
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and characterisation of bismacrocyclic DO3A-amide derivatives – an approach towards metal-responsive PARACEST agents†
Nevenka
Cakić
a,
Tatjana Ž.
Verbić
b,
Ratomir M.
Jelić
c,
Carlos
Platas-Iglesias
d and
Goran
Angelovski
*a
aMR Neuroimaging Agents, Max Planck Institute for Biological Cybernetics, Spemannstr. 41, 72076 Tübingen, Germany. E-mail: goran.angelovski@tuebingen.mpg.de; Fax: +49 7071 601 919; Tel: +49 7071 601 916
bDepartment of Analytical Chemistry, Faculty of Chemistry, University of Belgrade, Studentski trg 12-16, 11000 Belgrade, Serbia
cFaculty of Medicinal Science, University of Kragujevac, S. Markovića 69, 34000 Kragujevac, Serbia
dCentro de Investigaciones Científicas Avanzadas (CICA) and Departamento de Química Fundamental, Universidade da Coruña, Campus da Zapateira, Rúa da Fraga 10, 15008 A Coruña, Spain
First published on 26th February 2016
Abstract
Three new bismacrocyclic Ln3+ chelates consisting of triamide derivatives of cyclen with glycine, methyl and tert-butyl substituents (L1–3, respectively) linked to an acyclic EGTA-derived calcium chelator were synthesised as potential MRI contrast agents (EGTA – ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid). Eu3+ and Yb3+ complexes of L1–3 were investigated as chemical exchange saturation transfer (CEST) agents. Moderate to minor CEST effects were observed for Eu2L1, Eu2L2 and Yb2L2 complexes in the absence of Ca2+, with negligible changes upon addition of this metal ion. Luminescence steady-state emission and lifetime experiments did not reveal any changes in the coordination environment of the complexes, while the number of inner-sphere water molecules remained constant in the absence and presence of Ca2+. The protonation constants of Eu2L1 and Eu2L2 and stability constants of their complexes with Ca2+, Mg2+ and Zn2+ were determined by means of potentiometric titrations. The results show that the charge of the complex dramatically affects the protonation constants of the EGTA-binding unit. The stability constants of the complexes formed with Ca2+, Mg2+ and Zn2+ are several orders of magnitude lower than those of EGTA. These findings indicate that the nature of Ln3+ chelates and their charge are the main reasons for the observed results and weaker response of these EGTA-derived triamide derivatives compared to their tricarboxylate analogues.
Introduction
The continuous development of contrast agents (CAs) for magnetic resonance imaging (MRI) has provided a wide range of structurally different compounds with a range of diagnostic and therapeutic applications.1,2 The first generation and the most widely used MRI contrast agents are based on paramagnetic Gd3+ complexes or superparamagnetic iron–oxide nanoparticles (T1- and T2-shortening agents, respectively). Although these CAs remain extensively investigated in basic research and in clinical medicine, they exhibit certain limitations related to their lack of tissue specificity and response to the chemical environment. Therefore, alternative approaches for producing image contrasts that provide additional information are greatly appreciated, leading to the development of several methodologies based on novel types of CAs.3 Among these, the mechanism for altering MR contrast based on chemical exchange saturation transfer (CEST) has been recently established.4 This technique has been known and used in nuclear magnetic resonance (NMR) for more than four decades, however only lately has it attracted greater attention due to its capability to generate an MRI contrast on its own, and also due to its high sensitivity towards changes in the microenvironment.5 CEST imaging requires sufficiently slow exchange on the magnetic resonance time scale to allow selective irradiation of the protons of interest. The rate of exchange (kex) that occurs between the two magnetically distinct environments must not be greater than the difference in frequency between them (Δω), while several other physicochemical parameters can also affect CEST MR contrast mechanisms, including the relaxation rates of the two pools involved in chemical exchange, temperature and concentration.6 Furthermore, a methodology that exploits particular classes of paramagnetic lanthanide complexes for introducing tissue contrast via a CEST mechanism has also been developed. These complexes are specifically designed to shift exchangeable protons (–NH, –OH, –SH or bound water) further away from the bulk water allowing their distinct saturation, consequently reducing the intensity of the bulk water MR signal and hence producing the change in MRI contrast.7The dependence of CEST contrast on diverse factors, including those involving paramagnetic (PARACEST) agents, can be used for the detection of specific biological processes in tissues by means of MRI. Consequently, various molecular imaging probes responsive to particular molecular events – the so called bioresponsive or “smart contrast agents” (SCAs) – have been designed.2 In most of the cases, MRI signal changes are triggered by the variation in hydration number or rotation dynamics for the T1- and T2-based CA. On the other hand, signal differences produced by CEST agents are caused by the exchange rate and the chemical shift of the exchangeable proton pool, making CEST agents extremely sensitive to environmental changes, and leading to the fast development of responsive CEST agents. The most widely investigated probes of this class show response to pH and temperature, which allow a direct read-out of these relevant physiological parameters in the disease state.8 However, systems that provide responses to metabolites, biologically relevant ions, enzyme or redox activity have also been reported.6
The usage of SCAs in MRI for observing specific biological processes is an extremely promising and potentially very beneficial approach to study various functional processes at the molecular and cellular level. For example, successful monitoring of Ca2+ would be an extremely important step for the understanding of basic physiological processes in the brain. To date, there is a single report of PARACEST agents that provide responses to this metal ion. The lanthanide ion chelator consisted of the tetraamide derivative of cyclen and four imino(diacetate) moieties that were envisaged to interact with Ca2+. The corresponding Yb3+ and Eu3+ complexes were shown to provide CEST responses to Ca2+; however, Mg2+ induced similar CEST changes.9
On the other hand, excellent and selective responses to Ca2+ were previously obtained for a series of GdDO3A-based mono- and bismacrocyclic SCAs.10,11 In either of the cases, the organic molecule was comprised of two different moieties: a cyclen-based ring(s) appended with acetate arms for Gd3+ chelation and an acyclic EGTA-derived part (EGTA – ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid), as a high affinity and selective calcium chelator. The detailed studies on these systems revealed that Ca-induced alteration of the hydration number (q) was the major factor responsible for the longitudinal relaxivity (r1) change.10 The q alteration is the direct consequence of the change in coordination of the carboxylate groups in the EGTA-derived chelator in the major square antiprismatic (SAP) isomer, which flips from the Gd3+ coordination environment to Ca2+ upon its addition. Moreover, the amide groups of the EGTA-derived chelator are also expected to be in the vicinity of the lanthanide ion in the presence or absence of Ca2+.11
Having these insights on the specific coordination aspects that produce remarkable r1 changes on Gd-based Ca-responsive SCAs, we sought to investigate their structural analogues that could potentially serve as responsive PARACEST agents. However, polyamino polycarboxylate DOTA/DO3A-type ligands are not suitable for providing a CEST effect due to the rapid exchange of the coordinated water molecule, hence they should be converted into slow-exchanging species.1 This can be achieved by replacing the polyanionic arms of the ligand with the neutral ones, thereby decreasing the water exchange rate of the complex and making the agents suitable for PARACEST.12,13 The most commonly investigated ligands for this purpose are tetraamide derivatives of DOTA, especially DOTAM-gly and its derivatives, although DO3A and DO2A amide derivatives have also been reported.14 Thus, in an attempt to prepare Ca2+ responsive PARACEST agents we designed three different bismacrocyclic ligands, each of them bearing a standard EGTA-derived Ca-chelator coupled to amide-type macrocyclic chelators for the complexation of lanthanide metal ions. We varied the groups on the amide moieties aiming to investigate the effect of charge, hydrophobicity and steric hindrance. This resulted in the use of glycine, methyl and tert-butyl substituents (L1–3, respectively) to replace the six acetic moieties of the T1-responsive SCA (Chart 1). Upon their synthesis, various physicochemical aspects were investigated, including their CEST effect, hydration number assessment by means of time-resolved luminescence decay measurements, NMR studies and estimations of stability constants with endogenous metal ions by means of potentiometric titrations. CEST studies and luminescence lifetime measurements in the presence of Ca2+ were also carried out to assess the responsiveness of the synthesized agents to this metal ion.
