apoptosis using the MCF-7 cell line. (A) 50 bp step ladder, (B) DMSO control, (C) 1 (8 μM), (D) 1 (10 μM), (E) 2 (8 μM) and (F) 2 (10 μM) treated for 24 h. | ||
Optical microscopy imaging
Cell swelling and nuclear fragmentation were observed for both the cases upon treatment of complexes 1 and 2 on MCF-7 (ESI, Fig. S33†). Our earlier reports on 3 suggest that the observation is similar for all the three complexes.ICP-MS studies
To know the amount of ruthenium inside the cells we used the ICP-MS study. We obtained the ruthenium concentration inside the cell after 24 h incubation of the complex treated MCF-7 cells (with the respective complex) and the results show that the concentration of Ru inside the cells for complex 3 is more than that for 1 and 2 (Fig. 6). Complex 3 also happens to be the most lipophilic and slowest in hydrolysis among the three. | ||
| Fig. 6 Plot shows the amount of ruthenium present inside the cells (in ppb) with respect to the control experiment of complexes 1–3. Each data is the mean of three independent experiments. | ||
Caspase-7 activation
The caspase-7 activation study of all the complexes (1, 2 and 3) shows activation of caspase-7 in MCF-7 upon treatment of the respective complexes (Fig. 7). The colorimetric data based on the cleavage of the p-nitrophenol from the DEVD sequence shows that 1 and 2 have almost similar activities and the caspase-7 activity appears to be more in the case of complex 3 as the absorbance obtained is higher.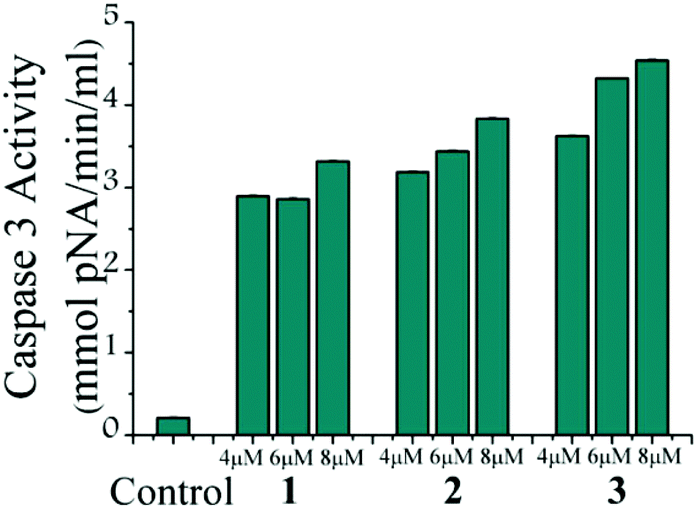 | ||
| Fig. 7 Representative diagram for caspase-7 activation of MCF-7 after 24 h treatment of 1–3 with three different concentrations (4, 6 and 8 μM) of each. Activities were shown in terms of pNA released. Error bars show standard deviations in data. | ||
Mitochondrial membrane potential change
When MCF-7 cells were treated with four different concentrations of 1–3, we observed a significant change in mitochondrial transmembrane potential (MMP). The change is maximum for complex 3 and minimum for 2 (Fig. 8, ESI, Fig. S30–S32†) using the same concentration of each complex although the dose dependence on IC50 is not the same for the complexes, rather complex 3 requires higher dosage to achieve IC50.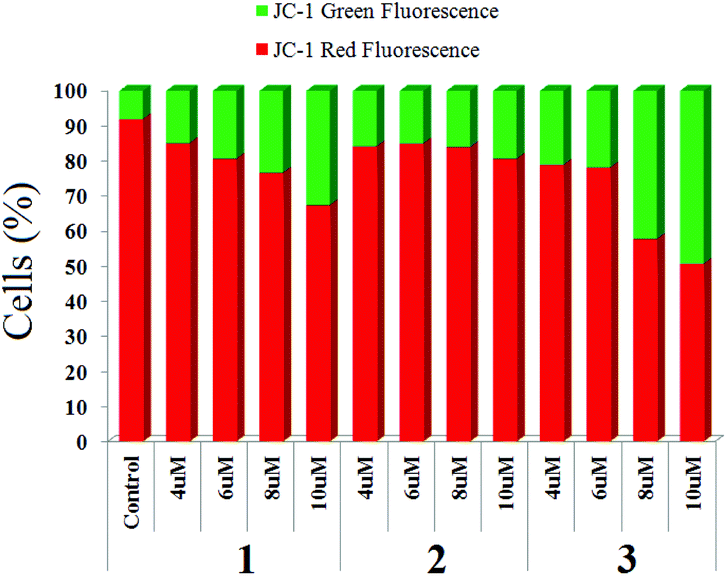 | ||
| Fig. 8 Representative diagram for change of mitochondrial membrane potential upon treatment of complexes (1–3) on MCF-7 stained by JC-1. | ||
Wound assay (migration)
The effects of 1–3 on cancer cell migration in the MCF-7 cell line were assessed using a scratch wound healing assay. From Fig. 9 it can be seen that the untreated control cells rapidly migrate over time, resulting in almost 45% healing in 12 h and >90% healing at 24 h of incubation. While for 1–3 the wide denuded area remained un-healed indicating the antiproliferative nature of the cells, attained by the treatment with the respective complexes (1–3).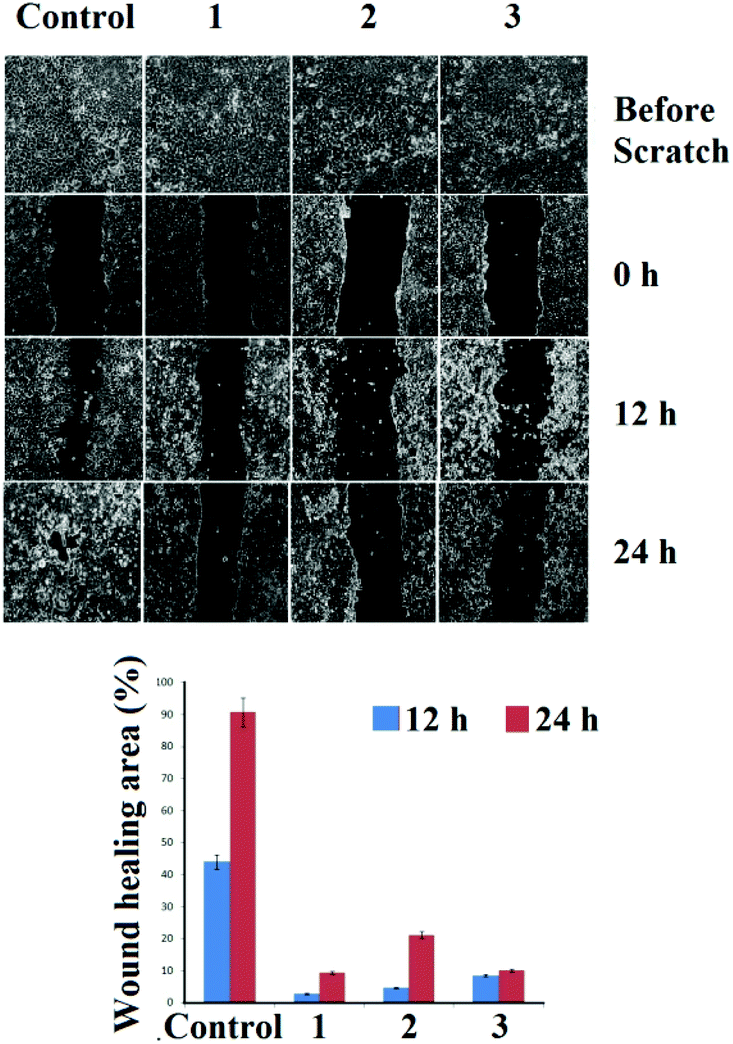 | ||
| Fig. 9 Analysis of migration of MCF-7 after treatment with the corresponding complexes. The images were taken after 12 h and 24 h of drug loading. | ||
Haemolysis assay
Interaction of the drug with the blood components particularly RBCs is an important and inevitable phenomenon, thus assessing the haemolysis becomes crucial in evaluating the blood compatibility of drugs.44,45 The results (ESI, Fig. S34 and Table S5†) show that compounds 1, 2 and 3 show good compatibility with RBCs. The haemolysis was found to be least for complex 1 it was ca. 1% at a concentration of 15 μM. For complex 2 at the same concentration the haemolysis was ca. 1.5% whereas it was the highest for complex 3 but less than 3% (ESI, Fig. S34†).Chick embryo angiogenesis assay (CEA)
The anti-angiogenic potential of the compounds was evaluated by the chick embryo angiogenesis (CEA) assay (Fig. 10). The results indicate the anti-angiogenic properties of 1–3 as degenerate and damaged blood vessels were observed on the chick embryo (affected zone is marked with white box and damaged blood vessels are marked with black arrows) after 4 h of exposure to 1–3. No new blood vessels were formed during the period of treatment. However the control shows healthy blood vessels and new blood vessels were formed during the 4 h incubation period.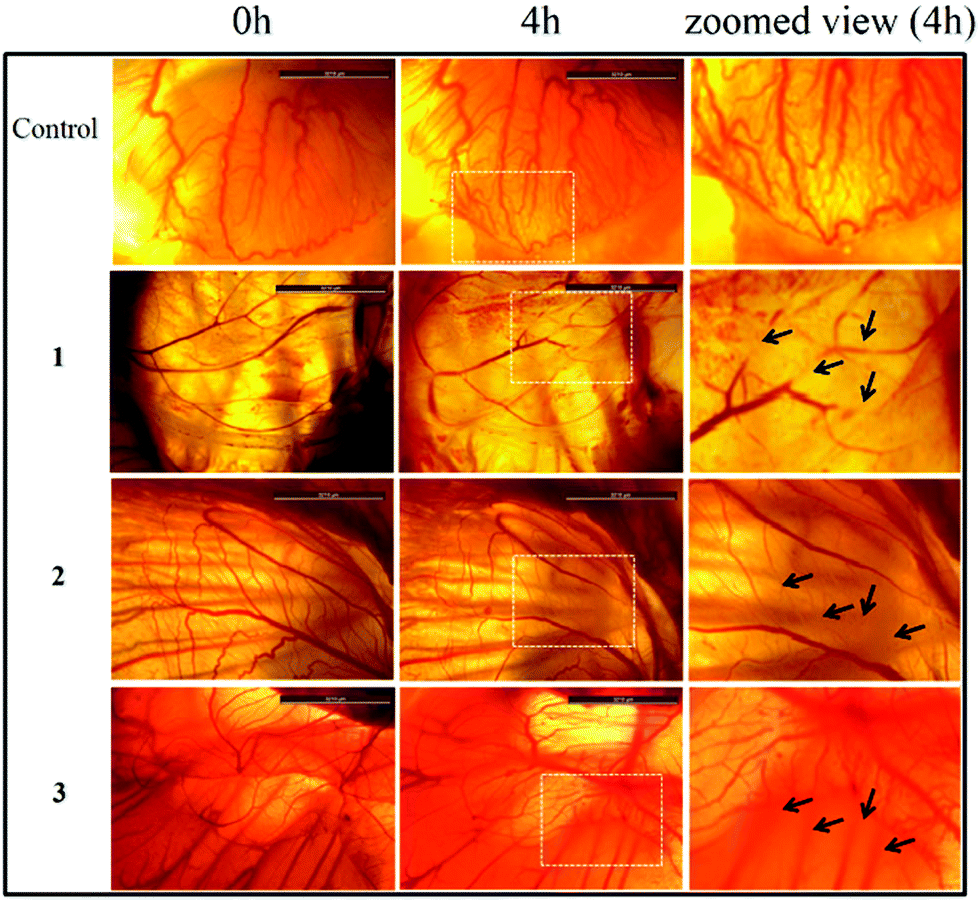 | ||
| Fig. 10 Ex ovo Chick Embryo Angiogenesis (CEA) assay depicting the anti-angiogenic potential of complexes 1–3. The zoomed area is of the dotted white box upon treatment after 4 h. | ||
Discussion
