
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA luminescent europium(III)–platinum(II) heterometallic complex as a theranostic agent: a proof-of-concept study†‡
Anirban
Chandra§
a,
Khushbu
Singh§
a,
Swati
Singh
b,
Sri
Sivakumar
b and
Ashis K.
Patra
*a
aDepartment of Chemistry, Indian Institute of Technology Kanpur, Kanpur 208016, Uttar Pradesh, India. E-mail: akpatra@iitk.ac.in
bDepartment of Chemical Engineering and Centre for Environmental Science and Engineering, Indian Institute of Technology Kanpur, Kanpur-208016, U.P., India
First published on 19th November 2015
Abstract
A luminescent heterometallic multifunctional theranostic Eu–Pt2 complex [{cis-PtCl2(DMSO)}2Eu(L)(H2O)] has been synthesized, possessing two therapeutic Pt-centers as covalent DNA binders and one emissive Eu3+-center which is sensitized by platinum-based metal-to-ligand charge-transfer excited states.
Multifunctional cancer theranostic agents with multiple therapeutic and diagnostic centers in a single platform have gained popularity in recent years.1–3 Recently, there are a few reports on heterometallic hairpin-shaped lanthanide–Pt2 complexes for DNA recognition and magnetic resonance imaging (MRI) based theranostic agents or for selectively delivering gadolinium to tumor cell nuclei.4 In this regard, theranostic Gd3+-based platinum complexes have been reported4c in which Gd3+ and conjugated cis-[Pt(NH3)2Cl]+-moieties act as MRI contrast and therapeutic centers, respectively. These complexes showed therapeutic efficiency along with improved MR imaging capability. However, their DNA-binding ability is limited because there are only two available DNA-crosslinking sites. In addition, the clinical use of platinum drugs is severely affected by drug resistance mediated by inadequate levels of platinum reaching critical target DNA.5 Notwithstanding this progress, it is highly desirable to increase the DNA binding sites that significantly enhance platinum content at target sites along with the tagging of a bright luminescent center as a diagnostic probe. To this end, we report the design of a multimodal targeted theranostic Eu–Pt2 conjugate possessing four DNA binding sites which can effectively target nuclear DNA along with a highly luminescent Eu3+ center to enable interference-free live-tracking of the drug using fluorescence microscopy. Luminescent lanthanide complexes were widely exploited in various bioassays since they offer unique photophysical properties like narrow emission bands, a large Stokes’ shift and long-lived excited state lifetimes.6,7 Since direct excitation of a Ln3+ f–f transition is very inefficient, chemists have designed a variety of chelating agents conjugated to a sensitizing organic chromophore called an antenna, which can transfer its excited state energy efficiently to the emissive Ln3+ ion leading to bright luminescence compared to the direct excitation of Ln3+ ions.8,9 Our approach exploits the advantages of transition metal complexes as they exhibit many desirable properties as a sensitizer than as an organic chromophore, such as tunable absorption bands, long-lived excited states which maximize the ET to Ln3+, excellent photochemical stability and kinetic inertness.10,11
Herein, we demonstrate a multifunctional Eu–Pt2 complex, [{cis-PtCl2(DMSO)}2Eu(L)(H2O)] (1) (Scheme 1), which has cytotoxic cis-[PtCl2(DMSO)] moieties that enable DNA binding, whereas the EuL unit acts as a luminescent reporter. Thus, a combination of a luminescent imaging probe and a conjugated therapeutic agent in a single hybrid 5d–4f complex can provide real-time feedback on drug delivery, distribution and target site localization in a non-invasive manner using fluorescence microscopy. Another key design feature of this complex is having four potential DNA cross-linking sites due to the presence of labile Pt–Cl bonds, thus a higher level of activated platinum will reach DNA, which is a possible way to lower drug resistance. Eu–Pt2 complex 1 was prepared in a sequential manner (Scheme S1, ESI‡) starting with a multidentate DTPA-bisamide ligand, H3L = N,N′′-bis(3-amidoquinolyl)diethylenetriamine-N,N′,N′′-triacetic acid, derived from the acylation of 3-aminoquinoline by DTPA-bis(anhydride). [Eu(L)(H2O)] was synthesized by reacting a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of the deprotonated ligand and Eu(NO3)3·6H2O in water. [{cis-PtCl2(DMSO)}2Eu(L)(H2O)] (1) was isolated after the reaction of EuL with freshly prepared cis-[Pt(DMSO)2Cl2] in a 1:2 molar ratio. Detailed ESI-MS studies of the Eu–Pt2 complex reveal an m/z of 714.52 corresponding to {M − 2Cl}2+ with a matching isotopic distribution profile, which can be unequivocally attributed to the formation of Eu–Pt2 along with other physicochemical data (Fig. S1, ESI‡). We observed dissociable chloride ligands in aqueous solution which is crucial for forming cross-links with base pairs in nuclear DNA.
:1 molar ratio of the deprotonated ligand and Eu(NO3)3·6H2O in water. [{cis-PtCl2(DMSO)}2Eu(L)(H2O)] (1) was isolated after the reaction of EuL with freshly prepared cis-[Pt(DMSO)2Cl2] in a 1:2 molar ratio. Detailed ESI-MS studies of the Eu–Pt2 complex reveal an m/z of 714.52 corresponding to {M − 2Cl}2+ with a matching isotopic distribution profile, which can be unequivocally attributed to the formation of Eu–Pt2 along with other physicochemical data (Fig. S1, ESI‡). We observed dissociable chloride ligands in aqueous solution which is crucial for forming cross-links with base pairs in nuclear DNA.
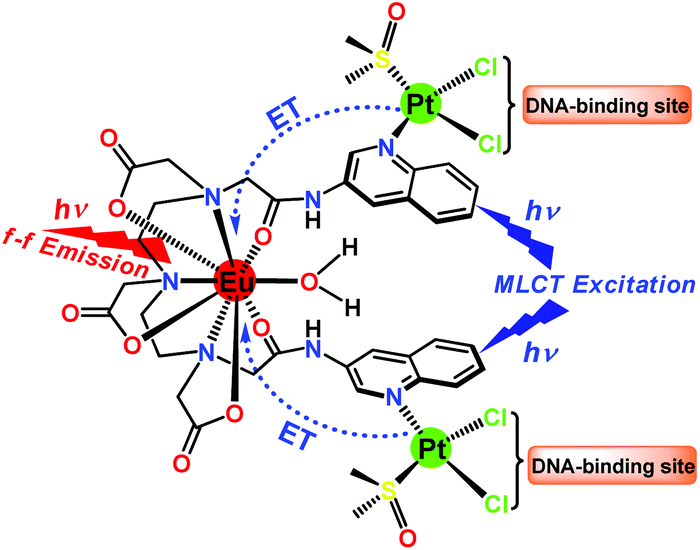 | ||
| Scheme 1 Molecular structure of Eu–Pt2 complex 1 showing a Pt → Eu energy transfer pathway upon MLCT photo-excitation leading to f–f emission from Eu3+ and dissociable Pt–Cl bonds as DNA-cross linking sites. | ||
The UV-vis spectrum of 1 exhibits a high energy band at 273 nm due to ligand centered π → π* transitions and a broad band ranging from 332–350 nm corresponding to π → π* MLCT transitions of the quinoline bound Pt2+ moiety (Fig. 1a). Here we have judiciously utilized this 3MLCT excited state as a means to populate the 5D0 emissive states of europium through efficient energy transfer. The 3MLCT → f energy transfer through such photosensitization is shown in few Eu–Pt complexes.12 The addition of cis-[Pt(DMSO)2Cl2] to EuL resulted in the appearance of a new band at 350 nm with an isobestic point at ∼270 nm, indicative of the formation of Eu–Pt2 complex 1 during titration (Fig. S2 in ESI‡). Upon excitation of the MLCT band, the Eu–Pt2 complex under time-gated mode displayed narrow emission bands spanning from 575–700 nm characteristic of the 5D0 → 7FJ (J = 0–4) f–f transitions of Eu3+ (Fig. 1b). The luminescence spectra demonstrate the efficient photosensitized energy transfer from the MLCT excited state of the quinoline bound Pt2+ moiety to the emissive 5D0 excited state of Eu3+ with an overall quantum yield (ϕoverall) of 0.04. Spectrophotometric titration of cis-[Pt(DMSO)2Cl2] with [Eu(L)(H2O)] at λex = 330 nm showed the formation of [{cis-PtCl2(DMSO)}2Eu(L)(H2O)] (1) with a gradual increase in europium centered emission until it reached a plateau at Eu/Pt = 0.5 (Fig. 1c). The decay rate of the emissive 5D0 excited state measured at 616 nm results in a monoexponential decay curve with a lifetime (τobs) of 0.65(±10%) ms in aqueous buffer medium, indicating the presence of a single chemical environment. The enhancement of the τobs in the presence of DNA (τobs = 0.89 (±10%) ms) indicates minimization of the nonradiative relaxation pathways in the DNA bound form of the Eu–Pt2 complex (Fig. 1d).
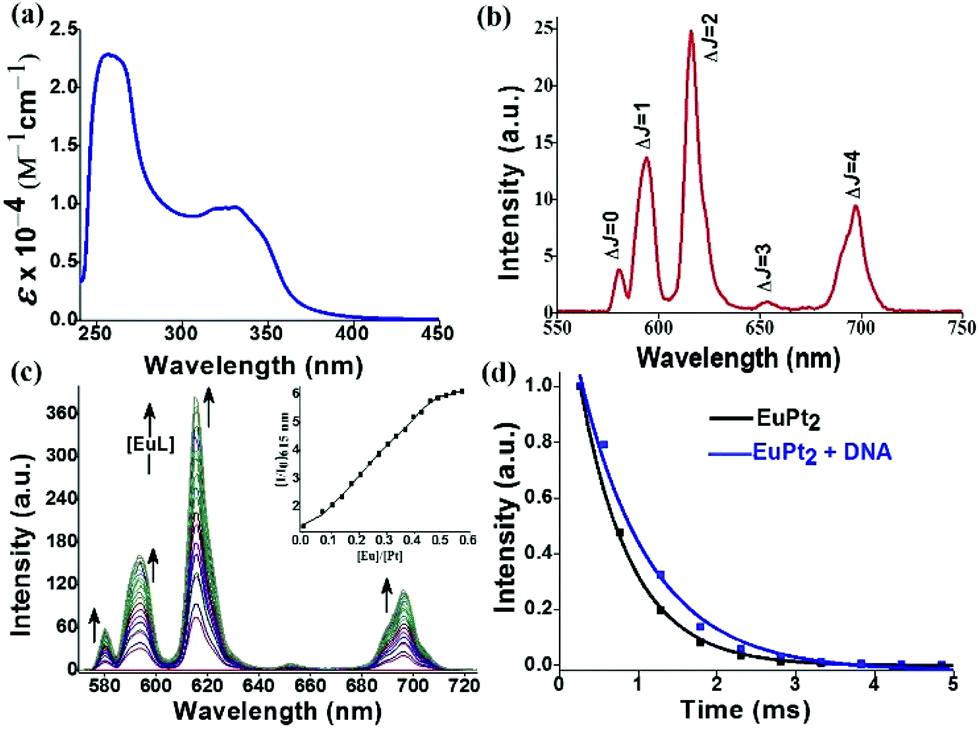 | ||
| Fig. 1 (a) UV-vis and (b) emission spectrum (λex = 330 nm) of Eu–Pt2 complex 1 in Tris-HCl buffer. (c) Evolution of the emission spectra with increasing [EuL] added to a solution of [PtCl2(DMSO)2] in DMF (λex = 330 nm), Inset: the plot of relative emission intensity at 615 nm vs. [Eu]/[Pt] ratio. (d) Luminescence decay profile from the 5D0 state of Eu3+ in complex 1 at λem = 616 nm (λex = 330 nm) (black) and in the presence of CT-DNA (blue) in Tris-HCl buffer (pH = 7.2). [1] = 90 μM, [DNA] = 450 μM, delay time and gate time = 0.1 ms. The solid lines are the best fits considering the single-exponential behaviour of the decay. | ||
Since DNA is the most important target for therapeutic platinum drugs, we attempted detailed binding studies of 1 with DNA. The hairpin-shaped complex 1 is activated by aquation through the substitution of the chloride ligands by water which generates a potent cation, [Eu(L)(H2O){cis-Pt(OH2)n/2(DMSO)}2]n+ (n = 1–4), which can readily cross-link with the nucleobases of ds-DNA (Scheme S2, ESI‡). The binding interaction of the Eu–Pt2 complex with calf-thymus DNA (CT-DNA) was studied using UV-vis titration, competitive displacement of ethidium bromide by fluorescence (Kapp = 4.9 × 105 M−1), circular dichroism (CD) and isothermal titration calorimetry (ITC) (Fig. S6–S8, ESI‡). The ITC titration plot suggests a biphasic sequential binding interaction13 of Eu–Pt2 with CT-DNA with an initial favorable exothermic binding event (K1 = 1.7 × 105 M−1, ΔH1 = −26.0 kcal mol−1), followed by a second exothermic event (K2 = 9.1 × 105 M−1, ΔH2 = −77.6 kcal mol−1) presumably due to successive sequential covalent cross-link formation with base pairs of duplex-DNA (Fig. 2a). The intrinsic binding constant (Kb = 1.5(±0.3) × 105 M−1) along with a hypochromism and bathochromic shift of the electronic spectral bands suggests a favorable binding interaction of the complex 1 with DNA due to EuPt2–DNA adduct formation, strong electrostatic interaction with activated {Eu–Pt2}n+ and favorable stacking interactions of the two planar –Pt(L) chromophores with the planar base pairs of ds-DNA as observed in similar hairpin shaped Eu–Pt2 complexes.4a,b Formation of such bis(bifunctional) platinum-DNA cross-links should induce major unrepairable structural distortion of the DNA double-helix. The significant decrease in ellipticity in the CD spectra of CT-DNA in the presence of 1 (Fig. 2b) also suggests unwinding of the DNA helix and major structural deformation of DNA induced by the Pt–DNA cross-links.14 This structural distortion of DNA could be beyond cellular DNA repair machinery and thereby inhibit transcription and replication, triggering cell-death pathways.5 The decrease in DNA migration rate in the presence of Eu–Pt2 compared to EuL in the gel electrophoretic mobility assay using SC pUC19 DNA also suggests unwinding of the supercoiled DNA helix by 1 (Fig. 2c).15 We have observed significant enhancement of the Eu-based luminescence intensity originating from the 5D0 → 7FJ transitions of Eu3+ upon the addition of DNA due to efficient energy transfer to Eu3+ and enhanced excited state lifetime in a hydrophobic environment created due to binding of the Eu–Pt2 complex with DNA (Fig. 2d). Serum albumin proteins constitute a major component in blood plasma proteins and play important roles in drug transport and metabolism. The interaction of the Eu–Pt2 complex with bovine serum albumin (BSA) studied using a tryptophan emission quenching experiment showed a high binding propensity (KBSA = 1.50 ± 0.03 × 105 M−1) desirable for efficient transport to the pathological site (Fig. S9, ESI‡).
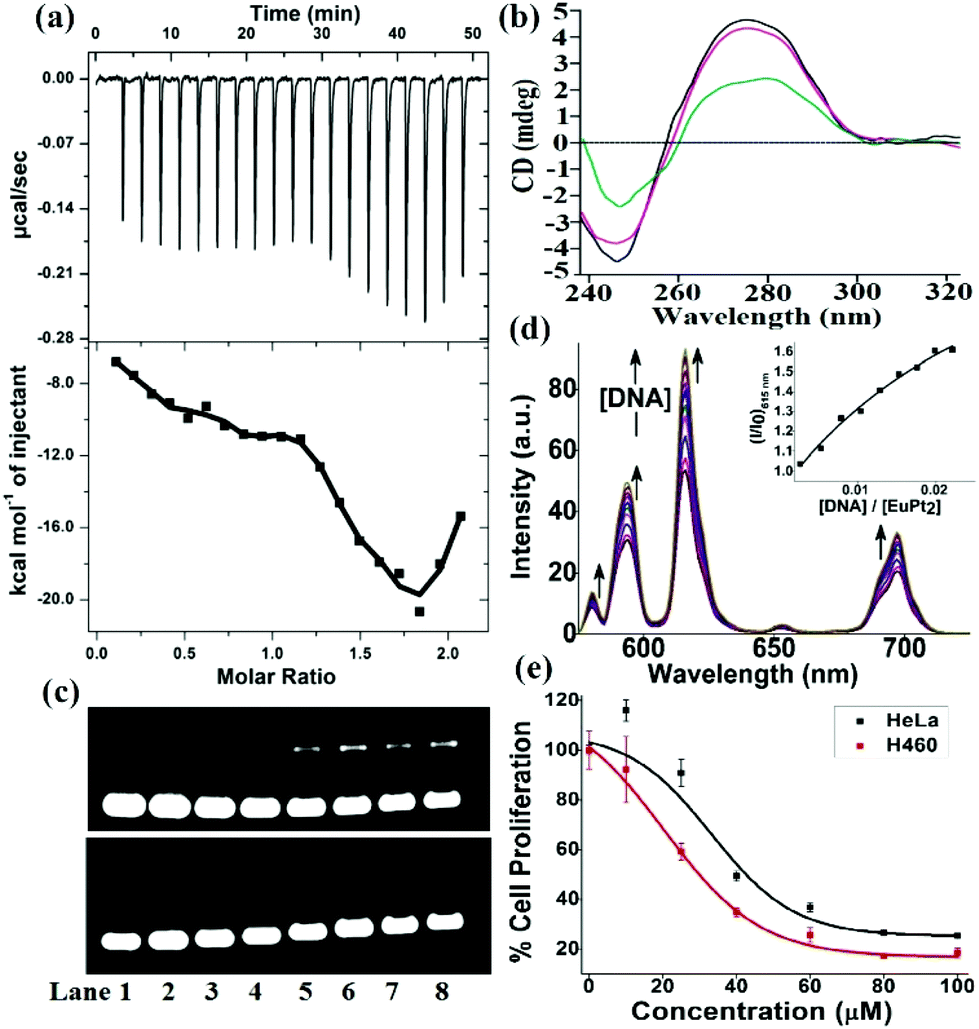 | ||
| Fig. 2 (a) ITC plot profile for the interaction of Eu–Pt2 (0.1 mM) with CT-DNA (0.01 mM) in 5 mM Tris-HCl/NaCl buffer (pH 8.5) at 30 °C. The top panel represents the raw data for 20 injections of 2 μL each. The bottom panel shows the amounts of heat evolved on interaction of the Eu–Pt2 complex with CT-DNA against the molar ratio of Eu–Pt2 complex to CT-DNA. Data were fitted by a sequential binding site model. (b) CD spectra of CT-DNA in the presence of EuL and Eu–Pt2 in Tris-HCl buffer medium. (c) Agarose gel electrophoresis of SC pUC19 DNA after incubation with EuL and Eu–Pt2 in 50 mM Tris-HCl buffer at 37 °C for 16 h. Lane 1: DNA control, lanes 2–8 (10, 15, 20, 40, 60, 80 and 100 μM complexes respectively). (d) Luminescence spectra of Eu–Pt2 (λex = 330 nm) with the addition of CT-DNA in 5 mM Tris-HCl buffer. Inset: the relative emission intensity (I/I0) of 1 at 615 nm vs. [DNA]/[Eu–Pt2] ratio. (e) Cell viability plots showing the cytotoxicity of the Eu–Pt2 complex with HeLa and H460 cells on 16 h incubation by MTT assay. | ||
To test our original ‘theranostic’ design, we performed an in vitro cytotoxicity assay using MTT on human cervical carcinoma HeLa and lung carcinoma H460 cell lines. The IC50 values for Eu–Pt2 complex 1 are 51.0 ± 1.05 μM in HeLa cells and 30.0 ± 1.27 μM in H460 cells (Fig. 2d). It exerts anticancer activity via extensive DNA-adduct formation through conjugated cis-[PtCl2(DMSO)] moieties with a similar mechanism to cisplatin.
The cellular internalization of the Eu–Pt2 complex was investigated to probe the diagnostic aspect of [Eu(L)(H2O)] utilizing the long luminescence lifetime and intrinsic luminescence from Eu3+ using confocal fluorescence microscopy (panel A, Fig. 3). The theranostic Eu–Pt2 complex showed significant cellular uptake within 4 h of incubation with the HeLa cells. Staining with nuclear staining dye Hoechst 33258 demonstrates both nuclear and cytosolic distribution of the complex (panel C, Fig. 3). The red spots observed in some nuclei originate from the luminescence of the Eu3+ reporter tag in the Eu–Pt2 theranostic conjugate (Fig. S11, ESI‡).
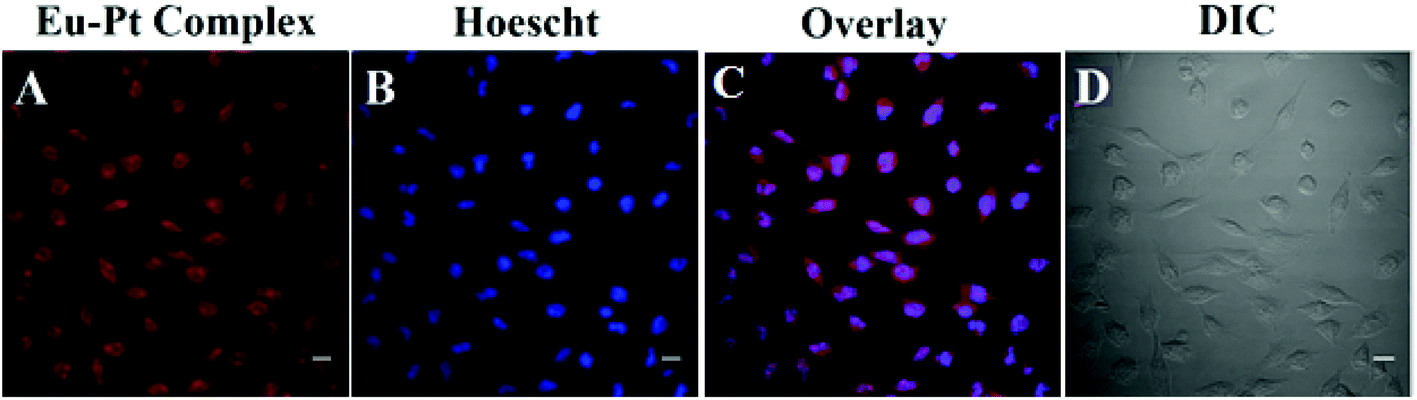 | ||
| Fig. 3 Confocal laser scanning fluorescence microscopic images of the HeLa cells treated with the Eu–Pt2 complex (25 μM) after 4 h of incubation and Hoechst 33258 dye (5 μg mL−1): (A) cells incubated with the Eu–Pt2 complex; (B) incubated with the Hoechst 33258 dye for staining nuclei, which show blue emission; (C) merged images showing nuclear and cytosolic localization of the complex; (D) DIC images. Scale bar = 20 μm. | ||
In conclusion, we have developed a luminescent multimodal heterometallic Eu–Pt2 theranostic system using sensitization and energy transfer from a conjugated Pt2+ based chromophore to Eu3+. The complex shows a strong binding propensity for DNA via the formation of Pt–DNA cross-links through four potential DNA binding sites. The complex exhibits cytotoxicity through DNA damage, and nuclear localization due to an Eu-based f–f transition is observed using fluorescence imaging. Thus, such systems will have great potential towards designing theranostic agents and delivery vehicles for cancer chemotherapy. Further studies are ongoing towards designing potent lanthanide based theranostic agents as efficient drug delivery platforms and understanding the mechanism of their action.
A.K.P. acknowledges SERB, Govt. of India and IIT Kanpur for funding. A.C. and K.S. acknowledge UGC and CSIR for their research fellowships.
Notes and references
- (a) H. Koo, M. S. Huh, I.-C. Sun, S. H. Yuk, K. Choi, K. Kim and I. C. Kwon, Acc. Chem. Res., 2011, 44, 1018–1028 CrossRef CAS PubMed; (b) T. Lammers, S. Aime, W. E. Hennink, G. Storm and F. Kiessling, Acc. Chem. Res., 2011, 44, 1029–1038 CrossRef CAS PubMed.
- (a) M. K. Yu, J. Park and S. Jon, Theranostics, 2012, 2, 3–44 CrossRef CAS PubMed; (b) J. D. Rocca, D. Liu and W. Lin, Acc. Chem. Res., 2011, 44, 957–968 CrossRef PubMed; (c) M. Liong, J. Lu, M. Kovochich, T. Xia, S. G. Ruehm, A. E. Nel, F. Tamanoi and J. I. Zink, ACS Nano, 2008, 2, 889–896 CrossRef CAS PubMed.
- A. S. Paraskar, S. Soni, K. T. Chin, P. Chaudhuri, K. W. Muto, J. Berkowitz, M. W. Handlogten, N. J. Alves, B. Bilgicer, D. M. Dinulescu, R. A. Mashelkar and S. Sengupta, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 12335–12340 CrossRef PubMed.
- (a) P. B. Glover, P. R. Ashton, L. J. Childs, A. Rodger, M. Kercher, R. M. Williams, L. D. Cola and Z. Pikramenou, J. Am. Chem. Soc., 2003, 125, 9918–9919 CrossRef CAS PubMed; (b) E. L. Crossley, J. B. Aitken, S. Vogt, H. H. Harris and L. M. Rendina, Angew. Chem., Int. Ed., 2010, 49, 1231–1233 CrossRef CAS PubMed; (c) Z. Zhu, X. Wang, T. Li, S. Aime, P. J. Sadler and Z. Guo, Angew. Chem., Int. Ed., 2014, 53, 13225–13228 CrossRef CAS PubMed; (d) H. Li, R. Lan, C.-F. Chan, L. Jiang, L. Dai, D. W. J. Kwong, M. H.-W. Lam and K.-L. Wong, Chem. Commun., 2015, 51, 14022–14025 RSC.
- (a) D. Wang and S. J. Lippard, Nat. Rev. Drug Discovery, 2005, 4, 307–320 CrossRef CAS PubMed; (b) L. Kelland, Nat. Rev. Cancer, 2007, 7, 573–584 CrossRef CAS PubMed.
- S. Cotton, Lanthanides and actinides, McMillan Physical Science Series, McMillan Education, London, 1991 Search PubMed.
- (a) J.-C. G. Bünzli, Chem. Rev., 2010, 110, 2729–2755 CrossRef PubMed; (b) J.-C. G. Bünzli and S. V. Eliseeva, Chem. Sci., 2013, 4, 1939–1949 RSC; (c) E. J. New, A. Congreve and D. Parker, Chem. Sci., 2010, 1, 111–118 RSC; (d) E. J. New, D. Parker, D. G. Smith and J. W. Walton, Curr. Opin. Chem. Biol., 2010, 14, 238–246 CrossRef CAS PubMed.
- (a) M. C. Hefferin, L. M. Matosziuk and T. J. Meade, Chem. Rev., 2014, 114, 4496 CrossRef PubMed; (b) A. J. Amoroso and S. J. A. Pope, Chem. Soc. Rev., 2015, 44, 4723–4772 RSC; (c) S. J. Butler, M. Delbianco, L. Lamarque, B. K. McMahon, E. R. Neil, R. Pal, D. Parker, J. W. Walto and J. M. Zwier, Dalton Trans., 2015, 44, 4791–4803 RSC.
- X. Wang, H. Chang, J. Xie, B. Zhaoa, B. Liu, S. Xu, W. Pei, N. Ren, L. Huang and W. Huang, Coord. Chem. Rev., 2014, 273/274, 201–212 CrossRef.
- (a) S. Faulkner, L. S. Natarajan, W. S. Perry and D. Sykes, Dalton Trans., 2009, 3890–3899 RSC; (b) M. D. Ward, Coord. Chem. Rev., 2010, 254, 2634–2642 CrossRef CAS; (c) F.-F. Chen, Z.-Q. Chen, Z.-Q. Bian and C.-H. Huang, Coord. Chem. Rev., 2010, 254, 972–990 CrossRef.
- (a) S. I. Klink, H. Keizer and F. C. J. M. van Veggel, Angew. Chem., Int. Ed., 2000, 39, 4319–4321 CrossRef CAS; (b) S. J. A. Pope, B. J. Coe, S. Faulkner, E. V. Bichenkova, X. Yu and K. D. Douglas, J. Am. Chem. Soc., 2004, 126, 9490–9491 CrossRef CAS PubMed; (c) E. Baggaley, D.-K. Cao, D. Sykes, S. W. Botchway, J. A. Weinstein and M. D. Ward, Chem. – Eur. J., 2014, 20, 8898–8903 CrossRef CAS PubMed.
- (a) P. Kadjane, C. P.-Iglesias, R. Ziessel and L. J. Charbonnière, Dalton Trans., 2009, 5688–5700 RSC; (b) R. Ziessel, S. Diring, P. Kadjane, L. Charbonnière, P. Retailleau and C. Philouze, Chem. – Asian J., 2007, 2, 975–982 CrossRef CAS PubMed; (c) T. K. Ronson, T. Lazarides, H. Adams, S. J. A. Pope, D. Sykes, S. Faulkner, S. J. Coles, M. B. Hursthouse, W. Clegg, R. W. Harrington and M. J. Ward, Chem. – Eur. J., 2006, 12, 9299–9313 CrossRef CAS PubMed.
- (a) N. Inukai, T. Kawai and J. Yuasa, Chem. – Eur. J., 2013, 19, 5938–5947 CrossRef CAS PubMed; (b) N. Inukai, T. Kawai and J. Yuasa, Chem. Commun., 2011, 47, 9128–9130 RSC; (c) J. Yuasa, T. Ohno, H. Tsumatori, R. Shiba, H. Kamikubo, M. Kataoka, Y. Hasegawa and T. Kawai, Chem. Commun., 2013, 49, 4604–4606 RSC; (d) C. P. Montgomery, E. J. New, D. Parker and R. D. Peacock, Chem. Commun., 2008, 4261–4263 RSC.
- W. C. Johnson, Circular Dichroism: Principles and Applications, ed. K. Nakanishi, N. Berova and R. W. Woody, VCH, New York, 1994, pp. 523–540 Search PubMed.
- M. V. Keck and S. J. Lippard, J. Am. Chem. Soc., 1992, 114, 3386–3390 CrossRef CAS.
Footnotes |
| † Dedicated to Professor Animesh Chakravorty on the occasion of his 80th birthday. |
| ‡ Electronic supplementary information (ESI) available: Experimental details; synthesis and characterization of compounds; ESI-MS analysis; DNA and BSA binding experiments; cytotoxicity and cellular internalization studies. See DOI: 10.1039/c5dt04470g |
| § A. C. and K. S. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |