
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nickel(II) complexes of N-CH2CF3 cyclam derivatives as contrast agents for 19F magnetic resonance imaging†
Jan
Blahut
a,
Petr
Hermann
a,
Andrea
Gálisová
b,
Vít
Herynek
b,
Ivana
Císařová
a,
Zdeněk
Tošner
c and
Jan
Kotek
*a
aDepartment of Inorganic Chemistry, Faculty of Science, Charles University (Univerzita Karlova), Hlavova 2030, 128 43 Prague 2, Czech Republic. E-mail: modrej@natur.cuni.cz; Fax: +420-221951253; Tel: +420-221951261
bDepartment of Radiodiagnostic and Interventional Radiology, Magnetic Resonance Unit, Institute for Clinical and Experimental Medicine, Vídeňská 1958/9, Prague 4, 140 21 Czech Republic
cNMR Laboratory, Faculty of Science, Charles University (Univerzita Karlova), Hlavova 2030, 128 43 Prague 2, Czech Republic
First published on 20th November 2015
Abstract
Kinetically inert Ni(II) complexes of N1,N8-bis(2,2,2-trifluoroethyl)cyclams with hydrogen atoms or phosphonic acid groups in the N4,N11-positions show significant 19F NMR relaxation rate enhancement useful for 19-fluorine MRI imaging.
Magnetic resonance imaging (MRI) is one of the most common techniques in molecular imaging. It is based on the detection of the NMR signal originating from water protons in a tissue. To increase its sensitivity, paramagnetic contrast agents (CAs) are often applied.1,2 They affect mainly the longitudinal (T1) relaxation time of the 1H signal, which leads to an increase in the intensity of the water proton MRI signal. However, essentially all tissues contain water and, thus, the background signal compromises the detection accuracy. This problem can be solved by using non-proton MRI and the 19F nucleus seems to be the most promising candidate.3–6 Natural monoisotopic 19F has an NMR resonance frequency close to that of 1H (40.08 MHz T−1 for 19F compared to 42.58 MHz T−1 for 1H) and exhibits sensitivity comparable to 1H (83%). Fluorine concentration in organisms is virtually zero and, therefore, the lack of background in fluorine-based images enables “hot-spot” imaging. The wider spectral range of the 19F nucleus (∼350 ppm) compared to 1H (∼10 ppm) is also beneficial for some applications. Moreover, only small hardware and software adjustments of standard 1H scanners are needed for 19F detection.6 This makes the nucleus very potent for e.g. cellular tracking of labelled cell cultures.7–11
However, the 19F nucleus present in organic molecules has usually a very long T1 relaxation time requiring a long delay between excitation pulses; this prolongs the total duration of imaging experiments to unrealistic lengths. Shortening of the T1 relaxation time can result in significant shortening of the experimental time. However, it is necessary to take into account the concomitant shortening of the transversal (T2 or 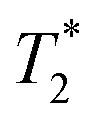 ) relaxation time, which leads to signal broadening and can result in very fast loss of signal intensity. It has been shown that the introduction of highly paramagnetic lanthanide(III) ions to the close vicinity of the fluorine atom(s) leads to significant shortening of the relaxation times,12 and the
) relaxation time, which leads to signal broadening and can result in very fast loss of signal intensity. It has been shown that the introduction of highly paramagnetic lanthanide(III) ions to the close vicinity of the fluorine atom(s) leads to significant shortening of the relaxation times,12 and the 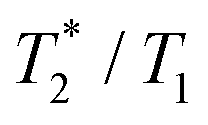 ratio is in the range of 0.3–0.9, which is suitable for MRI measurements.12c,13
ratio is in the range of 0.3–0.9, which is suitable for MRI measurements.12c,13
It is known that, despite the low overall electronic spin (S = 1) and magnetic momentum (μeff ∼ 3 B.M.) of Ni(II), this ion can induce a large paramagnetic chemical shift and relaxation enhancement comparable to that of lanthanide(III) ions with higher S and μ.14 Therefore, some Ni(II) complexes have been studied as MRI agents employable in the Chemical Exchange Saturation Transfer method.15 Here, we decided to study the 19F NMR relaxation properties of Ni(II) complexes. The Ni(II) ion fits perfectly in the cavity of 1,4,8,11-tetraazacyclotetradecane (cyclam) and cyclam derivatives are well-known to form Ni(II) complexes with high thermodynamic stability, especially with ligands having coordinating pendant arms enabling octahedral binding to the metal. The 2,2,2-trifluoroethyl side arm was chosen as a group containing a high number of equivalent fluorine atoms. Therefore, ligands 1 and H4te2p-tfe2 (Fig. 1) were suggested for testing the 19F NMR parameters of their complexes.
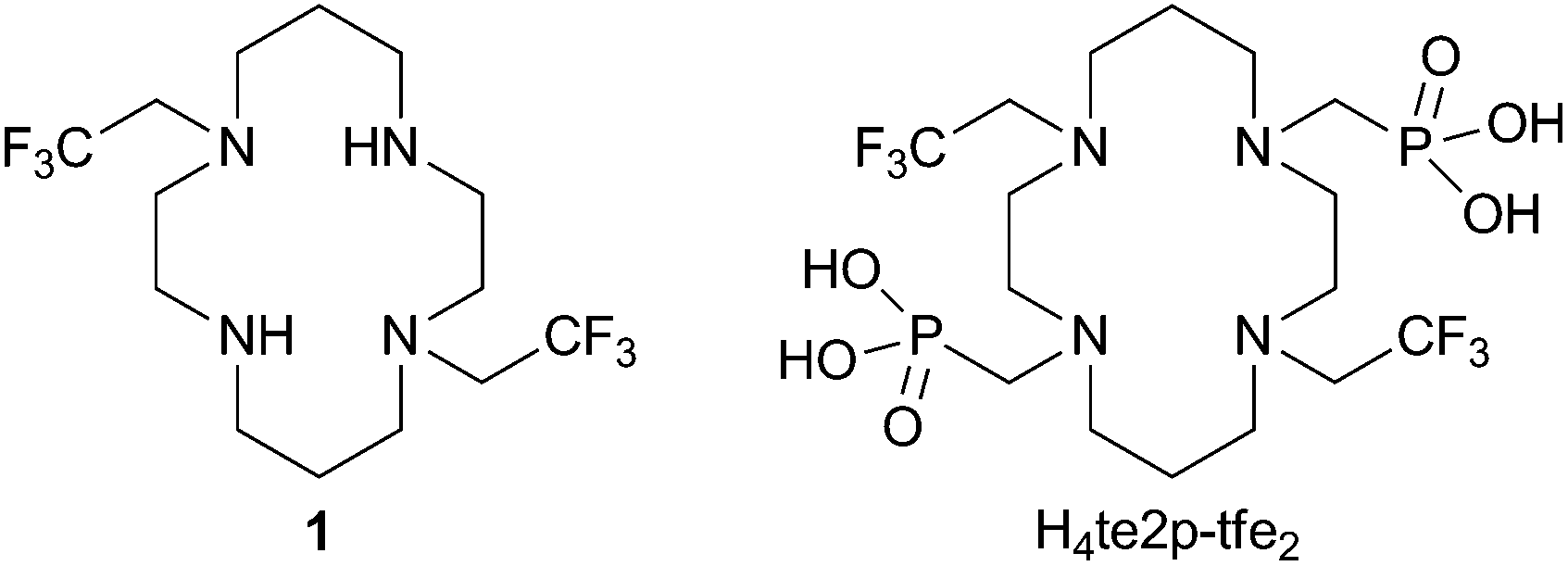 | ||
| Fig. 1 Ligands studied in this work. | ||
Acylation of 1,8-dibenzylcyclam with ethyl trifluoroacetate or trifluoroacetic anhydride yielded the corresponding bis(amide). It was followed by BH3 reduction16 in diglyme at elevated temperature and the benzyl protecting groups were removed by Pd/C hydrogenolysis to obtain ligand 1. The reaction17 of amine 1 in neat P(OEt)3 with CH2O led to the tetraethyl bis(methylenephosphonate) cyclam derivative. The ethylester groups were removed by transesterification with trimethylsilylbromide18 followed by the silylester hydrolysis to yield H4te2p-tfe2, which was isolated in a zwitterionic form after ion exchange chromatography. Synthetic details and results of a single-crystal X-ray diffraction study of 1·2HCl·2H2O and H4te2p-tfe2·4HBr·0.5H2O are given in the ESI (Fig. S5 and S6†).
Ligand 1 (in the form of a hydrochloride) reacts with Ni(II) salts in aqueous solutions to give a light greenish-blue precipitate. The structure of this compound was determined by a single-crystal X-ray study as cis-[Ni(1)(Cl)2] (see ESI Fig. S7†); the central ion is surrounded by four cyclam nitrogen atoms in the cis-V configuration19 with two-fold symmetry (dNi–N = 2.10 and 2.26 Å for secondary and tertiary amines, respectively) and cis-chloride anions coordinated with dNi–Cl = 2.42 Å.
However, the cis-[Ni(1)(Cl)2] complex shows extremely low solubility in all solvents. Thus, the presence of chloride ions had to be avoided during the preparation of a water-soluble complex. Therefore, ligand 1 in the form of a free base and Ni(ClO4)2 was used for further complex preparation. The course of the reaction in the H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMSO 1:6.5 mixture at 50 °C was followed by 19F NMR spectroscopy (Fig. S2†). Such a solvent mixture was used to keep the reaction mixture fully homogeneous right from the beginning as compound 1 is poorly soluble in water. The reaction proceeds (at 50 °C) through an intermediate (δF = −22.9 ppm) and is completed during 90 min to give the final complex with δF = −29.3 ppm (at 50 °C). On cooling to 25 °C, the signal shifts to −26.5 ppm. No further 19F NMR spectral changes were observed upon heating the solution at 100 °C for several days.
:DMSO 1:6.5 mixture at 50 °C was followed by 19F NMR spectroscopy (Fig. S2†). Such a solvent mixture was used to keep the reaction mixture fully homogeneous right from the beginning as compound 1 is poorly soluble in water. The reaction proceeds (at 50 °C) through an intermediate (δF = −22.9 ppm) and is completed during 90 min to give the final complex with δF = −29.3 ppm (at 50 °C). On cooling to 25 °C, the signal shifts to −26.5 ppm. No further 19F NMR spectral changes were observed upon heating the solution at 100 °C for several days.
To obtain an aqueous stock solution, prolonged heating (80 °C) of the suspension of ligand 1 with Ni(ClO4)2 in H2O:MeOH 1:1 (with subsequent evaporation of MeOH) was used. It led to the formation of a light blue aqueous solution of a single product with δF = −26.2 ppm. When the aqueous solution of the complex prepared in H2O:MeOH was mixed with the sample prepared in H2O:DMSO, only one symmetric signal in 19F NMR was observed revealing that the species formed in both experiments are identical complexes.
Despite a number of attempts, we were not able to crystallize this light blue product. However, red single-crystals of trans-[Ni(1)](ClO4)2 (Fig. 2) were obtained when the blue aqueous solution of the complex was saturated with NaClO4 and was left standing for a few weeks. In this complex, the cyclam ring is coordinated in the centrosymmetric trans-III configuration19 (dNi–N = 1.95 and 1.99 Å for secondary and tertiary amino groups, respectively). Consistent with the red colour, only very weak axial interaction with perchlorate anions located in distant positions (dNi–O = 2.83 Å) was observed.
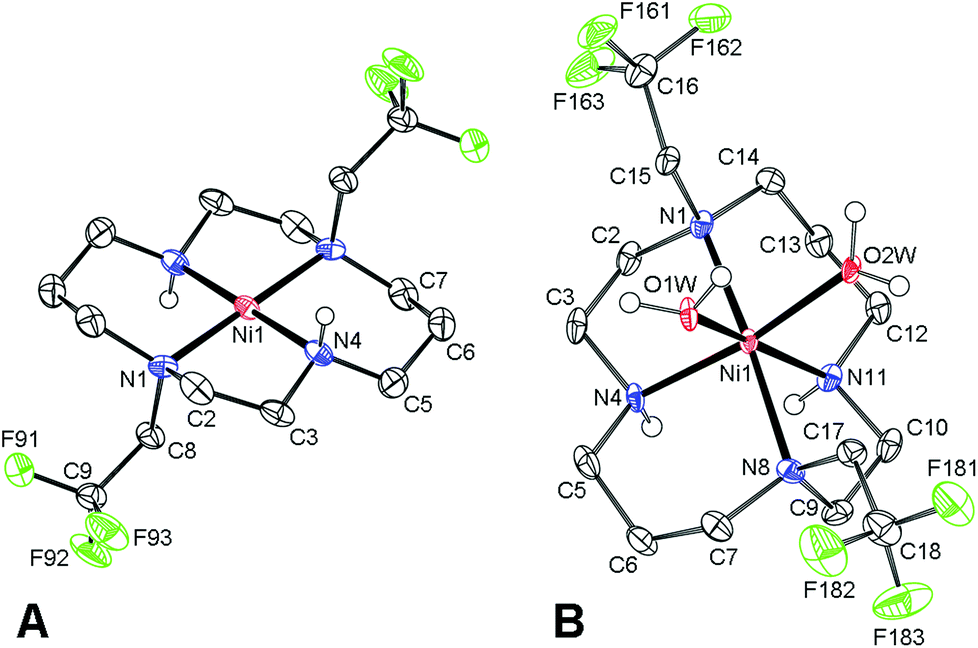 | ||
| Fig. 2 Structures of (A): trans-[Ni(1)]2+ and (B): cis-[Ni(1)(H2O)2]2+ complex cations found in the solid state structures of trans-[Ni(1)](ClO4)2 and cis-[Ni(1)(H2O)2](TsO)2, respectively. Carbon-bound hydrogen atoms are omitted for clarity. Thermal ellipsoids are shown at the 60% probability level. | ||
Dissolution of the red trans-[Ni(1)](ClO4)2 complex in water produced a light blue solution with δF = −19.3 ppm and the species slowly isomerizes with first-order kinetics (τ1/2 ∼3.5 h, 25 °C) to a species with δF −26.3 ppm (Fig. S3†). The final species is identical to the original Ni(II)–1 complex, as it was confirmed by 19F NMR after the addition of a standard. Taking into account the isomerism of metal ion–cyclam complexes,19 red trans-[Ni(1)](ClO4)2 with the trans-III configuration probably forms a blue hexacoordinated trans-[Ni(1)(H2O)2]2+ species upon dissolution, and this complex is rearranged in solution to the cis-[Ni(1)(H2O)2]2+ species with the cis-V cyclam conformation.
Typically, the trans-III isomer of cyclam complexes is considered to be the thermodynamically most stable one,20 and the preference of the cis-V cyclam conformation for the Ni(II)–1 complex is rather surprising. However, the higher stability of the cis-V isomer over the trans-III one was supported also by the isolation of cis-[Ni(1)(H2O)2](TsO)2 (Fig. 2). The structure shows Ni–N distances of 2.07 and 2.09 Å for secondary amino groups, and 2.22 and 2.25 Å for tertiary ones, respectively, with two water molecules coordinated with dNi–O = 2.07 and 2.10 Å.
Based on the data presented above, one can conclude that the reaction of ligand 1 with Ni(ClO4)2 leads to the formation of the cis-[Ni(1)(H2O)2](ClO4)2 complex. The geometries of Ni(II) coordination polyhedra found in the solid state structures are compiled in Table S2† and are comparable to other [Ni(L)(H2O)2] complexes of cyclam derivatives.21,22 The Ni–F distances found in all solid-state structures were in the range of 4.8–5.4 Å (Table S3†).
The complex of the second studied ligand, H4te2p-tfe2, was prepared by heating the ligand together with a slight excess of NiCl2 in aq. ammonia (pH 10) at 75 °C for 24 h; the excess of Ni(II) ions was removed by column chromatography. As mentioned above, the course of the reaction was followed by 19F NMR (Fig. S4†), which showed a fast drop in the concentration of the free ligand (δF = −68.3 ppm), and the formation of an intermediate (δF = −41.1 ppm) and its slower rearrangement to the final product (δF = −26.4 ppm). The time-dependence of intensities of all three signals could be satisfactorily fitted using a monoexponential function (Fig. S4†) and showed comparable rate constants for all three processes (see the ESI†). Such behaviour points to the presence of an equilibrium between the free ligand and the intermediate with an irreversible (rate-determining) reaction step leading to the formation of the final complex.
The final product was isolated in the form of light blue crystals, which were identified as (NH4){trans-[Ni(Hte2p-tfe2)]}·3.25H2O by single-crystal X-ray diffraction. Therefore, the complex species present in solution are expected to be trans-[Ni(Hnte2p-tfe2)]n−2 (n = 0, 1) depending on pH. The molecular structure of the complex anion is shown in Fig. 3, and geometric parameters of the coordination sphere of Ni(II) and Ni–F distances are listed in Tables S2 and S3.† The cyclam ring exhibits the trans-III configuration19 (dNi–N are ∼2.10 and 2.11 Å for amino groups bearing the methylenephosphonate pendant arms, and 2.22 and 2.23 Å for those substituted by trifluoroethyl groups) with the oxygen atoms of phosphonate groups occupying apical positions (Ni–O distances are 2.06 and 2.10 Å, respectively). The molecules of the complex are connected via short hydrogen bonds between the oxygen atoms of protonated and unprotonated phosphonate pendants (dO⋯O = 2.47 Å), forming infinite chains, similar to what was found for analogous complexes of cyclam-methylenephosphonate derivatives.23,24 Bonding distances and the overall molecular structure are very similar to those of Ni(II) complexes of analogous derivatives.22,24
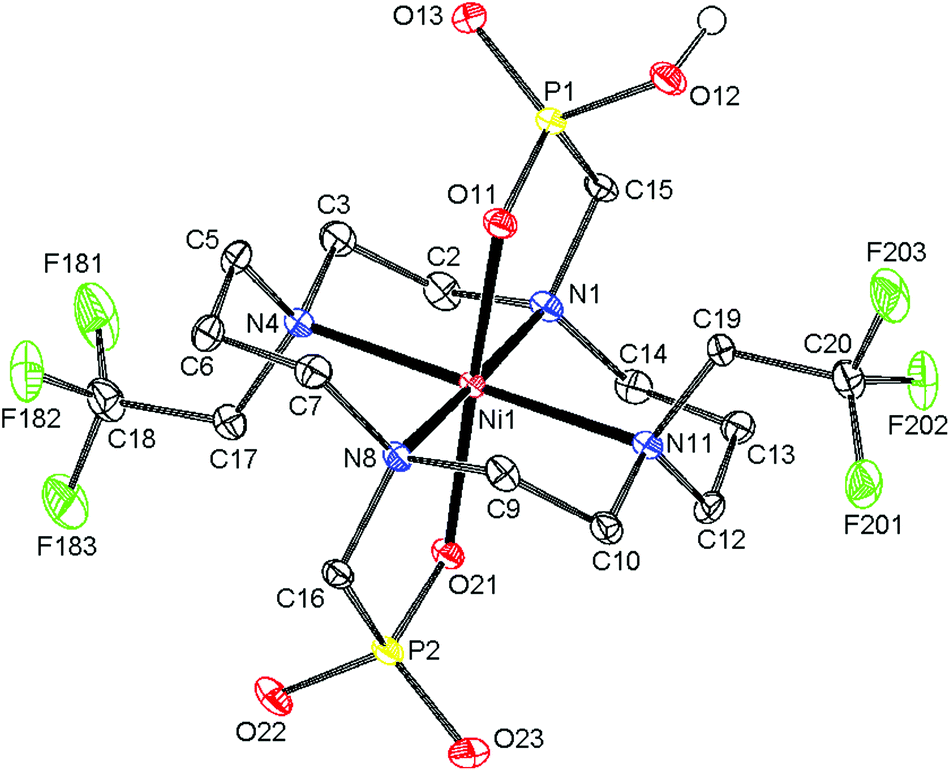 | ||
| Fig. 3 Molecular structure of the trans-[Ni(Hte2p-tfe2)]− anion found in the crystal structure of trans-(NH4)[Ni(Hte2p-tfe2)]·3.25H2O. Carbon-bound hydrogen atoms are omitted for clarity. Thermal ellipsoids are shown at the 60% probability level. | ||
The thermodynamics of complexing properties of the phosphonate H4te2p-tfe2 ligand was studied by potentiometry (see the ESI and Table S4†). The comparison of ligand stepwise protonation constants (logK1–4 10.86, 10.09, 5.60 and 4.73) with those of the N1,N8-dimethyl-N4,N11-bis(methylenephosphonate) analogue25 (logK1–4 11.47, 12.17, 7.20 and 6.33, Table S5†) points to significantly decreased ligand basicity caused by the presence of electron-withdrawing –CH2CF3 groups. Surprisingly, it affects not only the first two protonation constants corresponding to the ring amino groups but also those of the phosphonate moieties, probably as a result of a strong electron-withdrawing effect transferred through intramolecular hydrogen bonds, which are expected to have a geometry analogous to that found for related cyclam derivatives.25 Equilibration of the Ni(II)–H4te2p-tfe2 system is relatively slow and, therefore, the out-of-cell titration method had to be used. As the complexation mechanism is not fully straightforward (see above), the samples used for the out-of-cell titration were heated at 50 °C for 2 weeks to ensure quantitative rearrangement of the intermediate to the final trans isomer. The time required for equilibration was checked by 19F NMR of separate samples. The stability constant, logKNiL = 13.28, is about 2–7 orders of magnitude lower than the constants of complexes with related ligands (Table S5†),26,27 mainly as a consequence of the lower ligand basicity. The distribution diagram of the system (Fig. S8†) shows that full Ni(II) complexation by H4te2p-tfe2 is completed at pH 7 and the complex is present at this pH almost entirely in a fully deprotonated form.
For possible in vitro/in vivo utilization, kinetic inertness is a more important parameter than thermodynamic stability. Kinetic inertness is often tested in acidic solutions as acid-assisted complex dissociation. Thus, the decomposition of both studied complexes was examined in 1 M aq. HCl at 37 and 80 °C. Both complexes are decomposed relatively slowly by HCl at 37 °C (τ1/2 ∼8 and ∼10 h for cis-[Ni(1)(H2O)2]2+ and trans-[Ni(Hnte2p-tfe2)]n−2, respectively) but the decomplexation of the cis-[Ni(1)(H2O)2]2+ complex is substantially accelerated at the higher temperature (80 °C, τ1/2 ∼ 3 min compared to ∼5 h for trans-[Ni(Hnte2p-tfe2)]n−2, Table S6†), as the presence of apically coordinated pendant arms in the H4te2p-tfe2 complex enhances kinetic inertness. High inertness has been observed for several Cu(II) complexes with analogous cyclam-based ligands,28 and highly protonated species of several Ni(II)22,24 complexes of phosphonated cyclam derivatives have been isolated even in the solid state. These results suggest sufficient complex stability under physiological conditions and warrant the possible use of the Ni(II)–H4te2p-tfe2 complex in in vitro/in vivo applications.
As the trans-[Ni(te2p-tfe2)]2− complex is kinetically inert and promises reasonable stability in vivo, its 19F MRI-related parameters were investigated. Although the cis-[Ni(1)(H2O)2]2+ complex is not suitable for any in vivo application due to its low solubility in chloride-containing media, it was studied as well for comparative purposes as there are no related data reported in the literature at all. The 19F NMR relaxation measurement of both Ni(II) complexes at B0 = 7.05 T showed extreme shortening of 19F NMR T1 relaxation times by 2–3 orders of magnitude compared with the values observed for the free ligands, 1 and H4te2p-tfe2 (Table 1).
| Parameter | 1 | cis-[Ni(1)(H2O)2]2+ | H4te2p-tfe2 | trans-[Ni(te2p-tfe2)]2– |
|---|---|---|---|---|
a
T
1 was determined using inversion recovery pulse sequence; 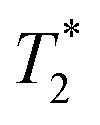 was determined from line-width using Lorentzian-shape fitting of the signal. was determined from line-width using Lorentzian-shape fitting of the signal.
|
||||
| B 0 = 7.05 T (300 MHz for 1H, 282 MHz for 19F) | ||||
| T 1(19F) | 0.8(3) s | 1.72(1) ms | 0.5(1) s | 2.8(7) ms |
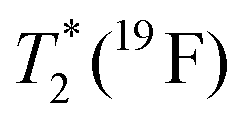
|
≈76 ms | ≈0.82 ms | ≈50 ms | ≈0.90 ms |
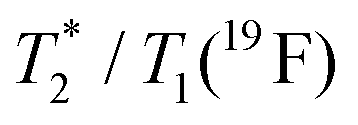
|
0.1 | 0.48 | 0.1 | 0.32 |
| r 1(1H) | — | 0.83(3) | — | 0.18(1) |
| B 0 = 4.70 T (200 MHz for 1H, 188 MHz for 19F) | ||||
| T 1(19F) | 0.82(1) s | 1.2(1) ms | 1.1(2) s | 4.2(1.1) ms |
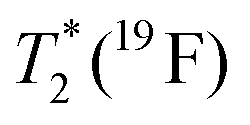
|
3.1(1) ms | 0.62(1) ms | 3.1(2) ms | 1.1(1) ms |
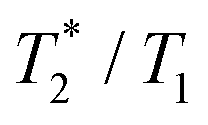
|
0.0038 | 0.52 | 0.0028 | 0.26 |
| r 1(1H) | — | 0.66(4) s–1 mM–1 | — | 0.12(2) s–1 mM–1 |
The suitability of the trans-[Ni(te2p-tfe2)]2−complex for 19F MRI was tested by phantom visualization at B = 4.7 T. At this magnetic field, the T1 of the complex is very short in the millisecond range with a still convenient 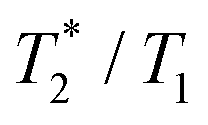 relaxation times ratio (Table 1). The observed relaxation times are even slightly shorter than those reported for studied Ln(III) complexes – in the cases of highly paramagnetic Tb(III), Dy(III) and Ho(III) complexes with the estimated Ln(III)–F distance lying in the range of 5–7 Å, the reported T1 is typically in the range of 7–11 ms at 4.7 T and room temperature.12c,13 The very short relaxation time of the Ni(II) complex required optimization of the fast pulse sequence – for the visualization of the complex, a fast gradient echo sequence with TE = 1.3 ms and TR = 3 ms was used. The slowly relaxing samples (containing the free ligand and trifluoroethanol used as a standard) were best measured using a long turbospin echo sequence employing TE = 40 ms and TR = 2000 ms. For localization of the samples, the 1H MRI scan (Fig. 4A) was also acquired. Fig. 4 shows the results of the MRI visualization. The brightness of aq. solution of the Ni(II) complex compared to aq. solutions of the free ligand and trifluoroethanol is caused by its paramagnetism, which shortens the T1 relaxation time of water protons (r1(complex) = 0.12 mm−1 s−1, 4.7 T, 25 °C). As each sample has a different 19F NMR chemical shift (δF −26 ppm, −68 ppm and −77 ppm for the complex, free ligand and trifluoroethanol, respectively), each signal can be excited separately. In the case of the fast sequence, only a negligible signal of the free ligand was detected, as virtually no diamagnetic sample relaxation occurs during the sequence time-scale. On the contrary, in the experiment employing the long sequence, no signal of the paramagnetic sample was found as its magnetization relaxes before the start of acquisition.
relaxation times ratio (Table 1). The observed relaxation times are even slightly shorter than those reported for studied Ln(III) complexes – in the cases of highly paramagnetic Tb(III), Dy(III) and Ho(III) complexes with the estimated Ln(III)–F distance lying in the range of 5–7 Å, the reported T1 is typically in the range of 7–11 ms at 4.7 T and room temperature.12c,13 The very short relaxation time of the Ni(II) complex required optimization of the fast pulse sequence – for the visualization of the complex, a fast gradient echo sequence with TE = 1.3 ms and TR = 3 ms was used. The slowly relaxing samples (containing the free ligand and trifluoroethanol used as a standard) were best measured using a long turbospin echo sequence employing TE = 40 ms and TR = 2000 ms. For localization of the samples, the 1H MRI scan (Fig. 4A) was also acquired. Fig. 4 shows the results of the MRI visualization. The brightness of aq. solution of the Ni(II) complex compared to aq. solutions of the free ligand and trifluoroethanol is caused by its paramagnetism, which shortens the T1 relaxation time of water protons (r1(complex) = 0.12 mm−1 s−1, 4.7 T, 25 °C). As each sample has a different 19F NMR chemical shift (δF −26 ppm, −68 ppm and −77 ppm for the complex, free ligand and trifluoroethanol, respectively), each signal can be excited separately. In the case of the fast sequence, only a negligible signal of the free ligand was detected, as virtually no diamagnetic sample relaxation occurs during the sequence time-scale. On the contrary, in the experiment employing the long sequence, no signal of the paramagnetic sample was found as its magnetization relaxes before the start of acquisition.
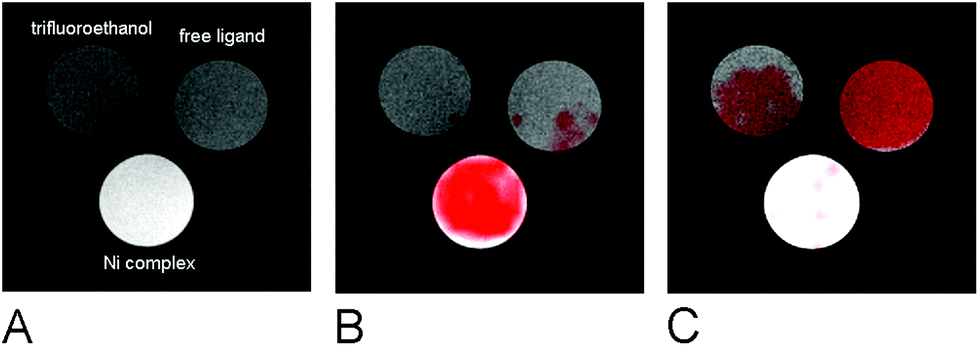 | ||
| Fig. 4 MRI study of phantoms containing trifluoroethanol, free H4te2p-tfe2 and the trans-[Ni(te2p-tfe2)]2− complex (cF = 0.004 M in all samples), B = 4.7 T, 25 °C, home-made 1H/19F surface single loop coil. (A) 1H MRI scan, gradient echo sequence, flip angle 30°, TE = 3.7 ms, TR = 100 ms, matrix 256 × 256. (B) Overlay of 1H MRI with 19F MRI; 19F MRI was optimized for the complex; acquired at δ = −26 ppm, gradient echo sequence, TE = 1.3 ms, TR = 3 ms, matrix 32 × 32 interpolated to 256 × 256. (C) Overlay of 1H MRI with 19F MRI; 19F MRI was optimized for the ligand; acquired at δ = –77 ppm, turbospin echo sequence, TE = 40 ms, TR = 2000 ms, matrix 32 × 32 interpolated to 256 × 256. | ||
The samples of free ligand 1 and cis-[Ni(1)(H2O)2](ClO4)2 show fully concordant behaviour (Fig. S9†).
In conclusion, transition metal ion complexes of fluorine-containing ligands can be considered a new class of 19F MRI contrast agents, as shown in the case of Ni(II). The presence of strongly complexing and electron donating phosphonates enhances the kinetic inertness of the studied complexes and compensates the disadvantageous coordination properties of fluorine-containing ligands. Relaxation parameters of the trans-[Ni(te2p-tfe2)]2− complex with fluorine atoms located about 5 Å from the Ni(II) centre are highly suitable for 19F MRI hot-spot imaging employing fast pulse sequences. As the Ni(II) complexes with coordinated water molecules exhibit useful water proton T1-relaxivity, properly designed compounds could be potentially used as dual 1H/19F MRI contrast agents.29
The work was supported by the Czech Science Foundation (P207-11-1437) and by the project of the Ministry of Health, Czech Republic, for development of research organization IN00023001 (Institutional support, Institute for Clinical and Experimental Medicine). We thank Z. Böhmová and J. Hraníček for potentiometric and AAS measurements, respectively.
References
- The Chemistry of Contrast Agents in Medical Magnetic Resonance Imaging, ed. A. Merbach, L. Helm and É. Tóth, Wiley, Hoboken, 2nd edn, 2013 Search PubMed.
- P. Caravan, J. J. Ellison, T. J. McMurry and R. B. Lauffer, Chem. Rev., 1999, 99, 2293–2352 CrossRef CAS PubMed; P. Hermann, J. Kotek, V. Kubíček and I. Lukeš, Dalton Trans., 2008, 3027–3047 RSC; C. F. G. C. Geraldes and S. Laurent, Contrast Media Mol. Imaging, 2009, 4, 1–23 CrossRef PubMed.
- J. Ruiz-Cabello, B. P. Barnett, P. A. Bottomley and J. W. M. Bulte, NMR Biomed., 2011, 24, 114–129 CrossRef CAS PubMed.
- J. C. Knight, P. G. Edwards and S. J. Paisey, RSC Adv., 2011, 1, 1415–1425 RSC.
- J.-X. Yu, R. R. Hallac, S. Chiguru and R. P. Mason, Prog. Nucl. Magn. Reson. Spectrosc., 2013, 70, 25–49 CrossRef CAS PubMed.
- I. Tirotta, V. Dichiarante, C. Pigliacelli, G. Cavallo, G. Terraneo and F. B. Bombelli, Chem. Rev., 2015, 115, 1106–1129 CrossRef CAS PubMed.
- S. Temme, F. Boenner, J. Schrader and U. Floegel, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2012, 4, 329–343 CrossRef CAS PubMed.
- E. T. Ahrens and J. Zhong, NMR Biomed., 2013, 26, 860–871 CrossRef CAS PubMed.
- Y. B. Yu, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2013, 5, 646–661 CrossRef CAS PubMed.
- D. Bartusik and B. Tomanek, Adv. Drug Delivery Rev., 2013, 65, 1056–1064 CrossRef CAS PubMed.
- H. Amiri, M. Srinivas, A. Veltien, M. J. van Uden, I. J. M. de Vries and A. Heerschap, Eur. J. Radiol., 2015, 25, 726–735 CrossRef PubMed.
- (a) P. K. Senanayake, A. M. Kenwright, D. Parker and S. K. van der Hoorn, Chem. Commun., 2007, 2923–2925 RSC; (b) A. M. Kenwright, I. Kuprov, E. De Luca, D. Parker, S. U. Pandya, P. K. Senanayake and D. G. Smith, Chem. Commun., 2008, 2514–2516 RSC; (c) K. H. Chalmers, E. De Luca, N. H. M. Hogg, A. M. Kenwright, I. Kuprov, D. Parker, M. Botta, J. I. Wilson and A. M. Blamire, Chem. – Eur. J., 2010, 16, 134–148 CrossRef CAS PubMed; (d) K. H. Chalmers, M. Botta and D. Parker, Dalton Trans., 2011, 40, 904–913 RSC; (e) P. Harvey, I. Kuprov and D. Parker, Eur. J. Inorg. Chem., 2012, 2015–2022 CrossRef CAS; (f) P. Harvey, K. H. Chalmers, E. De Luca, A. Mishra and D. Parker, Chem. – Eur. J., 2012, 18, 8748–8757 CrossRef CAS PubMed; (g) E. De Luca, P. Harvey, K. H. Chalmers, A. Mishra, P. K. Senanayake, J. I. Wilson, M. Botta, M. Fekete, A. M. Blamire and D. Parker, J. Biol. Inorg. Chem., 2014, 19, 215–227 CrossRef PubMed.
- K. H. Chalmers, A. M. Kenwright, D. Parker and A. M. Blamire, Magn. Reson. Med., 2011, 66, 931–936 CrossRef CAS PubMed.
- E. Belorizky, P. H. Fries, L. Helm, J. Kowalewski, D. Kruk, R. R. Sharp and P.-O. Westlund, J. Chem. Phys., 2008, 128, 052315 CrossRef PubMed.
- A. O. Olatunde, S. J. Dorazio, J. A. Spernyak and J. R. Morrow, J. Am. Chem. Soc., 2012, 134, 18503–18505 CrossRef CAS PubMed.
- W. Curran and R. Angier, J. Org. Chem., 1966, 31, 3867–3868 CrossRef CAS.
- X. Sun, M. Wuest, Z. Kovács, A. D. Sherry, R. Motekaitis, Z. Wang, A. E. Martell, M. J. Welch and C. J. Anderson, J. Biol. Inorg. Chem., 2003, 8, 217–225 CrossRef CAS PubMed.
- C. E. McKenna, M. T. Higa, N. H. Cheung and M.-C. McKenna, Tetrahedron Lett., 1977, 18, 155–158 CrossRef.
- B. Bosnich, C. K. Poon and M. Tobe, Inorg. Chem., 1965, 4, 1102–1108 CrossRef CAS.
- (a) K. R. Adam, I. M. Atkinson and L. F. Lindoy, Inorg. Chem., 1997, 36, 480–481 CrossRef CAS; (b) M. Zimmer, Coord. Chem. Rev., 2001, 212, 133–163 CrossRef CAS.
- M. A. Donnelly and M. Zimmer, Inorg. Chem., 1999, 38, 1650–1658 CrossRef CAS PubMed.
- J. Kotek, P. Vojtíšek, I. Císařová, P. Hermann and I. Lukeš, Collect. Czech. Chem. Commun., 2001, 66, 363–381 CrossRef CAS.
- J. Kotek, P. Hermann, I. Císařová, J. Rohovec and I. Lukeš, Inorg. Chim. Acta, 2001, 317, 324–330 CrossRef CAS.
- J. Havlíčková, H. Medová, T. Vitha, J. Kotek, I. Císařová and P. Hermann, Dalton Trans., 2008, 5378–5386 RSC.
- J. Kotek, P. Vojtíšek, I. Císařová, P. Hermann, P. Jurečka, J. Rohovec and I. Lukeš, Collect. Czech. Chem. Commun., 2000, 65, 1289–1316 CrossRef CAS.
- I. Svobodová, J. Havlíčková, J. Plutnar, P. Lubal, J. Kotek and P. Hermann, Eur. J. Inorg. Chem., 2009, 3577–3592 CrossRef.
- I. Svobodová, P. Lubal, J. Plutnar, J. Havlíčková, J. Kotek, P. Hermann and I. Lukeš, Dalton Trans., 2006, 5184–5197 RSC.
- J. Kotek, P. Lubal, P. Hermann, I. Císařová, I. Lukeš, T. Godula, I. Svobodová, P. Táborský and J. Havel, Chem. – Eur. J., 2003, 9, 233–248 CrossRef CAS PubMed.
- M. Wolters, S. G. Mohades, T. M. Hackeng, M. J. Post, M. E. Kooi and W. H. Backes, Invest. Radiol., 2013, 48, 341–350 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of studied compounds, single-crystal RTG diffraction data, details on potentiometric study, study of acid-assisted dissociation of prepared complexes, relaxation study, 1H/19F MRI visualization. CCDC 1430237–1430242. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt04138d |
| This journal is © The Royal Society of Chemistry 2016 |