Half sandwich RuII-complexes bearing the formulation [RuII(η6-p-cym)(L1)(Cl)](PF6) (1) and [RuII(η6-p-cym)(L2)(Cl)](PF6) (2) along with some new studies on our previously reported [RuII(η6-p-cym)(L3)(Cl)](PF6) (3) are discussed here. All the complexes are well characterized by NMR and single crystal X-ray structure. The X-ray structures of the complexes show that all of them have a chloride and one η6-bonded p-cymene ring. The geometric orientation rendered shows differences in terms of steric bulkiness in the vicinity of the metal centre (isopropyl to methyl to hydrogen). Complex 3 displays maximum steric hindrance and 1 shows the least. The hydrolysis results show that the solution half-life has increased with increase in steric hindrance (Table 1; t1/2 trend: 3 > 2 > 1) rendering better stability and changes the cytotoxicity pattern as discussed later (Table 2).The hydrolysis is also affected by the pH and concentration of the leaving anion (e.g. Cl−) as well as the overall ionic strength of a solution (Table 1). Although, the complexes hydrolyze faster in pH 6.7 but due to the steric hindrance the trend remains similar as mentioned earlier. The effect of chloride concentration change seems to be the least on the half-life of 1 at both the pH values. The trend in hydrolysis does not show uniform increase with increase in steric hindrance. However, the effect of better stabilization at higher chloride concentration is prominent. It is encouraging to find that at extracellular concentration of chloride (∼110 mM), all the three complexes are stable for at least 10 d (we did not probe beyond 10 d). In the case where there was no chloride added viz. the hydrolysis performed in 7:3 (v/v) D2O–DMSO-d6, by 1H NMR, showed that the complexes started to hydrolyze after 1–3 h but none of them lose any other ligand than Cl− up to 23 h (beyond which it was not probed) (Fig. 3). The ESI-MS data also supported the loss of chloride since we could obtain the peaks corresponding to the general formulation [RuII(η6-p-cym)(L-H)]+ at m/z values of 407.09, 436.18 and 490.15 for 1–3 respectively which matches well with the simulated values of 407.09, 436.13 and 490.18 for 1–3 respectively. The CT-DNA binding studies showed that the strongest interaction is exhibited by complex 2 (Kb = 1.11(3) × 105 M−1), although 2 is not the least hindered complex. This suggests that the presence of the ortho methyl groups on the benzene ring of the L2 leads to enhanced complex DNA interaction,46 which increases the DNA binding ability. The similarity in the DNA binding constants for the 12 h hydrolyzed sample and the fresh solution of complexes may be attributed to the slow hydrolysis, since the timescale of the DNA binding experiment is smaller (1.5 h). The p-cymene complexes of RuII show similar binding constants with DNA as reported by Garcia et al.47–50
Aquation is thought to be a major step which may be involved in activation of this type of metal complex. However, extracellular aquation may prevent the internalization of the complexes due to their di-positive charge and higher hydrophilicity. Since, the lipid bi-layered cell membrane has a hydrophobic interior and hydrophilic exterior surface, hence if a complex has to enter the cell through the diffusion pathway then the lipophilicity is an important parameter. The partition co-efficient values of 1 and 2 (0.42 and 0.66 respectively) are significantly less than 3 (logD = 3.2), which clearly tells us that the complexes (1 and 2) may not very efficiently cross the cell membrane by a simple diffusion process. However, the cytotoxicity studies described later shows that both 1 and 2 are in general more cytotoxic than 3. The cellular internalization of 3 is the highest in the series as per the ICP-MS studies, which signifies that the effective cytotoxic concentration is lower for 1 and 2. The results suggest that the steric hindrance may also prevent 3 from reacting with the relevant biomolecules resulting in relatively less cytotoxicity.
However, we have reported earlier that complex 3 is very effective in hypoxia under an excess concentration of GSH. The dose dependence in hypoxia is also better than the normoxic IC50 dosage as per the in vitro studies. Cytotoxicity data of all the three complexes show that 3 is the most effective one under hypoxia in the presence of excess GSH. Complexes 1 and 2 are very effective under normal circumstances without added excess GSH and also in hypoxia, except in MCF-7 for 2 (Table 2). When excess GSH is present in the surroundings, which may be thought of as a case similar to many resistant forms of cancer, than the dose dependence worsens for 1 and 2. The required dosage in the presence of ca. 750–1000 equivalent excess GSH escalates to almost double as compared to the general normoxia/hypoxia IC50 dose.
It is well known that A549 expresses higher GSH levels than normal lung fibroblasts.51 All the complexes also show poorest activity in A549 (Table 2). However, 3 requires the higher dosage but does exhibit better activity in hypoxia in the presence of excess extracellular GSH suggesting its resistance. However, it should be noted that inside the cell there is glutathione-S-transferase which catalyzes the conjugation of GSH to similar complexes for detoxification hence the higher dosage required. However, in hypoxia 3 seems to overcome that and shows better IC50 in the entire family which is encouraging.
Complex 1 may be considered to be the most effective candidate against HepG2 and MIA PaCa-2 followed by MCF-7. However, it is most susceptible to GSH deactivation. Complex 2 although showing a greater DNA binding constant is less effective than 1 in most of the cell lines except for MCF-7, where it shows the best activity in the series (Table 2). The cytotoxicity data of the complexes suggests that by the change of two ortho substitutions in L1–L3 (H in L1, methyl in L2 and isopropyl in L3) the activity of the complexes gets affected in terms of mechanism and pathways as described later. During our previous work, we found that the effect of hypoxia may also be applicable to other known potential RuII based anticancer agents41viz. the [(RuII(η6-p-cymene)(en)Cl)(PF6)] designed by Sadler et al.52 [(RuII(η6-p-cymene)(en)Cl)(PF6)] showed better activity in hypoxia against MCF-7 and A54941 but the required IC50 dosage is much higher for these cell lines. However, the encouraging part here is that RuII may have potential as a hypoxic active anticancer agent. Based on the higher degree of aquation at acidic pH, it may be speculated that complexes 1–3 have the potency to be more active inside tumors due to their lower interstitial pH.53
The activity comparison of complexes 1–3 against primary cell lines, primary human foreskin fibroblast (HFF-1) and mouse embryonic fibroblast (NIH 3T3) shows that complex 1 is more active against HepG2 and MIA PaCa-2 than HFF-1 and NIH 3T3 which is encouraging (Table 2). The clinical drug cisplatin is twice more cytotoxic to the primary cell line NIH 3T3 than any of the probed cancer cell lines in vitro. However, the in vitro cytotoxicity of complex 3 against NIH 3T3 is comparable to its activity against HepG2 or MIA PaCa-2, which is still better than the cytotoxicity pattern depicted by cisplatin in these cell lines.
The NMR studies to probe the binding of GSH by 1–3 due to the known inhibitory properties of GSH32 showed that under NMR conditions, where less excess of GSH is used (to obtain a good signal to noise ratio for both bound and unbound species), there is no binding of GSH by any of the complexes. However when we probed the GSH adduct formation by ESI-MS using more excess of GSH (around 25–100 equivalent excess) we found that with ca. 25 equivalents of excess GSH, there is GSH adduct formation by complexes 1 and 2. Under the same conditions of experimentation, the signal intensity of the GSH adduct is much higher for 1 as compared to 2. However using up to 100 equivalents of GSH we did not obtain any ESI-MS signal of the GSH adduct with 3 supporting our earlier reported NMR studies and the cytotoxicity data.41
The bulkiness of the ligands is in the order of 3 > 2 > 1 which corroborates well with the glutathione mediated deactivation under hypoxic conditions (1 > 2 > 3). Complex 3 having isopropyl, the most bulky group, appears to exhibit higher resistance to GSH. The slowest rate of hydrolysis may be the reason behind such activity. As a trend, complex 3 is least deactivated by GSH under hypoxia, followed by complexes 2 and 1. Complex 1 showed 77% and 100% increase in IC50 values in MCF7 and A549 respectively as a result of GSH mediated deactivation under hypoxia. The deactivation by extracellular glutathione is encouraging and suggests that steric hindrance exhibits a significant role against deactivation by GSH. The steric hindrance also affects the partition coefficient of the complexes rendering 3 more lipophilic hence ICP-MS studies show more internalization of 3 inside cells (Fig. 6) but yet 1 is more effective emphasizing that 1 may be a better anticancer agent for low GSH expressing hypoxic tumors.
The cell cycle inhibition studies show that there may be variation in the mechanistic pathway of action since, 1 and 2 arrest MCF-7 cells at the G2/M and S phases whereas 3 arrests in the G2/M and sub G1 phases. The arrest in the sub G1 phase is indicative of the apoptotic pathway for cytotoxicity by 3.54,55 However, it should be noted that apoptosis does not always render arrest of the sub G1 phase.56,57 The DNA ladder assay which provides a qualitative indication to apoptosis confirms that all the complexes follow the apoptotic pathway. In addition, the microscopy images also suggest apoptosis since there is membrane blebbing, cell swelling as well as nuclear fragmentation for all the complexes (ESI, Fig. S33†). Apoptotic cell killing involves extrinsic and intrinsic pathways. The intrinsic pathways lead to greater change in mitochondrial membrane potential. Complex 3 in spite of a poorer IC50 shows the highest change in MMP followed by complex 1 and the least change in MMP is observed with 2. Hence, the mitochondria mediated intrinsic apoptotic pathway may be least feasible in 2. In addition, the caspase activation trend by 2 and 1 although less than 3 is still high and suggests that perhaps extrinsic pathways of apoptosis are active for them. The overall trend of caspase-7 activation and MMP change shows that, it cannot be ruled out that there are multiple pathways active, especially in the case of 1 and 2.
Inhibition of migration of cancer cells is taken as a positive property to prohibit invasion and metastasis.58 The effects of 1–3 on cancer cell migration in the MCF-7 cell line showed that for complexes 1–3 the wide denuded area remained un-healed after 24 h of exposure indicating the antiproliferative nature of the cells, attained by the treatment of the respective drugs. Whereas, the untreated control cells rapidly migrated over time due to the metastatic property resulting in >90% healing. The chick embryo angiogenesis (CEA) assay is a well known angiogenesis assay.59,60 The results showed degenerate blood vessels with 1–3 treated embryos compared to the normal development of blood vessels in the control, which is indicative of the anti-angiogenic potential of the complexes.
Experimental section
Materials and methods
The solvents used for this work were dried and distilled as per the literature mentioned procedure.61 Aniline (Merck), 2,6-dimethylaniline (Sigma-Aldrich), imidazole-2-carboxaldehyde (Sigma-Aldrich), formic acid (Merck), ruthenium(III) chloride (Precious Metal Online, Australia), ammonium hexafluorophosphate (Sigma-Aldrich), ethidium bromide (SRL, India), agarose (molecular biology grade) (SRL, India), MTT [(3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide)] (USB) and other cell growth media and their supplements (Gibco), reduced L-glutathione (GSH) (Aldrich) were used without further purification. [RuII2(η6-p-cym)2(Cl)4] was synthesized using a known procedure.62 The spectroscopy grade solvents were used for spectroscopy and lipophilicity, and the NMR solvents were purchased from Cambridge Isotope Laboratories, Inc. (CIL). Fertilizable eggs were purchased from a recognized local poultry farm and incubated at 37 °C at the 60% humidity level to grow the embryo.SECOR India melting point apparatus was used to detect the melting point and the average value of three data are reported without standard deviation. A Perkin Elmer Lambda 35 spectrophotometer and a cary 300 UV-Visible spectrophotometer were used for UV-Visible experiments. FT-IR spectra were recorded on a Perkin-Elmer Spectrum RX I spectrometer in the solid state using KBr pellets. Either a JEOL ECS 400 MHz or a Bruker Avance III 500 MHz spectrometer was used to obtain 1H, proton decoupled 13C, HMQC and DEPT 135 NMR spectra, each spectrum was recorded at 25 °C and the values are reported in ppm. A Perkin-Elmer 2400 series II CHNS/O analyzer was used for elemental analysis. Electro-spray ionization mass spectra were recorded in +ve mode electrospray ionization using a Q-Tof micro™ (Waters) mass spectrometer. The recrystallization yields of isolated products are only reported. The synthesized ligands and complexes were dried in a vacuum and stored in desiccators in the dark under a nitrogen atmosphere. The synthetic procedure of 3 was recently reported by our group, hence not described here.41 The well known [RuII(η6-p-cym)(en)Cl](PF6) (C1) reported by Sadler et al. was synthesized according to the reported procedure and the analytically pure complex41 was used to study the cytotoxicity in normoxia and hypoxia due to its known toxicity profile.52 Human blood was collected from a volunteer and all the work was performed by following the institute ethical guidelines.
Synthesis
Synthesis of N-((1H-imidazol-2-yl)methylene)aniline (L1). L1 was synthesized using the above mentioned general procedure. Yield: 0.164 g (95%), Anal. Calc. for C10H9N3: C, 70.16; H, 5.30; N, 24.54; Found: C, 70.05; H, 5.33; N, 24.50%, mp: 126 °C, 1H NMR (400 MHz, CDCl3, 25 °C): δ 8.54 (s, 1H, CH
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 7.43 (t, J = 6.44 Hz, 2H, Ar–H), 7.33 (t, J = 5.36 Hz, 1H, Ar–H), 7.27 (d, J = 12.96 Hz, 2H, Ar–H), 7.25 (s, 2H, Imi-H) ppm (ESI, Fig. S1†); 13C NMR (100 MHz, CDCl3, 25 °C): δ 149.9 (imine-C), 149.5 (Imi-C), 145.2 (Ar–C), 129.5 (Imi-CH), 127.1 (Ar–CH), 121.1 (Ar–CH), 115.4 (Ar–CH) ppm (ESI, Fig. S2†); ESI-HRMS (methanol) m/z (calc.): 194.07 (194.07) [C10H9N3·Na+], FT-IR (KBr pellets, cm−1): 2914, 1625, 1440; UV-Vis: [CH3CN, λmax, nm (ε/dm3 mol−1 cm−1)]: 220 (6967), 296 (11262), 317 (11669).
N), 7.43 (t, J = 6.44 Hz, 2H, Ar–H), 7.33 (t, J = 5.36 Hz, 1H, Ar–H), 7.27 (d, J = 12.96 Hz, 2H, Ar–H), 7.25 (s, 2H, Imi-H) ppm (ESI, Fig. S1†); 13C NMR (100 MHz, CDCl3, 25 °C): δ 149.9 (imine-C), 149.5 (Imi-C), 145.2 (Ar–C), 129.5 (Imi-CH), 127.1 (Ar–CH), 121.1 (Ar–CH), 115.4 (Ar–CH) ppm (ESI, Fig. S2†); ESI-HRMS (methanol) m/z (calc.): 194.07 (194.07) [C10H9N3·Na+], FT-IR (KBr pellets, cm−1): 2914, 1625, 1440; UV-Vis: [CH3CN, λmax, nm (ε/dm3 mol−1 cm−1)]: 220 (6967), 296 (11262), 317 (11669).
Synthesis of N-((1H-imidazol-2-yl)methylene)-2,6-dimethylaniline (L2). L2 was synthesized using the above mentioned general procedure. Yield: 0.180 g (90%), Anal. Calc. for C12H13N3: C, 72.33; H, 6.58; N, 21.09; Found: C, 72.25; H, 6.62; N, 21.02%, mp: 121 °C, 1H NMR (400 MHz, CDCl3, 25 °C): δ 8.24 (s, 1H, CH
N), 7.13 (s, 2H, Imi-H), 7.08 (d, J = 6 Hz, 2H, Ar–H), 7.02 (t, J = 5.2 Hz, 1H, Ar–H), 2.13 (s, 6H, CH3) ppm (ESI, Fig. S3†); 13C NMR (100 MHz, CDCl3, 25 °C): δ 153.2 (imine-C), 135.8 (Imi-C), 128.6 (Ar–C), 128.4 (Imi-CH), 127.9 (Ar–CH), 125.3 (Ar–CH), 118.2 (Ar–CH), 18.4 (CH3) ppm (ESI, Fig. S4†); ESI-HRMS (methanol) m/z (calc.): 222.10 (222.10) [C12H13N3·Na+], FT-IR (KBr pellets, cm−1): 2910, 1638, 1442; UV-Vis: [CH3CN, λmax, nm (ε/dm3 mol−1 cm−1)]: 282 (18744), 323 (3871).
General procedure for synthesis of complexes 1 and 2
The complexes were synthesized according to a procedure similar to that reported by us earlier.41 Precisely, 2.4 mmol solution of the respective ligand in methanol (MeOH) was added to the methanolic solution of [RuII2(η6-p-cym)2(Cl)4] (0.612 g, 1 mmol) in the dark under a nitrogen atmosphere at room temperature. Then the reaction mixture was stirred for 10 h. NH4PF6 (0.326 g, 2 mmol) was added to the resultant reaction mixture and stirred for another 2 h. Evaporation of the reaction mixture led to an orange coloured mass which was washed twice with diethyl ether. The crude product was purified by crystallization from a dichloromethane solution layered with petroleum benzene.301), 385 (2962).
X-ray crystallography
Single crystals of 1 and 2 suitable for diffraction were coated with Fomblin oil and fixed on a loop and mounted on the goniometer under flow of nitrogen. The data were collected in a SuperNova, Dual, Cu at zero, Eos diffractometer, Agilent using a monochromatic Mo Kα source (λ = 0.71073 Å) using a detector distance of 75 mm at 100 K with exposure of 6 s. CrysAlisPro, Agilent Technologies, Version 1.171.37.31 (release 14-01-2014 CrysAlis171.NET) software was used for data collection and data reduction. All the structures were solved with Superflip63 structure solution programme Charge Flipping and refined with the ShelXL64 refinement package using least squares minimisation, using Olex2.65 All the hydrogen atoms were calculated and fixed using ShelXL after hybridization of all non-hydrogen atoms.66 Crystals data were deposited at https://deposit.ccdc.cam.ac.uk. The CCDC numbers are 1431153 for 1 and 1431154 for 2.The ORTEP diagrams were processed through the POV-ray with 50% probability level. All the non-hydrogen atoms were refined with anisotropic displacement parameters.
Lipophilicity
Distribution coefficient (logD) was determined using the standard shake-flask method42 for all ligands and complexes in the octanol–aqueous phosphate buffer (20 mM, pH 7.4) system. After overnight pre-equilibration of octanol and aqueous phosphate buffer (equal volume, 3 mL each)67 the solid samples (1.2 mg each) were added to the solvent mixture and shaken constantly for 10 hours at 37 °C on a shaking incubator to obtain full distribution between the two layers. Followed by this, all the tubes were centrifuged and left undisturbed for an hour. Aliquots of the aqueous and octanol layers were pipetted out separately and the absorbances were measured with a UV-Vis spectrophotometer using proper dilution. Each set was performed in triplicate, concentration of the substances in each layer was calculated using the respective molar extinction coefficients and the distribution coefficient values (logD) were obtained from the ratio.
Aquation studies: dependence on pH and chloride concentration
The pH dependent aquation of 1, 2 and 3 was measured by UV-Vis absorption spectroscopy at pH 7.4 and 6.7 (using 1% methanol–20 mM phosphate buffer in 40 mM or 4 mM NaCl) for 12 h. The hydrolysis of the complexes follows the pseudo-first order kinetics with respect to water and the half-life as well as the rate were obtained from monoexponential decay fitting at a constant wavelength with time.Hydrolysis of respective complexes was also monitored by 1H NMR by obtaining spectra at different intervals of time in DMSO-d6 and D2O mixtures at 25 °C (3:7 v/v).
Stability in saline solution
The stability of each complex in extracellular chloride concentration (110 mM) was observed by 1H NMR at 25 °C in D2O:DMSO-d6 (7:3 v/v), 110 mM saline solution. 2 mg of each complex was dissolved in 640 μL of 7:3 (v/v) D2O/DMSO-d6 solution containing 3.5 mg of NaCl and the spectra were recorded up to ten days.
