
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Alcohol- and water-soluble bis(tpy)quaterthiophenes with phosphonium side groups: new conjugated units for metallo-supramolecular polymers†
P.
Štenclová
,
K.
Šichová
,
I.
Šloufová
,
J.
Zedník
,
J.
Vohlídal
* and
J.
Svoboda
*
Department of Physical and Macromolecular Chemistry, Charles University in Prague, Faculty of Science, Hlavova 2030, Prague 2, CZ-128 40, Czech Republic. E-mail: jiri.vohlidal@natur.cuni.cz; jan.svoboda@natur.cuni.cz; Fax: +420 224919752; Tel: +420 221951310
First published on 2nd December 2015
Abstract
Bis(tpy)quaterthiophenes with symmetrically distributed two and four 6-bromohexyl side groups were prepared and modified by the reaction with triethylphosphine to give the corresponding ionic species. Both ionic and non-ionic bis(tpy)quaterthiophenes (unimers) were assembled with Zn2+ and Fe2+ ions to conjugated metallo-supramolecular polymers (MSPs), of which the ionic ones are soluble in alcohols and those derived from tetrasubstituted unimers are soluble even in water. The differences in assembly are specified between systems with (i) ionic and non-ionic unimers, (ii) Zn2+ and Fe2+ ion couplers, and (iii) methanol and water solvents. A substantial decrease in the stability of Fe-MSPs and a surprisingly high red shift of the luminescence band of Zn-MSPs were observed on going from methanol to aqueous solutions.
Introduction
Conjugated metallo-supramolecular polymers (MSPs) derived from α,ω-bis(tpy)oligoarylenes (tpy stands for 2,2′:6′,2′′-terpyridine-4′-yl end-group) are of interest as potential materials for devices with applications based on the light/electricity inter-conversion and non-linear optical phenomena (light-emitting devices, photovoltaic cells, etc.).1–16 However, an overwhelming majority of these MSPs suffer from low solubility, which makes their processing difficult. For example, MSPs derived from α,ω-bis(tpy)oligophenylenes have been processed from acids17,18 that surely are not optimal solvents.Chains of conjugated MSPs of this type are composed of molecules of bisterpyridines that are linked via facial and meridian coordination of their tpy end-groups to metal ions such as Ru2+, Fe2+, Zn2+ and Co2+ (generally Mt2+) ions. The tpy-Mt2+-tpy linkages are strictly linear and rigid.11,19–27 The enchained unimers are also quite rigid due to the delocalization of π-electrons. The rigidity of both of these constitutional units together with electrostatic repulsions of the main-chain Mt2+ cations favor extended conformations of MSP chains that are favorable for inter-chain attraction. Thus the above structural features can be regarded as the main reason for the low solubility of MSPs derived from α,ω-bis(tpy)oligoarylenes.
An increase in the solubility of the discussed conjugated MSPs has been achieved by introducing pendant alkyl groups into unimer units. However, this increase is insufficient.28 Further improvement in the solubility of MSPs can be achieved by introducing cationic pendant groups onto unimer building blocks. The markedly cationic character of MSP chains should reduce the inter-chain attraction and thus make MSPs more soluble mainly in polar solvents such as alcohols or even in water. Such solvents are perhaps the most desired for processing of conjugated MSPs.
