
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Isomerization of the osmium–tellurium cluster Os3(μ-TeR)2(CO)10: a kinetic and computational study†
E. K. S.
Shim
a,
W. K.
Leong
*a,
Y.-Z.
Li
a and
M. G.
Richmond
*b
aDivision of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, 21 Nanyang Link, Singapore 637371. E-mail: chmlwk@ntu.edu.sg; Tel: +65 6592 7577
bDepartment of Chemistry, University of North Texas, Denton, TX 76203, USA
First published on 8th March 2016
Abstract
The kinetics for the isomerization of the 50e cluster Os3(μ-TeTol-p)2(CO)10 (3), where the tellurides bridge two different Os–Os edges, to one in which the tellurides bridge the same open Os⋯Os edge (4) have been measured experimentally by 1H NMR spectroscopy. The determined activation parameters are ΔH‡ = 77 ± 9 kJ mol−1 and ΔS‡ = −12 ± 28 J mol−1 K. The conversion of 3 to 4 has been computationally investigated by electronic structure calculations using the model compound Os3(μ-TeMe)2(CO)10. The computed isomerization pathway is consistent with the kinetic data and the thermodynamic preference for the product stereoisomer that possesses a slipped, eclipsed conformation for the two p-tolyl groups.
Introduction
An interesting aspect of organometallic cluster chemistry is the possibility of fluxionality and isomerization. These could involve the ligand sphere or the metal core. Such processes involving heavier main group elements are expected to be less facile, often because of unfavourable factors such as their higher atomic mass and high inversion barrier. Few studies on the kinetics of such processes exist, partly because of the lack of suitable systems.1 A system suitable for such studies is the isomeric pair of di-telluride substituted clusters 3-Ph and 4-Ph, with the molecular formula Os3(μ-TePh)2(CO)10, which were obtained from the reaction of Os3(CO)10(NCCH3)2 (1) with diphenyl ditelluride (2-Ph).2 To our knowledge, that remains the only report on osmium–tellurium carbonyl clusters, in contrast to the plethora of reports on the chemistry of the congeners sulfur and selenium, and of their neighbour in the periodic table, antimony.3 In that study, it was shown that 3-Ph isomerized to 4-Ph in refluxing cyclohexane (Scheme 1).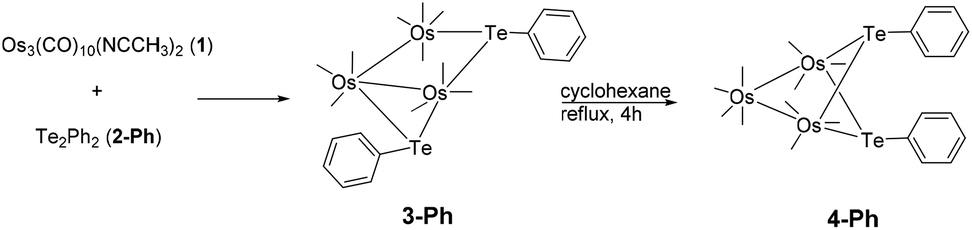 | ||
| Scheme 1 Synthesis and isomerization of Os3(μ-TePh)2(CO)10. | ||
Cluster 4-Ph adopts an unusual conformation in which the two phenyl rings are parallel to each other and stacked in a slipped, eclipsed orientation (A1). In principle, three different stereoisomers are possible, but the other two stereoisomers are not observed (Fig. 1). The important points that we wish to address in this report are: (i) the mechanism associated with the transformation of 3 to 4, and (ii) the relative stability of the three stereoisomers of 4 and their role, if any, in the isomerization reaction.