Interaction with CT DNA
Deoxyribonucleic acid sodium salt isolated from calf thymus (CT DNA) was dissolved in 50 mM Tris-NaCl solution at pH 7.4 and kept for 10 h at 4 °C. The UV-Visible spectrum showed two absorption bands at 260 nm and 280 nm and their absorption ratio was 1.9, which indicate that the DNA was apparently free from protein.68 The concentration of the DNA was determined from the absorption value at 260 nm (molar extinction coefficient is 6600 dm3 mol−1 cm−1)69 and the average value of the concentration was taken after three independent measurements of the same stock solution. The stock solution of the DNA was stored at 4 °C and used within four days.Interaction of 1 and 2 with CT DNA was measured with the help of UV-Vis spectroscopy in Tris-HCl buffer (50 mM, pH 7.4)–acetonitrile (99:1 v/v) media. Acetonitrile was used for the preparation of the stock solution of complexes. Spectroscopic titrations were carried out at room temperature (25 °C). The concentration of 1 and 2 for the binding experiments was fixed to 1 × 10−4 M and incubated for 12 h at 25 °C for pre-hydrolysis. In another set of experiments the concentrations were fixed to 5 × 105 and 4.4 × 105 M for 1 and 2 respectively and binding titration started instantaneously after preparing the complex solution. The change in absorbance at 311 and 300 nm for 1 and 2 respectively was monitored with subsequent addition of an aliquot of 5 μL (concentration of 1 × 10−2 M and 5.3 × 10−3 M for 12 h and 0 h of hydrolysis respectively) of CT DNA into the sample and also into the reference cuvette after 5 min of equilibration. The titration was continued until there was no significant change in absorbance for at least three successive additions.
Binding studies with reduced L-glutathione (GSH)
The binding studies of 1 and 2 with reduced L-glutathione were monitored by 1H NMR. The samples were prepared in a degassed D2O/DMSO-d6 (7:3 v/v) mixture at 25 °C under a nitrogen atmosphere to minimize the autooxidation of glutathione. Each experiment involved 0.002 mmol of complex and 0.004 mmol (2 equiv.) of GSH dissolved in the solution. The binding was studied up to 29 hours at different intervals of time although after 8 h, the total autooxidation process was completed.
Binding studies were also carried out using ESI mass spectrometry. Lower equivalents of GSH viz. up to 10 equiv. did not produce any m/z peak corresponding to the adduct with the respective complexes. Hence, higher amounts viz. at least 25 equiv. and up to 100 equivalents of GSH with the complex was incubated for half an hour at 37 °C under stirring conditions, diluted with water and ESI mass spectrometry was performed.
Cell lines and culture
The human breast adenocarcinoma (MCF-7), human lung adenocarcinoma (A549) and mouse embryonic fibroblast (NIH 3T3) were kindly donated from the Department of Biological Sciences, IISER Kolkata (originally purchased from ATCC); hepatocarcinoma (Hep G2) and human pancreatic carcinoma (MIA PaCa-2) were obtained from NCCS, Pune, and Primary Human Foreskin Fibroblast (HFF-1) was donated to us by Dr Rupak Dutta, Department of Biological Sciences, IISER Kolkata. Cell lines (MCF-7, A549, MIA PaCa-2) were maintained in the logarithmic phase in Dulbecco's Modified Eagle Medium (DMEM) while HFF-1 and Hep G2 were grown in Minimum Essential Medium (MEM). Both the media were supplemented with 10% fetal bovine serum (GIBCO) and antibiotics (100 units per ml penicillin and 100 mg per ml streptomycin). The cell culture conditions used was 95% humidity and 5% CO2 at a temperature of 37 °C. For the hypoxic cell culture the oxygen level was maintained at 1.5% and other aforementioned parameters were kept unaltered.Cell viability assay
MTT assay was used to determine the cytotoxicity of the compounds against the cancer cells based on cell viability as an indicator for the sensitivity of the cells to the individual compound. Briefly, exponentially grown cancer cells were seeded in a 96-well microplate (Nunc) at a density of 6 × 103 viable cells per well. Cells were incubated at 37 °C (5% CO2 atmosphere) for 48 h to resume exponential growth followed by removal and replacement of fresh media containing the desired concentration of test compounds. Each concentration was loaded in triplicate. The stock solutions of compounds were prepared in DMSO such that the concentration of DMSO in the well does not exceed 0.2%. After 48 h of subsequent incubation, compounds containing media were removed, followed by addition of 200 μL of fresh medium and 20 μL of 1 mg per ml MTT in PBS (pH 7.2) into each well. On incubation of the plates for 3 h at 37 °C under a humidified 5% CO2 atmosphere, MTT was allowed to form formazan crystals in metabolically active cells. Finally the media were removed after incubation and replaced with 200 μL of DMSO in each well that would solubilize the formazan crystals. The absorbance for each well was recorded at 515 nm using a BIOTEK ELx800 plate reader.IC50 values represent the drug concentration at which 50% cells are inhibited compared to control, which were calculated by fitting nonlinear curves in GraphPad Prism 5, constructed by plotting cell viability (%) versus log of drug concentration in μM or in nM. Each independent experiment was carried out in triplicate.
For the experiment under hypoxic conditions the oxygen percentage of the CO2 incubator (ESCO cell culture CO2 incubator, model: CCL-170T-8-UV) was maintained at 1.5%. Compounds were loaded in 96 well microplates in a level-II bio safety cabinet under atmospheric conditions which took ca. 10 minutes and then the drug loaded 96 well microplate was placed in the incubator programmed to attain 1.5% oxygen concentration. The incubator takes ca. 30–40 minutes to reach the 1.5% oxygen level.
Cell cycle analysis
MCF-7 cells (5 × 105 per plate) were seeded in a 90 mm dia petri-dish in 12 mL DMEM and incubated for 48 h at 37 °C under a 5% carbon dioxide atmosphere. After 48 h, media were removed, followed by addition of fresh media containing appropriate concentrations of test compounds and incubated for another 24 h under the same conditions as above. After 24 h of incubation with test compounds, cells were harvested by trypsinization and washed twice with cold 1× PBS (pH 7.2). Cells were fixed using 70% ethanol and stored at 4 °C for 12 h. In order to stain DNA for determining the cell cycle distribution, cell pellets were transferred and resuspended in PBS solution containing RNase (100 μg mL−1) and propidium iodide (55 μg mL−1). The resulting solution was then incubated for 30 min at 37 °C in the dark. Finally, homogenized cell samples were analyzed by flow-cytometry using a fluorescence-activated cell sorter (FACS) (FACS Calibur, Becton Dickinson, CA). The resulting DNA histograms were quantified using the CellQuestPro software (BD).DNA ladder assay for apoptosis detection
DNA ladder assay was used to detect the cellular apoptosis induced by the test compound. MCF-7 cells (5 × 105 per plate) were seeded in a 90 mm dia petri-dish in 12 mL DMEM and incubated for 48 h at 37 °C under a 5% carbon dioxide atmosphere. Subsequently the media were replaced with fresh media containing the required concentration of test compounds and incubated at 37 °C under a 5% carbon dioxide atmosphere for 24 h. Cells were then harvested by trypsinization followed by centrifugation at 2000 rpm for 5 minutes. Cells were subjected to PBS (pH = 7.2), washed and re-suspended in 500 μL lysis buffer [20 mM Tris-HCl (pH 7.4), 0.4 mM EDTA, 0.25% Triton-X 100] and incubated for 15 minutes at room temperature. Lysed cells were centrifuged at 14000 rpm for 10 minutes and the supernatant was collected. This was mixed with a 1:1 (v/v) phenol–chloroform mixture (1 mL) and the aqueous layer was carefully separated to which 55 μL of 5 M NaCl and 500 μL isopropanol were added respectively following incubation overnight at −20 °C. After that the solution was centrifuged at 14000 rpm and washed with 70% ice cold ethanol and dried in air which was again resuspended in 1× TE solution [10 mM Tris-HCl (pH 8.0), 1 mM EDTA] containing RNase (150 μg mL−1) and finally it was incubated at 37 °C for 20 minutes. The supernatant, containing isolated DNA was mixed with bromophenol blue dye and loaded in 1.6% agarose gel containing EtBr (1.0 μg mL−1) and run at 60 V for around 3 h in 1× TBE (Tris-borate-EDTA) buffer. Untreated cells were used as controls whereas a 50 bp DNA ladder was used to track the migration of bands and fragmentation sizes on the agarose gel.
Visible bands on the gel were observed and a picture taken on exposure to UV light through the gel documentation system of Bio-Rad.