The above approach has been recently tested on thiophene and bithiophene with tpy end-groups.29 In the present paper, we report the preparation and basic properties of α,ω-bis(tpy)quaterthiophenes with cationic side groups and related conjugated MSPs with Zn2+ and Fe2+ ion couplers. Since the studied MSPs show constitutional dynamics,30,31 they exist in solutions as short oligomeric chains composed of starting unimers. The term unimer proposed by Ciferri32 is used in the further text for a building block of MSPs.
Results and discussion
The prepared ionic as well as non-ionic unimers and their abbreviations are shown in Chart 1 together with the numbering of the central block positions and the marking of the rings used in the assignment of NMR spectra. The letter Q denotes the unimers with the quaterthiophene central block and the numbers behind it indicate the positions occupied by hexyl groups (suffix -H) or by hexyl groups capped with a 4-methoxyphenoxy group (suffix -A) or a bromine atom (suffix -Br) or a triethylphosphonium group (suffix -P+). MSPs are marked with the prefix PZn (polymers with Zn2+ ion couplers) or PFe (Fe2+ ion couplers) before the unimer label: for example, PZnQ27-Br denotes the MSPs formed by the assembly of Zn2+ ions and unimer Q27-Br that contains 6-bromohexyl groups attached to the quaterthiophene central block at positions 2 and 7; PFeQ45-P+ stands for the MSPs formed from Fe2+ ions and unimer Q45-P+ that contains two 6-(triethylphosphonium)hexyl groups attached to positions 4 and 5 of the central block, etc.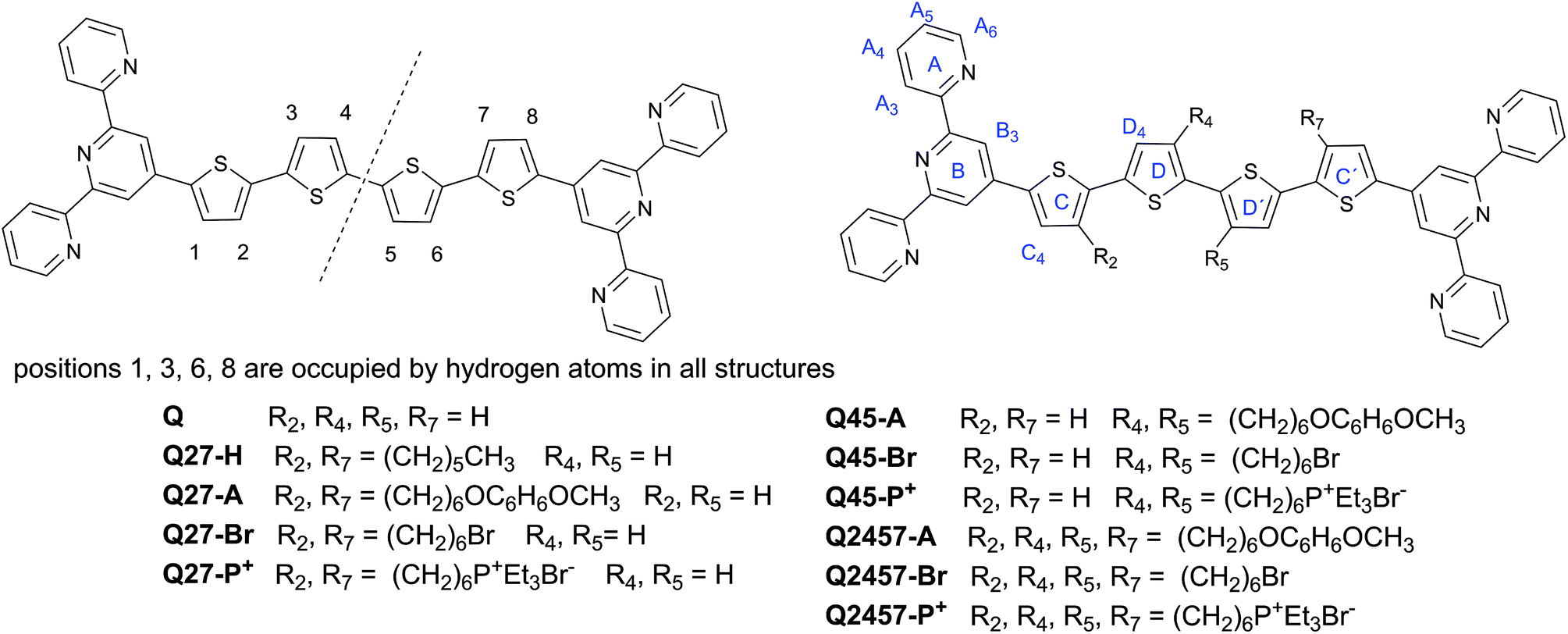 | ||
| Chart 1 Structures and codes of the prepared unimers. | ||
Synthesis and characterization of unimers and polymers
The reference unimers Q and Q27-H were prepared using the Suzuki–Miyaura coupling strategy (Scheme 1) and the conditions applied earlier.28,29,33Br-unimers were prepared using the strategy shown in Schemes 1 and 2a. The starting monomer 3-[6-(4-methoxyphenoxy)hexyl]thiophene was prepared using the procedure described elsewhere.34 The procedure starting from 3-(6-bromohexyl)thiophene used for the synthesis of ionic unimers with mono- (M) and bithiophene (B) central blocks29 was not so effective owing to low efficiency of purification of unimers with bromohexyl groups. Connecting tpy end-groups by Suzuki coupling (Scheme 1c) was accompanied by partial dehydrobromination of bromohexyl side groups promoted with tpy end-groups.29 Purification of short unimers could be done easily (for M) or feasibly (for B), but the purification of bis(tpy)quaterthiophenes was almost impossible.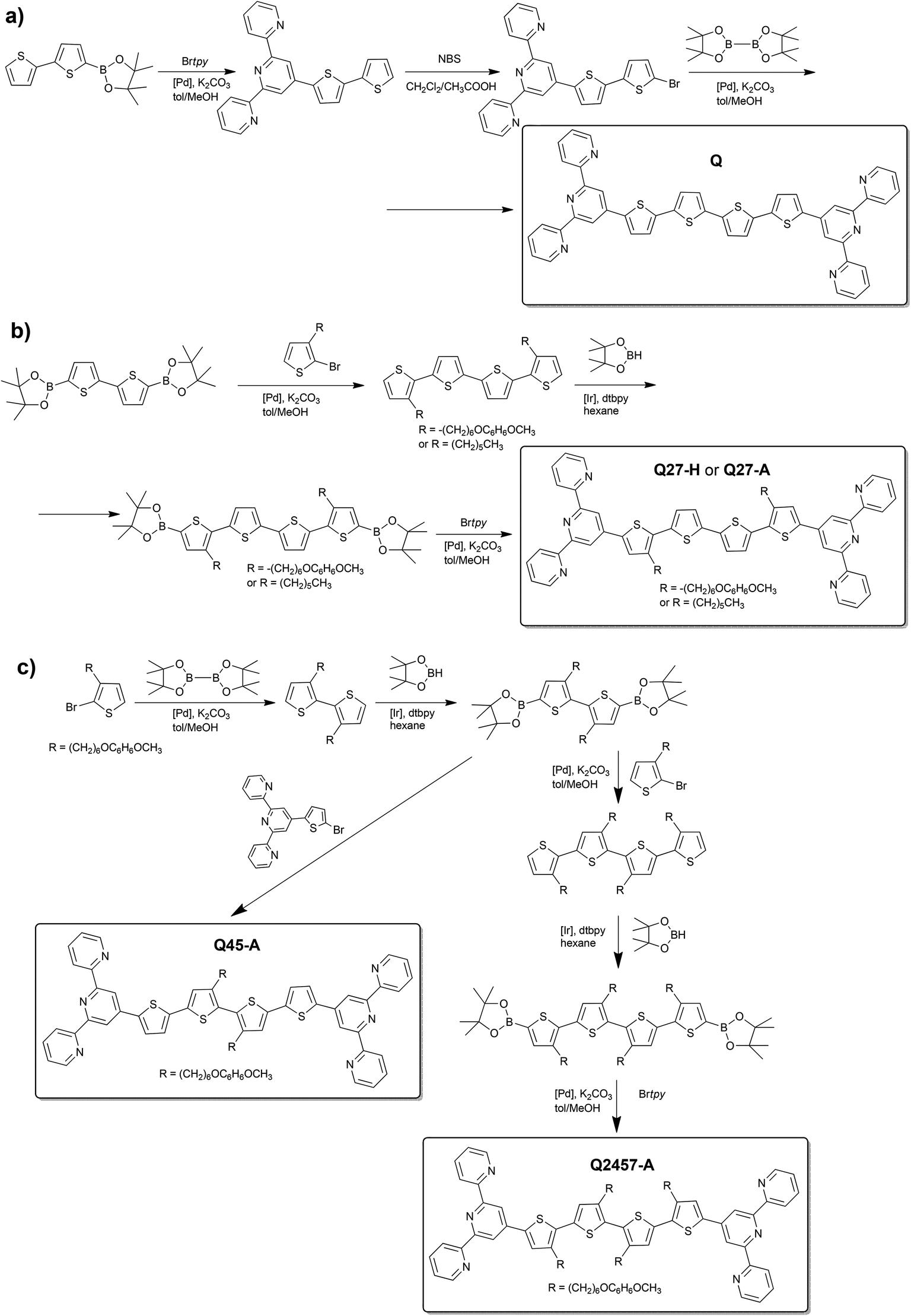 | ||
| Scheme 1 Synthesis pathways to the reference unimers Q and Q27-H and A-unimers. | ||
 | ||
| Scheme 2 Transformation of the type A-unimers into type Br- (a) and type P+-unimers (b). | ||
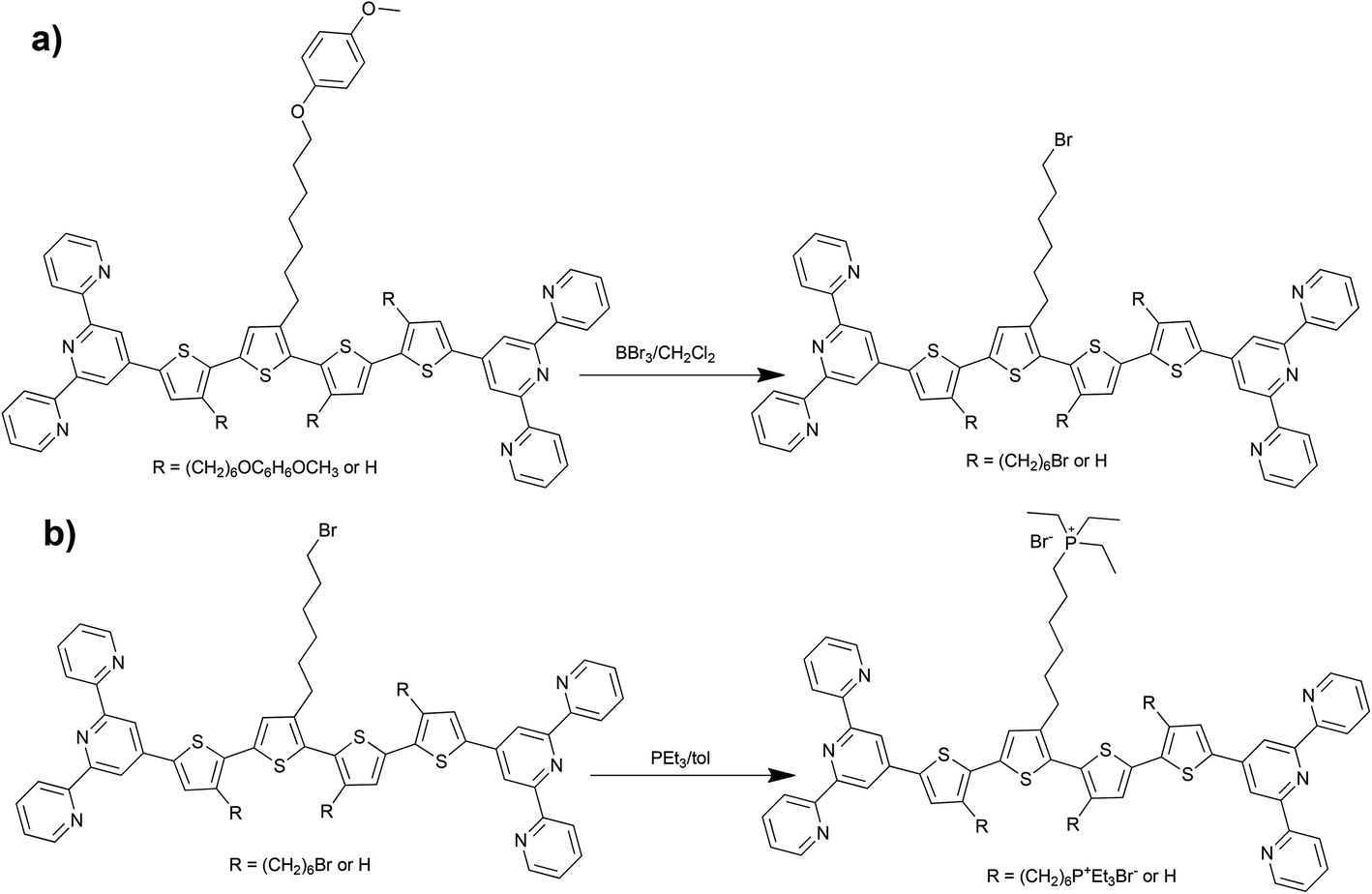
The use of new starting monomers avoided the above difficulties and, in addition, made the isolation of all intermediates as well as A-unimers much easier. The A-unimers were then allowed to react with BBr3 (Scheme 2a) to give the corresponding Br-unimers with bromohexyl side groups (yield 85–95%), which were finally treated with triethylphosphine to give the corresponding ionic P+-unimers (Scheme 2b). Excess PEt3 was easily removed by vacuum distillation and its oxide (POEt3) was washed away with toluene and ether. Solid products were isolated by centrifugation (yield of the ionization step was from 75 to 95%).
Solubility in methanol was the first evidence of successful transformation of the Br- into P+-unimers. The NMR spectra of modified unimers accordingly showed a 31P signal of P+-groups at around 39 ppm (38.93 for Q27-P+, 38.44 for Q45-P+, 39.99 for Q2457-P+), 1H signals of ethyl groups (part of P+) but no signal at 3.4 ppm that is typical of CH2-Br groups. Weak 1H signals at 5.93 ppm and 5.55–5.45 ppm were also observed indicating that some side chains contain terminal double bonds formed by dehydrobromination accompanying quaternization of Br-unimers. Complete removal of imperfect molecules from P+-unimers was not successful since they are soluble in alcohols.
TGA analyses of Br-unimers and P+-unimers showed thermal stability up to 205 °C.
Metallo-supramolecular polymers were simply prepared by mixing solutions of a given unimer and zinc(II) or iron(II) perchlorate in the metal ions to a unimer mole ratio of r = 1 (r = [Mt2+]/[unimer]). Br-unimers were assembled in the acetonitrile/chloroform mixed solvent (1/1 by vol.) while P+-unimers were assembled in methanol.
The solubility of the prepared unimers and polymers depends on the substitution of the unimer central block. The unsubstituted unimer Q is soluble in dichloromethane but poorly soluble in chloroform. Br-unimers are highly soluble in solvents such as THF, CHCl3, CH2Cl2, DMSO and the acetonitrile/chloroform mixture, which facilitates their isolation and purification. Unimers with two ionic groups are highly soluble in polar solvents such as methanol, ethanol and DMSO and sparingly soluble in water (complete dissolution to a colloidal solution takes a few weeks). Unimer Q2457-P+ carrying four ionic groups is easily soluble in water, which is quite unusual for this type of conjugated structure. Nevertheless, complete dissolution of Q2457-P+ to the molecular level takes time on the day scale as can be seen from the time development of the UV/vis spectrum of its aqueous solution shown in Fig. S1, ESI.† MSPs show similar solubility to the corresponding unimers.
Vibrational spectra of unimers and polymers
The IR spectra of unimers show each the bands of stretching modes (νCC, νCN) of tpy end-groups (1500–1620 cm−1), the quaterthiophene central block (1370–1500 cm−1), aromatic νCH modes (3000–3100 cm−1) and the main out-of-plane modes (ρCH) of aromatic moieties (790 and 658 cm−1) at nearly identical positions (Fig. S2, ESI†). Differences due to different substitutions of the central blocks are mainly seen in the fingerprint region from 850 to 1350 cm−1. The presence of hexyl chains is mainly observable in the C–H stretching region (2800–3000 cm−1). The most intensive bands characteristic of vibrations of hexyl groups in the fingerprint region (1466 and 1379 cm−1) are overlapped by the ring stretching modes of the quaterthiophene central blocks. Only a small shift of the band maximum from 1459 cm−1 (for Q) to 1466 cm−1 (for Q27-H) and a new shoulder at 1384 cm−1 for Q27-H are observable. The broad spectral band at 3400 cm−1 observed for all P+-unimers is due to the presence of hydrogen bonded water molecules in these unimers.The off-resonance Raman spectra of unimers show strong stretching bands of quaterthiophene blocks but weak bands of tpy end-groups. Their spectral patterns reflect differences in the substitution of quaterthiophene blocks (Fig. S3, ESI†). The Raman spectra of Zn-polymers were disturbed by strong fluorescence but spectra of non-fluorescent Fe-polymers were well measurable. The bands characteristic of tpy groups35 occur at 1610 cm−1 (νs), 1290 cm−1 (δip) and 1038 cm−1 (breathing mode) while the bands of quaterthiophene blocks36–38 occur in the region 1380–1520 cm−1 (Fig. S4a–S7a, ESI†). Deconvolution of the latter band using the OMNIC software gave robust results, showing that this band is composed of at least five bands (Fig. S4b–S7b, ESI†) whose intensities depend on the positions of side groups. The band at 1472 cm−1 should also be attributed to transitions in tpy groups.35
Optical spectra of unimers and polymers
The solution UV/vis absorption spectra of unimers (ESI, Fig. S8a†) show: (i) a flat band at 280–284 nm mainly contributed by transitions in tpy end groups, and (ii) a band with the apex at a wavelength λA from 381 nm (Q2457-P+) to 441 nm (Q) belonging to transitions from HOMO that is spread over thiophene rings and central rings of tpy groups.28,33 The value of λA (see Table 1) decreases (i) with increasing distortion of the quaterthiophene central block (see Table 2), and (ii) on going from the non-ionic (-Br, -H) to ionic unimer (-P+) of the same type. Absorption maxima in the spectra of unimer thin films (ESI, Fig. S8b†) are not in such good correlation with the chain distortion, which reflects the importance of the molecular packing effect or the electronic effect of substituents. An exceptionally high λA of Q2457-Br thin films (500 ± 10 nm) was obtained. The fact that Q2457-P+ shows much lower λA can be ascribed to the bulkiness of P+Et3 groups and the effect of bromine counterions.| Sample | UV/vis absorption | Luminescence | Stokes shift | |||
|---|---|---|---|---|---|---|
| λ A, nm | λ F, nm (Φ, %) | ν, cm−1 | ||||
