
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnique anisotropic optical properties of a highly stable metal–organic framework based on trinuclear iron(III) secondary building units linked by tetracarboxylic linkers with an anthracene core†
A. V.
Vinogradov
*a,
V. A.
Milichko
a,
H.
Zaake-Hertling
b,
A.
Aleksovska
b,
S.
Gruschinski
b,
S.
Schmorl
b,
B.
Kersting
b,
E. M.
Zolnhofer
c,
J.
Sutter
c,
K.
Meyer
c,
P.
Lönnecke
b and
E.
Hey-Hawkins
*b
aITMO University, St. Petersburg, 197101, Russian Federation. E-mail: vinogradoffs@gmail.com
bLeipzig University, Faculty of Chemistry and Mineralogy, Institute of Inorganic Chemistry, D-04103 Leipzig, Germany. E-mail: hey@uni-leipzig.de
cFriedrich-Alexander University Erlangen – Nürnberg (FAU), Department of Chemistry & Pharmacy, Inorganic Chemistry, D-91058 Erlangen, Germany
First published on 12th February 2016
Abstract
A highly stable metal–organic framework, [{Fe3(ACTBA)2}X·6DEF]n (1; X = monoanion), based on trinuclear iron(III) secondary building units connected by tetracarboxylates with an anthracene core, 2,6,9,10-tetrakis(p-carboxylatophenyl)anthracene (ACTBA), is reported. Depending on the direction of light polarisation, crystals of 1 exhibit anisotropic optical properties with birefringence Δn = 0.3 (λ = 590 nm).
Introduction
Metal–organic frameworks (MOFs) are an attractive class of porous solids with potential applications in gas storage and separation, energy conversion and catalysis.1–5 All of these versatile physical and chemical properties or niche applications have been shown to be heavily reliant on their unique structures. On the other hand, Fe-based MOFs have attracted wide scientific attention owing to their low toxicity and magnetic properties, medical and biological functions (drug release, imaging) and application in lithium-ion batteries and hydrocarbon separations.6–8 However, most recent research has concentrated on the exploration of Cu- and Zn-based MOFs,1a while studies on Fe-based MOFs are relatively scarce.5 The main reason is the difficulty in controlling methodology and experimental conditions, as iron salts exhibit a strong tendency to undergo hydrolysis resulting in stable hydrated iron oxide and precipitation instead of crystallisation.9The research presented here is focused on a tetratopic anthracene-based ligand, namely, 2,6,9,10-tetrakis(p-carboxylatophenyl)anthracene (referred to as anthracene tetrabenzoate (ACTBA), Fig. 1), as conjugated or fused aromatic compounds may exhibit interesting optical features, acting as charge donors or acceptors. Furthermore, with iron ions, such compounds are capable of exhibiting unique optical properties due to strong spatial anisotropy on the nanoscale, as was discovered for the first time in this class of materials described here. This feature opens up broad prospects for the use of MOF single crystals in managing photoexcitation under the influence of light.10
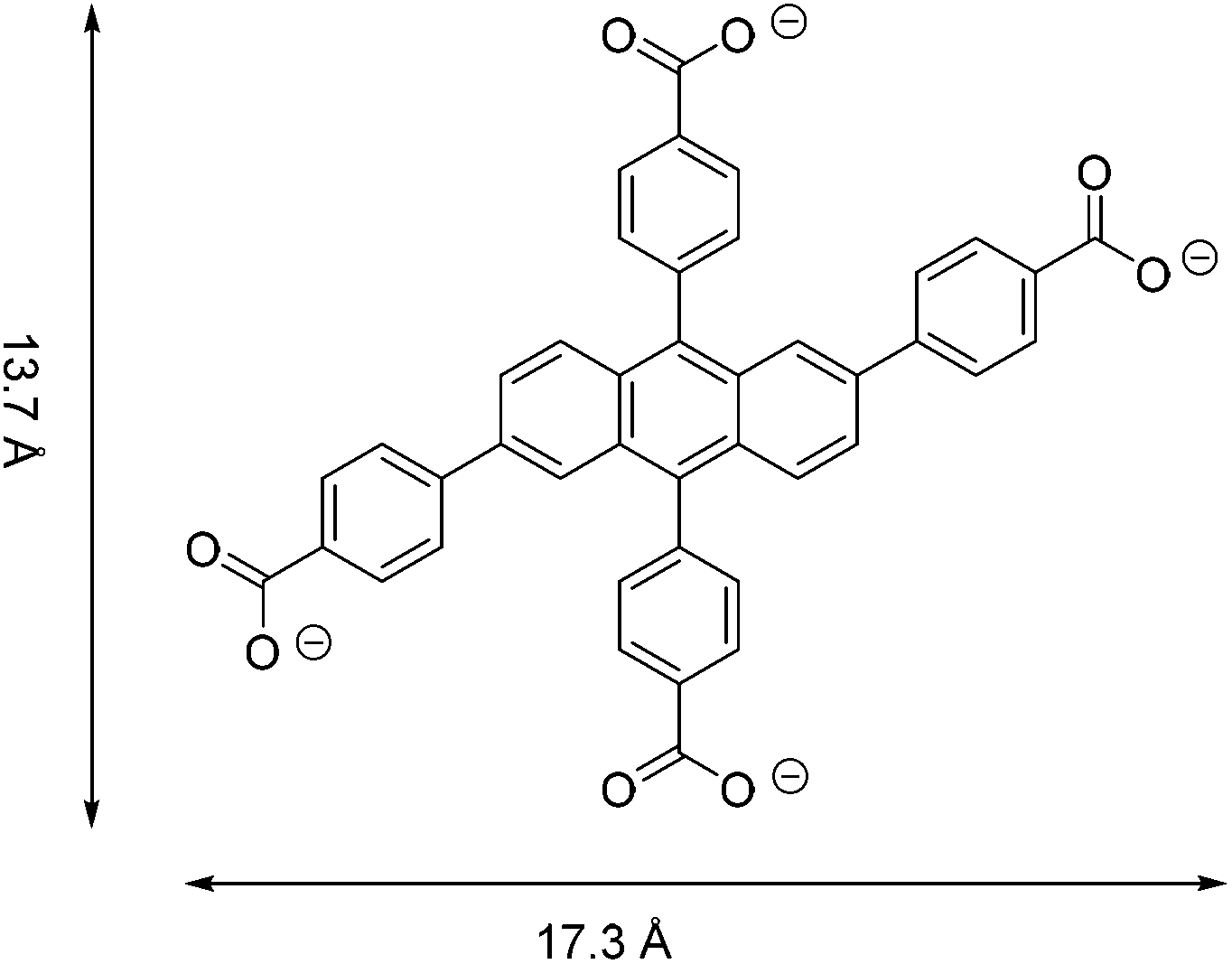 | ||
| Fig. 1 The tetracarboxylate linker, ACTBA. | ||
Results and discussion
Single crystals of [{Fe3(ACTBA)2}X·6DEF]n (1; X = monoanion) were obtained by solvothermal synthesis from FeCl3·6H2O and the corresponding acid of the linker, H4-ACTBA, in diethylformamide (DEF). The colour of the crystals ranges from dark yellow to brown, depending on their thickness and the angle of incidence of a light beam. Compound 1 crystallises in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (see ESI, Table S1†) forming a porous network consisting of Fe3(RCO2)8 as a secondary building unit (Fig. 2).
(see ESI, Table S1†) forming a porous network consisting of Fe3(RCO2)8 as a secondary building unit (Fig. 2).
 | ||
| Fig. 2 The SBU Fe3(RCO2)8. | ||
The central iron atom (Fe2) is located on a crystallographic inversion centre and exhibits perfect octahedral coordination, whereas the coordination environment of Fe1/Fe1′ is best described as distorted tetrahedral if the two chelating carboxylato groups are regarded as occupying one coordination site. The eight carboxylato groups belong to eight separate ACTBA linkers and exhibit different bonding modes: four are μ2,κ1:κ1 (between Fe1 or Fe1′ and Fe2), two are μ2,κ2:κ1 (κ2 at Fe1 or Fe1′ and κ1 at Fe2), and two are κ2 (at Fe1 and Fe1′). With respect to the planar anthracene core, the four phenylene substituents exhibit different angles between the ring planes, which range from 40.0 to 73.8° (see ESI, Fig. S11 and Table S2†). The resulting structure (Fig. 3) is highly porous and exhibits channels along the a, b and c axes (Fig. 4) as well as in other directions (see ESI, Fig. S4†). Viewed along the a axis, two different pores are observed, labelled A1 (ca. 6.4 × 4.0 Å) and A2 (ca. 5.8 × 4.9 Å); the largest pores are observed along the b axis (B, ca. 12.0 × 7.5 Å), and the smallest along the c axis (C, ca. 7.2 × 2.6 Å) (Fig. 4; the pore dimensions were obtained from space-filling models based on van der Waals radii).
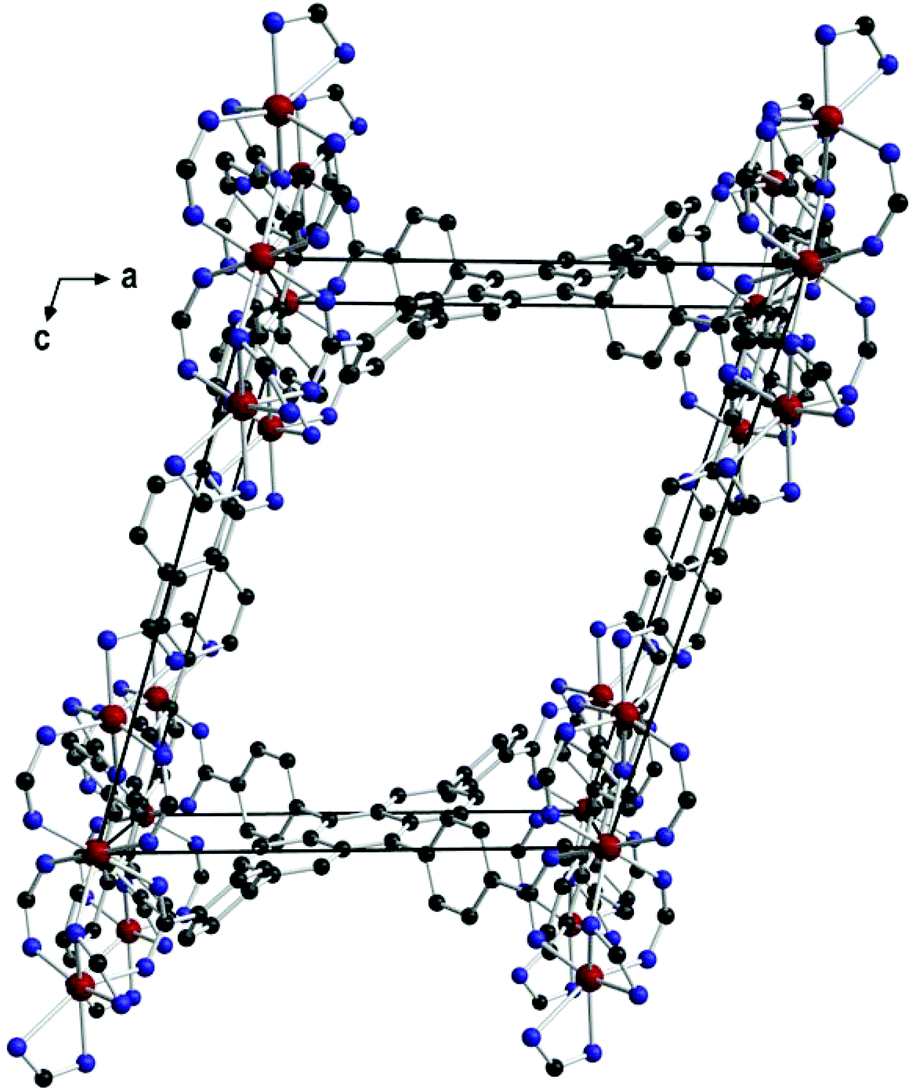 | ||
| Fig. 3 View along the b axis in 1 showing one pore (B) and the connectivity of the linkers. | ||
 | ||
| Fig. 4 Views along the a, b and c axes in 1 showing the different pores. | ||
All attempts to locate solvent molecules inside the pores failed due to their high disorder. Therefore, these molecules were removed with the SQUEEZE routine implemented in PLATON; the electron count suggests the presence of six poorly defined DEF molecules in the unit cell. Furthermore, as magnetic measurements and Mößbauer spectroscopy indicate the presence of three FeIII cations, an anion X [possibly hydroxide or formate (from decomposition of DEF) besides 10% chloride] must be present for charge neutrality.
TG-DTA analysis was carried out in the range of 30–700 °C and showed 1 to be stable up to 350 °C (see ESI, Fig. S9†). The thermal stability is comparable to that of MIL-53,11 whereby the high stability may be due to the high valence of the metal ions and the connectivity of the SBU rods.12,13 Also, the recently reported MOFs ROD-6 and ROD-7, based on Mn or In and 1,3,6,8-tetrakis(p-carboxylatophenyl)pyrene as linker, exhibit similar stability.12
The permanent porosity of an activated sample was confirmed by the N2 sorption isotherm at 77 K (see ESI, Fig. S2†). The microporosity was assessed by using the Langmuir and BET theories for surface area analysis and the Dubinin–Astakhov (DA) method for pore size distribution (see ESI, Fig. S3†). The BET and Langmuir surface areas are 554 and 715 m2 g−1, respectively, which were calculated from the nitrogen physisorption data in the relative pressure range p/p0 = 0.06–0.30. The total pore volume of 1 based on this method is 0.164 cm3 g−1 associated with an average pore radius of 5.2 Å (DA). The data obtained are in accord with the pore volumes calculated by using the program package Platon.14 However, in the structure determination, the SQUEEZE analysis revealed a total volume of 1.978 nm3 for the solvent-accessible area of the unit cell. This corresponds to a pore volume of 0.56 cm3 g−1, which is 3.5 times higher than the value that was deduced from the Barrett–Joyner–Halenda (BJH) analysis.
In order to understand the electronic structure of the trinuclear SBU in 1, microcrystalline samples were studied by VT-SQUID, VT-CW X-band EPR and zero-field 57Fe Mößbauer spectroscopy at 77 K (see ESI, Fig. S6†). The SQUID magnetisation measurement (see ESI, Fig. S5†), recorded in the temperature range 2–300 K, revealed a reproducible, strongly temperature-dependent magnetic moment over the entire temperature range. The magnetic moment at 2 K was determined to be 3.9 μB, and thus indicates an Stot = 3/2 ground state for the SBU. With increasing temperature, the magnetic moment increases steadily to give a value of approximately 11 μB at room temperature. The zero-field Mößbauer spectrum of 1, recorded at 77 K (see ESI, Fig. S6†), shows one symmetric quadrupole doublet with an isomer shift δ of 0.52 mm s−1 and a quadrupole splitting ΔEQ of 0.70 mm s−1. These Mößbauer parameters are characteristic for high-spin iron(III) ions (Fe3+, d5, S = 5/2) in an O-donor ligand environment. However, a single Mößbauer feature appears to be inconclusive and not in agreement with the trinuclear SBU, as the Fe centres in 1 have significantly different ligand environments. The outer Fe ions (Fe1, Fe1′) are best described as being situated in a tetrahedral coordination sphere, while the central ion (Fe2) is clearly octahedrally coordinated. Accordingly, one would expect two Mößbauer signals with an intensity ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. In principle, if both Fe sites were high-spin S = 5/2, they could have similar Mößbauer parameters. With that said, the observation of a relatively sharp (ΓFWHM = 0.57 mm s−1) and symmetric doublet still would not be anticipated. In addition, the formulation of three magnetically coupled high-spin Fe ions cannot result in an electronic ground state Stot of 3/2, as suggested by SQUID magnetisation. An EPR spectroscopic study remains equally inconclusive. CW X-band EPR spectra of two batches, recorded at 8 and 6 K (see ESI, Fig. S7 and S8†), show very similar features with identical g values. A signal at g = 8.47 and 4.13 is indicative for an S = 5/2 ion, for which the expected high-field third resonance is broadened into the baseline or hidden underneath the second, rhombic signal centred at g = 1.81, 2.00 and 2.18. Although these signals are remarkably reproducible, their intensities are not and, furthermore, they are not in the expected integer ratio of 2:1 to each other. Accordingly, a temperature-dependent EPR study (6–290 K, see ESI, Fig. S8†) was carried out. Clearly, with increasing temperature, the low-field feature at g ≈ 8.5 and 4 loses signal intensity, while the resonance at g ≈ 2 shifts to slightly lower fields, resulting in a single, broad resonance centred at g = 2.64, 1.97 and 1.90. The coalescence temperature of about 80–100 K could explain the single quadrupole doublet observed in the 57Fe Mößbauer spectrum (recorded at 77 K). In conclusion, the magnetochemical study (see ESI†) suggests that the electronic structure of 1 cannot be described as an electronically isolated, trinuclear, molecular Fe moiety. Instead, the spectroscopic data suggest that the SBUs engage in intermolecular interactions, most likely in addition to intramolecular magnetic superexchange. Accordingly, a simple interpretation of the electronic structure of supramolecular 1 on a molecular level (the SBU) is severely hampered by spin–spin interactions, as is most obviously seen in the VT EPR study. This leads to complicated magnetic phenomena, detailed interpretation of which is beyond the scope of this work.
:1. In principle, if both Fe sites were high-spin S = 5/2, they could have similar Mößbauer parameters. With that said, the observation of a relatively sharp (ΓFWHM = 0.57 mm s−1) and symmetric doublet still would not be anticipated. In addition, the formulation of three magnetically coupled high-spin Fe ions cannot result in an electronic ground state Stot of 3/2, as suggested by SQUID magnetisation. An EPR spectroscopic study remains equally inconclusive. CW X-band EPR spectra of two batches, recorded at 8 and 6 K (see ESI, Fig. S7 and S8†), show very similar features with identical g values. A signal at g = 8.47 and 4.13 is indicative for an S = 5/2 ion, for which the expected high-field third resonance is broadened into the baseline or hidden underneath the second, rhombic signal centred at g = 1.81, 2.00 and 2.18. Although these signals are remarkably reproducible, their intensities are not and, furthermore, they are not in the expected integer ratio of 2:1 to each other. Accordingly, a temperature-dependent EPR study (6–290 K, see ESI, Fig. S8†) was carried out. Clearly, with increasing temperature, the low-field feature at g ≈ 8.5 and 4 loses signal intensity, while the resonance at g ≈ 2 shifts to slightly lower fields, resulting in a single, broad resonance centred at g = 2.64, 1.97 and 1.90. The coalescence temperature of about 80–100 K could explain the single quadrupole doublet observed in the 57Fe Mößbauer spectrum (recorded at 77 K). In conclusion, the magnetochemical study (see ESI†) suggests that the electronic structure of 1 cannot be described as an electronically isolated, trinuclear, molecular Fe moiety. Instead, the spectroscopic data suggest that the SBUs engage in intermolecular interactions, most likely in addition to intramolecular magnetic superexchange. Accordingly, a simple interpretation of the electronic structure of supramolecular 1 on a molecular level (the SBU) is severely hampered by spin–spin interactions, as is most obviously seen in the VT EPR study. This leads to complicated magnetic phenomena, detailed interpretation of which is beyond the scope of this work.
From a physical point of view, a central feature of the crystal structure of 1 are the ordered FeIII cations forming atomically thin metal-ion chains and defined nanoscale channels. Objects with such structural features have strongly anisotropic physical properties,15 for instance, optical or conductive properties. At best, they may have unique metamaterial properties16a due to the combination of metallic (Fe chains) and dielectric elements (channels) in a single crystal. The latter is enhanced by the fact that all single elements are nanoscale and can be considered as a quantum system. In turn, this is essential for quantum metamaterials.16b Basically, classical optical studies on MOF conglomerates and arrays strongly dominate over specific studies on single crystals of MOFs, which are mostly dedicated to luminescence.17 Therefore, studies of the anisotropy of the optical properties of 1 were carried out. The transmission/reflection spectra (experimental setup described in ESI, Fig. S10†) obtained for a single crystal at two orthogonal polarisations (E1 and E2) were normalised by the spectra acquired under the same conditions from the slide in DEF (Fig. 5). Here, the transmission spectra T were recalculated to optical densities OD (OD = −lgT), which in turn can be considered as characteristic of absorption A of radiation in a crystal of length L (A = 2.3 OD/L).
 | ||
| Fig. 5 Reflectance spectra and optical density of a single crystal of 1 irradiated perpendicular to its surface. Red and blue curves correspond to reflection/absorption of electromagnetic waves with electric vectors E which are perpendicular (E1) or parallel (E2) to the longest side of the crystal, as shown in the inset. The oscillations in the reflectance spectra indicate interference in thin films, and the calculated optical thickness of the crystal shown is 10 μm. The slope of the optical density curves supports different band gaps of 1 depending on light polarisation (2.7 eV for E1 and 2.4 eV for E2). | ||
For single crystals of 1 it can be concluded that the different amplitudes and shapes of the reflectance/absorption spectra for light with orthogonal polarisations (E1 and E2) indicate a strong anisotropy of the optical properties (Fig. 5): (i) different band gap for light with orthogonal polarisation (2.7 eV versus 2.4 eV), and (ii) anisotropy of the refractive index n. The refractive indices of 1 in the case of normal incidence of light on a homogeneous dielectric film (i.e., single crystal of 1)18 can be obtained from the following expression:
| R = [(n1n2 − n2)/(n1n2+n2)]2 |
Also a higher value of birefringence can be achieved in nanostructured semiconductors22 or liquid crystals.23 However, the former is valid for the IR range due to small band gaps, and the latter has a restriction on ultrafast operation.
Polarised transmittance and reflectance spectroscopy was carried out for 13 single crystals of 1 with lateral dimension from 2 to 20 μm showing consistent anisotropic optical properties. The observed optical anisotropy directly results from the crystal structure of 1. Due to the charge-transfer properties of the anthracene derivative (ACTBA)24 and metal-ion chains ordered in different spatial directions, crystals with a structural period of less than 1 nm have anisotropic permittivity and metallic features in the visible spectral range. As a result, the interaction between polarised optical radiation and a single crystal of 1 results in the above-mentioned properties.
Conclusions
In conclusion, the novel highly porous MOF reported here based on a tetracarboxylic linker with an anthracene core (ACTBA) and an Fe3(RCO2)8 SBU reveals giant optical anisotropy arising from metal-ion chains ordered in different spatial directions allowing the single crystals to change their reflectance/absorption depending on the direction of the polarisation vector. We expect that this study, performed for the first time on individual microcrystals, will motivate investigations of other anisotropic properties of single crystals of MOFs and will thus open up unprecedented new applications.Experimental
General methods
:1) were added via cannula to a mixture of 2,6,9,10-tetrabromoanthracene (1 eq.), boronic acid (in total 5.8 to 6.6 eq. of 1-COOMe-4-B(OH)2-C6H4, starting with 3 eq. and adding additional portions of 1.2 eq. after 3, 6 and 9 days), base (7.5 eq. Na2CO3) and catalyst ([Pd(PPh3)4], 1.5 mol%) under nitrogen. The mixture was refluxed for 10–12 days. Additional boronic acid was added after every third day. Solvent was removed on a rotary evaporator. Purification can be achieved by column chromatography using n-hexane/ethyl acetate (8:1) with a rising gradient of ethyl acetate as eluent on silica. Me4-ACTBA is soluble in warm and polar organic solvents as well as in aromatic solvents. M.p.: decomp. without melting. Analytical data for Me4-ACTBA: 1H-NMR (CDCl3, δ in ppm): 8.34 (d, 8.2 Hz, 4 H, presumably pos. 3′/5′ or 3′′/5′′), 8.07 (d, 8.2 Hz, 4 H, presumably pos. 3′/5′ or 3′′/5′′), 7.87 (d, ca. 1.3 Hz, 2 H, 1,5-pos.), 7.74 (d, ca. 9.1 Hz, 2 H, 4,8-pos.), 7.67–7.64 (m/overlap, 6 H, presumably pos. 3,7 and 2′/6′ or 2′′/6′′), 7.59 (d, 8.2 Hz, 4 H, presumably pos. 2′/6′ or 2′′/6′′), 4.04 (s, 6 H, Me), 3.92 (s, 6 H, Me); it is assumed that positions 1,3 and 5,7 couple via4JH–H; coupling constants in this area are hard to obtain with certainty. Related compounds with a similar substitution pattern (e.g. 2,6-dibromo-9,10-anthraquinone) show a distinct 4JH–H for these positions. MS (ESI+, DCM/MeOH; [m/z]): 715.5 [M + H]+, 737.2 [M + Na]+. IR (![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/char_e0e1.gif) in cm−1): 3437 (br s, H2O), 3080–2970 (several w), 2953 (w), 2847 (w), 2380–2320 (several w), 2078 (br w), 1724 (vs), 1680 (m/w–w), 1653 (m/w–w), 1608 (s), 1570–1500 (several w), 1456 (m/w–w), 1436 (m), 1404 (w), 1378 (w), 1279 (br, vs), 1192 (m/w), 1180 (m/w), 1113 ((br) s), 1019 (m/w), 951 (m/w–w), 916 (w), 889 (w), 858 (m/w–w), 819 (m/w–w), 771 (m), 737 (w), 709 (m/w), 640–580 (several w), 530–418 (several w). Found C, 74.99%; H, 4.68%; calcd for C46H34O8 × 1/3 DCM (DCM from workup): C, 74.89%; H, 4.70%.
in cm−1): 3437 (br s, H2O), 3080–2970 (several w), 2953 (w), 2847 (w), 2380–2320 (several w), 2078 (br w), 1724 (vs), 1680 (m/w–w), 1653 (m/w–w), 1608 (s), 1570–1500 (several w), 1456 (m/w–w), 1436 (m), 1404 (w), 1378 (w), 1279 (br, vs), 1192 (m/w), 1180 (m/w), 1113 ((br) s), 1019 (m/w), 951 (m/w–w), 916 (w), 889 (w), 858 (m/w–w), 819 (m/w–w), 771 (m), 737 (w), 709 (m/w), 640–580 (several w), 530–418 (several w). Found C, 74.99%; H, 4.68%; calcd for C46H34O8 × 1/3 DCM (DCM from workup): C, 74.89%; H, 4.70%.
H4-ACTBA was obtained by saponification by adding aqueous NaOH and refluxing overnight. Dioxane was removed on a rotary evaporator. If necessary, more water was added. The warm mixture was filtrated and the solid washed with water. The basic filtrate (clear yellow solution) was poured slowly into ice/water/HCl (aq) under stirring. The product precipitated as a bright yellow powder. The product was isolated by filtration, washed with hot water and dried in vacuum at 140 °C for several hours yielding a yellow powder. Typical yields: 80–86% (4.9 to 12.3 g). The product is not soluble in aliphatic solvents, slightly soluble in alcohols at room temperature (esp. iPrOH), better at elevated temperature. M.p.: decomp. without melting. Analytical data for H4-ACTBA: 1H-NMR (DMSO-d6, d in ppm): 7.67 (d, ca. 2 H, ca. 1.5 Hz), 7.70–7.73 (m(?), ca. 6 H), 7.74 (s, ca. 2 H), 7.84 (s, 2 H, probably 1,5-position, possibly d, 1.5 Hz), 7.87 (probably dd, 2 H, 9 Hz and small d of ca. 1.5 Hz, likely 3,7-position because of possible 4JH–H with 1,5-position), 8.02 (d, 4 H, ca. 8 Hz), 8.28 (d, 4 H, ca. 8 Hz); COOH not observed. The overlapping signals from 7.67 to 7.74 ppm integrate to ca. 10 H. MS (ESI−, MeOH; [m/z]): 328.0 [M − 2H]2−, 657.1 [M − H]−, 679.0 [M − 2H + Na]−. IR ( in cm−1): 3456 (br s, H2O), 2964 (m), 1691 (br s), 1609 (s), 1568 (w), 1409 (br m), 1262 (m/s), 1178 (w/m), 1103 (m), 1019 (w/m), 951 (w), 860 (w), 801 (m), 708 (w). Found C, 75.17%; H, 4.02%; calcd for C42H26O8 × 0.5H2O: C, 75.55%; H, 4.08%.
in cm−1): 3438 (br, H2O), 2965 (m), 1691 (m), 1658 (m), 1652 (m), 1607 (br s), 1558–1502 (br, several absorptions, m), 1404 (br s), 1262 (s), 1178 (m), 1098 (br s), 1019 (s), 950 (m/w), 862 (m/w), 803 (s), 709 (m/w), 626 (w), ca. 502 (several absorptions, w).
Crystal structure determination
The X-ray data were collected on a Gemini-S CCD diffractometer (Agilent Technologies) using Mo-Kα radiation (λ = 0.71073 Å), ω-scan rotation. Data reduction was performed with CrysAlis Pro,27 including the program SCALE 3 ABSPACK28 for empirical absorption correction. Structure solution with SHELXS-2013 (direct method). Anisotropic refinement of all non-hydrogen atoms with SHELXL-2014.29 Hydrogen atoms are calculated on idealised positions. Solvent molecules were removed with the SQUEEZE routine implemented in PLATON.14 Figures were drawn with Diamond v 3.2i. Structural data of 1: C114H110ClFe3N6O22 ({Fe3(ACTBA)2}X·6DEF), M = 2119.07, triclinic, space group P, a = 13.3774(7) Å, b = 17.0481(7) Å, c = 17.4322(7) Å, α = 118.491(4)°, β = 99.961(4)°, γ = 100.101(4)°, V = 3285.1(3) Å3, Z = 1, ρcalcd = 1.071 Mg m−3, μ = 0.407 mm−1, Θmax. = 26.37°, R(all data) = 0.0817, Rw(all data) = 0.1772, T = 130(2) K, 13416 independent reflections, 466 parameters, no restraints, maximal residual electron density 0.618 e Å−3. CCDC 1062660 (1) contains the supplementary crystallographic data for this paper.
Acknowledgements
We gratefully acknowledge financial support from Leipzig University, the DAAD (postdoctoral grant for A. V. V., doctoral grant to A. A. (GSSP)) and the ERASMUS+ mobility program, the Russian Government, Ministry of Education (research was made possible due to funds provided aiming at maximising ITMO University's competitive advantage among world's leading educational centres) and Russian Foundation for Basic Research (grant no. 16-37-60073 mol_a_dk).References
- (a) W. Xuan, C. Zhu, Y. Liu and Y. Cui, Chem. Soc. Rev., 2012, 41, 1677–1695 RSC; (b) A. Vinogradov, H. Zaake-Hertling, E. Hey-Hawkins, A. V. Agafonov, G. A. Seisenbaeva, V. G. Kessler and V. V. Vinogradov, Chem. Commun., 2014, 50, 10210–10213 RSC.
- C. Volkringer, D. Popov, T. Loiseau, N. Guillou, G. Férey, M. Haouas, F. Taulelle, C. Mellot-Draznieks, M. Burghammer and C. Riekel, Nat. Mater., 2007, 6, 760–764 CrossRef CAS PubMed.
- S. Kitagawa, Y. Kubota, R. V. Belosludov, T. C. Kobayashi, H. Sakamoto, T. Chiba, M. Takata, Y. Kawazoe and Y. Mita, Nature, 2005, 436, 238–241 CrossRef PubMed.
- M. Sindoro, N. Yanai, A.-Y. Jee and S. Granick, Acc. Chem. Res., 2014, 47, 459–469 CrossRef CAS PubMed.
- (a) R. J. Kuppler, D. J. Timmons, Q.-R. Fang, J.-R. Li, T. A. Makal, M. D. Young, D. Yuan, D. Zhao, W. Zhuang and H.-C. Zhou, Coord. Chem. Rev., 2009, 253, 3042–3066 CrossRef CAS; (b) S. Bhattacharjee, J. S. Choi, S. T. Yang, S. B. Choi, J. Kim and W. S. Ahn, J. Nanosci. Nanotechnol., 2010, 10, 135–141 CrossRef CAS PubMed; (c) S. Ma, J. M. Simmons, D. Sun, D. Yuan and H.-C. Zhou, Inorg. Chem., 2009, 48, 5263–5268 CrossRef CAS PubMed.
- (a) M. Murugesu, R. Clérac, W. Wernsdorfer, C. E. Anson and A. K. Powell, Angew. Chem., Int. Ed., 2005, 44, 6678–6682 ( Angew. Chem. , 2005 , 117 , 6836–6840 ) CrossRef CAS PubMed; (b) T. Liu, Y. J. Zhang, Z. M. Wang and S. Gao, J. Am. Chem. Soc., 2008, 130, 10500–105001 CrossRef CAS PubMed; (c) Z. M. Zhang, Y. G. Li, S. Yao, E. B. Wang, Y. H. Wang and R. Clérac, Angew. Chem., Int. Ed., 2009, 48, 1581–1584 ( Angew. Chem. , 2009 , 121 , 1609–1612 ) CrossRef CAS PubMed.
- (a) P. Horcajada, T. Chalati, C. Serre, B. Gillet, C. Sebrie, T. Baati, J. F. Eubank, D. Heurtaux, P. Clayette, C. Kreuz, J. S. Chang, Y. K. Hwang, V. Marsaud, P. N. Bories, L. Cynober, S. Gil, G. Férey, P. Couvreur and R. Gref, Nat. Mater., 2009, 9, 172–180 CrossRef PubMed; (b) S. R. Miller, D. Heurtaux, T. Baati, P. Horcajada, J. M. Greneche and C. Serre, Chem. Commun., 2010, 46, 4526–4528 RSC.
- (a) G. Combarieu, M. Morcrette, F. Millange, N. Guillou, J. Cabana, C. P. Grey, I. Margiolaki, G. Férey and J. M. Tarascon, Chem. Mater., 2009, 21, 1602–1611 CrossRef; (b) G. Férey, F. Millange, M. Morcrette, C. Serre, M. L. Doublet, J. M. Greneche and J. M. Tarascon, Angew. Chem., Int. Ed., 2007, 46, 3259–3263 ( Angew. Chem. , 2007 , 119 , 3323–3327 ) CrossRef PubMed; (c) R. Canioni, C. Roch-Marchal, F. Sécheresse, P. Horcajada, C. Serre, M. Hardi-Dan and Y. K. Hwang, Mater. Chem., 2011, 21, 1226–1233 RSC; (d) S. Bauer, C. Serre, T. Devic, P. Horcajada, J. Marrot, G. Férey and N. Stock, Inorg. Chem., 2008, 47, 7568–7576 CrossRef CAS PubMed.
- M. H. Zeng, X. L. Feng and X. M. Chen, Dalton Trans., 2004, 2217–2223 RSC.
- S. H. Chae, H.-C. Kim, Y. S. Lee, S. Huh, S.-J. Kim, Y. Kim and S. J. Lee, Cryst. Growth Des., 2015, 15, 268 CAS.
- T. Loiseau, C. Serre, C. Huguenard, G. Fink, F. Taulelle, M. Henry, T. Bataille and G. Férey, Chem. – Eur. J., 2004, 10, 1373–1382 CrossRef CAS PubMed.
- R.-J. Li, M. Li, X.-P. Zhou, D. Li and M. O'Keeffe, Chem. Commun., 2014, 50, 4047–4049 RSC.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- http://www.chem.gla.ac.uk/~louis/software/platon/ CrossRef CAS PubMed; A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- I. B. Martini, I. M. Craig, W. C. Molenkamp, H. Miyata, S. H. Tolbert and B. J. Schwartz, Nat. Nanotechnol., 2007, 2, 647–652 CrossRef CAS PubMed.
- (a) A. Poddubny, I. Iorsh, P. Belov and Y. Kivshar, Nat. Photonics, 2013, 7, 948–957 CrossRef CAS; (b) N. I. Zheludev and Y. S. Kivshar, Nat. Mater., 2012, 11, 917–924 CrossRef CAS PubMed.
- (a) Y. Tang, W. He, Y. Lu, J. Fielden, X. Xiang and D. Yan, J. Phys. Chem. C, 2014, 118, 25365–25373 CrossRef CAS; (b) W. W. Lestari, P. Lönnecke, M. B. Sárosi, H. Cerqueira Streit, M. Adlung, C. Wickleder, M. Handke, W.-D. Einicke, R. Gläser and E. Hey-Hawkins, CrystEngComm, 2013, 15, 3874–3884 RSC; (c) W. W. Lestari, P. Lönnecke, H. Cerqueira Streit, M. Handke, C. Wickleder and E. Hey-Hawkins, Eur. J. Inorg. Chem., 2014, 1775–1782 CrossRef CAS; (d) W. W. Lestari, H. Cerqueira Streit, P. Lönnecke, C. Wickleder and E. Hey-Hawkins, Dalton Trans., 2014, 43, 8188–8195 RSC; (e) W. W. Lestari, P. Lönnecke, H. Cerqueira Streit, F. Schleife, C. Wickleder and E. Hey-Hawkins, Inorg. Chim. Acta, 2014, 421, 392–398 CrossRef CAS; (f) Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815–5840 RSC.
- M. Born and E. Wolf, Principles of Optics, 4th edn, 1970, p. 63 Search PubMed.
- G. Ghosh, Opt. Commun., 1999, 163, 95–102 CrossRef CAS.
- W. L. F. Armarego and C. L. L. Chai, Purification of Laboratory Chemicals, 6th edn, Butterworth-Heinemann (Elsevier), 2009 Search PubMed.
- M. Horie, T. Sassa, D. Hashizume, Y. Suzaki, K. Osakada and T. Wada, Angew. Chem., Int. Ed., 2007, 46, 4983–4986 ( Angew. Chem. , 2007 , 119 , 5071–5074 ) CrossRef CAS PubMed.
- S.-H. Yang, M. L. Cooper, P. R. Bandaru and S. Mookherjea, Opt. Express, 2008, 16, 8306–8316 CrossRef PubMed.
- K. Okano, O. Tsutsumi, A. Shishido and T. Ikeda, J. Am. Chem. Soc., 2006, 128, 15368–15369 CrossRef CAS PubMed.
- (a) Z. Wang and S. Hi, J. Serb. Chem. Soc., 2008, 73, 1187–1196 CrossRef CAS; (b) http://www.photonics.com/Category.aspx?CatID=19950 .
- P. G. Del Rosso, M. F. Almassio, M. Bruno and R. O. Garay, Tetrahedron Lett., 2010, 51, 6730–6733 CrossRef CAS.
- N. Miyaura, K. Yamada and A. Suzuki, Tetrahedron Lett., 1979, 20, 3437–3440 CrossRef.
- CrysAlis Pro, Oxford Diffraction Ltd, Oxfordshire, U.K., 2010 Search PubMed.
- SCALE3 ABSPACK , Oxford Diffraction Ltd., Oxfordshire, U.K., 2010.
- SHELXL: G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1: X-ray powder diffraction, determination of the surface area, different views of the 3D structure, magnetic susceptibility, Mößbauer spectroscopy, EPR measurements, DTA-TG, optical properties. CCDC 1062660. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt00390g |
| This journal is © The Royal Society of Chemistry 2016 |