Optical microscopy imaging
MCF-7 cells were seeded at 15 × 103 per well in a 6-well plate and were grown with 3 mL of DMEM. Existing media were replaced after 48 h of incubation and the cells were further incubated with fresh medium containing the required concentrations of the metal complex for 24 h. After removal of the drug containing media, the cells were treated using 4% paraformaldehyde solution in 1× PBS (pH 7.2) and incubated at 4 °C for half an hour. The cells were then washed two times with 1× PBS (pH 7.2) and stained with DAPI (1 μg mL−1) for a few seconds. The cells were washed several times using 1× PBS (pH 7.2). The optical microscopy images of MCF-7 cells were acquired using an OLYMPUS IX 81 epifluorescence inverted microscope at 60× magnification. Both DIC and fluorescence microscopy images were taken and processed using OLYMPUS Cell P software. Merged images were produced for better understanding.Ruthenium accumulation study in cancer cells
Briefly, 1 × 106 MCF-7 cells were seeded in each 90 mm dia petri-dish and incubated for 48 h. After incubation, equimolar concentrations (3 μM) of each complex were added and a further 24 h of drug treatment was allowed. Following this, the cells were washed with PBS (pH 7.2), treated with trypsin-EDTA, counted and 1 × 106 number of cells were collected as cell pellets using centrifugation. Cell pellets were digested overnight in concentrated nitric acid (70% v/v) at 65 °C; the resulting solutions were diluted using double-distilled water to give a final concentration of 5% HNO3 and the amount of Ru taken up by the cells was determined by ICP-MS (Thermo Scientific XSERIES2). The standard solutions of ruthenium was freshly prepared and analyzed while analyses for the samples were carried out in triplicates and the standard deviations were calculated.Caspase-7 activation
A quantitative estimation of caspase activity was studied using MCF-7 cells by using a colorimetric Caspase Assay kit from Sigma. The technique principally detects the free chromophore (p-nitroaniline, pNA), following its cleavage from the Ac-DEVD-pNA substrate. Though the substrate Ac-DEVD-pNA is cleaved by both caspase-3 and caspase-7, yet this experiment would indicate the presence of caspase-770–74 since MCF-7 cells are already known not to have caspase-3.75 Briefly, 1 × 106 MCF-7 cells were seeded in a 90 mm dia petri-dish and incubated for 48 h followed by treatment with test compounds with an additional incubation of 24 h. Then the cells were harvested, lysed and treated as per the kit manufacturer's (Sigma) standard protocol. All the samples were tested in triplicate including the standard curve of pNA and data were recorded after 24 h of incubation with cell lysate at 405 nm using an ELISA plate reader.
The data is graphically represented as the absolute concentration of pNA released (nmol min−1 ml−1) in the y-axis and concentration of the treated drugs in the x-axis. Each concentration has been performed in triplicate.
Detection of mitochondrial membrane potential
1 × 106 MCF-7 cells were seeded in each 90 mm dia petri-dish and incubated for 48 h. Media were then replaced with fresh media containing the desired sub IC50 concentrations (4, 6 and 8 μM) of each compound. After further incubation of 24 h, the cells were harvested by removing the media and subsequently washed with 1× PBS solution. The entire washings were collected in falcon tubes and centrifuged at 2500 rpm for 10 minutes. Supernatants were discarded and the pellets were washed with 1× PBS solution. Washed cells were suspended in the 1× PBS buffer containing 10 μg mL−1 of JC-1 and supplemented with 10% FBS followed by incubation for 30 minutes at 37 °C under dark conditions. Finally the supernatant was removed by centrifugation and the cell pellet was further re-suspended in 1× PBS buffer. The stained cells were analyzed using a BD Biosciences FACS Calibur flow cytometer measuring the red and green fluorescence intensities.Wound assay (migration)
To evaluate the effect of drugs against cancer cell migration, the wound healing assay was used as a tool.76,77 The migration capacities of drug treated cells were compared to the control in MCF-7 cell lines. Briefly, 1 × 106 cells were seeded in each well of a 6 well plate and incubated at 37 °C in a humidified incubator with 5% CO2. Sufficient time was provided for the formation of a uniform monolayer in the plate. The monolayer of the cells was then carefully scratched using a sterile microtip to form a denuded area across the diameter of each well. Each well was carefully washed with 1× PBS followed by replenishment of fresh medium and addition of a sub IC50 concentration (6 μM) of 1–3. The experiments were carried out in duplicate. The images were captured on a Phase contrast microscope (OLYMPUS IX 81 epifluorescence inverted microscope) at variable time points (0 h i.e. initial, 12 h and 24 h). The wound area was quantified using ImageJ software.Haemolysis assay
The haemolysis assay was performed following the standard method.44 Briefly, blood was collected in EDTA containing vials, followed by centrifugation at 3000 rpm for 10 min. Erythrocytes were subsequently washed with cold PBS (pH 7.4) 3 times by centrifugation at 3000 rpm for 10 min. The cells were re-suspended in the same buffer to a final concentration of 20% (v/v) and stored at 4 °C. Various concentrations of 1, 2 and 3 were prepared in DMSO and diluted in PBS such that the final concentration of compounds remains 5, 10 and 15 μM in the reaction vial, while the final DMSO concentration is kept at 0.2%. The reaction vial contains 2% (final concentration) cell suspension and the required concentrations of compounds. The vials were incubated at 37 °C for 1 h in a shaking water bath. After incubation the vials were centrifuged at 3000 rpm for 10 min and the supernatants were collected. The release of haemoglobin in the supernatant was assessed spectrophotometrically at 540 nm. Positive control in which complete haemolysis takes place was achieved using 0.2% (final concentration) Triton X-100. Appropriate DMSO containing PBS was used as a negative control. The experiments were run in triplicate and the percentages of haemolysis were calculated as follows:| Haemolysis percentage (%) = [(As − An)/(Ap − An)] × 100 |
Chick embryo angiogenesis assay (CEA)
The chick embryo angiogenesis assay (CEA) was used to investigate the anti-angiogenic activity of 1–3 using a reported protocol.76 To briefly mention the used procedure, specific pathogen free (SPF) fertile chicken eggs were obtained from a recognized local poultry farm which was incubated at 37 °C under a humid atmosphere. After the fourth day of incubation, the shells of the eggs were cautiously broken using forceps and placed on a sterile petri-dish. Precautions were taken to prevent puncture of any of the blood vessels while transferring. Stock solutions of complexes (1–3) were prepared in 10% DMSO–PBS (v/v) solution and further diluted with PBS to achieve a complex concentration of 10 μM (final DMSO concentration 0.2%). Sterile filter paper discs (6 mm dia) were soaked in a solution of the respective complexes and placed in 3 different positions (10 μM; 10 μL) over the generating blood vessels. Sterile filter paper discs (6 mm dia) soaked with 10 μL of 0.2% DMSO in PBS were also used in triplicate as control. Images of blood vessels were captured using a camera attached to a stereomicroscope (Leica M80) at initial (i.e. 0 h) and after 4 h of incubation.Statistical analysis
All the IC50 data are expressed as mean ± standard deviation of three independent experiments carried out in each cell line and in each experiment each concentration was assayed in triplicate. The statistical analyses were performed using Graph pad prism® software 5.0 with student's t-test. Differences with *P < 0.05 were considered statistically significant.Conclusions
In summary, we have shown that design of RuII-p-cymene complexes (1–3) by variation of the steric hindrance in the ligands (L1–L3) may result in the change of cytotoxicity and mechanism of action towards various cancer cells. Faster hydrolysis does lead to higher cytotoxicity but it appears that the pathway of action also changes as found from the cell cycle arrest, MMP change and caspase activation data of the complexes. All our complexes are stable against 110 mM chloride concentration. None of the complexes lose the arene moiety up to 24 h or more. All the complexes arrest the cell cycle in the G2/M phase. However, 1 and 2 also exhibit arrest in the S-phase whereas 3 shows significant arrest in the sub G1 phase. Although the lipophilicity of complexes 1 and 2 is low, yet they exhibit higher cytotoxicity in spite of lower accumulation inside cells. The cellular toxicity data suggests that higher steric hindrance can prohibit deactivation by GSH and also alter the pathway of action. Hence by proper tuning of the steric effect it may be possible to avoid direct inhibition by cellular thiols, in RuII–arene complexes, in those forms of cancers where GSH is overexpressed. It is encouraging to see that none of the complexes exhibit binding to GSH until they are incubated with at least 25 equivalent excess GSH and 3 shows no GSH adduct even up to 100 equivalents of GSH. Complex 3 shows the maximum change in the MMP and highest caspase-7 activation, suggesting an intrinsic pathway of apoptosis involving the mitochondria may be active. Whereas in the case of 1 and 2, other extrinsic pathways may be playing a significant role in cell killing, especially in complex 2 which has the lowest MMP change but higher activation of caspase than 1. In addition complexes 1–3 exhibit potential anti-metastatic and anti-angiogenic activity.Abbreviations
| GSH | Glutathione |
| MMP | Mitochondrial membrane potential |
| ppm | Parts per million |
Acknowledgements
We sincerely acknowledge DST for financial support vide project no SB/S1/IC-02/2014. We also thank IISER Kolkata for the annual research funding and infra-structural support, including NMR, single crystal X-ray, microscopy and FACS facilities. K. P. and S. K. thank UGC, S. C. thanks IISER Kolkata for providing the research fellowship. We sincerely thank Prof. Subhransu Pan, West Bengal University of Animal and Fishery Sciences for useful discussions on the CEA assay.Notes and references
- E. Meggers, Curr. Opin. Chem. Biol., 2007, 11, 287–292 CrossRef CAS PubMed.