 | ||
| Chart 1 Chemical structures of ligands L1–3 investigated in this work. | ||
Results and discussion
Synthesis of the ligands
The desired ligands were prepared according to a convenient six-step procedure (Scheme 1). The synthesis commenced from the commercially available cyclen, which was monoalkylated with benzyl(3-bromopropyl)carbamate 1 to give the building block 2. The installation of different amide substituents was accomplished by alkylation of 2 with particular halogenides 3a–c in acetonitrile. The primary amines 5a–c were obtained by reductive hydrogenation of 4a–c in ethanol using 10% Pd on carbon as a catalyst. Further coupling of the obtained amides 6a–c with amine 7 led to the protected bismacrocyclic ligands 8a–c, which were treated with hydrochloric or formic acid to afford the desired ligands L1–3. Finally, bimetallic Eu3+ or Yb3+ complexes were prepared by mixing the ligands with the corresponding LnCl3 salt while maintaining the pH value between 6 and 7.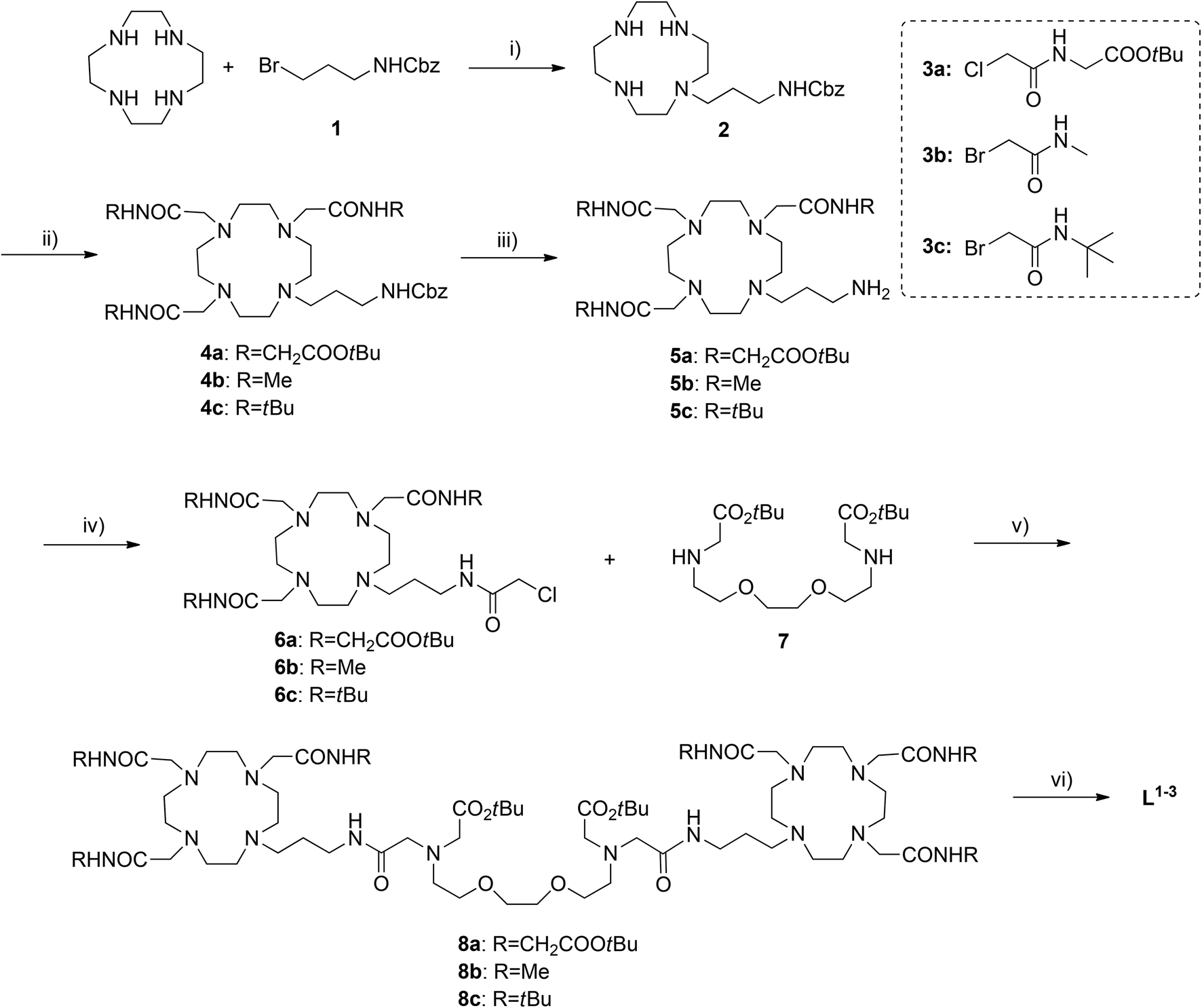 | ||
| Scheme 1 Synthesis of the ligands L1–3. Reagents and conditions: (i) Et3N, CH2Cl2, RT, 59%. (ii) 3a–c, K2CO3, CH3CN, 70 °C, 66–83%. (iii) H2, Pd/C, EtOH, RT, 92–96%. (iv) (ClCH2CO)2O, Et3N, CH3CN, RT, 74–82%. (v) K2CO3, KI, CH3CN, 70 °C, 67–79%. (vi) HCl or HCOOH, RT, 86–93%. | ||
CEST properties
All the Z-spectra had been initially acquired at 25 °C with identical concentrations of the complexes (15 mM per Ln3+). Eu2L1 exhibited a weak CEST effect at 57 ppm, which is attributed to the proton exchange between the Eu-bound and bulk water. Temperature enhancement to 37 °C had a noticeable influence on the CEST effect resulting in an almost double increase in intensity and an upfield shift of 3 ppm (Fig. 1a and b). Previously it was shown that PARACEST agents are more suitable for noninvasive MRI thermometry methods than those depending on T1 relaxation-time changes, chemical shift or the diffusion coefficient of bulk water. In the former case a strong linear dependence of the chemical shift of the bound water pool with temperature was observed (∼0.5 ppm °C−1), while methods based on the determination of diffusion coefficients present low temperature sensitivity (∼0.01 ppm °C−1).15 The CEST effect observed for Eu2L1 is in good correlation with these findings, showing a shift of 3 ppm for the increase in temperature of 12 °C (∼0.25 ppm °C−1). However, the addition of Ca2+ (up to 10 equiv.) did not provoke a marked change in the CEST effect at any of the investigated temperatures. This result suggests that Ca2+ addition does not trigger important changes in the coordination environment of the paramagnetic Eu3+ centre, unlike the Gd-based Ca-sensitive systems containing acetate pendant arms. | ||
| Fig. 1 The CEST spectra of 15 mM complexes (irradiation time 3 s, B1 = 25 μT) recorded in the absence (red) and presence (black) of 1 equiv. of Ca2+ for: (a) Eu2L1, recorded at 25 and 37 °C, showing signals at 57 ppm and 54 ppm, respectively; (b) Eu2L1 (magnified CEST effects, conditions provided under (a); (c) Yb2L2, insets show magnified signals originating from bound water (230 ppm) and amide (∼−24 ppm) protons; (d) Eu2L2 the inset shows the magnified signal originating from bound water (53 ppm). | ||
The Z-spectra were also recorded using solutions of the complexes with chelators L2–3, which possess a net positive charge. For the ligand with N-methylamide groups, both Eu2L2 and Yb2L2 complexes showed a very weak CEST effect upon saturation of the resonances corresponding to the coordinated water molecule (∼5% and 1% at 53 ppm and 230 ppm, respectively). Similar to Eu2L1, a very small quenching of the CEST signal was observed upon Ca2+ addition in both of these complexes (Fig. 1c and d). Moreover, the Yb2L2 complex provides a weak CEST effect at an offset frequency of ∼−24 ppm that remains nearly unaffected upon addition of Ca2+. This CEST effect is attributed to exchangeable NH protons, and is in agreement with previous findings that revealed CEST peaks due to amide protons in Yb3+ complexes in the range −15 to −29 ppm.16–18 Finally, the Z-spectra of the complexes with bulky tert-butyl substituents Eu2L3 and Yb2L3 did not show any CEST effect in the presence or absence of Ca2+ (data not shown).
Apparently, the acquired Z-spectra and obtained CEST properties indicated that the prepared DO3A-amide Eu3+ and Yb3+ complexes likely have different coordination characteristics from their carboxylic Gd3+ analogues. The strongest CEST signal was obtained for the DO3AM-gly-type derivative (Eu2L1), suggesting that the polarity of the side arms and the overall charge of the complexes play important roles in determining the exchange rate of the coordinated water molecule responsible for the CEST signal. Previous reports showed that the introduction of bulky groups into the amide side arms of DOTAM derivatives accelerates water exchange in a favourable way for the CEST effect,19 although the opposite effects were also obtained.20 In this study the CEST effect gradually decreases on the DO3AM-type bismacrocyclic derivatives towards the less polar and bulky substituents resulting in the preferred order CH2COOH > CH3 > C(CH3)3. However, the absence of any response to Ca2+ (its addition did not cause observable reductions or increases in CEST effects on Eu2L1–2 or Yb2L2) suggested that the coordination environment of the paramagnetic ions in these triamide systems has been changed compared to their tricarboxylic analogues, requiring further investigation.
Luminescence experiments
The hydration states in the presence and absence of Ca2+ can provide a good indication of potential changes in the environment of the paramagnetic ion upon Ca2+ addition. Thus, the luminescence emission lifetimes of Eu2L1–3 were recorded in H2O and D2O, and the hydration numbers (q) were calculated according to a previously established methodology (Table 1).21 The obtained results show that the q number stays constant, within the experimental error, upon calcium addition to all the three investigated complexes. This behaviour is opposite that of DO3A-based analogue SCA systems, which showed an increase in the number of inner-sphere water molecules upon Ca2+ addition.10,11 Further, the findings indicate that Eu2L1 and Eu2L3 are monohydrated complexes, while the less sterically hindered complex Eu2L2 displays an equilibrium between dihydrated and monohydrated species. The absence of prominent CEST effects upon Ca2+ addition to aqueous solutions of Eu2L1–3 can certainly be correlated with hydration states remaining constant, indicating that the addition of Ca2+ does not provoke significant changes in the coordination environment of the lanthanide ion.| Complex | Without Ca2+ | +Ca2+ (1 equiv.) | ||||
|---|---|---|---|---|---|---|
| k H2O (ms−1) | k D2O (ms−1) | q | k H2O (ms−1) | k D2O (ms−1) | q | |
| Eu2L1 | 1.58 | 0.66 | 1.08 | 1.56 | 0.66 | 1.05 |
| Eu2L2 | 2.16 | 0.71 | 1.71 | 2.17 | 0.73 | 1.70 |
| Eu2L3 | 1.76 | 0.75 | 1.18 | 1.76 | 0.79 | 1.13 |
The luminescence steady-state emission spectra of Eu2L1–3 in H2O were also recorded. Similarly to the decay experiments, addition of Ca2+ did not produce any significant change in the intensity or shape of the major 5D0 → 7F0–4 transitions (Fig. S1 in the ESI†). However, further splitting of the signals due to the 5D0 → 7F1 and 5D0 → 7F4 transitions and change in the intensity ratios of 5D0 → 7F1 and 5D0 → 7F2 transitions at 590 and 615 nm were observed at basic pH for Eu2L1–2, suggesting the change in polarisability of the axial donor and the local symmetry at the metal centre (Fig. S2 in the ESI†). The major cause for these spectral alterations was apparently a change in protonation states of amines from the EGTA-derived chelator and inner-sphere water molecules (see below).
NMR studies
The 1H NMR spectra of Eu2L1–3 complexes were recorded in D2O solutions at pD ∼ 8.0 (Fig. 2). They present relatively broad resonances that spread over the range ∼−20 to 30 ppm due to the paramagnetic shifts induced by the metal ion. The spectrum of Eu2L1 points to the presence of at least three different isomers in solution. It is well-known that DOTA-like complexes may exist in solution as two different isomers providing either a square-antiprismatic (SAP) or a twisted-square antiprismatic (TSAP) coordination around the lanthanide(III) ion. These isomers differ either in the orientation of the pendant arms of the macrocycle, which is often denoted as Δ or Λ, or the conformation of the cyclen moiety [(δδδδ) or (λλλλ)].22,23 In Eu3+ complexes of DOTA and DO3A derivatives the signals of the pseudo-axial protons on the cyclen rings are usually found between 24 and 45 ppm in the square antiprismatic (SAP) isomer and below 25 ppm in the twisted-square antiprismatic (TSAP) isomer.24–29 In the case of Eu2L1, one of the species present in solution shows signals in the range +15 to −20 ppm, while the second isomer with a smaller population display signals due to the macrocyclic axial protons at lower fields, one clearly visible at ∼19 and a broader one at 27 ppm. These results point to the presence of two nine coordinate species with SAP and TSAP coordination around the metal ion, the overall population being dominated by the TSAP form (ca. 90%). This ratio of species is completely inverse to that found for the responsive Gd-DO3A complexes where only the SAP isomer changes its hydration upon binding with Ca2+.11 Moreover, a third set of signals dominated by two resonances at ca. 7.5–8.5 ppm is also observed. The spectrum of Eu2L2 is dominated by the latter signals, which on the basis of the hydration number of this complex can be assigned to a complex DO3A-type species containing two coordinated water molecules. Similarly to Eu2L1, the spectrum of Eu2L3 displays signals due to the monohydrated SAP and TSAP isomers, again with the minor SAP isomer representing ca. 30% of the population of the TSAP isomer. Additionally, the species attributed to the bis-hydrated complex are also present, which is likely the reason why luminescence lifetime measurements provide hydration numbers slightly higher than one (Table 1). | ||
| Fig. 2 1H NMR spectra of Eu2L1–3 recorded in D2O solutions (pD ∼ 8.0, 25 °C). The asterisks indicate the most shifted resonances of axial protons of the macrocycle in the SAP form. The insets show enhancements of the 15–30 ppm regions obtained for Eu2L1 and Eu2L3. | ||
Potentiometric titrations
To further characterize Eu2L1–2 complexes, their protonation constants, as well as stability constants of the complexes formed with Ca2+, Mg2+ and Zn2+ were determined by means of potentiometric titrations (Fig. S3 and S4 in the ESI†).| pEu + qH + rL ⇄ EupHqLr; βp,q,r (L = L1 or L2) | (1) |
 | (2) |
In order to study speciation in the three-component systems Eu–H–L or Eu–OH–L (L = L1 or L2), it was necessary to characterize the binary equilibria, i.e., hydrolysis of Eu3+ and the ligands’ proteolytic equilibria. The equilibrium constants of Eu3+ hydrolysis were taken from the literature,30 and the ligand protonation constants calculated using the ADMET Predictor software31 (Table S1 in the ESI†), showing good agreement with previously published values for DOTAM-type systems.32 The equilibrium constants of the complexes were determined using the Hyperquad 2008 software (using ionic product value pKw = 13.77).33 Species distribution diagrams were plotted according to calculated constants using the HySS software.34
The equilibrium constants of Eu–H–L complexes were determined by acid–base potentiometric titrations (Tables 2 and S2 in the ESI†). Analysis of the potentiometric titration data was performed to find the model that gives the best fit to the experimental data (statistical parameters which determine the quality of fit are provided in Table S2 in the ESI†). The calculations revealed the formation of [Eu2(HnL)] (n = 1, 2, 3) as well as [Eu2(L)] complexes. The formation of [Eu2L(OH)2] complexes was also noticed. The obtained protonation constants were compared to those previously published for ethylenediaminetetraacetic acid (EDTA), EGTA and EGTA–bisamide (Table 2),35 while the experimentally determined stability constants with standard deviations are provided in the ESI (Table S2†).
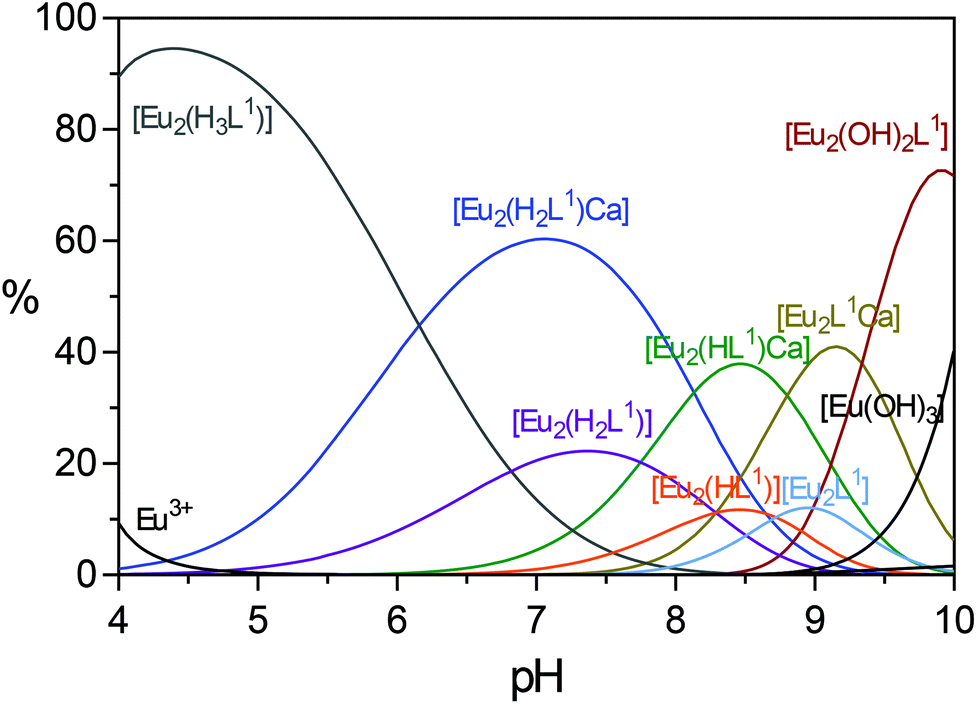
The first two protonation constants (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) KH1 and logKH2) stand for protonation of the amine nitrogen atoms of the EGTA-derived part. These values are quite different for Eu2L1 and Eu2L2. The complex Eu2L1 displays a higher basicity similar to EDTA and especially EGTA; this can be explained by the overall charge as the presence of six carboxylate groups on the macrocycles in Eu2L1 neutralize the positive charge of two bound Eu3+ ions. This can also be the reason for the relatively high logKH3. On the other hand, the highly positively charged Eu2L2 behaves similar to the EGTA–bisamide, having one neutral and one acidic amine nitrogen, respectively, and a lower logKH3 value compared with Eu2L1. Distribution diagrams of species in the Eu2L1–2 system, for the concentration ratio [L]:[Eu] = 1:2, indicate this different behaviour (Fig. 3 and S5 in the ESI†). The dominating complex species at pH < 5 is [Eu2(H3L)], a complex in which Eu3+ ions are coordinated to the ligand L1 or L2 and the EGTA part is triprotonated. Consequently, the species [Eu2(H3L1)] and [Eu2(H3L2)] have a maximum concentration at pH 4.7 and 4.2, respectively. As pH values increase, the [Eu2(H3L)] complex releases protons forming [Eu2(H2L)], [Eu2(HL)], and [Eu2(L)], and finally the [Eu2L(OH)2] complex. The complex [Eu2(L1)] starts to form at pH 7 and reaches the maximal concentration at pH 8.9 (Fig. 3), while the complex [Eu2(L2)] starts to form at pH 5 and reaches the maximal concentration at pH 8.7 (Fig. S5 in the ESI†). Moreover, the hydroxo complex [Eu2L(OH)2] in both cases starts to form around pH 8, and reaches the maximum concentration around pH 9.5. The hydroxide Eu(OH)3 begins to form around pH 8.5, and its concentration increases with the further increase of pH.
KH1 and logKH2) stand for protonation of the amine nitrogen atoms of the EGTA-derived part. These values are quite different for Eu2L1 and Eu2L2. The complex Eu2L1 displays a higher basicity similar to EDTA and especially EGTA; this can be explained by the overall charge as the presence of six carboxylate groups on the macrocycles in Eu2L1 neutralize the positive charge of two bound Eu3+ ions. This can also be the reason for the relatively high logKH3. On the other hand, the highly positively charged Eu2L2 behaves similar to the EGTA–bisamide, having one neutral and one acidic amine nitrogen, respectively, and a lower logKH3 value compared with Eu2L1. Distribution diagrams of species in the Eu2L1–2 system, for the concentration ratio [L]:[Eu] = 1:2, indicate this different behaviour (Fig. 3 and S5 in the ESI†). The dominating complex species at pH < 5 is [Eu2(H3L)], a complex in which Eu3+ ions are coordinated to the ligand L1 or L2 and the EGTA part is triprotonated. Consequently, the species [Eu2(H3L1)] and [Eu2(H3L2)] have a maximum concentration at pH 4.7 and 4.2, respectively. As pH values increase, the [Eu2(H3L)] complex releases protons forming [Eu2(H2L)], [Eu2(HL)], and [Eu2(L)], and finally the [Eu2L(OH)2] complex. The complex [Eu2(L1)] starts to form at pH 7 and reaches the maximal concentration at pH 8.9 (Fig. 3), while the complex [Eu2(L2)] starts to form at pH 5 and reaches the maximal concentration at pH 8.7 (Fig. S5 in the ESI†). Moreover, the hydroxo complex [Eu2L(OH)2] in both cases starts to form around pH 8, and reaches the maximum concentration around pH 9.5. The hydroxide Eu(OH)3 begins to form around pH 8.5, and its concentration increases with the further increase of pH.
 | ||
| Fig. 3 Distribution diagram of Eu3+–L1 species at [L]:[Eu] = 1:2 concentration ratio; total Eu3+ concentration 0.5 mM; I = 0.1 M (NaCl), t = 25 ± 1 °C. | ||
| pEu + qH + rL + mM ⇄ EupHqLrMm; βp,q,r,m | (3) |
 | (4) |
These stability constants were also determined by acid–base potentiometric titrations. The obtained values were compared with those previously reported for EDTA, 2,2′-oxybis-(ethylamine)-N,N,N′,N′-tetraacetic acid (OBETA) and EGTA (Table 3),36–38 while the final set of complexes existing in the studied aqueous solutions and experimentally determined stability constants with standard deviations and statistical parameters showing the quality of fit are provided in the ESI (Table S3†). Finally, the corresponding distribution diagrams of the species in the Eu3+–L–M systems (L = L1 or L2 and M = Ca2+, Mg2+ or Zn2+) for the concentration ratio [Eu]:[L]:[M] = 2:1:1 are also plotted (Fig. 4 and S6–10 in the ESI†).
 | ||
| Fig. 4 Distribution diagram of Eu3+–L1–Ca species at [Eu]:[L1]:[Ca] concentration ratio = 2:1:1; total Eu3+ concentration 0.5 mM; I = 0.1 M (NaCl), t = 25 ± 1 °C. | ||
| L1 | L2 | EDTAb | OBETAc | EGTAb | |
|---|---|---|---|---|---|
| a I = 0.1 M (NaCl), t = 25 ± 1 °C. b I = 0.1 M (KCl for EDTA, KNO3 for EGTA), ref. 36 and 37. c I = 0.1 M (KNO3), ref. 38. | |||||
| Ca2+ | 4.60 | 4.24 | 10.61 | 9.77 | 10.86 |
| Mg2+ | 4.08 | 3.38 | 8.69 | 7.95 | 5.28 |
| Zn2+ | 6.93 | 5.59 | 16.50 | 15.00 | 12.60 |
| logKEu2CaL/logKEu2MgL |
logKCaL/logKMgL |
||||
| 1.13 | 1.25 | 1.22 | 1.23 | 2.06 | |
| logKEu2ZnL/logKEu2CaL |
logKZnL/logKCaL |
||||
| 1.51 | 1.32 | 1.56 | 1.54 | 1.16 | |
The stability constants obtained for Eu2L1,2 with the investigated metal ions were several orders of magnitude lower than that for the well-studied EDTA, OBETA or EGTA chelators with the same metals. There might be a few reasons for such behaviour. First, the binding affinity of bisamide–bisacid chelators (Eu2L1,2) is expected to be weaker than in the case of tetraacid chelators (EDTA, OBETA or EGTA) due to the reduction in the number of negatively charged carboxylate chelating groups. Next, the additional positive charge on the macrocyclic ring (especially valid for Eu2L2) induces the repulsion between the Eu2L1,2 complex and the positively charged metal complex. This phenomenon can also be confirmed by the differences in stability constants between Eu2L1 and Eu2L2, where the former exhibits slightly higher values for all three metal ions due to its lower net positive charge when compared to the latter complex. Finally, the reduced flexibility of the EGTA-derived chelator in Eu2L1,2 due to the presence of bulky appended macrocycles may prevent efficient wrapping around the metal ion compared with the case of flexible EDTA, OBETA or EGTA chelators.
Furthermore, the obtained stability constants also indicate a decrease in the selectivity of Eu2L1,2 for Ca2+, Mg2+ and Zn2+. This phenomenon can be easily followed by comparing the ratio of stability constants for a single chelator with two different metals (Table 3). As it can be seen, the ratio of logKEu2CaL/logKEu2MgL or logKEu2ZnL/logKEu2CaL for both Eu2L1,2 is comparable to analogous ratios obtained for EDTA and OBETA despite much lower logKML values for Eu2L1,2 and the structural similarity of the chelating site to EGTA. This loss of selectivity of Eu2L1,2 for Ca2+vs. Mg2+ could be explained by the same reasons that lead to the drop in logKEu2ML values. Namely, the reduced flexibility of Eu2L1,2 compared to EGTA prevents better recognition and size match of the EGTA-derived chelator with Ca2+ than with Mg2+, while the increase in the positive charge of Eu2L1,2 additionally impairs binding to metal ions and their recognition.
Despite the considerably lower stability constant values for the three investigated metals, the distribution diagrams indicate that heteronuclear complexes are the major species at the physiological pH (Fig. 4 and S6–10 in the ESI†). The heteronuclear complexes [Eu2(HnL1)Ca] (n = 0, 1 or 2) are probably formed by the following reaction:
| [Eu2(HnL1)] + Ca2+ ⇄ [Eu2(HnL1)Ca]; n = 0, 1 or 2. |
Consequently, the dominating complex at lower pH values is [Eu2(H3L1)], with a maximal concentration at pH around 4.5 (Fig. 4). Similar assumptions can be made according to the obtained distribution diagrams of other heteronuclear complexes, [Eu2(HnL1)Mg] and [Eu2(HnL1)Zn] (Fig. S6 and S7 in the ESI†). The protonated heteronuclear complex, [Eu2(H2L1)Ca] has a maximum concentration at pH around 7 and is fairly stable (Table S3 in the ESI†). The formation of the complex [Eu2(HL1)Ca] starts at around pH 6 and reaches the maximum concentration at pH 8.2. The complex [Eu2(L1)Ca] starts to form at pH 7 and reaches the maximum concentration around pH 9.
Conclusions
In this work we synthesised three different bismacrocyclic DO3A-amide derivatives appended with the EGTA-derived chelator. The amides had glycine, methyl and tert-butyl as substituents resulting in ligands L1–3, respectively. The paramagnetic complexes of L1–3 were prepared and various aspects of their physicochemical behaviour were investigated. Eu2L1 exhibited a greater CEST effect than Eu2L2 and Yb2L2 complexes, while Eu2L3 and Yb2L3 showed the absence of any CEST effect, suggesting that the polarity of substituents and the overall charge of the complexes play an essential role in the existence of the CEST effect. Addition of Ca2+ led to negligible changes in the CEST effects in the investigated complexes. The luminescence steady-state emission and lifetime measurements confirmed the insensitivity of Eu2L1–3 towards Ca2+ as the inner-sphere hydration of the complexes remained intact in the presence and absence of Ca2+. These findings were in line with the results from 1H NMR experiments which indicated the presence of different mono- or bis-hydrated isomers of Eu2L1–3 in solution. However, the analysis of NMR data revealed a switch of the SAP/TSAP isomer population for DO3A-amide complexes when compared to the previously investigated responsive Gd-DO3A complexes. In DO3A-amides, the TSAP isomer appeared to be the major species, which does not change the coordination environment around the Ln3+ ion (i.e. inner-sphere hydration) upon complexation with Ca2+. It has been shown that the population of the TSAP isomer increases upon increasing the steric hindrance for the coordination of the pendant arms to the lanthanide(III) ion.39 Thus, it is likely that the presence of the amide substituents in Eu2L1–3 introduces some steric hindrance that favors the TSAP isomer over the SAP one. Finally, the results from potentiometric titrations showed a great dependence of protonation constants on the net charge of the complexes Eu2L1–2. They bind Ca2+ with reduced affinity and with a lower selectivity towards Mg2+ or Zn2+ compared to the original EGTA chelator, which is a consequence of their positive charge and reduced flexibility of the binding site.The results presented here suggest that replacement of carboxylate for amide groups in macrocycles changes the coordination to lanthanide metal ions and results in alterations in their behaviour. Consequently, the interaction with metal ions such as Ca2+ is dramatically affected, converting the Ca-sensitive DO3A-derived bioresponsive T1 agent into non-sensitive DO3A-amide-derived potential CEST agents. Obviously, further adjustments in the microenvironment around the paramagnetic metal are required to obtain efficient CEST agents, prior or post interactions with the desired metal ion. Thus, the present work provides an important set of information that will aid better design of future bioresponsive agents for CEST MRI.
Experimental section
General remarks
Commercially available reagents and solvents were used without further purification. Compounds 1,403a,173b,413c19 and 710 were synthesised according to previously published procedures. Purification of the synthesised compounds was performed using silica gel 60 (0.03–0.2 mm) from Carl Roth (Germany). Standard 0.1 M HCl and NaOH solutions were prepared from ampoules (Titrisol, Merck, Darmstadt, Germany) and potentiometrically standardised. Standard 5 mM Ca2+, Mg2+, and Zn2+ solutions were prepared from appropriate salts and standardised by titration with an EDTA solution. Acros Organics buffers were used for electrode calibration (phthalate pH 4.00, phosphate pH 7.00, and carbonate pH 10.00). All NMR spectra were acquired on a Bruker Avance III 300 MHz, processed using TopSpin 2.1 (Bruker GmbH), and analysed with TopSpin 2.1 or ACD/SpecManager 9.0 (Advanced Chemistry Development, Inc.). The concentration of the complexes was determined using the bulk magnetic susceptibility shift (BMS).42 ESI-HRMS were performed on a Bruker BioApex II ESI-FT-ICR, equipped with an Agilent ESI-Source, measured via flow injection analysis. ESI-LRMS were performed on an ion trap SL 1100 system (Agilent, Germany). Luminescence lifetime measurements were performed by using a Quanta-MasterTM 3-PH fluorescence spectrometer from Photon Technology International, Inc., (Monmouth Junction, NJ, USA). Potentiometric titrations were performed by using a Metrohm Basic Titrino 794 (Herisau, Switzerland) equipped with an InLab Micro electrode (Mettler-Toledo International Inc., Columbus, Ohio, USA).
Synthetic procedures. Benzyl(3-(1,4,7,10-tetraazacyclododecan-1-yl)propyl)carbamate (2)
Compound 2 was synthesised according to the previously described procedure for monoalkylation of cyclen,43 using 1 as the alkylating agent.Compound 2: isolated yield: 59%. 1H NMR (CDCl3, 300 MHz), δ (ppm): 7.38–7.27 (m, 5H), 5.06 (s, 2H), 3.24–3.18 (br, 2H), 2.73–2.67 (br, 4H), 2.65–2.59 (br, 4H), 2.54–2.40 (br, 10H), 1.72–1.63 (m, 2H). 13C NMR (CDCl3, 75 MHz), δ (ppm): 156.6, 136.9, 128.3, 128.0, 127.8, 66.2, 52.1, 51.5, 47.0, 46.0, 45.1, 39.3, 27.5. ESI-HRMS: for C19H33N5O2+: calc. 364.2707 [M + H]+, found 364.2711.
General procedure for the synthesis of 4a–4c
3a, 3b or 3c (3.2 equiv.) was added in an already prepared suspension of 2 (1.0 equiv.) and K2CO3 (4.0 equiv.) in anhydrous acetonitrile. The reaction mixture was stirred at 65 °C for 18 h. After cooling, the product mixture was filtered and the solvent was removed under reduced pressure. The residue was dissolved in dichloromethane and washed twice with water. The organic layer was dried over Na2SO4 and then evaporated under reduced pressure to give crude 4a, 4b or 4c. The compounds were purified by column chromatography over silica (eluent MeOH in CH2Cl2) to give 4a–c as amorphous solids.General procedure for the synthesis of 5a–5c
10% Pd/C was added to a solution of 4a (3.00 g, 3.4 mmol), 4b (3.70 g, 6.4 mmol) or 4c (3.10 g, 4.4 mmol) in ethanol (50 mL). The resulting mixture was stirred at room temperature under a hydrogen atmosphere (3 bar) for 18 hours. The reaction was then filtered over Celite® to remove the catalyst and the solvent was removed by rotary evaporation. The residue was dissolved in CH2Cl2 (50 mL) and the precipitate was removed by filtration. The organic layer was dried over Na2SO4 and then evaporated to give 5a (2.40 g, 94%), 5b (2.60 g, 92%) or 5c (2.40 g, 96%).General procedure for the synthesis of 6a–6c
5a, 5b or 5c (1 equiv.) and Et3N (1.5 equiv.) were dissolved in anhydrous acetonitrile. The solution was cooled to 0 °C under a nitrogen atmosphere and a solution of chloroacetic anhydride (1.3 equiv.) in acetonitrile was added dropwise. The reaction was stirred at 0 °C for an additional 3 h. The solvent was evaporated from the reaction mixture and the crude product was purified by column chromatography over silica gel (eluent MeOH in CH2Cl2) to give 6a, 6b or 6c.General procedure for the synthesis of 8a–8c
The reaction flask was charged with 6a, 6b or 6c (1 equiv.), 7 (0.4 equiv.), K2CO3 (1.7 equiv.), KI (1 equiv.) and anhydrous acetonitrile and the reaction mixture was heated at 70 °C overnight under a nitrogen atmosphere. The inorganic salts were removed by filtration and the solvent was evaporated to dryness. The crude product was purified by column chromatography over silica gel using 8% MeOH in CH2Cl2 as the eluent to give 8a, 8b or 8c.General procedure for the synthesis of L1–3
Bismacrocycle 8a, 8b or 8c was dissolved in formic acid (3 mL) and the solution was heated at 60 °C for 18 h (for potentiometric titrations, L1–2 were prepared by mixing 8a,b with 4 N HCl (3 mL) in dioxane at RT for 4 hours). After the solution was cooled, formic acid was removed on the rotary evaporator. The residue was dissolved in a minimal amount of water, added dropwise to cooled acetone (−20 °C) and stored in the freezer overnight. The acetone was decanted from the solid material and the crude products L1–3 were obtained by drying in a vacuum.General procedure for the preparation of Ln3+ complexes
The Ln3+ complexes of L2 and L3 were prepared by mixing the respective ligand and the LnCl3 solutions in a 2:1 (Ln3+:L) molar ratio. The exact amount of ligand was determined by elemental analysis. The solution was stirred at RT for 48 h. The pH value was adjusted to 7.0–7.5 using 1 M NaOH. The absence of a free ion (Yb3+ or Eu3+) was verified by colorimetric assay using xylenol orange.
In the case of negatively charged L1, EuCl3 was added in slight excess (>2 equiv.). The mixture was stirred for 48 h at RT maintaining the pH at 7.0–7.5, using 1 M NaOH. The resulting solution was treated with Chelex 100 to remove the excess Eu3+ and the absence of free Eu3+ was verified by colorimetric assay using xylenol orange.
Luminescence lifetime experiments
The lifetime measurements were performed on a QuantaMasterTM 3 PH fluorescence spectrometer from Photon Technology International, Inc. The measurements were performed in H2O and D2O (25 °C) at 5 mM Eu3+ concentration. The Eu3+ ion was directly excited at 395 nm, and the emission intensity at 615 nm was measured with a 10 μs resolution. The excitation and emission slits were set to 15 and 5 nm bandpass, respectively. Data sets are an average of 25 scans, and each reported value is the mean of three independent measurements. The obtained curves are fitted to a first-order exponential decay with r2 = 0.99. The q values were calculated using eqn (5).21qEu = 1.2 × [kH2O − kD2O − 0.25 + 0.075nO![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHN], n = 3 CHN], n = 3 | (5) |
Potentiometric titrations
Experiments were performed at t = 25 ± 1 °C, with a constant argon flow, using a 794 Basic Titrino automatic titrator (Metrohm, Herisau, Switzerland) equipped with an InLab Micro electrode (Mettler-Toledo International Inc., Columbus, Ohio, USA). The electrode–pH-meter system was calibrated by means of a strong acid–strong base titration in 0.1 M NaCl, using GLEE – GLass Electrode Evaluation software;44 standard potential (E0 = 383.6 ± 0.2 mV) and slope (57.4 ± 0.1 mV) are obtained as mean values of four titrations. Hyperquad 2008 software was used to calculate protonation and stability constant values as the mean values of four titrations.33Acknowledgements
The financial support of the Max-Planck Society and the European COST CM1006 Action is gratefully acknowledged.Notes and references
- M. M. J. Modo and J. W. M. Bulte, Molecular and cellular MR imaging, CRC Press, Taylor & Francis Group, Boca Raton, London, New York, 2007 Search PubMed.
- A. E. Merbach, L. Helm and É. Tóth, The chemistry of contrast agents in medical magnetic resonance imaging, Wiley, Chichester, 2013 Search PubMed.
- E. Terreno, D. D. Castelli, A. Viale and S. Aime, Chem. Rev., 2010, 110, 3019–3042 CrossRef CAS PubMed.
- A. D. Sherry and M. Woods, Annu. Rev. Biomed. Eng., 2008, 10, 391–411 CrossRef CAS PubMed.
- P. C. M. van Zijl and N. N. Yadav, Magn. Reson. Med., 2011, 65, 927–948 CrossRef CAS PubMed.
- D. V. Hingorani, A. S. Bernstein and M. D. Pagel, Contrast Media Mol. Imaging, 2015, 10, 245–265 CrossRef CAS PubMed.
- M. Woods, E. W. C. Donald and A. D. Sherry, Chem. Soc. Rev., 2006, 35, 500–511 RSC.
- D. Delli Castelli, E. Terreno and S. Aime, Angew. Chem., Int. Ed., 2011, 50, 1798–1800 CrossRef CAS PubMed.
- G. Angelovski, T. Chauvin, R. Pohmann, N. K. Logothetis and É. Tóth, Bioorg. Med. Chem., 2011, 19, 1097–1105 CrossRef CAS PubMed.
- G. Angelovski, P. Fouskova, I. Mamedov, S. Canals, E. Toth and N. K. Logothetis, ChemBioChem, 2008, 9, 1729–1734 CrossRef CAS PubMed.
- P. Kadjane, C. Platas-Iglesias, P. Boehm-Sturm, V. Truffault, G. E. Hagberg, M. Hoehn, N. K. Logothetis and G. Angelovski, Chem. – Eur. J., 2014, 20, 7351–7362 CrossRef CAS PubMed.
- M. Woods, D. E. Woessner, P. Y. Zhao, A. Pasha, M. Y. Yang, C. H. Huang, O. Vasalitiy, J. R. Morrow and A. D. Sherry, J. Am. Chem. Soc., 2006, 128, 10155–10162 CrossRef CAS PubMed.
- J. R. Slack and M. Woods, J. Biol. Inorg. Chem., 2014, 19, 173–189 CrossRef CAS PubMed.
- S. Viswanathan, Z. Kovacs, K. N. Green, S. J. Ratnakar and A. D. Sherry, Chem. Rev., 2010, 110, 2960–3018 CrossRef CAS PubMed.
- N. McVicar, A. X. Li, M. Suchy, R. H. E. Hudson, R. S. Menon and R. Bartha, Magn. Reson. Med., 2013, 70, 1016–1025 CrossRef CAS PubMed.
- S. Aime, D. Delli Castelli, F. Fedeli and E. Terreno, J. Am. Chem. Soc., 2002, 124, 9364–9365 CrossRef CAS PubMed.
- M. M. Ali, M. Woods, E. H. Suh, Z. Kovacs, G. Tircso, P. Y. Zhao, V. D. Kodibagkar and A. D. Sherry, J. Biol. Inorg. Chem., 2007, 12, 855–865 CrossRef CAS PubMed.
- S. Aime, A. Barge, D. D. Castelli, F. Fedeli, A. Mortillaro, F. U. Nielsen and E. Terreno, Magn. Reson. Med., 2002, 47, 639–648 CrossRef CAS PubMed.
- T. Mani, G. Tircso, O. Togao, P. Zhao, T. C. Soesbe, M. Takahashi and A. D. Sherry, Contrast Media Mol. Imaging, 2009, 4, 183–191 CrossRef CAS PubMed.
- S. Aime, A. Barge, A. S. Batsanov, M. Botta, D. Delli Castelli, F. Fedeli, A. Mortillaro, D. Parker and H. Puschmann, Chem. Commun., 2002, 1120–1121 RSC.
- A. Beeby, I. M. Clarkson, R. S. Dickins, S. Faulkner, D. Parker, L. Royle, A. S. de Sousa, J. A. G. Williams and M. Woods, J. Chem. Soc., Perkin Trans. 2, 1999, 493–503 RSC.
- S. Aime, M. Botta, M. Fasano, M. P. M. Marques, C. F. G. C. Geraldes, D. Pubanz and A. E. Merbach, Inorg. Chem., 1997, 36, 2059–2068 CrossRef CAS PubMed.
- D. Parker, R. S. Dickins, H. Puschmann, C. Crossland and J. A. K. Howard, Chem. Rev., 2002, 102, 1977–2010 CrossRef CAS PubMed.
- C. Adair, M. Woods, P. Y. Zhao, A. Pasha, P. M. Winters, G. M. Lanza, P. Athey, A. D. Sherry and G. E. Kiefer, Contrast Media Mol. Imaging, 2007, 2, 55–58 CrossRef CAS PubMed.
- M. Regueiro-Figueroa, K. Djanashvili, D. Esteban-Gomez, T. Chauvin, E. Toth, A. de Blas, T. Rodriguez-Blas and C. Platas-Igleslas, Inorg. Chem., 2010, 49, 4212–4223 CrossRef CAS PubMed.
- M. Polasek, J. Kotek, P. Hermann, I. Cisarova, K. Binnemans and I. Lukes, Inorg. Chem., 2009, 48, 466–475 CrossRef CAS PubMed.
- F. A. Dunand, R. S. Dickins, D. Parker and A. E. Merbach, Chem. – Eur. J., 2001, 7, 5160–5167 CrossRef CAS.
- S. Aime, M. Botta and G. Ermondi, Inorg. Chem., 1992, 31, 4291–4299 CrossRef CAS.
- P. Caravan, J. J. Ellison, T. J. McMurry and R. B. Lauffer, Chem. Rev., 1999, 99, 2293–2352 CrossRef CAS PubMed.
- W. Hummel, U. Berner, E. Curti, F. J. Pearson and T. Thoenen, Radiochim. Acta, 2002, 90, 805–813 CrossRef CAS.
- ADMET Predictor, Simulations Plus, Inc., Lancaster, CA, USA, ver. 7.2, 2015 Search PubMed.
- A. Pasha, G. Tircso, E. T. Benyo, E. Brucher and A. D. Sherry, Eur. J. Inorg. Chem., 2007, 4340–4349 CrossRef CAS PubMed.
- P. Gans, A. Sabatini and A. Vacca, Talanta, 1996, 43, 1739–1753 CrossRef CAS PubMed.
- L. Alderighi, P. Gans, A. Ienco, D. Peters, A. Sabatini and A. Vacca, Coord. Chem. Rev., 1999, 184, 311–318 CrossRef CAS.
- S. Aime, A. Barge, M. Botta, L. Frullano, U. Merlo and K. I. Hardcastle, J. Chem. Soc., Dalton Trans., 2000, 3435–3440 RSC.
- L. Tei, Z. Baranyai, M. Botta, L. Piscopo, S. Aime and G. B. Giovenzana, Org. Biomol. Chem., 2008, 6, 2361–2368 CAS.
- C. De Stefano, A. Gianguzza, D. Milea, A. Pettignano and S. Sammartano, J. Alloys Compd., 2006, 424, 93–104 CrossRef CAS.
- R. Negri, Z. Baranyai, L. Tei, G. B. Giovenzana, C. Platas-Iglesias, A. C. Benyei, J. Bodnar, A. Vagner and M. Botta, Inorg. Chem., 2014, 53, 12499–12511 CrossRef CAS PubMed.
- M. Purgel, Z. Baranyai, A. de Blas, T. Rodriguez-Blas, I. Banyai, C. Platas-Iglesias and I. Toth, Inorg. Chem., 2010, 49, 4370–4382 CrossRef CAS PubMed.
- K. Dhingra, P. Fouskova, G. Angelovski, M. E. Maier, N. K. Logothetis and E. Toth, J. Biol. Inorg. Chem., 2008, 13, 35–46 CrossRef CAS PubMed.
- O. Vasalatiy, P. Y. Zhao, M. Woods, A. Marconescu, A. Castillo-Muzquiz, P. Thorpe, G. E. Kiefer and A. D. Sherry, Bioorg. Med. Chem., 2011, 19, 1106–1114 CrossRef CAS PubMed.
- D. M. Corsi, C. Platas-Iglesias, H. van Bekkum and J. A. Peters, Magn. Reson. Chem., 2001, 39, 723–726 CrossRef CAS.
- J. Massue, S. E. Plush, C. S. Bonnet, D. A. Moore and T. Gunnlaugsson, Tetrahedron Lett., 2007, 48, 8052–8055 CrossRef CAS.
- P. Gans and B. O'Sullivan, Talanta, 2000, 51, 33–37 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Luminescence emission spectra, equilibrium constants and distribution diagrams. See DOI: 10.1039/c5dt04625d |
| This journal is © The Royal Society of Chemistry 2016 |