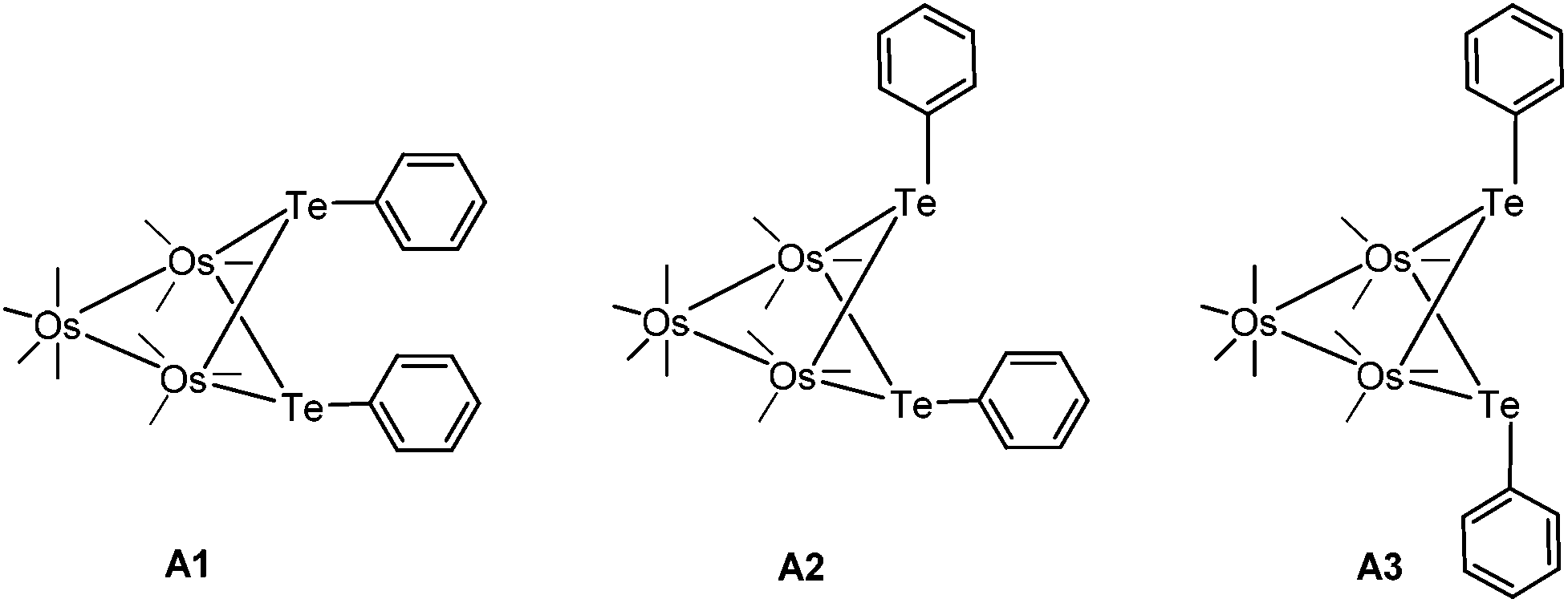 | ||
| Fig. 1 Different stereoisomers of 4–Ph. | ||
Results and discussion
In order to study the isomerization, we prepared the p-tolyl analogs Os3(μ-Te-p-tolyl)2(CO)10, 3-Tol-p and 4-Tol-p, using the same synthetic route previously reported;2 the singlet 1H resonances for the methyl groups would enable the straightforward monitoring of the isomerization by NMR spectroscopy. The structure of 4-Tol-p was also confirmed via a single-crystal X-ray diffraction study to have the tolyl groups arranged as in the isomer A1; the ORTEP plot depicting its molecular structure, together with selected bond parameters, is given in Fig. 2. A variable-temperature 1H NMR experiment conducted up to 90 °C showed no evidence of isomerization or the presence of any of the other isomers, thus suggesting that the stereoisomer A1 is thermodynamically the most stable.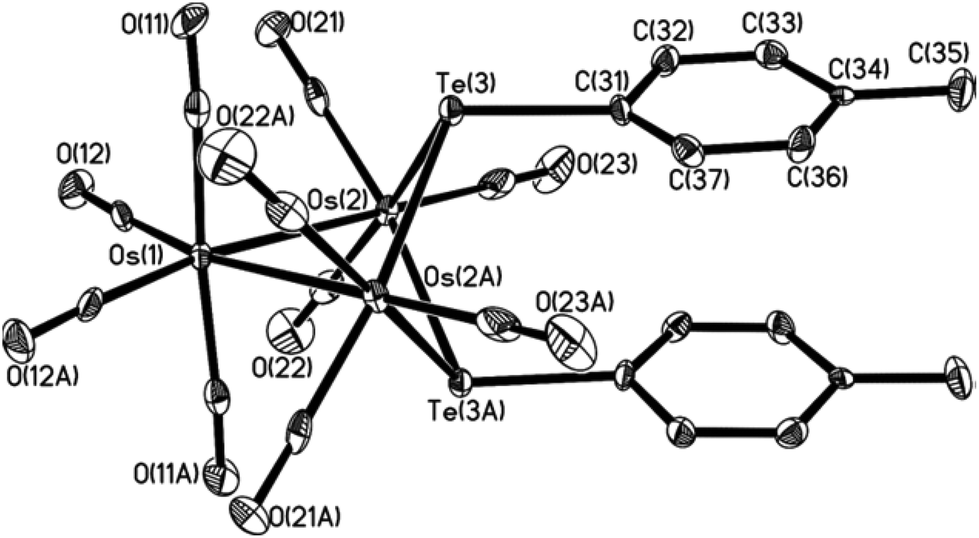 | ||
| Fig. 2 ORTEP diagram of cluster 4-Tol-p. Thermal ellipsoids are drawn at the 50% probability level. All H atoms have been omitted for clarity. Selected bond lengths (Å) and angles (°): Os1–Os2 = 2.9343(4); Os2–Te3 = 2.7330(6); Os2–Te3A = 2.7375(6); Os2⋯Os2A = 3.607; Os2–Te3–Os2A = 82.509(15); Os2–Os1–Os2A = 75.859(14). | ||
This is also supported by a computational study, using density functional theory (DFT), on the three possible isomers. To facilitate the computations and the subsequent study on the mechanism of the isomerization of 3 → 4, we replaced the p-tolyl groups in 4-Tol-p with methyl groups; the optimized structures are depicted in Fig. 3. The lowest energy isomer is A1 where the two methyl groups adopt an equatorial-like disposition with respect to the Os3 core and oriented similar to the aryl groups in the solid-state structures of clusters 4-Ph and 4-Tol-p. The bond distances and angles in the optimized structure of A1 are in good agreement with the solid-state structures of 4-Ph and 4-Tol-p. Isomer A2 lies 7.0 kJ mol−1 above A1, and the destabilization can be attributed to the proximity of the axial methyl group to the Os(CO)4 moiety. The energetic penalty for two axial methyl groups is severe, with A3 lying 40.5 kJ mol−1 above A1.
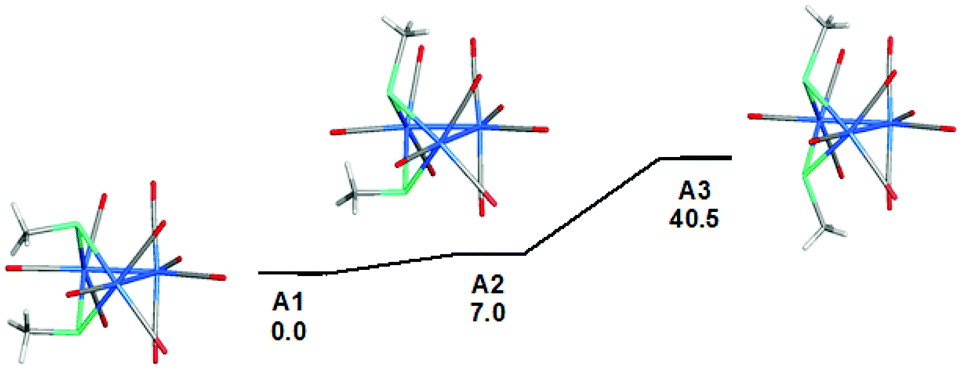 | ||
| Fig. 3 Geometry-optimized M06 structures and ground-state energy ordering for the stereoisomeric Os3(μ-TeMe)2(CO)10 clusters A1–A3. The reported free energies are in kJ mol−1 relative to A1. | ||
The charge distribution and bonding in species A1–A3 were also examined, and the data are consistent with our published results on related 50e clusters (Table S2†).4 In brief, the osmium atoms and the μ-TeMe ligands have opposing natural charges (∼−1.5 and 0.8, respectively). The formal Os–Os and Os–Te single bonds are characterized by Wiberg bond indices (WBI) of ∼0.41 and 0.76, respectively, while the values for the corresponding nonbonding vectors are ∼0.11 and 0.06, respectively. Elongation of the Os1–Os3 vector in A1–A3 follows from the Polyhedral Skeletal Electron Pair (PSEP) theory, the bonding in the cluster core being derived from seven skeletal electron pairs.5
The kinetics for the isomerization of 3-Tol-p to 4-Tol-p were followed by monitoring the methyl resonances in the 1H NMR spectrum; the former shows two distinct resonances at 2.379 and 2.362 ppm while the latter shows a single resonance at 2.289 ppm. The rate constants at five different temperatures were determined (Fig. 4), and an Eyring plot gave the kinetic parameters as ΔH‡ = 77 ± 9 kJ mol−1 and ΔS‡ = −12 ± 28 J mol−1 K, which corresponds to a ΔG‡ of 80 ± 17 kJ mol−1 at 298 K.
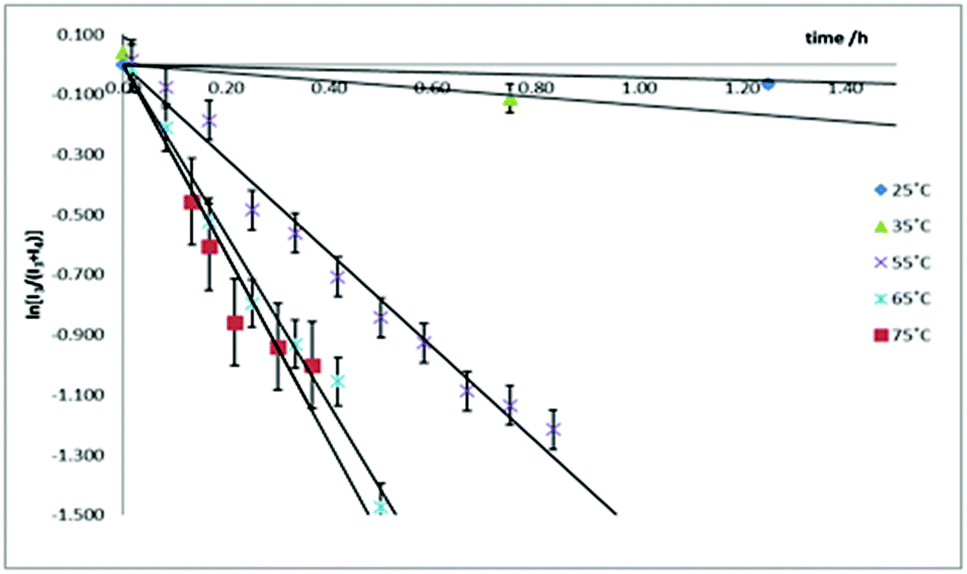 | ||
| Fig. 4 Plots of ln[I3/(I3 + I4)] against time at various temperatures. The two lowest temperature plots have been truncated, and all the plots shifted to a common origin. | ||
This isomerization reaction was investigated computationally with the methyl analog of cluster 3, viz. B. The geometry-optimized structure of B (Fig. S7†) is in good agreement with the experimentally determined structure of 3-Ph.2 The proposed pathway from B to the final product A1 is depicted in Scheme 2, together with the free energy change (in kJ mol−1) for each step; the potential energy surface for the isomerization is depicted in Fig. 5.
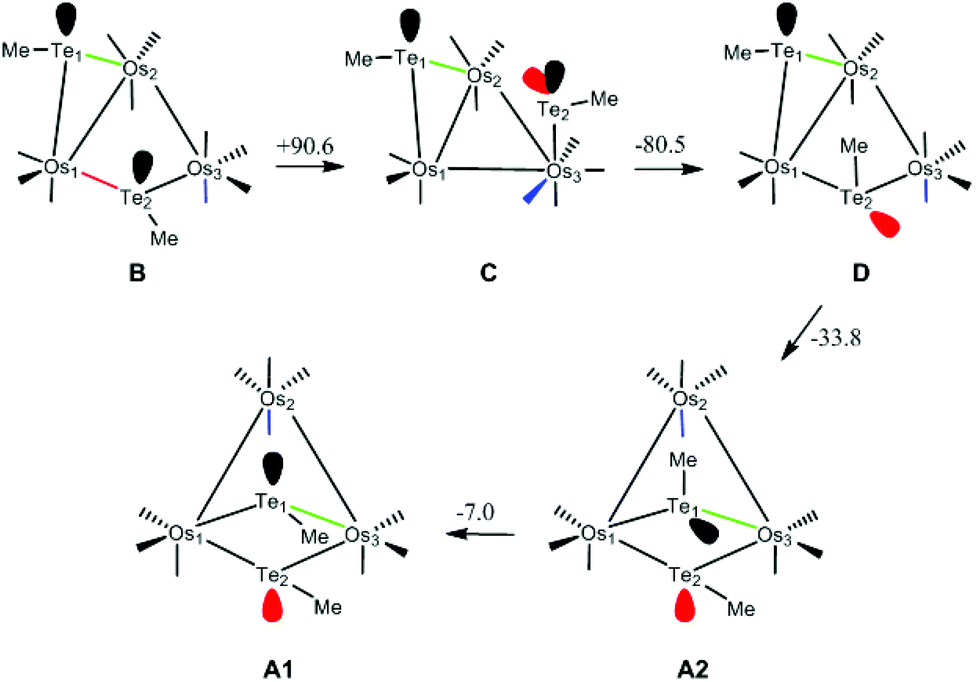 | ||
| Scheme 2 Isomerization of B to A1 with atom labels and the stereochemically active lone pair(s) at each tellurium centre highlighted. Energy values given for each step are ΔG (kJ mol−1). | ||
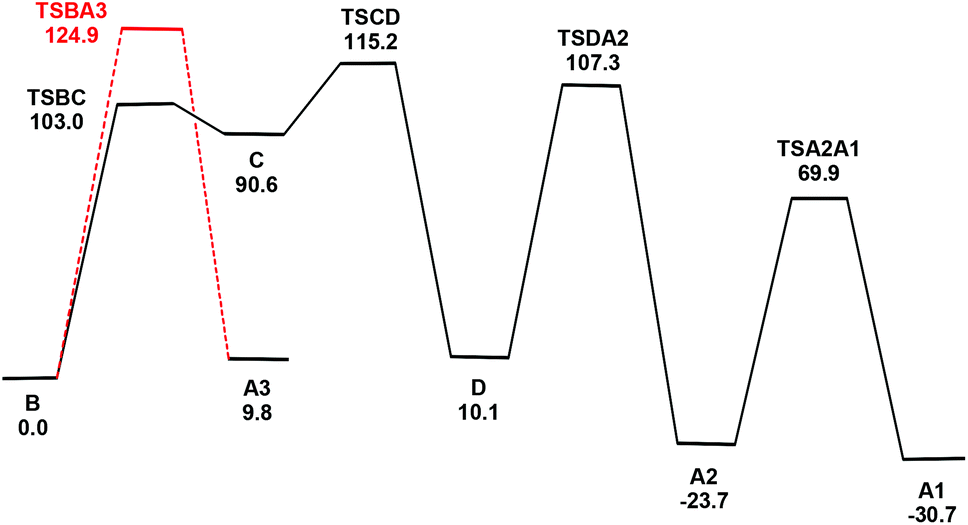 | ||
| Fig. 5 Potential energy surface for the isomerization of B to A1 and the conversion of B to A3. Energy values are ΔG in kJ mol−1 with respect to B. | ||
The first step involves the conversion of B to an isomer Dvia the intermediate species C which contains an η1-TeMe group. While the bridging Te2 moiety that spans the non-bonded Os1⋯Os3 vector in B contains a single stereochemically-active lone pair, the tellurium centre in the η1-TeMe group in C possesses two stereochemically-active lone pairs. Species C lies 90.6 kJ mol−1 above B, and points to the thermodynamic preference of a bridging TeMe moiety over the η1 mode.6 The transition state TSBC for the conversion from B to C has been located and lies 103.0 kJ mol−1 above B; the internuclear distance between the non-bonded Os1 and Os3 atoms in B contracts by 1.054 Å as a bond develops between them.
Rotation of the η1-TeMe ligand in C, coupled with expansion of the adjacent Os1–Os2 bond and reformation of the μ-TeMe ligand using the original lone pair on Te2 in B, furnishes D. This transformation corresponds formally to an exchange of the electron pair in the Os1–Te2 bond with the lone pair on the Te2 centre.7 The transition state TSCD for this step lies 115.2 kJ mol−1 above B and represents the rate-limiting step for the overall process. The stereoisomers B and D differ in the disposition of their methyl groups. The methyl group on the Te2 centre in D is in close contact with the Te1 centre and the adjacent axial CO ligand on the Os(CO)4 moiety. Accordingly, D lies 10.1 kJ mol−1 above B. We have also checked the possible inversion at the Te2 centre that would give D directly from B; this leads to an increase in energy above 165 kJ mol−1 and without a feasible transition state. This rise in energy can be attributed to the eclipsing interactions between the methyl group and the two adjacent equatorial CO groups as the former migrates to its final orientation in D.
The conversion of D to A2 proceeds via a merry-go-round of five CO groups and the μ-Te1 ligand, with the latter migrating across the non-tellurium-ligated Os2–Os3 vector (Scheme 3).8 The optimized structure of the transition state TSDA2, which lies 107.3 kJ mol−1 above B, clearly shows the presence of the capping μ3-Te1 and bridging μ2-CO groups; the Wiberg bond indices for the Os1–Te1 (0.79), Os2–Te1 (0.44), and Os3–Te1 (0.50) vectors are consistent with the face-capping TeMe moiety.
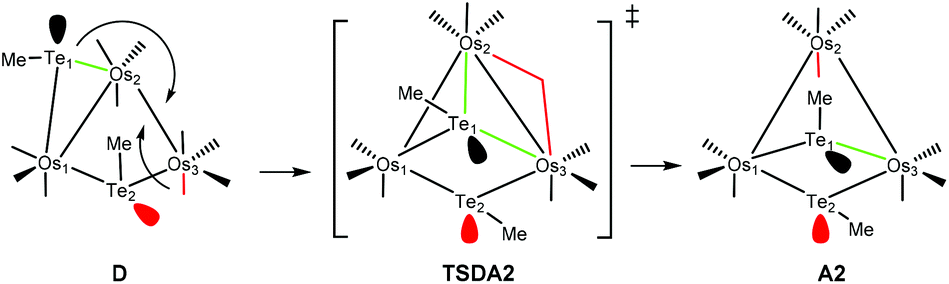 | ||
| Scheme 3 Conrotation of the CO and Te1 groups about the Os2–Os3 vector in D through the transition state TSDA2 to furnish A2. | ||
The final step in the reaction involves a formal inversion of the Te1 centre in A2, through the transition state TSA2A1. The energy barrier for this inversion is relatively low, lying only 46.2 kJ mol−1 above A2. The sum of the angles around the Te1 (ca. 360°) and Te2 (ca. 293°) centres in TSA2A1 are consistent with the inversion at Te1. The overall reaction of B → A1 is exergonic by 30.7 kJ mol−1.
Migration of the Te1 moiety across the Os2–Os3 bond in B to give A3 was also examined. This process, which involves a merry-go-round of CO and the Te1 ligands, is analogous to that for the D → A2 transformation. The transition state TSBA3 for this presents a barrier of 124.9 kJ mol−1, which is 9.7 kJ mol−1 higher than the rate-limiting step (TSCD) in the four-step pathway outlined above. While not evaluated computationally due to the unfavorable thermodynamics associated with TSBA3, a stepwise inversion sequence at each tellurium centre in A3 is expected to yield A2 and subsequently A1.
Conclusions
In this work, we have investigated the polyhedral rearrangement attendant in the conversion of cluster 3 to 4 by NMR spectroscopy and determined the activation parameters for the process. The reaction pathway has been examined computationally with the model cluster Os3(μ-TeMe)2(CO)10, and the rate-limiting step is shown to involve a merry-go-round in which a μ-TeMe ligand migrates across an Os–Os bond to an open Os⋯Os edge in the cluster, coupled with the permutation of five CO ligands around the same Os–Os bond.Experimental
All reactions and manipulations were carried out under an argon atmosphere using standard Schlenk techniques. All solvents employed in this study were distilled over an appropriate drying agent under argon before use. Infrared (IR) spectra were recorded in a solution IR cell with NaCl windows and a path length of 0.1 mm, at a resolution of 2 cm−1. 1H NMR spectra were recorded on a Bruker AV300 at 300 MHz while variable temperature 1H NMR spectra were recorded on a JEOL ECA 400 at 400 MHz, referenced to the residual proton resonance of the solvent. Compounds 1 and 2 were synthesized according to literature methods.2,9Kinetics measurements
Cluster 3-Tol-p (8 mg, 0.0062 mmol) was dissolved in C6D6 and 1,3,5-trimethoxybenzene was added as an internal standard. The solutions were placed in an oil bath set at various temperatures (25 °C, 35 °C, 55 °C or 75 °C) and the conversion of 3-Tol-p was monitored via1H NMR spectroscopy. The mass balance was checked by plotting (I3 + I4) against the reaction time (t), where I3 and I4 are the scaled, integrated intensities of 3-Tol-p and 4-Tol-p, respectively. The rate constant (k) at each temperature was obtained from a plot of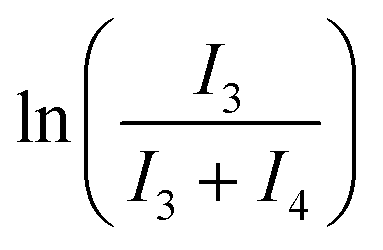 against t. An Eyring plot was then obtained to obtain ΔH‡ and ΔS‡.
against t. An Eyring plot was then obtained to obtain ΔH‡ and ΔS‡.
X-ray crystal structure determination
A crystal of 4-Tol-p was mounted on a quartz fiber. X-ray data were collected on a Bruker AXS APEX system, using Mo Kα radiation, with the SMART suite of programs.10 The data were processed and corrected for Lorentz and polarization effects with SAINT,11 and for absorption effects with SADABS.12 Structural solution and refinement were carried out with the SHELXTL suite of programs.13 The structure was solved by direct methods, followed by difference maps to complete the structure for all the non-hydrogen atoms. The structure was found to be a racemic twin. Organic hydrogen atoms were placed in the calculated positions and refined with a riding model. All non-hydrogen atoms were generally given anisotropic displacement parameters in the final model.Computational studies
All DFT calculations were carried out with the Gaussian 09 package of programs,14 using M06 as the DFT functional.15 The osmium and tellurium atoms were described with the Stuttgart–Dresden effective core potential and SDD basis set,16 and the 6-31G(d′) basis set was employed for all the remaining atoms.17All the reported geometries were fully optimized, and analytical second derivatives were evaluated at each stationary point to verify whether the geometry was an energy minimum (positive eigenvalues) or a transition structure (one negative eigenvalue). Unscaled vibrational frequencies were used to make zero-point and thermal corrections to the electronic energies. The resulting free energies are reported in kJ mol−1 relative to the specified standard. All transition states on the potential energy surface were evaluated by intrinsic reaction coordinate (IRC) calculations, in order to establish the reactant and product species associated with each transition-state structure. The natural charges and Wiberg bond indices were computed using Weinhold's natural bond orbital (NBO) program, as executed by Gaussian 09.18 Geometry-optimized structures have been drawn with the JIMP2 molecular visualization and manipulation program.19
Acknowledgements
This work was supported by Nanyang Technological University, the Ministry of Education (Research Grant No. M4011158), and the Robert A. Welch Foundation (Grant B-1093-MGR). Computational resources through the High-Performance Computing Services and CASCaM at the University of North Texas are acknowledged. We thank Dr Rakesh Ganguly for assistance with the X-ray crystallographic data collection and processing and Prof. Michael B. Hall (TAMU) for providing us a copy of his JIMP2 program, which was used to prepare the geometry-optimized structures reported here.Notes and references
- Some kinetic studies carried out on osmium clusters: (a) W. H. Watson, B. Poola and M. G. Richmond, J. Organomet. Chem., 2006, 691, 4676–4685 CrossRef CAS; (b) L. J. Pereira and W. K. Leong, J. Organomet. Chem., 2006, 691, 1941–1944 CrossRef CAS; (c) J. Cooke and J. Takats, Organometallics, 1995, 14, 698–702 CrossRef CAS; (d) J. B. Keister, U. Frey, D. Zbinden and A. E. Merbach, Organometallics, 1991, 10, 1497–1501 CrossRef CAS; (e) L. J. Farrugia, Organometallics, 1989, 8, 2410–2417 CrossRef CAS; (f) S. J. Heyes, C. M. Dobson, M. A. Gallop, B. F. G. Johnson and J. Lewis, Inorg. Chem., 1991, 30, 3850–3856 CrossRef CAS; (g) R. D. Adams, M. P. Pompeo and W. Wu, Inorg. Chem., 1991, 30, 2899–2905 CrossRef CAS.
- J. Zhang and W. K. Leong, J. Chem. Soc., Dalton Trans., 2000, 1249 RSC.
- Reports on osmium–sulfur, selenium, and antimony clusters: (a) O. Reyes-López, S. A. Sánchez-Ruiz, A. Flores-Parra, M. A. Leyva, R. J. Alvarez-Méndez, A. Vela and M. J. Rosales-Hoz, J. Organomet. Chem., 2014, 772–773, 248–257 CrossRef; (b) R. D. Adams and J. A. Belinski, Organometallics, 1992, 11, 2488–2493 CrossRef CAS; (c) B. F. G. Johnson, J. Lewis, P. G. Lodge, P. R. Raithby, K. Henrick and M. McPartlin, J. Chem. Soc., Chem. Commun., 1979, 719–720 RSC; (d) J. Arce, A. Karam, Y. De Sanctis, R. Machado, M. V. Capparelli and J. Manzur, Inorg. Chim. Acta, 1997, 254, 119–130 CrossRef; (e) J. Arce, R. Machado, C. Rivas, Y. De Sanctis and A. J. Deeming, J. Organomet. Chem., 1991, 419, 63–75 CrossRef.
- (a) E. C. Morrison, W. K. Leong and J. Tan, J. Cluster Sci., 2007, 18, 753–763 CrossRef CAS; (b) J. C. Sarker, K. M. Uddin, M. S. Rahman, S. Ghosh, T. A. Siddiquee, D. A. Tocher, M. G. Richmond, G. Hogarth and S. E. Kabir, Inorg. Chim. Acta, 2014, 409, 320–329 CrossRef CAS; (c) L. Yang, V. N. Nesterov, X. Wang and M. G. Richmond, J. Cluster Sci., 2014, 26, 93–109 CrossRef.
- D. M. P. Mingos and D. J. Wales, Introduction to Cluster Chemistry, Prentice Hall, Englewood Cliffs, NJ, 1990 Search PubMed.
- Preference for μ-TeR over η1-TeR moieties in polynuclear clusters is strengthened by the numerous structural examples of the former coordination mode extant in the current Cambridge Crystallographic Database (CSD 5.36). To our knowledge, no examples exist for a polynuclear cluster with a pendant η1-TeR ligand(s).
- For reports on the related terminal-bridge-terminal exchange behaviour of ancillary sulfides (SR2) at polynuclear clusters, see: (a) R. D. Adams, B. Captain, W. Fu and P. J. Pellechia, Inorg. Chem., 2003, 42, 3111–3118 CrossRef CAS PubMed; (b) D. A. Hrovat, E. Nordlander and M. G. Richmond, Organometallics, 2012, 31, 6608–6613 CrossRef CAS.
- X. Zhang, S. Kandala, L. Yang, W. H. Watson, X. Wang, D. A. Hrovat, W. T. Borden and M. G. Richmond, Organometallics, 2011, 30, 1253–1268 CrossRef CAS.
- (a) J. N. Nicholls, M. D. Vargas, A. J. Deeming and S. E. Kabir, in Inorganic Syntheses, John Wiley & Sons, Inc., 1990, ch. 58, pp. 232–235 Search PubMed; (b) N. Petragnani and H. A. Stefani, in Tellurium in Organic Synthesis (Second Edition), ed. N. P. A. Stefani, Academic Press, London, 2007, pp. 9–113 Search PubMed.
- SMART version 5.628, Bruker AXS Inc., Madison, Wisconsin, USA, 2001 Search PubMed.
- SAINT+ version 6.22a, Bruker AXS Inc., Madison, Wisconsin, USA, 2001 Search PubMed.
- G. M. Sheldrick, SADABS, 1996 Search PubMed.
- SHELXTL version 5.1, Bruker AXS Inc., Madison, Wisconsin, USA, 1997 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- (a) Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed; (b) Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2007, 120, 215–241 CrossRef.
- (a) M. Dolg, U. Wedig, H. Stoll and H. Preuss, J. Chem. Phys., 1987, 86, 866–872 CrossRef CAS; (b) S. P. Walch and C. W. Bauschlicher, J. Chem. Phys., 1983, 78, 4597–4605 CrossRef CAS.
- (a) G. A. Petersson and M. A. Al-Laham, J. Chem. Phys., 1991, 94, 6081–6090 CrossRef CAS; (b) G. A. Petersson, A. Bennett, T. G. Tensfeldt, M. A. Al-Laham, W. A. Shirley and J. Mantzaris, J. Chem. Phys., 1988, 89, 2193–2218 CrossRef CAS.
- (a) A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS; (b) K. B. Wiberg, Tetrahedron, 1968, 24, 1083–1096 CrossRef CAS.
- JIMP2, version 0.091, a free program for the visualization and manipulation of molecules: (a) M. B. Hall and R. F. Fenske, Inorg. Chem., 1972, 11, 768–775 CrossRef CAS; (b) J. Manson, C. E. Webster and M. B. Hall, Texas A&M University, College Station, TX, 2006, http://www.chem.tamu.edu/jimp2/index.html.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1421965 for 4-Tol-p. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt00588h |
| This journal is © The Royal Society of Chemistry 2016 |