- C. G. Hartinger and P. J. Dyson, Chem. Soc. Rev., 2009, 38, 391–401 RSC.
- G. Suess-Fink, Dalton Trans., 2010, 39, 1673–1688 RSC.
- M. A. Jakupec, M. Galanski, V. B. Arion, C. G. Hartinger and B. K. Keppler, Dalton Trans., 2008, 183–194 RSC.
- V. Brabec and O. Novakova, Drug Resist. Updates, 2006, 9, 111–122 CrossRef CAS PubMed.
- A. Levina, A. Mitra and P. A. Lay, Metallomics, 2009, 1, 458–470 RSC.
- E. S. Antonarakis and A. Emadi, Cancer Chemother. Pharmacol., 2010, 66, 1–9 CrossRef CAS PubMed.
- A. Bergamo, C. Gaiddon, J. H. M. Schellens, J. H. Beijnen and G. Sava, J. Inorg. Biochem., 2012, 106, 90–99 CrossRef CAS PubMed.
- J. M. Rademaker-Lakhai, D. Van Den Bongard, D. Pluim, J. H. Beijnen and J. H. M. Schellens, Clin. Cancer Res., 2004, 10, 3717–3727 CrossRef CAS PubMed.
- C. G. Hartinger, S. Zorbas-Seifried, M. A. Jakupec, B. Kynast, H. Zorbas and B. K. Keppler, J. Inorg. Biochem., 2006, 100, 891–904 CrossRef CAS PubMed.
- M. Pongratz, P. Schluga, M. A. Jakupec, V. B. Arion, C. G. Hartinger, G. Allmaier and B. K. Keppler, J. Anal. At. Spectrom., 2004, 19, 46–51 RSC.
- P. Schluga, G. Hartinger Christian, A. Egger, E. Reisner, M. Galanski, A. Jakupec Michael and K. Keppler Bernhard, Dalton Trans., 2006, 1796–1802 RSC.
- M. Matczuk, M. Przadka, S. S. Aleksenko, Z. Czarnocki, K. Pawlak, A. R. Timerbaev and M. Jarosz, Metallomics, 2014, 6, 147–153 RSC.
- M. J. Clarke, Coord. Chem. Rev., 2003, 236, 209–233 CrossRef CAS.
- A. D. Kelman, M. J. Clarke, S. D. Edmonds and H. J. Peresie, J. Clin. Hematol. Oncol., 1977, 7, 274–288 CAS.
- R. Trondl, P. Heffeter, C. R. Kowol, M. A. Jakupec, W. Berger and B. K. Keppler, Chem. Sci., 2014, 5, 2925–2932 RSC.
- M. J. Clarke, S. Bitler, D. Rennert, M. Buchbinder and A. D. Kelman, J. Inorg. Biochem., 1980, 12, 79–87 CrossRef CAS PubMed.
- Y. Han, Q. Luo, X. Hao, X. Li, F. Wang, W. Hu, K. Wu, L. Shuang and P. J. Sadler, Dalton Trans., 2011, 40, 11519–11529 RSC.
- J. Reedijk, Chem. Rev., 1999, 99, 2499–2510 CrossRef CAS PubMed.
- K. Mitra, S. Patil, P. Kondaiah and A. R. Chakravarty, Inorg. Chem., 2015, 54, 253–264 CrossRef CAS PubMed.
- M. Groessl, Y. O. Tsybin, C. G. Hartinger, B. K. Keppler and P. J. Dyson, JBIC, J. Biol. Inorg. Chem., 2010, 15, 677–688 CrossRef CAS PubMed.
- R. Fernandez, M. Melchart, A. Habtemariam, S. Parsons and P. J. Sadler, Chem. – Eur. J., 2004, 10, 5173–5179 CrossRef CAS PubMed.
- S. M. Smalley Keiran, R. Contractor, K. Haass Nikolas, N. Kulp Angela, G. E. Atilla-Gokcumen, S. Williams Douglas, H. Bregman, T. Flaherty Keith, S. Soengas Maria, E. Meggers and M. Herlyn, Cancer Res., 2007, 67, 209–217 CrossRef CAS PubMed.
- J. Reedijk, Platinum Met. Rev., 2008, 52, 2–11 CrossRef CAS.
- W.-H. Ang, A. Casini, G. Sava and P. J. Dyson, J. Organomet. Chem., 2011, 696, 989–998 CrossRef CAS.
- C. A. Smith, A. J. Sutherland-Smith, B. K. Keppler, F. Kratz and E. N. Baker, JBIC, J. Biol. Inorg. Chem., 1996, 1, 424–431 CrossRef CAS.
- S. H. van Rijt, A. J. Hebden, T. Amaresekera, R. J. Deeth, G. J. Clarkson, S. Parsons, P. C. McGowan and P. J. Sadler, J. Med. Chem., 2009, 52, 7753–7764 CrossRef CAS PubMed.
- T. Bugarcic, O. Novakova, A. Halamikova, L. Zerzankova, O. Vrana, J. Kasparkova, A. Habtemariam, S. Parsons, P. J. Sadler and V. Brabec, J. Med. Chem., 2008, 51, 5310–5319 CrossRef CAS PubMed.
- A. Habtemariam, M. Melchart, R. Fernandez, S. Parsons, I. D. H. Oswald, A. Parkin, F. P. A. Fabbiani, J. E. Davidson, A. Dawson, R. E. Aird, D. I. Jodrell and P. J. Sadler, J. Med. Chem., 2006, 49, 6858–6868 CrossRef CAS PubMed.
- F. Wang, A. Habtemariam, E. P. L. van der Geer, R. Fernandez, M. Melchart, R. J. Deeth, R. Aird, S. Guichard, F. P. A. Fabbiani, P. Lozano-Casal, I. D. H. Oswald, D. I. Jodrell, S. Parsons and P. J. Sadler, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 18269–18274 CrossRef CAS PubMed.
- H. Chen, J. A. Parkinson, S. Parsons, R. A. Coxall, R. O. Gould and P. J. Sadler, J. Am. Chem. Soc., 2002, 124, 3064–3082 CrossRef CAS PubMed.
- F. Wang, J. Xu, A. Habtemariam, J. Bella and P. J. Sadler, J. Am. Chem. Soc., 2005, 127, 17734–17743 CrossRef CAS PubMed.
- S. Chatterjee, S. Kundu, A. Bhattacharyya, C. G. Hartinger and P. J. Dyson, JBIC, J. Biol. Inorg. Chem., 2008, 13, 1149–1155 CrossRef CAS PubMed.
- F. Wang, H. Chen, J. A. Parkinson, P. d. S. Murdoch and P. J. Sadler, Inorg. Chem., 2002, 41, 4509–4523 CrossRef CAS PubMed.
- J. Estrela, A. Ortega and E. Obrador, Crit. Rev. Clin. Lab. Sci., 2006, 43, 143–181 CrossRef CAS PubMed.
- M. L. O'Brien and K. D. Tew, Eur. J. Cancer, Part A, 1996, 32A, 967–978 CrossRef.
- N. Traverso, R. Ricciarelli, M. Nitti, B. Marengo, A. L. Furfaro, M. A. Pronzato, U. M. Marinari and C. Domenicotti, Oxid. Med. Cell. Longevity, 2013, 972913 Search PubMed.
- M. Benlloch, A. Ortega, P. Ferrer, R. Segarra, E. Obrador, M. Asensi, J. Carretero and J. M. Estrela, J. Biol. Chem., 2005, 280, 6950–6959 CrossRef CAS PubMed.
- Y. Lin, Y. Huang, W. Zheng, F. Wang, A. Habtemariam, Q. Luo, X. Li, K. Wu, P. J. Sadler and S. Xiong, J. Inorg. Biochem., 2013, 128, 77–84 CrossRef CAS PubMed.
- F. Wang, J. Xu, K. Wu, S. K. Weidt, C. L. MacKay, P. R. R. Langridge-Smith and P. J. Sadler, Dalton Trans., 2013, 42, 3188–3195 RSC.
- K. Purkait, S. Karmakar, S. Bhattacharyya, S. Chatterjee, S. K. Dey and A. Mukherjee, Dalton Trans., 2015, 44, 5969–5973 RSC.
- C. Zhang, Y. Wang and F. Wang, Bull. Korean Chem. Soc., 2007, 28, 1183–1186 CrossRef CAS.
- M. M. Compton, Cancer Metastasis Rev., 1992, 11, 105–119 CrossRef CAS PubMed.
- H.-W. An, S.-L. Qiao, C.-Y. Hou, Y.-X. Lin, L.-L. Li, H.-Y. Xie, Y. Wang, L. Wang and H. Wang, Chem. Commun., 2015, 51, 13488–13491 RSC.
- B. Naeye, H. Deschout, M. Roding, M. Rudemo, J. Delanghe, K. Devreese, J. Demeester, K. Braeckmans, S. C. De Smedt and K. Raemdonck, Biomaterials, 2011, 32, 9120–9127 CrossRef CAS PubMed.
- S. Ramakrishnan, E. Suresh, A. Riyasdeen, M. A. Akbarsha and M. Palaniandavar, Dalton Trans., 2011, 40, 3245–3256 RSC.
- N. Busto, J. Valladolid, C. Aliende, F. A. Jalon, B. R. Manzano, A. M. Rodriguez, J. F. Gaspar, C. Martins, T. Biver, G. Espino, J. M. Leal and B. Garcia, Chem. – Asian J., 2012, 7, 788–801 CrossRef CAS PubMed.
- J. Valladolid, C. Hortigueela, N. Busto, G. Espino, A. M. Rodriguez, J. M. Leal, F. A. Jalon, B. R. Manzano, A. Carbayo and B. Garcia, Dalton Trans., 2014, 43, 2629–2645 RSC.
- M. Martinez-Alonso, N. Busto, F. A. Jalon, B. R. Manzano, J. M. Leal, A. M. Rodriguez, B. Garcia and G. Espino, Inorg. Chem., 2014, 53, 11274–11288 CrossRef CAS PubMed.
- N. Busto, M. Martinez-Alonso, J. M. Leal, A. M. Rodriguez, F. Dominguez, M. I. Acuna, G. Espino and B. Garcia, Organometallics, 2015, 34, 319–327 CrossRef CAS.
- A. Russo, W. DeGraff, N. Friedman and J. B. Mitchell, Cancer Res., 1986, 46, 2845–2848 CAS.
- R. E. Morris, R. E. Aird, P. d. S. Murdoch, H. Chen, J. Cummings, N. D. Hughes, S. Parsons, A. Parkin, G. Boyd, D. I. Jodrell and P. J. Sadler, J. Med. Chem., 2001, 44, 3616–3621 CrossRef CAS PubMed.
- B. A. Webb, M. Chimenti, M. P. Jacobson and D. L. Barber, Nat. Rev. Cancer, 2011, 11, 671–677 CrossRef CAS PubMed.
- W. Xia, S. Spector, L. Hardy, S. Zhao, A. Saluk, L. Alemane and N. L. Spector, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 7494–7499 CrossRef CAS.
- M. Kajstura, H. D. Halicka, J. Pryjma and Z. Darzynkiewicz, Cytometry, Part A, 2007, 71A, 125–131 CrossRef CAS PubMed.
- I. Romero-Canelon, L. Salassa and P. J. Sadler, J. Med. Chem., 2013, 56, 1291–1300 CrossRef CAS PubMed.
- R. Pettinari, C. Pettinari, F. Marchetti, B. W. Skelton, A. H. White, L. Bonfili, M. Cuccioloni, M. Mozzicafreddo, V. Cecarini, M. Angeletti, M. Nabissi and A. M. Eleuteri, J. Med. Chem., 2014, 57, 4532–4542 CrossRef CAS PubMed.
- P. Friedl and K. Wolf, Nat. Rev. Cancer, 2003, 3, 362–374 CrossRef CAS PubMed.
- P. Nowak-Sliwinska, C. M. Clavel, E. Paunescu, M. T. te Winkel, A. W. Griffioen and P. J. Dyson, Mol. Pharmaceutics, 2015, 12, 3089–3096 CrossRef CAS PubMed.
- A. K. Barui, V. Veeriah, S. Mukherjee, J. Manna, A. K. Patel, S. Patra, K. Pal, S. Murali, R. K. Rana, S. Chatterjee and C. R. Patra, Nanoscale, 2012, 4, 7861–7869 RSC.
- D. D. Perrin and W. L. F. Armarego, Purification of Laboratory Chemicals, 3rd edn, 1988 Search PubMed.
- M. A. Bennett, T. N. Huang, T. W. Matheson and A. K. Smith, Inorg. Synth., 1982, 21, 74–78 CrossRef CAS.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Int. Union Crystallogr., Crystallogr. Symp., 1991, 5, 145–157 CAS.
- J. Sangster and A. D. Pelton, J. Phys. Chem. Ref. Data, 1987, 16, 509–561 CrossRef CAS.
- J. Marmur, J. Mol. Biol., 1961, 3, 208–218 CrossRef CAS.
- M. E. Reichmann, C. A. Rice, C. A. Thomas and P. Doty, J. Am. Chem. Soc., 1954, 76, 3047–3053 CrossRef CAS.
- X.-H. Yang, T. L. Sladek, X. Liu, B. R. Butler, C. J. Froelich and A. D. Thor, Cancer Res., 2001, 61, 348–354 CAS.
- U. Janicke Reiner, Breast Cancer Res. Treat., 2009, 117, 219–221 CrossRef CAS PubMed.
- R. U. Janicke, M. L. Sprengart, M. R. Wati and A. G. Porter, J. Biol. Chem., 1998, 273, 9357–9360 CrossRef CAS PubMed.
- H. Kurokawa, K. Nishio, H. Fukumoto, A. Tomonari, T. Suzuki and N. Saijo, Oncol. Rep., 1999, 6, 33–37 CAS.
- D. Twiddy, G. M. Cohen, M. MacFarlane and K. Cain, J. Biol. Chem., 2006, 281, 3876–3888 CrossRef CAS PubMed.
- N. A. Thornberry, T. A. Rano, E. P. Peterson, D. M. Rasper, T. Timkey, M. Garcia-Calvo, V. M. Houtzager, P. A. Nordstrom, S. Roy, J. P. Vaillancourt, K. T. Chapman and D. W. Nicholson, J. Biol. Chem., 1997, 272, 17907–17911 CrossRef CAS PubMed.
- P. Nagababu, A. K. Barui, B. Thulasiram, C. S. Devi, S. Satyanarayana, C. R. Patra and B. Sreedhar, J. Med. Chem., 2015, 58, 5226–5241 CrossRef CAS PubMed.
- G. V. M. Sharma, A. Ramesh, A. Singh, G. Srikanth, V. Jayaram, D. Duscharla, J. H. Jun, R. Ummanni and S. V. Malhotra, MedChemComm, 2014, 5, 1751–1760 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Table for crystal refinement parameters, NMR, and UV-visible spectra of hydrolysis, plots of cell viability, cell cycle, JC-1. CCDC 1431153 and 1431154. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt04781a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |