
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTriphos derivatives and diphosphines as ligands in the ruthenium-catalysed alcohol amination with NH3†
N.
Nakagawa
a,
E. J.
Derrah
a,
M.
Schelwies
b,
F.
Rominger
c,
O.
Trapp
c and
T.
Schaub
*ab
aCatalysis Research Laboratory (CaRLa), Im Neuenheimer Feld 584, D-69120 Heidelberg, Germany. E-mail: thomas.schaub@basf.com
bSynthesis & Homogeneous Catalysis, BASF SE, Carl-Bosch-Straße 38, D-67056 Ludwigshafen, Germany
cOrganisch-Chemisches Institut, Ruprecht-Karls-Universität Heidelberg, Im Neuenheimer Feld 270, D-69120 Heidelberg, Germany
First published on 10th February 2016
Abstract
The ruthenium-triphos and diphosphine-catalysed amination of alcohols with ammonia is reported. Various types of triphos derivatives with electron-donating functional group were synthesized and used as ligands in the Ru-catalysed alcohol amination with NH3. The triphos derivatives are effective for the formation of primary amines. On the other hand, if hemilabile diphosphines as tridentate ligands are used, mixtures of secondary-along with primary amines are obtained. It was found that even simple diphosphines can be used as ligands for the selective formation of the secondary amines. The diphosphine system allows a new entry to the Ru-catalysed formation of secondary amines.
Introduction
Amines are important building blocks in the manufacture of pharmaceuticals and agrochemicals.1 Intensive research has focused on the selective formation of primary, secondary and tertiary amines.2 One of the most straightforward and environmentally-friendly methods is the amination of alcohols using cheap and abundant ammonia (NH3), which represents an atom efficient route towards this valuable class of organic compounds, along with water as the sole by-product.3 The amination of alcohols with ammonia is performed with heterogeneous catalysts on industrial scale. Using heterogeneous catalysts, it is in many cases difficult to control the selectivity as harsh reaction conditions are required.1c During the last decade, the amination of alcohols has been further developed with a series of homogeneous catalysts.4 In particular, ruthenium catalysts were identified to exhibit a high performance for the selective formation of primary amines5 and secondary amines.6 The combination of a ruthenium pre-catalyst and the tridentate phosphine ligand 1,1,1-tris(diphenylphosphinomethyl)ethane 1 (= triphos) was first published as one of the candidates in the alcohol amination by Beller and co-workers7a and investigated in detail by our laboratory (Scheme 1).7b We disclosed that the catalytic active species is a cationic ruthenium-triphos complex through experimental and computational investigations. Our previous report on this ruthenium catalysis described only the use of the commercial available triphos 1 and effects of other phosphine derivatives have still remained undisclosed and undeveloped. We herein report the synthesis and evaluation of triphos derivatives and the corresponding ruthenium complexes in order to gain an insight how variations of the triphos scaffold influence the ruthenium-catalysed amination of alcohols with NH3. Our approach was to alter the coordination sphere of triphos-type Ru-complexes in order to change the selectivities in the corresponding amination reactions. | ||
| Scheme 1 Ruthenium-triphos-catalysed amination of 1-octanol with ammonia.7b | ||
Results and discussion
Ligands
To evaluate the ligand influence on the catalytic performance of the ruthenium-catalysed amination of 1-octanol with NH3, the following triphos derivatives were used: triphos 1, the newly synthesised phenyl-p-tolyl-mixed triphos derivative 2, and the all-p-tolyl triphos derivative 3 (obtained via a modified procedure).8 We also examined diphosphines with an electron-donating functional group such as a pyridyl substituted derivative 4, which is potentially hemilabile ligand,9 as well as oxygen-substituted ones 510 and 6 (Fig. 1). | ||
| Fig. 1 Triphos 1, triphos derivatives 2 and 3, and diphosphines with electron-donating functional groups 4–6 used in this work. | ||
Synthesis of triphos derivatives via modified protocol
C 3V symmetrical triphos-type ligands such as 3 can be obtained using 1,1,1-tris(chloromethyl)ethane as a starting material. A route to C1 symmetric variants of the ligand with three different phosphine donors was reported by Huttner and Helmchen.8 However, since we were only interested altering one of the three donor groups of triphos, we chose 7 as key intermediate10a in our synthesis (Scheme 2). The substitution reaction of CH2C(CH2OMs)2(CH2Br) 7 with an excess amount of lithium diphenylphosphide, followed by borane protection afforded a monomesyl diphosphine–borane compound 6-PG in 64% yield (Scheme 2a). After deprotection with DABCO, the intermediate 6 was obtained in 66% yield. We repeated this sequence using lithium di(p-tolyl)phosphide with monomesylate 6 and obtained the unsymmetrical triphos–borane 2-PG. The structure of 2-PG was confirmed by X-ray analysis (Fig. 2). Deprotection of 2-PG afforded the unsymmetrical triphos 2 in 27% overall yield. The same protocol was applied to the synthesis of all-p-tolyl triphos 3 (51% overall yield, Scheme 2b).11 | ||
| Scheme 2 Optimised synthetic protocol for triphos derivatives. | ||
 | ||
| Fig. 2 ORTEP drawing of the borane-adduct 2-PG where all non-hydrogen atoms are represented by Gaussian ellipsoids at the 50% probability level. All hydrogen atoms have been omitted for clarity. | ||
Synthesis of ruthenium complexes
Ruthenium complexes of the triphos derivatives 1–3 were prepared according to the reported protocol for Ru-triphos complex A (Scheme 3).12 When using 2 as a ligand, a mixture of three isomers is formed. 31P-NMR indicates that the three isomers differ in the position of the tolyl moiety. Elemental analysis confirms the composition of the mixture of B-1, B-2, and B-3. In case of the all-tolyl substituted ligand 3, only one complex (C) is formed like in the synthesis of A. The structure of C was determined by X-ray analysis (Fig. 3). The distance of Ru–P bonds in C is the range of 2.27–2.38 Å which is similar like in the reported structure for RuHCl(CO)(triphos) A.13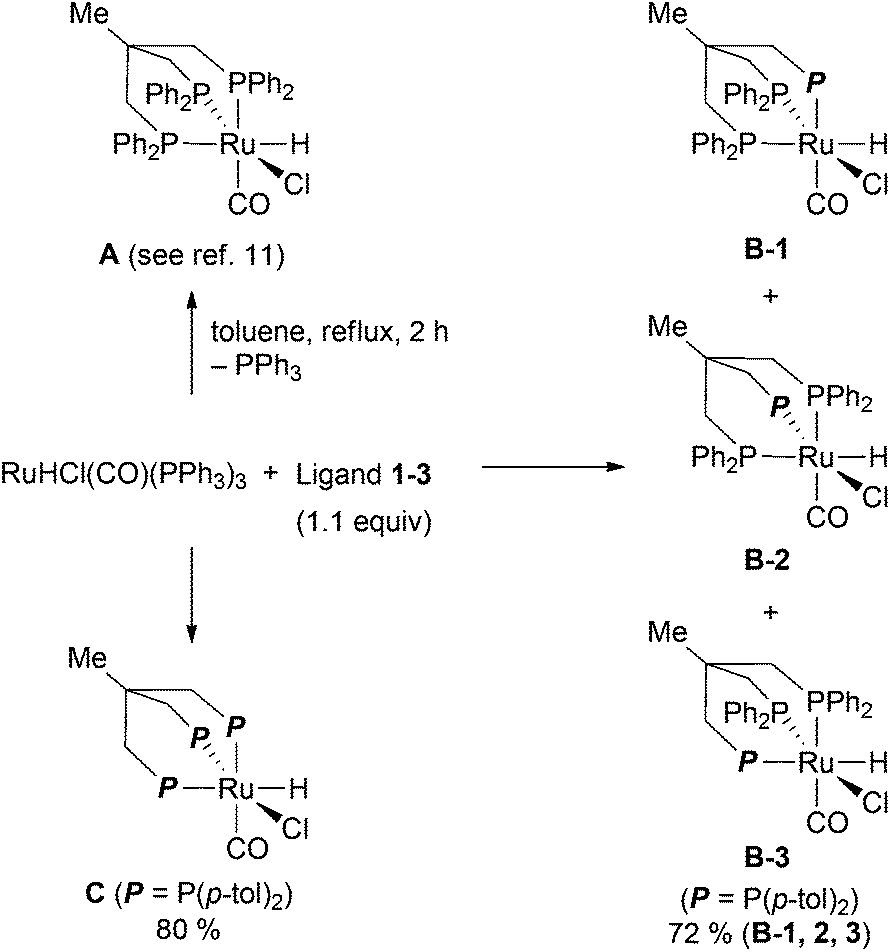 | ||
| Scheme 3 Synthesis of triphos-type ruthenium complexes A–C. | ||
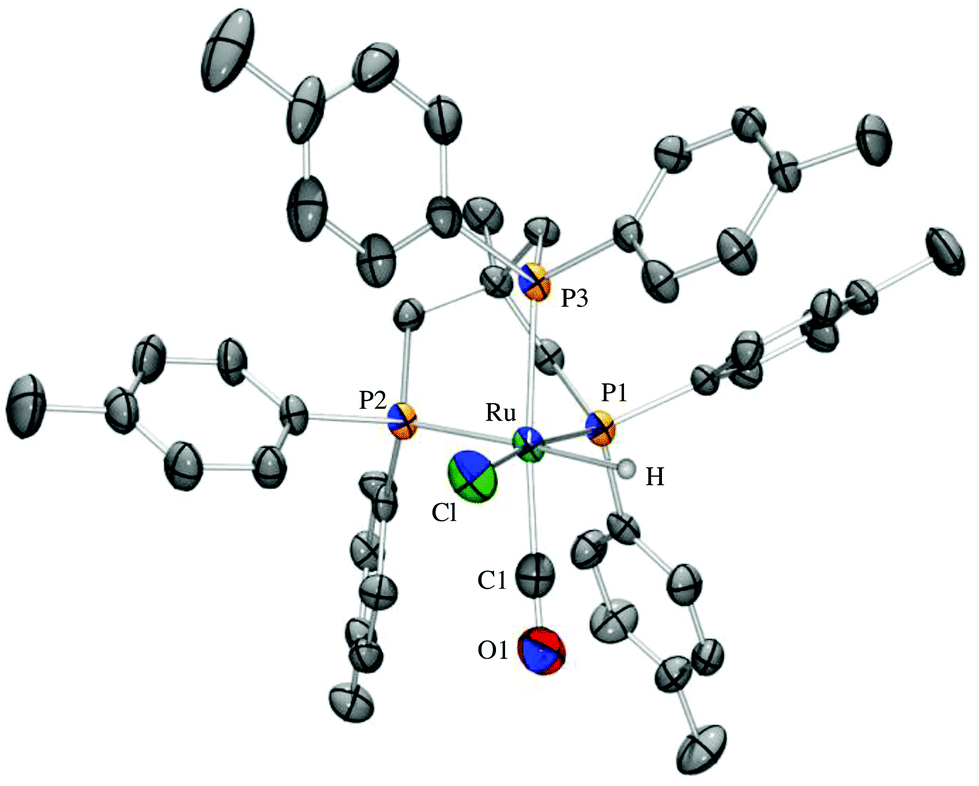 | ||
| Fig. 3 ORTEP drawing of the ruthenium complex C where all non-hydrogen atoms are represented by Gaussian ellipsoids at the 50% probability level and all hydrogen atoms have been removed for simplicity. Selected bond lengths (Å) and angles (°) of C: Ru–P1 = 2.2740(12); Ru–P2 = 2.3847(12); Ru–P3 = 2.3840(13); Ru–H = 1.88(3); Ru–Cl = 2.5000(13); P1–Ru–P2 = 88.31(4); P2–Ru–P3 = 86.94(4); P3–Ru–P1 = 88.67(4). | ||
We also conducted the preparation of the corresponding ruthenium complexes with pyridyl substitued diphosphine 4.9 Using this ligand, a mixture of three complexes was obtained: the two isomers D-1 and D-2 as well as the dichloro ruthenium species D-3 (Scheme 4). The structure of D-3 was confirmed by X-ray analysis (Fig. 4). Regarding the structure of D-3, the carbonyl ligand is located trans to the pyridyl moiety. Although the reaction pathway towards D-3 is not confirmed, we propose that the chlorine atom on D-3 was derived from the CH2Cl2 solvent through the chlorine abstraction by ruthenium hydride species. Under these conditions, selective formation and complete separation of the three isomers turned out to be difficult. In addition, we failed to obtain the ruthenium–pyridyl diphosphine complex selectively in toluene as solvent. Therefore, we used the pyridyl diphoshine 4 directly in combination with [Ru(PPh3)3(CO)HCl] in the amination reaction to generate the desired complexes in situ (vide infra).
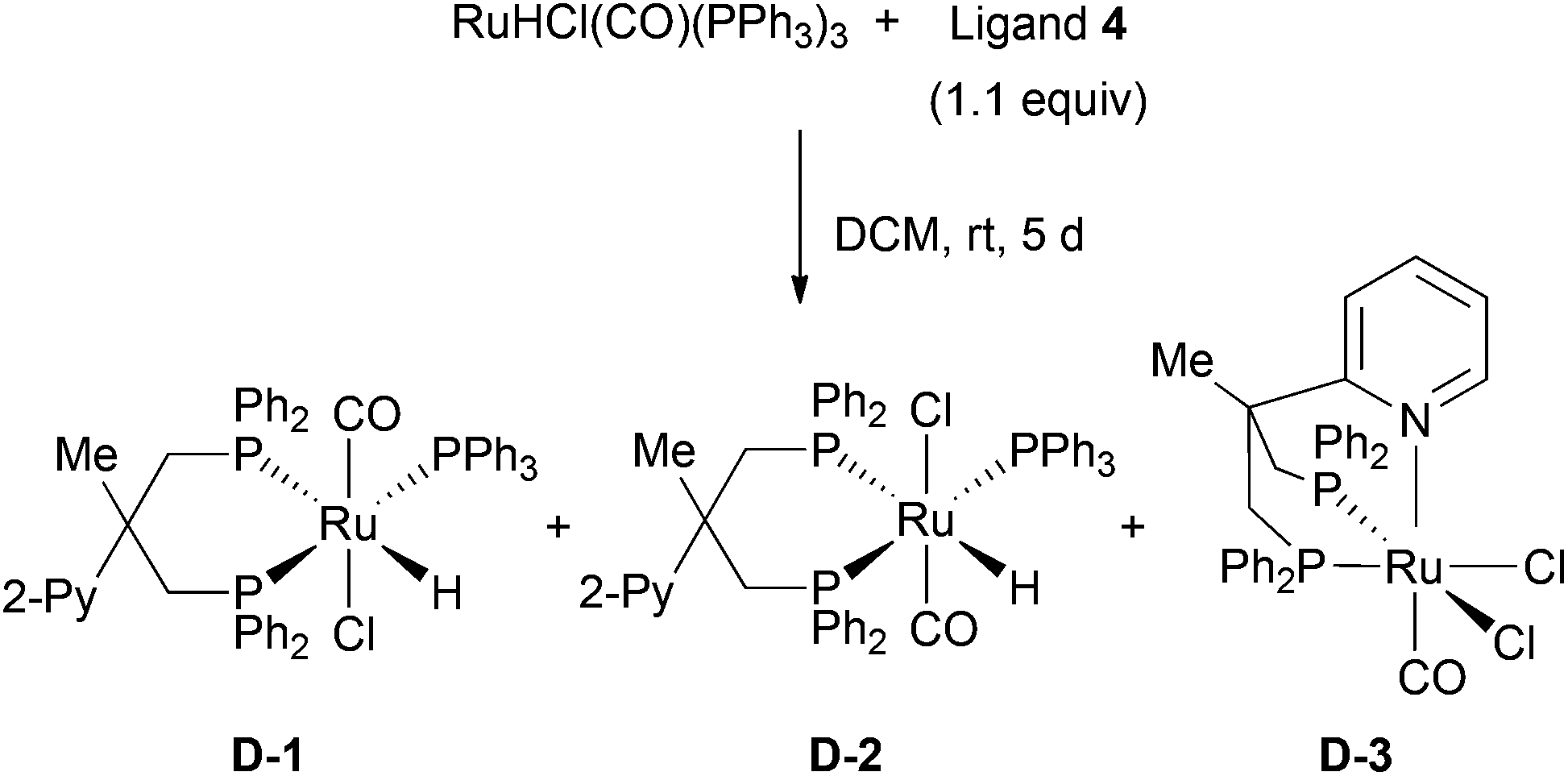 | ||
| Scheme 4 Preparation of pyridyl diphosphine ruthenium complexes D-1, D-2, and D-3. | ||
 | ||
| Fig. 4 ORTEP drawing of the dichloro ruthenium complex D-3 where all non-hydrogen atoms are represented by Gaussian ellipsoids at the 50% probability level. All hydrogen atoms and coordinating solvent have been omitted for clarity. Selected bond lengths (Å) and angles (°) of D-3: Ru–P1 = 2.2970(14); Ru–P2 = 2.2844(15); Ru–N = 2.178(4); Ru–Cl1 = 2.4314(13); Ru–Cl2 = 2.4775(14); P1–Ru–P2 = 87.65(5); P1–Ru–N = 83.76(11); P2–Ru–N = 90.25(11). | ||
We were also successful to synthesise a novel hydroxyl-containing ruthenium complex E from 510 (Scheme 5). The structure of E was confirmed by X-ray analysis (Fig. 5). One PPh3 ligand from the precursor remains on ruthenium which is consistent with reported RuHCl(CO)PPh3(diphosphine) complexes.5e,g The hydroxyl group orients to the opposite direction from the ruthenium centre, thus it seems to be an unsuitable coordinating group to achieve a tridentate coordination mode. In order to facilitate a coordination of the oxygen atom, the ruthenium complex E was treated with potassium tert-butoxide (Scheme 5). The red powder obtained was assigned as the alkoxo ruthenium complex F by 1H and 31P NMR spectra as well as LIFDI mass spectroscopy. It is conceivable that under a basic ammonia atmosphere of the alcohol amination the alkoxo complex F will be formed in situ from E.
 | ||
| Scheme 5 Synthesis of hydroxyl-containing diphosphine ruthenium complex E and reaction between complex E and tBuOK. | ||
 | ||
| Fig. 5 ORTEP drawing of hydroxyl-containing diphosphine ruthenium complex E where all non-hydrogen atoms are represented by Gaussian ellipsoids at the 50% probability level. All hydrogen atoms and coordinating solvent have been omitted for clarity. Selected bond lengths (Å) and angles (°) of E: Ru–P1 = 2.4279(10); Ru–P2 = 2.3586(10); Ru–P3 = 2.3806(11); Ru–H = 1.62(5); Ru–Cl = 2.4725(11); P1–Ru–P2 = 94.38(3); P3–Ru–P1 = 100.04(4). | ||
Catalytic investigations
In order to investigate the ligand effects, the ruthenium-catalysed amination of 1-octanol with NH3 was conducted for the above described ligands 1–6 (Table 1). As previously reported by our group,7 the ruthenium-triphos complex A showed a high catalytic performance towards octylamine 8 along with small amounts of the secondary and tertiary amines 9 and 10 (entry 1). Note that it is unnecessary to use the isolated ruthenium catalyst A: the in situ prepared ruthenium-triphos catalyst showed the same activity and selectivity as A (entry 2). In the absence of any additional phosphine, the amination products 8 to 10 were not obtained (entry 3). When the p-tolyl-substituted triphos derivatives 2 and 3 were used instead of 1, it was found that the selectivity towards the primary amine is slightly shifted to dioctylamine 9 (entries 4 and 5). The use of the pyridyl diphosphine 4, a potentially hemilabile ligand, led to the formation of dioctylamine 9 (entry 6). The amination reactions using the hydroxyl–diphosphine complex E and its alkoxo complex F were sluggish and the dioctylamine 9 is formed in good selectivity (entries 7 and 8). These results can be interpreted as the complex E probably converts into the alkoxo complex F through deprotonation of the hydroxyl group under the basic conditions of the amination. When the mesyl diphosphine 6, which is the synthetic intermediate of the ligands 2 and 3, was used as a ligand, dioctylamine 9 is formed in good selectivity (entry 9). Probably the mesyl group (OMs) will be displaced by the amino group (NH2) via nucleophilic substitution under the ammonia atmosphere of the amination reaction.11 To our surprise, even simpler diphosphines, such as dpppdmp (1,3-bis(diphenylphosphino)-2,2-dimethylpropane, Ph2PCH2C(CH3)2CH2PPh2) and dppp (1,3-bis(diphenylphosphino)propane, Ph2PCH2CH2CH2PPh2), were found to be good ligands for the preferable formation of dioctylamine 9 (entries 10 and 11).14,15 Finally, the highly selective formation of dioctylamine 9 was achieved with dppp at a higher substrate concentration (entries 12 and 13).|
|
||||||
|---|---|---|---|---|---|---|
| Entrya | Ru-phosphine catalyst | GC area ratio (%) | Selectivity of mono![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :di (%) :di (%) |
|||
| Mono | di | tri | SM recov. | |||
| a 1-Octanol (3.0 g, 23.6 mmol) and 17 mL of toluene was used. b 0.22 mol% of ligand was used. c dppdmp = 1,3-bis(diphenylphosphine)-2,2-dimethylpropane. d dppp = 1,3-bis(diphenylphosphino)propane. e 6 mL of toluene was used as a solvent. f p(NH3) = 4 bar (∼52 mmol). g The calibrated GC yields against hexadecane as an internal standard. | ||||||
| 1 | A | 96 | 3 | <1 | 1 | 97:3 |
| 2b | RuHCl(CO)(PPh3)3 + 1 | 93 | 5 | <1 | 2 | 95:5 |
| 3 | RuHCl(CO)(PPh3)3 | <1 | <1 | <1 | 99 | — |
| 4 | B | 90 | 8 | <1 | 1 | 92:8 |
| 5 | C | 86 | 11 | 1 | 2 | 88:12 |
| 6 | RuHCl(CO)(PPh3)3 + 4 | 14 | 66 | 3 | 17 | 18:82 |
| 7 | E | 10 | 47 | 2 | 41 | 17:83 |
| 8 | F | 9 | 51 | 2 | 38 | 15:85 |
| 9 | RuHCl(CO)(PPh3)3 + 6 | 10 | 88 | <1 | 1 | 10:90 |
| 10 | RuHCl(CO)(PPh3)3 + dppdmpc | 11 | 86 | 2 | 1 | 11:89 |
| 11 | RuHCl(CO)(PPh3)3 + dpppd | 35 | 63 | <1 | 1 | 36:64 |
| 12e | RuHCl(CO)(PPh3)3 + dpppd | <1 | 95 | 4 | 1 | <1:>99 |
| 13e,f | RuHCl(CO)(PPh3)3 + dpppd | 5 | 87 | 6 | 2 | 8:92 |
| [5]g | [88]g | [5]g | [2]g | |||

The reduced amount of toluene significantly affected the formation of the secondary amine. In order to get more insights in the reaction, we examined the reaction profile by analysing the reaction mixture at several reaction times (1–8 h; Fig. 6).16 The obtained curves showed that there is no significant induction period, and that the amount of octylamine 8 and dioctylamine 9 increases as time elapses without decomposing or interconverting into the other products. We also confirmed the formation of small amount of octyloctan-1-imine 11, which is assumed to be the intermediate before dioctylamine 9 is formed. The imine 11 is propably reactive to a Ru–H species to produce dioctylamine 9 through hydrogenation.
 | ||
| Fig. 6 Reaction profile for the Ru-dppp-catalysed amination of octanol with NH3. Reaction conditions: in a premix autoclave (60 mL stainless steel), 1-octanol (3.0 g, 24 mmol) (◆), p(NH3) = 4 bar (0.9 g, 53 mmol of NH3), RuHCl(CO)(PPh3)3 (45 mg, 0.047 mmol), dppp (21 mg, 0.052 mmol), and toluene (6 mL). Observed products: octylamine 8 (▲), dioctylamine 9 (■), and octyloctan-1-imine 11 (●). Trioctylamine 10 was observed in 2–4% GC area ratio in each reaction time. | ||
The possible mechanism is described in Scheme 6. As we postulated in the previous report,7 the selective mono-alkylation of ammonia using ruthenium-triphos complexes involves the formation of cationic ruthenium-triphos intermediates as active species (Scheme 6a). Among the triphos derivatives, the different ratio between the primary and secondary amines can originate from the difference of electron negativity of P atom. In the case of hemilabile diphosphines and simple diphosphines, the formation of the secondary amine is preferred under the same reaction conditions. This observation can be explained by considering a different mechanism. As Vogt and co-workers disclosed the mechanism on the amination of cyclohexanol using xantphos,5g the dissociation of the remaining PPh3 can play an important role (Scheme 6b). Although the mechanism of dialkylation is still unclear, it is possible to consider that dissociation of PPh3 and chloride can provide more vacant sites on the ruthenium than in the triphos systems. The transient ketone or imine can coordinate at the vacant site and incorporate in the further alkylation. When octylamine 8 as a starting material is used instead of 1-octanol, the formation of the dioctylamine 9 was only observed in small amounts (eqn (1)). Therefore, it is conceivable that the ruthenium–diphosphine complex possesses a poor ability on dehydrogenation of the primary amine 8. Presumably, in situ generated ketone or imine (from the starting alcohol) are the reactants for the dilakylation.
 | (1) |
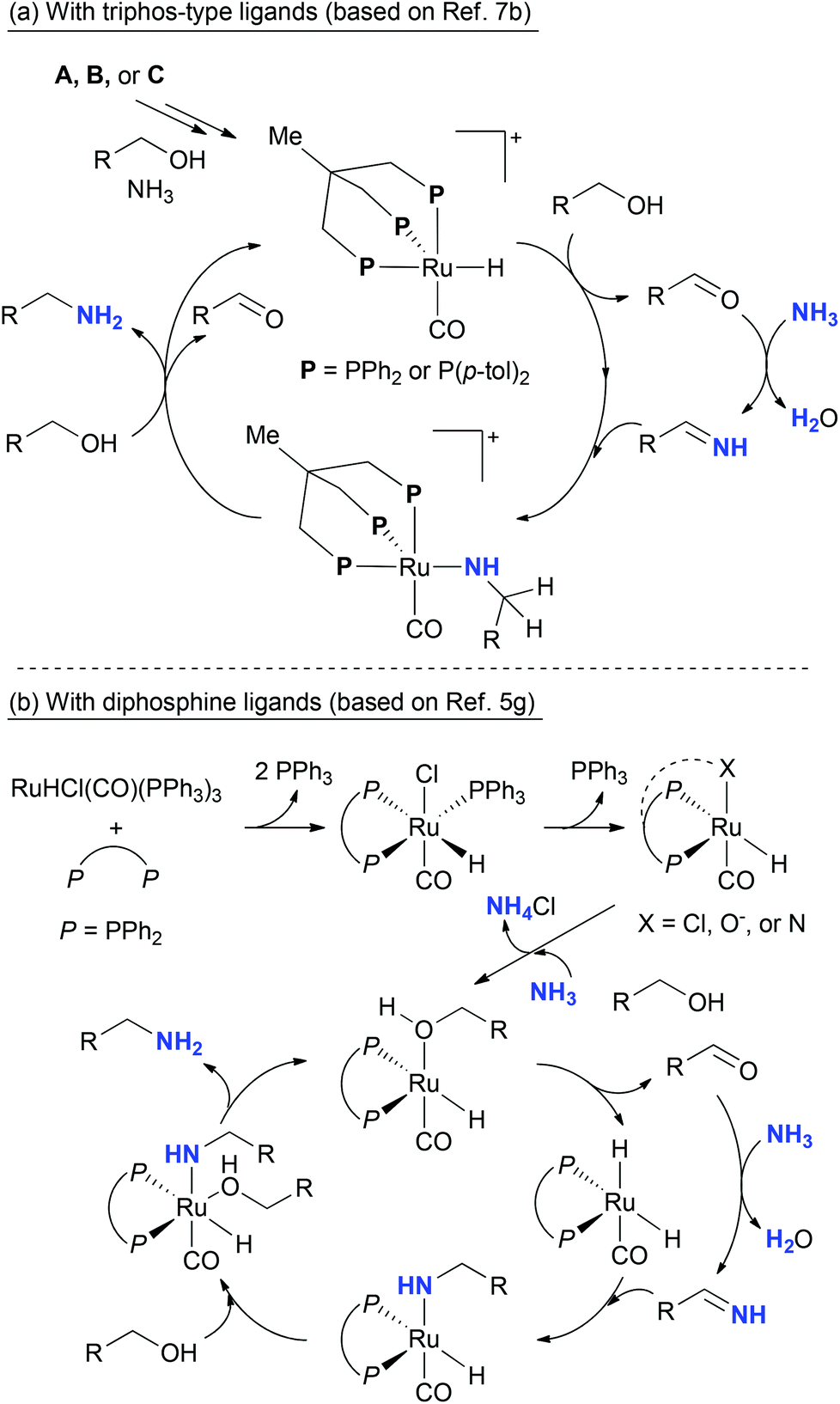 | ||
| Scheme 6 Simplified possible catalytic cycles. | ||
Conclusions
A series of ruthenium-complexes bearing triphos derivatives and diphosphines with the electron-donating functional group were prepared and examined for the amination of primary alcohols with NH3. Deviating the P-substituents on the triphos scaffold slightly affects the ratio of mono- and dialkylamines, but these triphos derivatives are basically effective for the formation of the primary amines. The reaction mechanism of mono-alkylation can involve the formation of the cationic ruthenium complex based on our previous investigations. On the other hand, the use of hemilabile diphosphines leads to a mixture of the primary and secondary amines. It seems to be clearer after these results, that the origin of high selectivities towards the primary amines is related to the very stable ligand spheres of Ru/triphos and the Milstein type Ru/pincer systems.5a,h The selective formation of the secondary amine was finally realized by using simple dppp as the ligand. The reaction mechanism of the ruthenium–diphosphine system can be influenced by dissociation of attaching PPh3 which is different to the triphos-based systems. So far, the preparation of secondary amines from alcohols with NH3 using homogeneous catalysts has been achieved by Milstein and co-worker with Ru catalyst,6 or by Fujita, Yamaguchi, and co-workers as well as in previous work from our laboratory using Ir catalysts.17 Compared with these studies, our current diphosphine system offers a simple protocol for the preparation of diamines with commercially available catalysts.Experimental
General considerations
All reactions were carried out under a positive pressure of argon in an MBraun glovebox or using standard Schlenk line techniques. All nondeuterated solvents were dried using an MBraun SPS-800 solvent purification system and degassed prior to use. 3-Methyl-3-oxetanemethanol (a precursor of 7), and 1-octanol were purchased from Aldrich and distilled prior to use. Di-p-tolyl and di-phenylphosphines were purchased from ABCR, RuHCl(CO)(PPh3)3 was supplied by BASF and used without further purification. All other products were purchased from Aldrich and used without further purification. Liquid reagents including deuterium solvents were distilled prior to use, and all others were used without further purification. 1H, 13C{1H}, and 31P{1H} NMR spectra were recorded on a Bruker Avance 200, 400, or 600 MHz spectrometer. 1H and 13C chemical shifts are reported relative to residual solvent signals of CD2Cl2 (5.32 and 54.0 ppm) and THF-d8 (5.38 and 67.21 ppm). 31P{1H} chemical shifts are referenced to an external 85% solution of phosphoric acid. The 13C NMR data were assigned by HSQC and HMBC spectra. FAB and HR mass spectrometry was measured at the Mass Spectrometry Facility (Institute of the Organic Chemistry, University Heidelberg). Gas chromatography was performed on an Agilent 6890N modular GC base equipped with a split-mode capillary injection system and a flame ionization detector using a BGB-5 capillary column (Agilent 122-1033; 30 m × 0.32 mm × 0.25 μm; He flow 1.0 mL min−1, program: initial 50 °C for 2 min, ramp 6 °C min−1, 300 °C for 10 min). Starting materials and products had the following retention times: octylamine (tR = 14.54 min), 1-octanol (tR = 15.16 min), dioctylamine (tR = 31.34 min), and trioctylamine (tR = 41.71 min). Elemental analysis were performed in the “Mikroanalytisches Laboratorium der Chemischen Institute der Universität Heidelberg”. X-ray structures were solved by derect methods and refined against F2 with a full-matrix least squares algorithm by using the SHELXTL (version 2014/7) software package.18 Intensities were corrected for Lorentz and polarisation effects.19 CCDC 1440322 (2-PG), 1440323 (C), 1440324 (D-3), and 1440325 (E) contain the supplementary crystallographic data for this paper.Synthesis of H3CC(CH2OMs)2(CH2Br) (7)
A 100 mL round-bottom Schlenk flask was charged with 25 mL dioxane and 3-methyl-3-oxetanemethanol (5.45 g, 53 mmol). Aqueous HBr (48%, 7.2 mL, 64 mmol) was slowly added over a 5–7 min period to give a light yellow solution that was slightly warm to the touch. The solution was refluxed for 3.5 h and the solvent was removed under vacuum (60 °C) to give H3CC(CH2OH)2(CH2Br) (9.38 g, 51 mmol, 96% yield) as an orange-brown solid. The product was used in the next step without further purification.1H NMR (200.1 MHz, CD2Cl2, δ): 3.63 (s, 4H, CH2OH), 3.54 (s, 2H, CH2Br), 2.95 (s, 2H, OH), 0.91 (s, 3H, CH3).
A 500 mL round-bottom Schlenk flask was charged with H3CC(CH2OH)2(CH2Br) (4.08 g, 22.3 mmol), triethylamine (4.96 g, 0.49 mol) and 200 mL CH2Cl2. The colourless solution was cooled in an ice bath and neat methanesulfonyl chloride (5.36 g, 47 mmol) was added dropwise to give a colourless solution and white precipitate. The solution was stirred for 2 h at 0 °C. The solution was concentrated to ∼100 mL under vacuum and water (100 mL) was added. The organic layer was extracted and washed with another 100 mL of water, dried over MgSO4, and the organic solvent was removed under vacuum to obtain H3CC(CH2OMes)2(CH2Br) 7 (6.14 g, 18 mmol, 81% yield) as a thick yellowish oil.
Synthesis of H3CC(CH2PPh2·BH3)2(CH2OMes) (6-PG)
A 500 mL round-bottom Schlenk flask was charged with HPPh2 (7.99 g, 43 mmol) and 60 mL THF. The colourless solution was cooled in an ice bath and 2.5 M n-BuLi in hexane (19 mL, 47 mmol) was added drop wise via syringe to give a red/orange solution. The solution was stirred for 30 min before being transferred to a 100 mL addition funnel affixed to a 500 mL round-bottom Schlenk flask charged with H3CC(CH2OMes)2(CH2Br) (7.28 g, 21 mmol) in 150 mL THF. The solution was cooled to −40 °C and the LiPPh2-solution was added slowly over a 2 h period. The temperature was maintained at −40 °C for 3 h before being slowly warmed up to room temperature and the orange mixture was stirred overnight. A solution of BH3·SMe2 (35 mL, 71 mmol) in THF (2.0 M) was slowly added to the light yellow reaction mixture. The resulting mixture was stirred for 2 h and the solvent was removed under vacuum. 250 mL dietyhlether (250 mL) and 250 mL water were added to the white solid. The organic layer was separated and washed with 250 mL water, dried over MgSO4 and the organic solvent was removed under vacuum. The white solid was loaded onto a silica column (10 × 4 cm) and elucidated with a 70:30 mixture of ether and petroleum ether. The first fraction was discarded. The second fraction was collected to give H3CC(CH2PPh2·BH3)2(CH2OMes) 6-PG (7.76 g, 14 mmol, 65% yield) as white powder after the solvent was removed under vacuum.
31P{1H} NMR (81.0 MHz, CD2Cl2, δ): 8.9 (br, 2P); 11B{1H} NMR (128.3 MHz, CD2Cl2, δ): −37.5 (br, 2B); 1H NMR (399.9 MHz, CD2Cl2, δ): 7.78–7.73 (m, 4 H, Ho), 7.65–7.60 (m, 4H, Ho), 7.51 (m, 12H, Hm and Hp), 4.28 (s, 2H, CH2OMs), 3.02–2.94 (m, 2H, CH2P), 2.92 (s, 3H, SCH3), 2.52–2.45 (m, 2H, CH2P), ∼1.15 (broad in baseline, 6H, BH3), 0.86 (s, 3H, CH3); 13C{1H} NMR (100.6 MHz, CD2Cl2, δ): 132.7 (d, 2JCP = 9 Hz, Co), 132.2 (d, 2JCP = 9 Hz, Co), 131.8 (d, 4JCP = 2 Hz, Cp), 131.7 (d, 4JCP = 2 Hz, Cp), 130.8 (d, 1JCP = 33 Hz, Ci), 130.2 (d, 1JCP = 32 Hz, Ci), 129.4 (d, 3JCP = 2 Hz, Cm), 129.3 (d, 3JCP = 2 Hz, Cm), 76.7 (t, 3JCP = 5 Hz, CH2OMs), 40.1 (s, C), 37.6 (s, SCH3), 34.9 (d, 1JCP = 6 Hz, CH2P), 34.6 (d, 1JCP = 6 Hz, CH2P), 23.5 (t, 3JCP = 4 Hz, CH3).
HR-MS (FAB): [M]+ − BH4; theoretical C30H34BO3P2S: 547.1797; experimental 547.1813.
Elemental Analysis Calc. (Found): C 64.08% (64.5%), H 6.81% (6.95%).
Synthesis of H3CC(CH2PPh2)2(CH2OMes) (6)
A 100 mL Teflon caped Schlenk flask was charged with H3CC(CH2PPh2·BH3)2(CH2OMes) (3.16 g, 5.6 mmol), DABCO (1.89 g, 17 mmol) and 20 mL toluene. The headspace was evacuated and the flask was heated at 80 °C for 2 h under static vacuum. The solution was filtered through a small plug of silica (3 cm) on a glass filter frit and washed with toluene (3 × 100 mL). The solvent was removed from the supernatant to give H3CC(CH2PPh2)2(CH2OMes) 6 (1.57 g, 2.9 mmol, 52% yield) as a thick white oil.31P{1H} NMR (81.0 MHz, CD2Cl2, δ): −27.2 (s); 1H NMR (200.1 MHz, CD2Cl2, δ): 7.47–7.38 (m, 8 H, CH), 7.35–7.27 (m, 12 H, CH), 4.11 (s, 2H, CH2OMs), 2.70 (s, 3H, SCH3), 2.51–2.31 (m, 2H, CH2P), 1.00 (s, 3H, CH3).
Synthesis of H3CC(CH2PPh2·BH3)2(CH2P(p-tol)2·BH3) (2-PG)
A 100 mL round-bottom Schlenk flask was charged with HP(p-tol)2 (0.344 g, 2.1 mmol) and 25 mL THF. The colorless solution was cooled in an ice bath and 2.5 M n-BuLi in hexane (0.89 mL, 2.2 mmol) was slowly added via syringe to give a red/orange solution. The solution was stirred for 30 min before drop wise addition to a 25 mL THF solution of H3CC(CH2PPh2)2(CH2OMes) (845 mg, 1.58 mmol) in a 100 mL round-bottom Schlenk flask cooled to −40 °C. After the addition the solution was slowly warmed to room temperature and stirred overnight. The reaction mixture was additionally heated to reflux for 2 h. A solution of BH3·SMe2 (2.84 mL, 5.69 mmol) in THF (2.0 M) was slowly added to the reaction mixture. The resulting mixture was stirred overnight and the volatile compounds were removed under vacuum. 100 mL Et2O and 100 mL water were added to the white solid. The organic layer was separated and the aqueous layer was extracted with Et2O for 2 times. The combined organic layer was washed with 100 mL water for 2 times, and then dried over MgSO4. After the removal of Et2O under vacuum, the white residue in 20 mL Et2O was diffused into hexane to give a white powder. The obtained powder was filtered off and washed 2 times with 10 mL hexane. After drying in vacuum, H3CC(CH2PPh2·BH3)2(CH2P(p-tol)2·BH3) 2-PG (0.710 g, 1.02 mmol, 65% yield) was obtained as a white powder.31P{1H} NMR (81.0 MHz, CD2Cl2, δ): 9.2 (br, 2P), 7.4 (br, 1P); 1H NMR (200.1 MHz, CD2Cl2, δ): 7.66–7.37 (m, 24 H, CH), 7.24–7.19 (m, 4 H, CH), 2.93–2.82 (m, 6H, CH2P), 2.36 (s, 6H, CH3), ∼1.17 (broad in baseline, 9H, BH3), 0.79 (s, 3H, CH3).
Elemental Analysis Calc. (Found): C 74.39% (73.38%), H 7.55% (7.55%).
Synthesis of H3CC(CH2PPh2)2[CH2P(p-tol)2] (2)
A 100 mL Teflon caped Schlenk flask was charged with H3CC(CH2PPh2·BH3)2[CH2P(p-tol)2·BH3] (11a, 0.689 g, 0.99 mmol), DABCO (0.390 g, 3.47 mmol), and 20 mL toluene. The headspace was evacuated and the flask was heated at 80 °C for 2 h under static vacuum. The solution was filtered through a small plug of silica (3 cm) on a glass filter frit and washed with toluene (2 × 100 mL). The solvent was removed from the supernatant to give H3CC(CH2PPh2)2[CH2P(p-tol)2] 2 (0.663 g, 0.99 mmol, quantitative yield) as a thick white oil.31P{1H} NMR (81.0 MHz, CD2Cl2, δ): −28.8 (t, 4JPP = 2.6 Hz, 1P) −26.2 (d); 1H NMR (200.1 MHz, CD2Cl2, δ): 7.38–7.19 (m, 26 H, CH), 7.10–7.06 (m, 2 H, CH), 2.43–2.34 (m, 6H, CH2P), 2.31 (s, 6H, CH3), 0.93 (s, 3H, CH3).
Synthesis of RuHCl(CO){H3CC(CH2PPh2)2[CH2P(p-tol)2]}} (B)
A round-bottom Schlenk flask was charged with H3CC(CH2PPh2)2(CH2P(p-tol)2)) (0.663 g, 1.0 mmol), RuHCl(CO)(PPh3)3 (0.881 g, 0.92 mmol) and 40 mL toluene. The colorless solution with an off white precipitate was heated at reflux for 2 h to give a light yellow precipitate. The solution was filtered through a filter frit, the yellow residue washed three times with 10 mL toluene and dried in vacuum to give RuHCl(CO){H3CC(CH2PPh2)2[CH2P(p-tol)2]}} B (0.544 g, 0.66 mmol, 72% yield) as a isomeric mixture.31P{1H} NMR (81.0 MHz, CD2Cl2, δ): 48.5 (dd, 2JPP = 40 Hz, 2JPP = 18 Hz, 1P), 48.4 (dd, 2JPP = 40 Hz, 2JPP = 18 Hz, 1P), 46.2 (dd, 2JPP = 40 Hz, 2JPP = 17 Hz, 1P), 13.5 (dd, 2JPP = 40 Hz, 2JPP = 32 Hz, 1P), 13.3 (dd, 2JPP = 40 Hz, 2JPP = 32 Hz, 1P), 12.2 (dd, 2JPP = 40 Hz, 2JPP = 32 Hz, 1P), 0.79 (dd, 2JPP = 32 Hz, 2JPP = 18 Hz, 1P), 0.58 (dd, 2JPP = 32 Hz, 2JPP = 18 Hz, 1P), −1.4 (dd, 2JPP = 32 Hz, 2JPP = 18 Hz, 1P); 1H NMR (200.1 MHz, CD2Cl2, δ): 7.83–6.53 (m, 28H, CH), 2.36–2.19 (m, 12H, CH2 and CH3), 1.54–1.51 (m, 3H, CH3), −5.68 to −6.37 (m, 1H, RuH).
IR (KBr): 512, 697, 739, 837, 1093, 1434, 1483, 1923 (m, νRu–H), 1970 (vs, νCO), 2918, 2954, 3051 cm−1.
Elemental Analysis Calc. (Found): C 64.58% (64.16%), H 5.42% (5.32%).
Improved synthesis of H3CC[CH2P(p-tol)2]3 (3)
A 500 mL round-bottom Schlenk flask was charged with HP(p-tol)2 (3.06 g, 14 mmol) and 80 mL THF. The colorless solution was cooled in an ice bath and 2.5 M n-BuLi in hexanes (6 mL, 15 mmol) was slowly added via syringe to give a red/orange solution. The solution was stirred for 30 min before drop wise addition to a THF (100 mL) solution of H3CC(CH2OMes)2(CH2Br) (7, 1.21 g, 3.6 mmol) in a 500 mL round-bottom Schlenk flask cooled to −40 °C. The resulting mixture was slowly warmed to room temperature, then heated at reflux overnight. A solution of BH3·SMe2 (8.0 mL, 16 mmol) in THF (2.0 M) was slowly added to the colorless solution. The resulting mixture was stirred for 2 h and the solvent was removed under vacuum. 100 mL Ether and 100 mL water were added to the white solid. The organic layer was separated and washed with 150 mL water, dried over MgSO4 and the organic solvent was removed under vacuum. The residue was suspended in 20 mL diethylether, 20 mL hexane was added, the product filtered off and washed two times with 20 ml hexane. After drying in vacuum H3CC[CH2P(p-tol)2·BH3]3 (3-PG, 1.92 g, 2.6 mmol, 72% yield) was obtained as a white powder. A 100 mL Teflon caped Schlenk flask was charged with H3CC[CH2P(p-tol)2·BH3]3 (3-PG, 1.92 g, 2.6 mmol), DABCO (1.58 g, 14.1 mmol), and 20 mL toluene. The headspace was evacuated and the flask was heated at 80 °C for 2 days under static vacuum. The solution was filtered through a small plug of silica (3 cm) on a glass filter frit and the product extracted from the residue by washing it twice with 100 mL toluene. The solvent as removed from the combined organic layers to obtain a thick oil. After trituration of the oil with diethylether, a white powered was formed, which was filtered of and dried in vacuum to give H3CC(CH2P(p-tol)2)33 (1.03 g, 0.18 mmol, 72% yield; 51% yield overall) as a sticky white solid. All spectroscopic data was consistent with those reported in the literature.8Synthesis of RuHCl(CO){H3CC[CH2P(p-tol)2]3} (C)
A round-bottom Schlenk flask was charged with H3CC(CH2P(p-tol)2)3 (500 mg, 0.71 mmol), RuHCl(CO)(PPh3)3 (612 mg, 0.64 mmol) and 40 mL toluene. The colorless solution with an off white precipitate heated at reflux for 2 h to give a light yellow precipitate. 10 mL pentane was added to the reaction mixture, the preticipate was filtered off via a sinter frit and washed three times with 10 mL pentane. After drying in vacuum, RuHCl(CO){H3CC[CH2P(p-tol)2]3} C (499 mg, 0.57 mmol, 80% yield) was obtained as a yellow powder.31P{1H} NMR (81.0 MHz, CD2Cl2, δ): 46.5 (dd, 2JPP = 40 Hz, 2JPP = 18 Hz, 1P), 12.6 (dd, 2JPP = 40 Hz, 2JPP = 32 Hz), −0.8 (2JPP = 32 Hz, 2JPP = 18 Hz); 1H NMR (200.1 MHz, CD2Cl2, δ): 7.73–6.58 (m, 24 H, CH), 2.52–2.08 (m, 15 H, CH2 and CH3), 1.50–1.46 (m, 3H, CH3), −6.02 (ddd, 2JHP = 93.6 Hz, 2JHP = 18.8 Hz, 2JHP = 14.9 Hz, 1H, RuH).
IR (KBr): 521, 558, 624, 713, 735, 804, 838, 1020, 1092, 1190, 1397, 1440, 1499, 1599, 1895 (m, νRu–H), 1979 (vs, νCO), 2866, 2920, 2948, 3019 cm−1.
Elemental Analysis Calc. (Found): C 65.93% (65.75%), H 5.99% (6.01%).
Reaction of RuHCl(CO)(PPh3)3 with H3CC(CH2PPh2)2(2-pyridyl)
A round-bottom Schlenk flask was charged with H3CC(CH2PPh2)2(2-pyridyl) 4 (160 mg, 0.31 mmol, 1.2 equiv.), (PPh3)3RuHCl(CO) (250 mg, 0.26 mmol, 1 equiv.) and dichloromethane (40 mL). The light yellow solution was stirred at room temperature for 5 days to light yellow solution. The solution was filtered to remove a small amount of white precipitate and the volume was reduced to 2 mL. A fine yellow precipitate was obtained with the addition of hexane (∼20 mL). The yellow solid contained a mixture of D-1, D-2, and D-3. Recrystallization by slow solvent diffusion of pentane (20 mL) into a concentrated DCM solution (2 mL) gave a small amount of 21 (∼20 mg) in reasonable purity (∼95%). A second recrystallization attempt of the same sample gave a mixture of D-1, D-2 and D-3. Single crystal of D-3 were isolated from the sample and used for X-ray diffraction analysis.MS (LIFDI, crude mixture): m/z (%): 896.2 (100%, D-1,2+–Cl), 669.0 (25%, D-3+–Cl).
D-1: 31P{1H} NMR (121.4 MHz, CD2Cl2, δ): 44.9 (dd, 2JPP = 280 Hz, 2JPP = 20 Hz), 28.6 (dd, 2JPP = 280 Hz, 2JPP = 20 Hz), 3.85 (t, 2JPP = 20 Hz); 1H NMR (300.1 MHz, CD2Cl2, δ): −6.55 (ddd, 2JHP = 18 Hz, 2JHP = 22 Hz, 2JHP = 108 Hz), all other peaks are overlapping with those of D-2 and D-3.
D-2: 31P{1H} NMR (121.4 MHz, CD2Cl2, δ): 45.9 (dd, 2JPP = 230 Hz, 2JPP = 20 Hz), 34.2 (dd, 2JPP = 230 Hz, 2JPP = 20 Hz), 9.8 (t, 2JPP = 20 Hz); 1H NMR (300.1 MHz, CD2Cl2, δ): −5.19 (dt, 2JHP = 17 Hz, 2JHP = 92 Hz), all other peaks are overlapping with those of D-1 and D-3.
D-3: 31P{1H} NMR (121.4 MHz, CD2Cl2, δ): 37.9 ppm (s); 1H NMR (300.1 MHz, CD2Cl2, δ): all peaks are overlapping with those of D-1 and D-2.
Synthesis of RuHCl(CO)(PPh3)[H3CC(CH2PPh2)2(CH2OH)] (E)
In a glovebox, a round-bottom Schlenk flask was charged with H3CC(CH2PPh2)2(CH2OH) 5 (0.502 g, 1.1 mmol), RuHCl(CO)(PPh3)3 (952 mg, 1.0 mmol) and 40 mL toluene. The colorless solution with an off white precipitate heated at reflux for 2 h, then the volume of toluene was reduced under vacuum to ca. 10 mL. In the glovebox, 10 mL pentane was added to the reaction mixture, forming a pale yellow powder. The precipitate was filtered off via a sinter frit and washed three times with 10 mL pentane. After drying in vacuum, RuHCl(CO)(PPh3)[H3CC(CH2PPh2)2(CH2OH)] E (0.610 g, 0.69 mmol, 69% yield) was obtained as a pale yellow powder. The 31P NMR indicated that the isolated compounds were the mixture of regioisomers of E.IR (KBr): 517, 695, 742, 806, 839, 1054, 1091, 1158, 1188, 1312, 1434, 1481, 1586, 1924 (vs, νCO and νRu–H), 2928, 3053 cm−1.
Elemental Analysis Calc. (Found): C 65.19% (62.58%), H 5.24% (5.15%).
31P{1H} NMR (162.1 MHz, CD2Cl2, δ): 44.2 (dd, 2JPP = 282 Hz, 2JPP = 20 Hz), 43.6 (dd, 2JPP = 282 Hz, 2JPP = 20 Hz), 27.4 (dd, 2JPP = 282 Hz, 2JPP = 20 Hz), 26.9 (dd, 2JPP = 282 Hz, 2JPP = 20 Hz), 3.0 (t, 2JPP = 20 Hz), 0.4 (t, 2JPP = 20 Hz). All peaks are derived from the regioisomers of E; 1H NMR (400.3 MHz, CD2Cl2, δ): 8.05–6.80 (m, 35H, CH), 3.30–0.18 (m, 9H, CH2 and CH3), −6.73 (ddd, 2JHP = 109 Hz, 2JHP = 23 Hz, 2JHP = 17 Hz, 1H, RuH), −6.83 (ddd, 2JHP = 109 Hz, 2JHP = 23 Hz, 2JHP = 17 Hz, 1H, RuH). All peaks are derived from the regioisomers of E.
Reaction and RuHCl(CO)(PPh3)[H3CC(CH2PPh2)2(CH2OH)] (E) with t-BuOK
In a glovebox, a round-bottom Schlenk flask was charged with RuHCl(CO)(PPh3)[H3CC(CH2PPh2)2(CH2OH)] E (0.114 g, 0.13 mmol), t-BuOK (14.4 mg, 0.13 mmol) and 5 mL toluene, forming a red solution at room temperature in 2 h. All volatile compounds were removed under vacuum, then 0.5 mL toluene and 5 mL pentane was added to a red powder. The resulting suspension was passed through a pad of celite, then a red filtrate was dried under vacuum to give alkoxo ruthenium complex F (81.3 mg, 0.096 mmol, 75% yield) as a deep red powder. The 1H and 31P NMR spectra of the obtained powder suggested that it was constituted of mainly alkoxo ruthenium complex F with some impurities including small amounts of the unreacted E.Characteristic peaks of F are described below:
31P{1H} NMR (243.0 MHz, d8-THF, δ): 35.7 (dd, 2JPP = 276 Hz, 2JPP = 20 Hz), 33.5 (dd, 2JPP = 276 Hz, 2JPP = 20 Hz), 13.7 (dd, 2JPP = 20 Hz); 1H NMR (600.2 MHz, d8-THF, δ): −4.65 (ddd, 2JHP = 113 Hz, 2JHP = 26 Hz, 2JHP = 13 Hz, 1H, RuH). CH3 peaks from t-Bu group were not observed in 1H NMR spectra.
IR (KBr): 515, 695, 742, 836, 999, 1092, 1186, 1434, 1480, 1586, 1896 (m, νRu–H), 1910 (vs, νCO), 2674, 2754, 2921, 2951, 3051 cm−1.
MS (LIFDI): m/z (%): 848.01 (100%, F+).
A representative procedure for catalyst screening described in Table 1
Entry 1: In an argon-filled glovebox, to a premex autoclave (60 mL, stainless steel) equipped with a magnetically coupled propeller blade stirrer was added RuHCl(CO)(triphos) A (36 mg, 0.047 mmol), toluene (17 mL), and 1-octanol (3.0 g, 24 mmol), then the autoclave was sealed. After introducing NH3 gas (7–8 bar) at room temperature, the autoclave was heated to 165 °C for 15 h with vigorous stirring (700–800 rpm) without reintroducing NH3 After cooling to the room temperature, the pressurized NH3 gas in the autoclave was released in a fume hood. An aliquot of the reaction mixture was taken for the GC analysis, and the conversion of 1-octanol and the yield of amine products were calculated based on GC area %.Entry 2: RuHCl(CO)(PPh3)3 (45 mg, 0.047 mmol) and triphos (33 mg, 0.052 mmol) were used.
Entry 3: RuHCl(CO)(PPh3)3 (45 mg, 0.047 mmol) was used.
Entry 4: RuHCl(CO){H3CC(CH2PPh2)2[CH2P(p-tol)2]} (38 mg, 0.047 mmol) was used.
Entry 5: RuHCl(CO){H3CC[CH2P(p-tol)2]3} (40 mg, 0.047 mmol) was used.
Entry 6: RuHCl(CO)(PPh3)3 (45 mg, 0.047 mmol) and CH3C (CH2PPh2)2(2-pyridyl) 4 (26 mg, 0.046 mmol) were used.
Entry 7: RuHCl(CO)(PPh3)[H3CC(CH2PPh2)2(CH2OH)] E (41 mg, 0.047 mmol) was used.
Entry 8: The proposed alkoxo-ruthenium complex F (20 mg, 0.023 mmol), toluene (8.5 mL), and 1-octanol (1.5 g, 12 mmol) were used.
Entry 9: RuHCl(CO)(PPh3)3 (45 mg, 0.047 mmol) and CH3C (CH2PPh2)2(CH2OMs) 6 (28 mg, 0.052 mmol) were used.
Entry 10: RuHCl(CO)(PPh3)3 (45 mg, 0.047 mmol) and dppdmp (23 mg, 0.052 mmol) were used.
Entry 11: RuHCl(CO)(PPh3)3 (45 mg, 0.047 mmol) and dppp (21 mg, 0.052 mmol) were used.
Entry 12: RuHCl(CO)(PPh3)3 (45 mg, 0.047 mmol), dppp (21 mg, 0.052 mmol), and toluene (6 mL) were used.
Entry 13: RuHCl(CO)(PPh3)3 (45 mg, 0.047 mmol), dppp (21 mg, 0.052 mmol), toluene (6 mL), and p(NH3) = 4 bar (53 mmol of NH3) were used.
Acknowledgements
CaRLa (Catalyst Research Laboratory) is co-financed by Ruprechts-Karls-University Heidelberg (Heidelberg University) and BASF SE.Notes and references
- (a) B. R. Brown, The Organic Chemistry of Aliphatic Nitrogen Compounds, Oxford University, New York, 1994 Search PubMed; (b) S. A. Lawrence, Amines: Synthesis, Properties and Applications, Cambridge University Press, Cambridge, 2005 Search PubMed; (c) P. Roose, K. Eller, E. Henkes, R. Rossbacher and H. Höke, Amines, Aliphatic: Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2015 Search PubMed.
- R. N. Salvatore, C. H. Yoon and K. W. Jung, Tetrahedron, 2001, 57, 7785 CrossRef CAS.
- (a) J. Kim, H. J. Kim and S. Chang, Eur. J. Org. Chem., 2013, 3201 CrossRef CAS; (b) J. L. Klinkenberg and J. F. Hartwig, Angew. Chem., Int. Ed., 2011, 50, 86 CrossRef CAS PubMed; (c) S. Bähn, S. Imm, L. Neubert, M. Zhang, H. Neumann and M. Beller, ChemCatChem, 2011, 3, 1853 CrossRef.
- Recent reviews, see: (a) S. Bähn, S. Imm, L. Neubert, M. Zhang, H. Neumann and M. Beller, ChemCatChem, 2011, 3, 1852 Search PubMed; (b) C. Gunanathan and D. Milstein, Chem. Rev., 2014, 114, 12024 CrossRef CAS PubMed; (c) K. Shimizu, Catal. Sci. Technol., 2015, 5, 1412 RSC; (d) Q. Yang, Q. Wang and Z. Yu, Chem. Soc. Rev., 2015, 44, 2305 RSC.
- (a) C. Gunanathan and D. Milstein, Angew. Chem., Int. Ed., 2008, 47, 8661 CrossRef CAS PubMed; (b) D. Pingen, C. Müller and D. Vogt, Angew. Chem., Int. Ed., 2010, 49, 8130 CrossRef CAS PubMed; (c) S. Imm, S. Bähn, L. Neubert, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 8126 CrossRef CAS PubMed; (d) G. Walther, J. Deutsch, A. Martin, F. E. Baumann, D. Fridag, R. Franke and A. Köckritz, ChemSusChem, 2011, 4, 1052 CrossRef CAS PubMed; (e) W. Baumann, A. Spannenberg, J. Pfeffer, T. Haas, A. Köckritz, A. Martin and J. Deutsch, Chem. – Eur. J., 2013, 19, 17702 CrossRef CAS PubMed; (f) D. Pingen, O. Diebolt and D. Vogt, ChemCatChem, 2013, 5, 2905 CrossRef CAS; (g) D. Pingen, M. Lutz and D. Vogt, Organometallics, 2014, 33, 1623 CrossRef CAS; (h) X. Ye, P. N. Plessow, M. K. Brinks, M. Schelwies, T. Schaub, F. Rominger, R. Paciello, M. Limbach and P. Hofmann, J. Am. Chem. Soc., 2014, 136, 5923 CrossRef CAS PubMed.
- E. Balaraman, D. Srimani, Y. D. Posner and D. Milstein, Catal. Lett., 2015, 145, 139 CrossRef CAS.
- (a) S. Imm, S. Bähn, M. Zhang, L. Neubert, H. Neumann, F. Klasovsky, J. Pfeffer, T. Haas and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 7599 CrossRef CAS PubMed; (b) E. J. Derrah, M. Hanauer, P. N. Plessow, M. Schelwies, M. K. da Silva and T. Schaub, Organometallics, 2015, 34, 1872 CrossRef CAS; (c) T. Schaub, B. Buschhaus, M. K. Brinks, M. Schelwies, R. Paciello, J. P. Melder and M. Merger, WO2012119927, 2012 Search PubMed.
- (a) H. Heidel, G. Huttner and G. Helmchen, Z. Naturforsch., B: Chem. Sci., 1993, 48, 1681 CrossRef CAS; (b) A. Muth, O. Walter, G. Huttner, A. Asam, L. Zsolnai and C. Emerich, J. Organomet. Chem., 1994, 468, 149 CrossRef CAS.
- S. Doherty, E. G. Robins, M. Nieuwenhuyzen, P. A. Champkin and W. Clegg, Organometallics, 2002, 21, 4147 CrossRef CAS.
- (a) T. Seitz, A. Muth and G. Huttner, Chem. Ber., 1994, 127, 1837 CrossRef CAS; (b) Y.-T. Hsieh, C.-M. Cheng, M.-S. Peng and T.-S. Liu, J. Chem. Soc., Dalton Trans., 1994, 3499 RSC.
- Attempts to install other diorganophosphino moiety or heteroatom such as P(o-tolyl)2, P(p-F-C6H4)2, PCy2, PiPr2, and NH2, were unsuccessful by using our approach.
- K.-M. Sung, S. Huh and J.-M. Jun, Polyhedron, 1999, 18, 469 CrossRef CAS.
- RuHCl(CO)(triphos) A was characterised by X-ray analysis, see: S. Savourey, G. Lefèvre, J.-C. Berthert, P. Thuéry, C. Genre and T. Contat, Angew. Chem., Int. Ed., 2014, 53, 10466 CrossRef CAS PubMed.
- Preparation and characterization of RuHCl(CO)(dppp)(PPh3) from RuHCl(CO)(PPh3)3 and dppp was reported, see: (a) A. Santos, J. López, J. Montoya, P. Noheda, A. Romero and A. M. Echavarren, Organometallics, 1994, 13, 3605 CrossRef CAS; (b) S. Huh, Y. Cho and J.-M. Jun, Polyhedron, 1994, 13, 1887 CrossRef CAS.
- Cyclohexanol and ammonia undergo the selective nomo-alkylation with dppp ligand. See ref. 5e.
- In Fig. 6, P(NH3) = 4.1–4.5 bar was used, which corresponded to approximately 53 mmol of NH3 in the autoclave.
- (a) R. Yamaguchi, S. Kawagoe, C. Asai and K. Fujita, Org. Lett., 2008, 10, 181 CrossRef CAS PubMed; (b) R. Kawahara, K. Fujita and R. Yamaguchi, J. Am. Chem. Soc., 2010, 132, 15108 CrossRef CAS PubMed; (c) S. Wöckel, M. Schelweis, M. K. Brinks, F. Rominger, P. Hofmann and M. Limbach, Org. Lett., 2013, 15, 266 CrossRef PubMed.
- Program SADABS 2012/1 for absorption correction, see: G. M. Sheldrick, Bruker Analytical X-Ray Division, Madison, Wisconsin, 2012 Search PubMed.
- L. Gonzáles-Sebastián, M. Flores-Alamo and J. J. García, Organometallics, 2012, 31, 8200 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1440322–1440325. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt04870b |
| This journal is © The Royal Society of Chemistry 2016 |