| Solution | Film | Solution | Film | Solution | Film | |
| Q | 441 | 425 | 514, 546 (30%) | 645 (<1%) | 4350 | 7450 |
| Q27-H | 425 | 460 | 554 (26%) | 630 (1%) | 5500 | 5300 |
| Q27-Br | 425 | 455 | 554 (31%) | 630 (1%) | 5500 | 6000 |
| Q27-P+ | 419 | 455 | 550 (18%) | ∼650 (<1%) | 5700 | 7300 |
| Q45-Br | 397 | 425 | 530 (14%) | 610 (1%) | 6300 | 7000 |
| Q45-P+ | 393 | 410 | 536 (11%) | 550 (1%) | 6800 | 6200 |
| Q2457-Br | 386 | 500 | 536 (14%) | 560, 603 (3%) | 7250 | 3600 |
| Q2457-P+ | 381 | 410 | 536 (10%) | 560 (1%) | 7600 | 6400 |
Q2457-P+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
400 | 555 | 6980 | |||
| Zn polymers | ||||||
| P Zn Q | 486 | 500 | 656 | ∼690 (1%) | 5350 | 5050 |
| P Zn Q27-H | 468 | 490 | 673 | ∼640 (2%) | 6500 | 5250 |
| P Zn Q27-Br | 470 | 510 | 675 | ∼710 (1%) | 6450 | 5450 |
| P Zn Q27-P + | 483 | 500 | 550 | ∼705 (<1%) | 2500 | 5700 |
| P Zn Q45-Br | 447 | 455 | 673 | 585 (3%) | 7500 | 4850 |
| P Zn Q45-P + | 426 | 430 | 536 | 660 (1%) | 4800 | 8050 |
| P Zn Q2457-Br | 432 | 430 | 668 | 590 (3%) | 8200 | 5800 |
| P Zn Q2457-P + | 439 | 440 | 552 | 625 (1%) | 4650 | 6450 |
| P Zn Q2457-P + | 462a | 720a | 7750a | |||
| Fe polymers | ||
|---|---|---|
| UV/vis absorption | ||
| λ A, nm, (λMLCT, nm) | ||
| Solution | Film | |
| a Data from aqueous solution. b Shoulder. | ||
| P Fe Q | 395, 471 (603) | 475 (621) |
| P Fe Q27-H | 437 (601) | 460 (622) |
| P Fe Q27-Br | 387, 452 (601) | 465 (621) |
| P Fe Q27-P + | 386, 436 (593) | 465 (611) |
| P Fe Q45-Br | 405 (598) | 410 (613) |
| P Fe Q45-P + | 399 (593) | 410 (609) |
| P Fe Q2457-Br | 384, 423sh, (594) | 395b (609) |
| P Fe Q2457-P + | 384 (591) | 395b (604) |
| P Fe Q2457-P + | 400 (607)a | |
| Ground state | Excited state | |||||
|---|---|---|---|---|---|---|
| δ BC | δ CD | δ DD′ | δ BC | δ CD | δ DD′ | |
| a Values are not available for the first 720 hours. | ||||||
| Q | 15.8 | 12.5 | 0.7 | 0.0 | 0.0 | 0.0 |
| Q27-H | 17.3 | 28.6 | 1.0 | 0.0 | 0.0 | 0.0 |
| Q27-Br | 13.9 | 22.8 | 1.0 | 0.5 | 3.2 | 1.3 |
| Q27-P+·Br− | 17.3 | 32.4 | 16.3 | |||
| Q45-Br | 14.6 | 18.5 | 58.9 | 1.4 | 3.1 | 23.7 |
| Q45-P+·Br− | 18.0 | 15.4 | 63.5 | |||
| Q2457-Br | 19.6 | 40.8 | 97.5 | |||
| Q2457-P+·Br− | 15.8 | 27.4 | 124.4 | |||
In the spectra of Zn-polymers, the absorption band of transitions involving quaterthiophene blocks is red shifted by about 35–65 nm compared to its position λA for the unimer in the solution spectra and by about 20–75 nm in thin films (Table 1). The only but great exception is the spectrum of PZnQ2457-Br thin films that surprisingly shows a blue shift of λA of about −70 nm, which is obviously due to the exceptionally high value of λA of the unimer Q2457-Br.
The spectra of Fe-polymers contain a new band belonging to transitions in the metal-to-ligand charge transfer (MLCT) complex which is typical of tpy-Fe-tpy linkages35,39 (see Table 1 and Fig. S8c and S8d in the ESI†). In Fe-polymers, this band is significantly contributed by transitions involving neighboring oligothiophene blocks.29
Luminescence spectra of unimers in solutions (ESI, Fig. S9a†) show higher similarity than their UV/vis spectra (λF around 550 nm; lowered values of about 535 nm are actually given by different band shapes). This is obviously due to the fast transition of excited unimer molecules to nearly coplanar conformations with quinoidal rings, from which the light emission takes place.36 Minor differences are nevertheless seen: unimers with less distorted chains (Q, Q27-H, Q27-Br and Q27-P+) show a better resolved vibrational structure than unimers with more distorted chains (Q45-Br, Q45-P+, Q2457-Br and Q2457-P+). The luminescence spectra of unimers and Zn-polymer thin films are shown in ESI, Fig. S9b–d† and the band wavelengths are summarized in Table 1, and the luminescence lifetimes are presented in ESI, Table S1.† Fe-polymers do not show luminescence.28,29,36
Assembly of unimers to metallo-supramolecular polymers in solutions
The assembly in solutions was monitored by UV/vis and luminescence spectroscopy, viscometry and size exclusion chromatography (SEC). A chloroform/acetonitrile mixed solvent (1/1 by volume) was used for Br-unimers while methanol and water were used for P+-unimers. For spectroscopic studies, a set of solutions of a constant unimer concentration (2 × 10−5 M) and a stepwise increasing metal ions to unimer mole ratio (r from 0 to 3) was prepared for each Mt2+/unimer system and solutions were allowed to equilibrate for 24 hours before monitoring the spectra. The SEC and viscometric measurements were done with solutions of the concentration of 5 × 10−4 M.Spectral changes accompanying the assembly of unimers with Mt2+ ions showed three stages differing in the development trend, similarly to the related systems studied recently.21,25,28,29,40 The UV/vis spectra obtained for systems of composition ratios r from 0 to ca. 0.5 (the first stage of assembly) showed up to three isosbestic points (see examples in Fig. 1a and 2a and a complete set of the spectra in ESI, Fig. S10 and S11†), which indicates the transformation of the unimer species into another well-defined species. Regarding the stoichiometry, the new species should be a dimer species unimer-Mt2+-unimer.
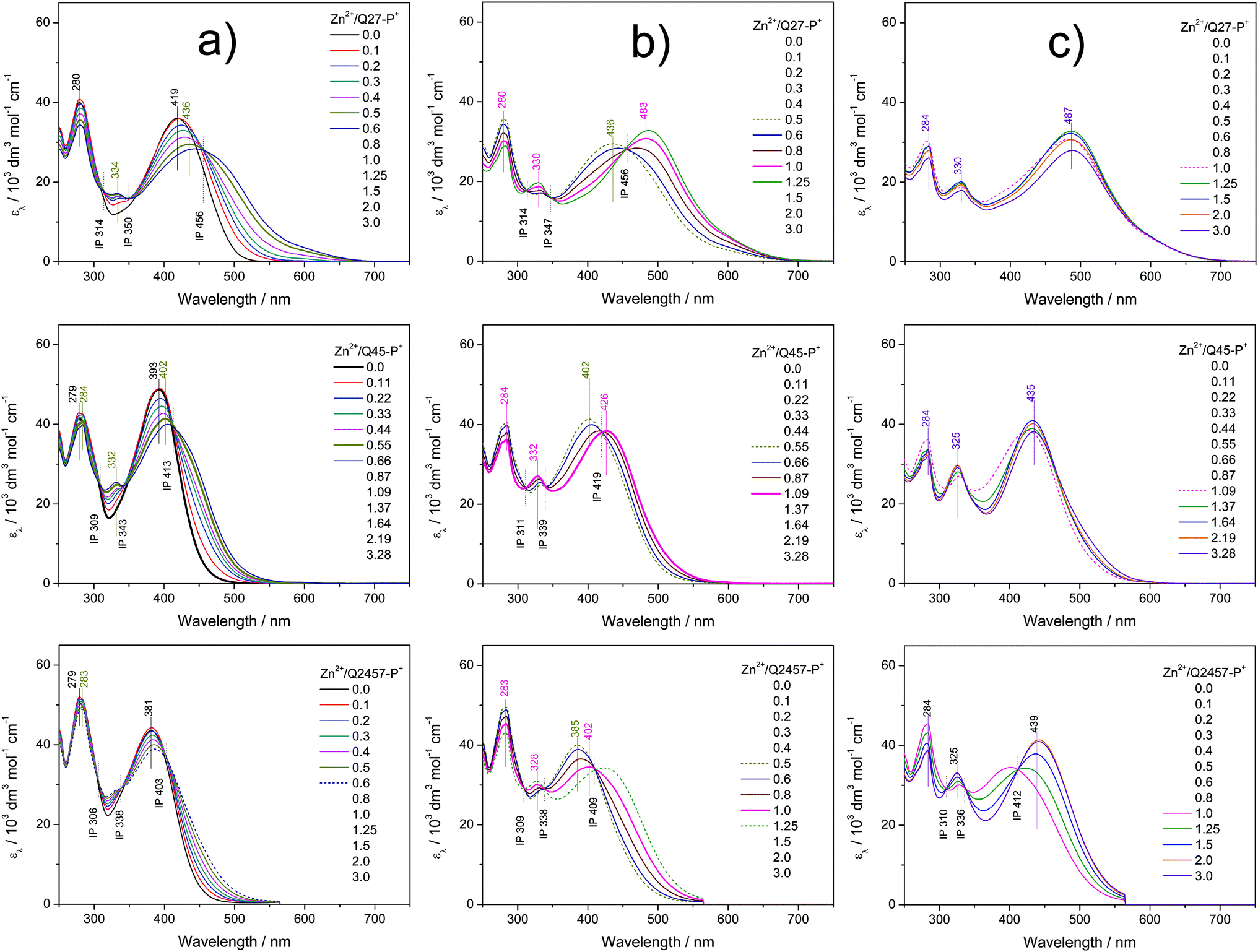 | ||
| Fig. 1 Changes in UV/vis spectra accompanying the titration of ionic unimers with Zn2+ ions. Initial unimer concentration 2 × 10−5 M in methanol, room temperature. Column (a) shows the first stage of assembly, column (b) shows the second stage and column (c) shows the third stage; see the text. | ||
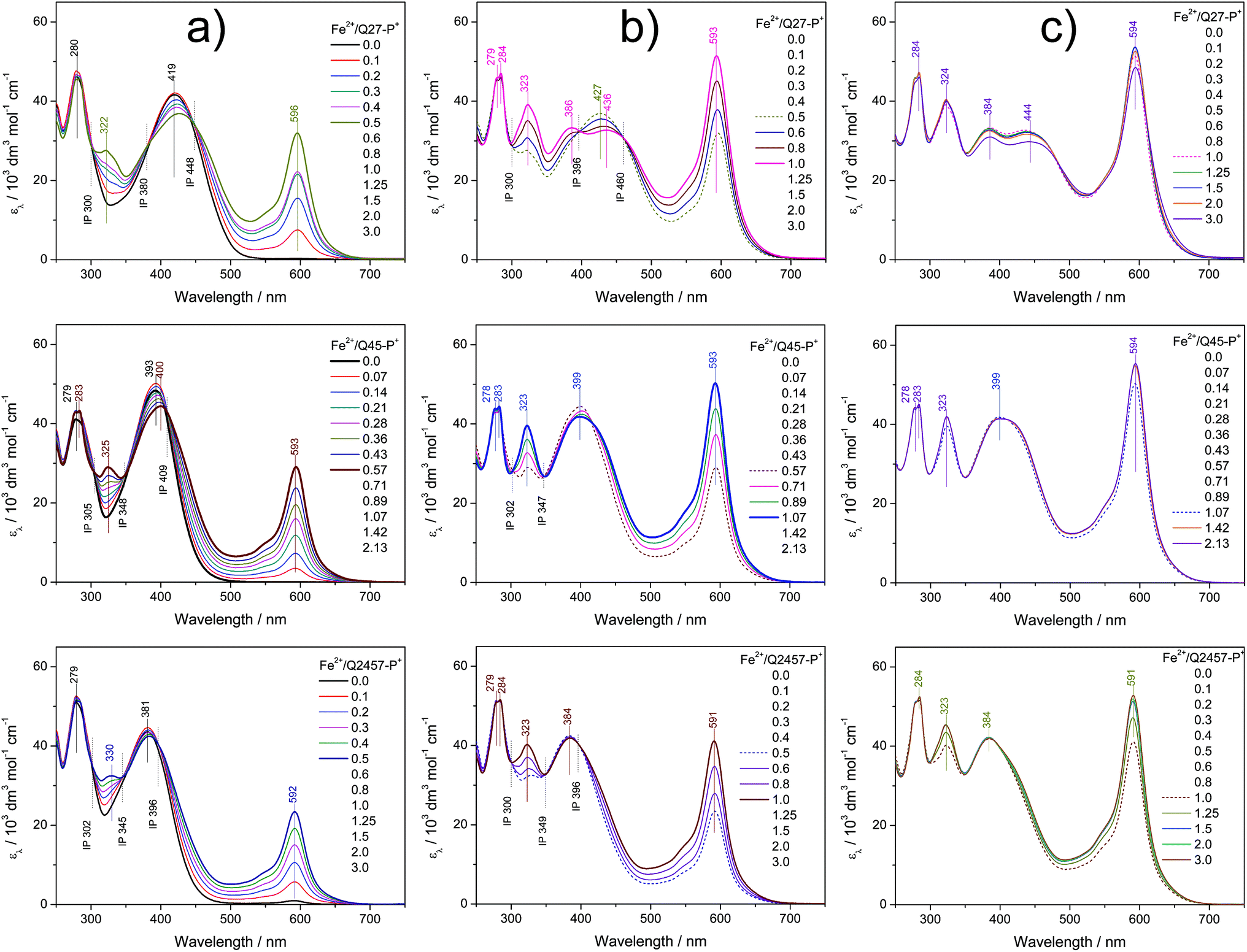 | ||
| Fig. 2 Changes in UV/vis spectra accompanying the titration of ionic unimers with Fe2+ ions. Initial unimer concentration 2 × 10−5 M in methanol, room temperature. Column (a) shows the first stage of assembly, column (b) shows the second stage and column (c) shows the third stage; see the text. | ||
The spectra obtained for systems with ratios r from ca. 0.6 to 1 also show isosbestic points but at different wavelengths (Fig. 1b and 2b). This indicates that the systems entered the second stage of assembly in which longer polymer chains are formed. As can be seen from ESI, Fig. S10 and S11,† the absorption bands characteristic of free unimers and dimers disappear while the band of enchained unimer units fully develops in the case of systems with non-ionic Br-unimers. These systems then enter the third stage of assembly (r > 1), where spectral changes are quite low and can be attributed to the end-capping of polymer chains with the metal ions and partial depolymerization of the polymer chains to the shorter also end-capped ones (Fig. 1c and 2c). The reaction of (tpy)2Zn2+ species with Zn2+ ions giving two (tpy)Zn2+ species has been reported for mono- as well as bis(tpy) species.18,29,41,42 The reaction of (tpy)2Fe2+ species with Fe2+ ions giving two (tpy)Fe2+ species was reported only for metallo-supramolecular polymers.18,29,43
The spectral changes in the second stage of assembly of ionic P+-unimers are less progressive than those in the case of Br-unimers or even incomplete, which indicates lowered thermodynamic stability (i.e., stability constants) of ionic polymers in methanol. The lowered stability of ionic Fe-polymers is also seen from the changes in the position and intensity of the MLCT band (at around 595 nm) that is not fully developed at the ratio r ≅ 1 (Fig. 2b). The UV/vis spectral patterns indicate that the ionic polymers acquire their maximum length at the ratios r of about 1.5 or higher in methanol solutions.
The changes in luminescence spectra accompanying the assembly of ionic unimers with Zn2+ ions are shown in Fig. 3. The complexation is manifested by the disappearance of the unimer emission band and the creation of a new band red shifted by about 130 nm. Unlike the case of shorter ionic polymers derived from bis(tpy) mono- and bithiophenes,29 the new emission band is much less intense than the band of free unimers. A similar luminescence attenuation is also exhibited by systems with non-ionic polymers. This shows that the prolongation of the unimer central oligothiophene block increases the efficiency of non-radiative paths of the decay of excited states in Zn-polymers.
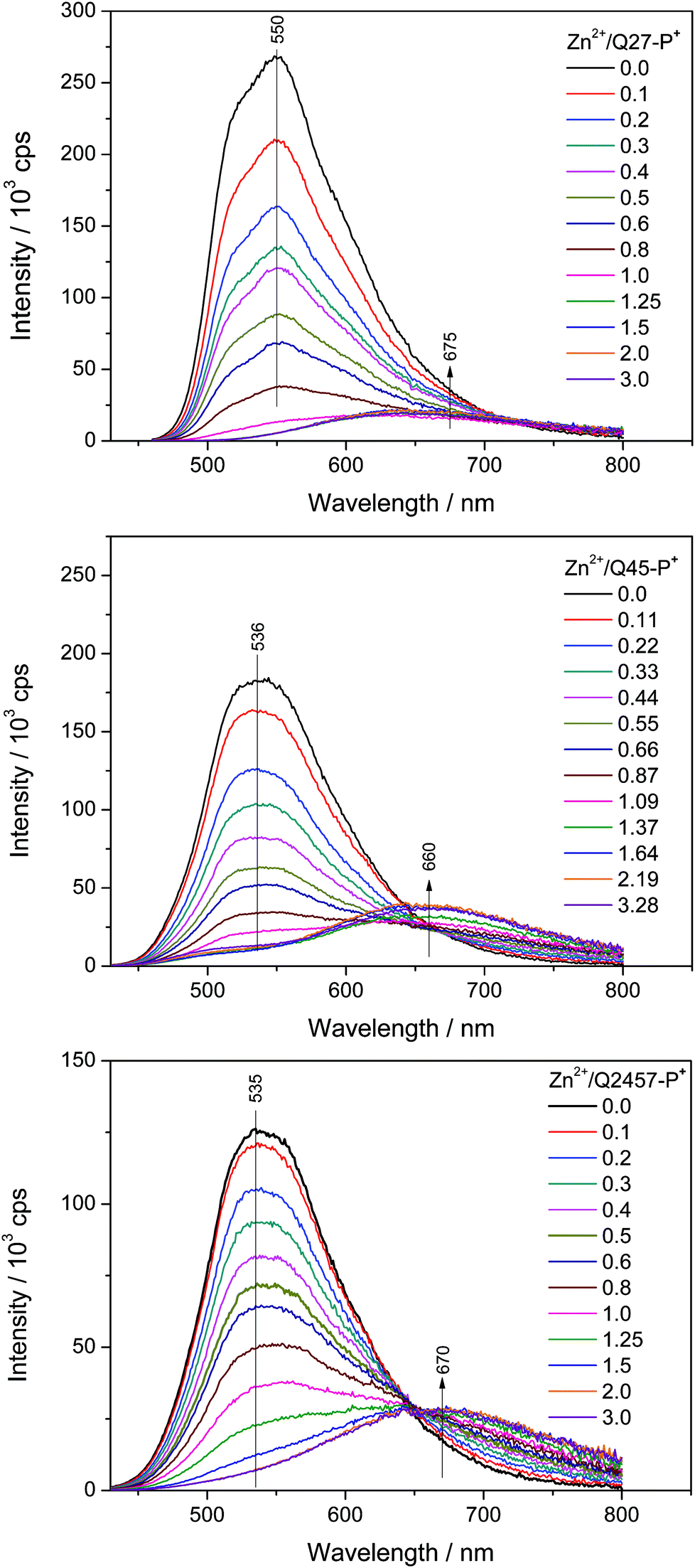 | ||
| Fig. 3 Changes in photoluminescence spectra accompanying the titration of ionic unimers with Zn2+ ions. Initial unimer concentration 2 × 10−5 M in methanol, room temperature. | ||
Unlike the systems with Zn2+ ions, those with Fe2+ ions show a monotonous luminescence quenching with increasing ratios r up to ca. 0.6, at which the luminescence disappears (for example see ESI, Fig. S12†). This behaviour, which is exhibited by other systems with bis(tpy)Fe2+ species, is attributed to the fact that the lowest excited state of bis(tpy)Fe2+ species, the d–d triplet state, is close to the ground state.44 As the d–d triplet state easily depletes higher excited states and potential phosphorescence from the d–d state is spin forbidden, its decay by non-radiative transitions is unambiguously preferred in accord with the energy gap law.8,45
The molar mass distribution of Br-polymers in CHCl3/CH3CN (1:1) solutions was examined using an SEC system equipped with a diode-array UV/vis detector (DAD). (Analysis of ionic polymers failed owing to the strong adsorption of their chains inside SEC columns.) Mixed solutions of a Br-unimer (0.5 mM) and Zn2+ or Fe2+ ions (r from 0 to 2.0) equilibrated for one day were injected into the SEC system. The SEC records of systems with Zn2+ ions showed nothing but the peak of free unimers, which proves rapid dissociation of the Zn-polymer chains upon multifold dilution of their solution inside SEC columns. In contrast, the systems with Fe2+ ions provided SEC records typical of covalent polymers (Fig. 4 and ESI, S13†), which demonstrates very slow constitutional dynamics of Fe-polymers in the used solvent. Similar results were recently obtained for MSPs of shorter bis(tpy)thiophenes.29
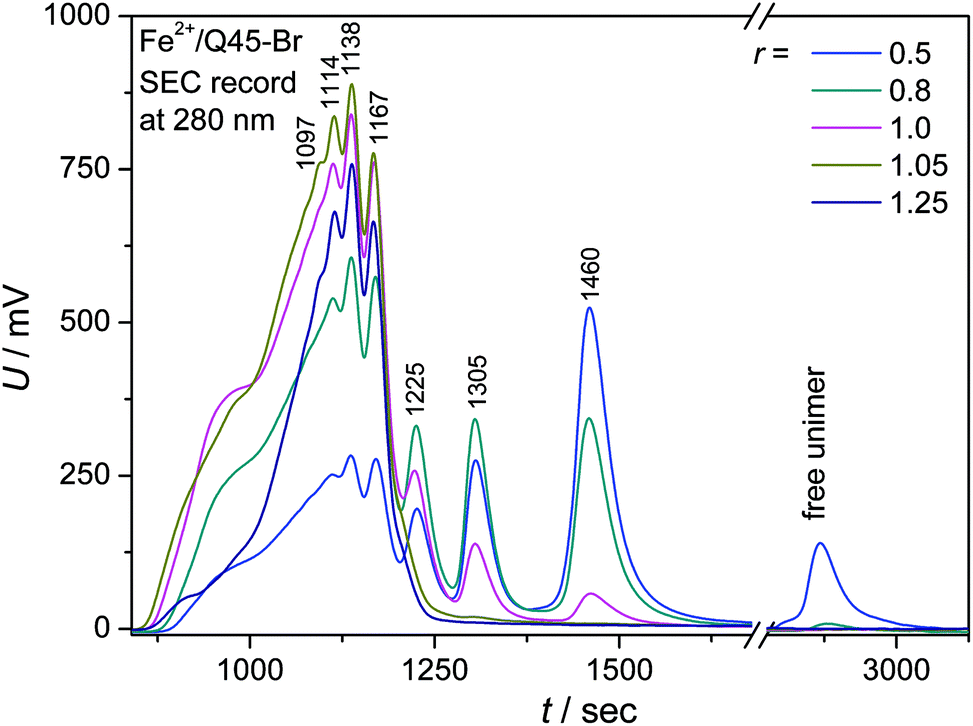 | ||
| Fig. 4 The SEC records of the Fe2+/Q45-Br systems of different compositions. | ||
Well resolved SEC records were obtained only for systems with a composition ratio r < 1 (Fig. 4). Systems with r ≥ 1 gave poorly resolved SEC records, in which the area under the elution peak decreased with increasing value of r. This indicates retention of longer chains in SEC columns. The detained polymer chains, obviously end-capped with Fe2+ ions, had to be additionally washed out of the columns with 2,2′-bipyridine. The UV/vis spectral pattern of SEC fractions showed a perfect development with the elution time (tel): a pattern typical of long polymer chains was observed for the first eluted SEC fractions while that typical of the dimers for the last fraction (Fig. 5a and ESI, Fig. S14a†). Differences are also seen when comparing the spectra of fractions of dimers formed in systems of different compositions (Fig. 5b and ESI, Fig. S14b†). These differences can be attributed to the end-capping of their molecules with Fe2+ ions. However, these differences are substantially smaller than those observed for the Fe-polymers formed from unimers with mono- and bithiophene central blocks.29
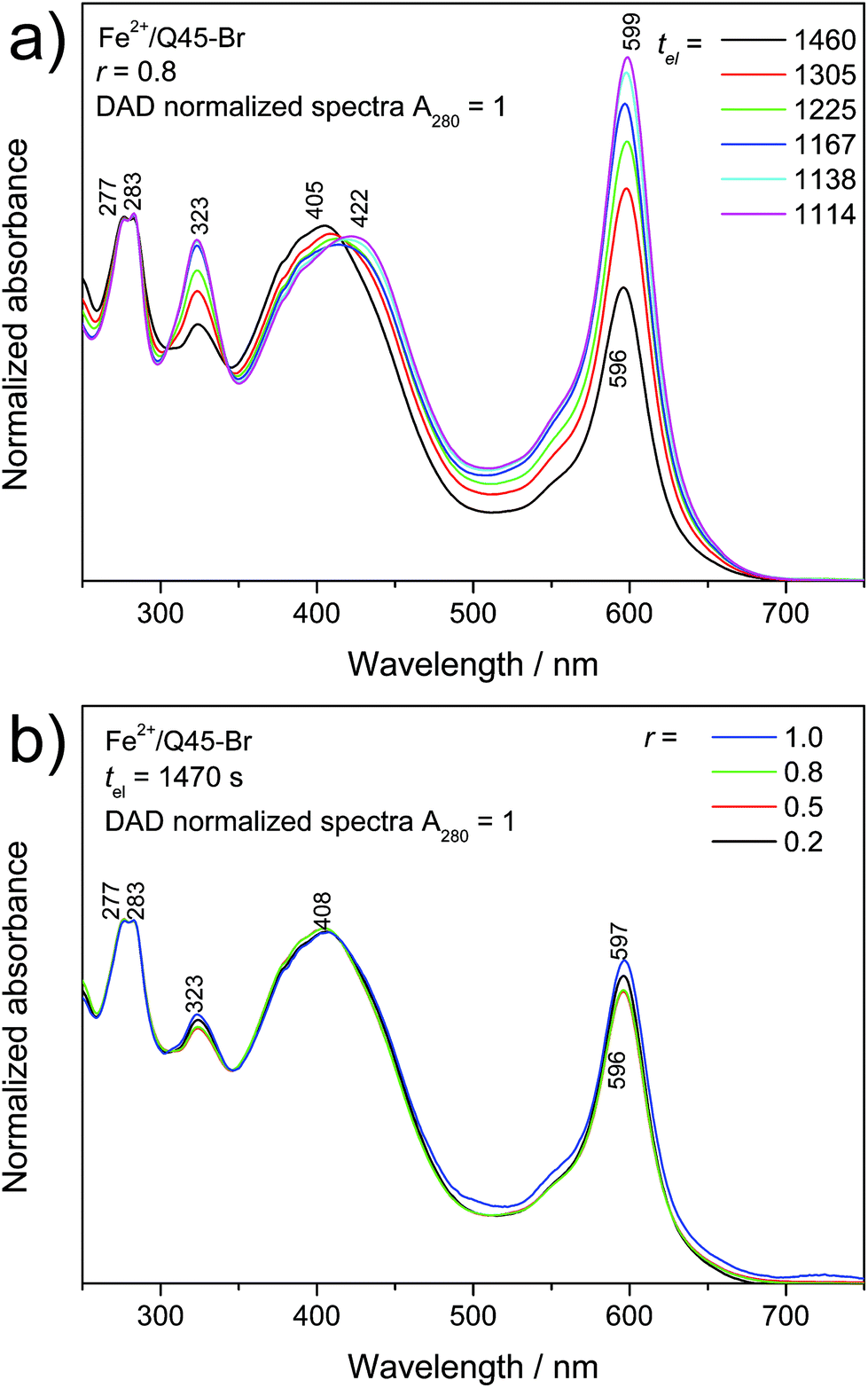 | ||
| Fig. 5 The UV/vis DAD spectra of SEC fractions of the Fe2+/Q45-Br system (r = 0.8) eluted at different elution times tel (a) and comparison of the spectra of the last fraction (dimers; tel = 1470 s) of systems of composition r from 0.2 to 1.0 (b). | ||
The presence of higher fractions in solutions containing a stoichiometric lack of Fe2+ ions (r = 0.2 and 0.5) can be explained by the transiently locally increased concentration of the ions and unimers during mixing of their solutions. The formation of polymer chains is most likely a kinetically controlled process which, on mixing twenty five times more concentrated solutions (0.5 mM instead of 0.02 mM), shall be ca. 625 times accelerated. Thus it can give rise to a significant number of longer chains that do not dissociate during the SEC analysis thanks to their slow constitutional dynamics. Thus the degree of polymerization, X, of Fe-polymer chains in solution could be estimated. If the peak eluted at tel = 1460 s (see Fig. 4) is ascribed to dimers, the peak with tel = 1305 s to trimers, and so on, one can still resolve the peak of heptamers at tel = 1114 s. Calculations based on this peak assignment provide the weight-average degree of polymerization equal to ca. 7 for Fe-polymer in the solution with r ≅ 1 (Table 3). In addition, it is seen from Fig. 4 that the stoichiometric excess of Fe2+ ions in solution results in the formation of shorter chains. The latter is supported by the results of viscometric measurements, which also indicate shortening of the polymer chains in the presence of excess Fe2+ ions (ESI, Fig. S15†).
| r | X n | X w | Đ |
|---|---|---|---|
| 0.2 | 3.07 | 4.23 | 1.36 |
| 0.5 | 3.24 | 4.77 | 1.47 |
| 0.8 | 4.43 | 6.02 | 1.36 |
| 1.0 | 6.13 | 7.23 | 1.18 |
| 1.05 | 6.79 | 7.62 | 1.12 |
| 1.25 | 6.26 | 6.84 | 1.09 |
Assembly of Q2457-P+ in water
The water-soluble unimer Q2457-P+ has been assembled with metal ions also in aqueous solutions. Since molecular dissolution of this unimer in water takes a long time a month-old solution of Q2457-P+ was used in these experiments. As can be seen from Fig. 6, the optical spectral changes accompanying the assembly in water substantially differ from those observed for assembly in methanol.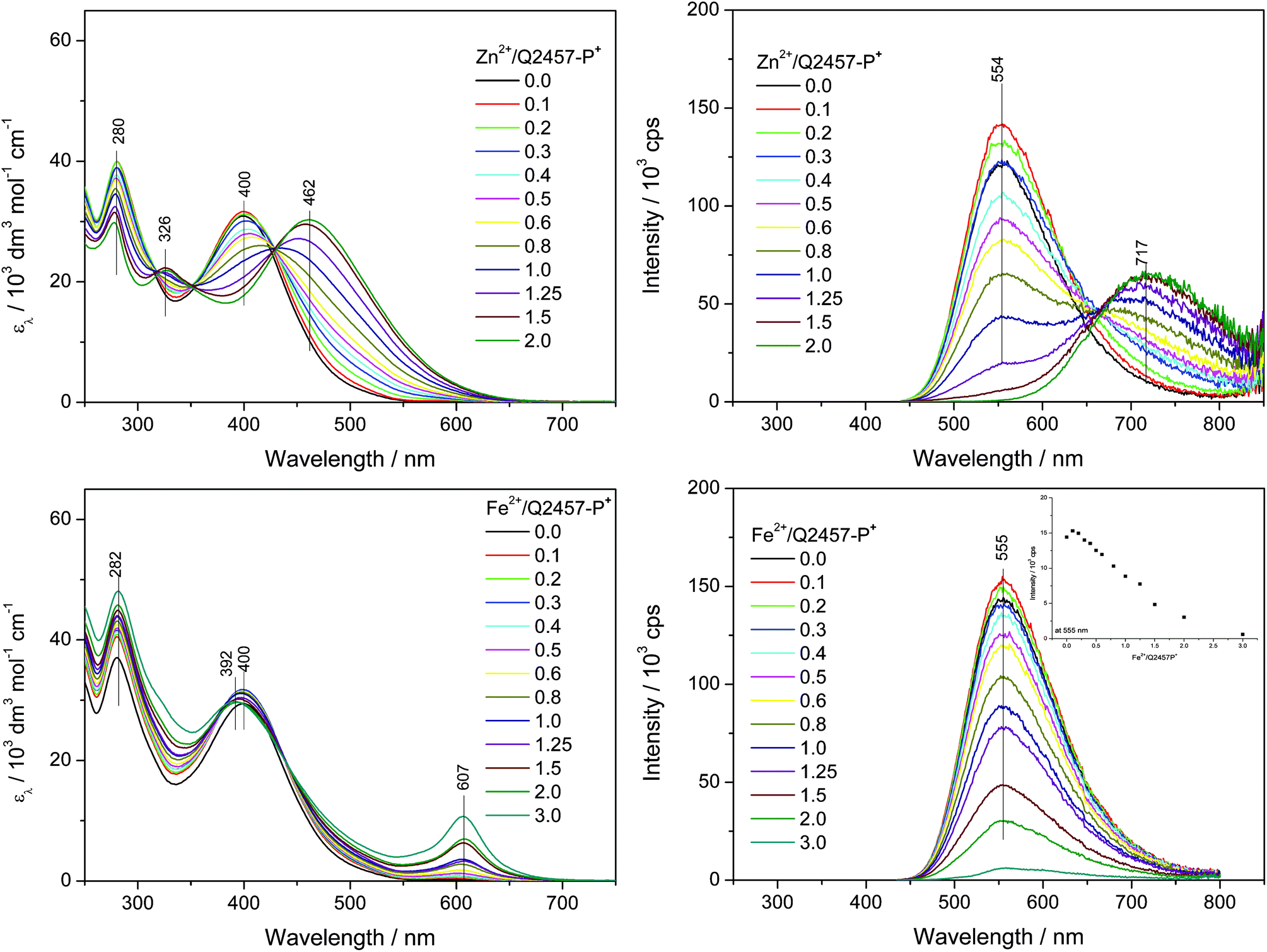 | ||
| Fig. 6 Changes in UV/vis (left column) and photoluminescence (right column) spectra accompanying the titration of ionic unimers with Zn2+ or Fe2+ ions. Initial unimer concentration 2 × 10−5 M in water, room temperature. | ||
(i) The absorption maxima of Q2457-P+ (λA = 400 nm) and its Zn-polymer (r = 2, λA = 462 nm) as well as the luminescence maximum of the unimer (λF = 555 nm) are red shifted by about ca. 20 nm compared to their positions in methanol solutions, which indicates that the free as well as enchained unimer species acquire more planar conformations in water than in methanol. This can be attributed to the substantial increase in the solvent permittivity, which, in accord with the Coulomb law, reduces repulsive ionic interactions among neighbouring P+Et3 groups as well as their attractive interactions with counterions.
(ii) The luminescence emission band observed for Zn-polymer (λF = 720 nm) is enormously red shifted (about 168 nm) compared to the band for methanol solution. The Stokes shift for PZnQ2457-P+ in water (7750 cm−1) is much higher than the shift in methanol (4650 cm−1, Table 1), which proves the much higher extent of conformational relaxation of excited states in aqueous compared to methanol solutions.
(iii) The UV/vis spectra for assembly of Q2457-P+ with Zn2+ ions show a single set of isosbestic points and a fluent course of changes up to r = 2. Luminescence spectra indicate the presence of free unimers in solution with r equal to at least 1.5. These features consistently indicate a lowered stability and increased constitutional dynamics of PZnQ2457-P+ in aqueous solutions.
(iv) Surprisingly, in accord with the last mentioned observations, the UV/vis spectra for assembly of Q2457-P+ with Fe2+ ions show small changes and a weak MLCT band and the luminescence spectra show emission even at the composition ratio r = 3. It should be stressed here that no new emission band occurs; only reluctant luminescence quenching with increasing r is observed. The observed spectral changes indicate that the chains of a highly ionic Fe-polymer are, in aqueous solution, less stable than the chains of its Zn-counterpart. This observation represents a flip in the stability of Fe- and Zn-polymers derived from bis(tpy)quaterthiophenes compared to perhaps all the data reported so far.41,46–49 The reduced stability of the ionic Fe-polymer in water is obviously associated with the inhibition of the MLCT process by water. The solvent dependence of the MLCT is well known.50,51
Conclusions
The synthesis strategy developed here enables preparation of bis(tpy)quaterthiophenes with two or four side groups symmetrically distributed along the quaterthiophene central block. The modification of side groups enabled preparation of ionic unimers that are soluble in green solvents such as alcohols or even in water.Optical spectral patterns of dissolved unimers and the corresponding polymers depend primarily on the distribution of side groups along the quaterthiophene central block and only secondarily on the nature of side-chain-capping groups. The effect of the latter is more apparent in the solid state spectra since the capping groups significantly influence molecular packing.
During the assembly of unimers with metal ions the development of UV/vis spectra with increasing ratio r conclusively indicates that the P+-unimers assemble with metal ions less readily than the Br-unimers. Besides, in water, Q2457-P+ assembles with Zn2+ ions considerably less progressively (with rising r) than in methanol and, with Fe2+ ions, still much less readily, showing only a very weak MLCT band but significant luminescence of the free unimer even at the ratio r = 3. The solvation effect is thus obvious. A red shift of the luminescence band of PZnQ2457-P+ by about ca. 170 nm on going from methanol to aqueous solution is observed. Such a big shift indicates much higher conformational freedom of PZnQ2457-P+ chains in aqueous compared to methanol solutions.
The SEC study of the non-ionic polymers proved that the constitutional dynamics of Zn-polymers is fast and that of Fe-polymers is very slow in the chloroform/acetonitrile mixed solvent, which is in accord with observations of other authors on related systems. However, the results obtained here on assembly of the ionic unimers in aqueous solutions anticipate faster constitutional dynamics of Fe-polymers compared to Zn-polymers.
Experimental section
Materials
2,2′-Bithiophene-5-boronic acid pinacol ester, 2,2′-bithiophene-5,5′-diboronic acid bis(pinacol) ester, thiophene-2-boronic acid pinacol ester, 3,3′′′-dihexyl-2,2′:5′,2′′:5′′,2′′′-quaterthiophene, bis(pinacolato)diboron (B2pin2), 4,4,5,5-tetramethyl-1,3,2-dioxaborolane (HBpin), 4,4′-di-tert-butyl-2,2′-dipyridyl (dtbpy), bis(1,5-cyclooctadiene)di-μ-methoxydiiridium(I) ([Ir(OMe)(COD)]2), boron tribromide (BBr3), triethylphosphine (PEt3, 1.0 M in THF), N-bromosuccinimide (NBS), [1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene](3-chloropyridyl)palladium(II) dichloride (PEPPSI-IPr), zinc perchlorate hexahydrate, iron(II) perchlorate hydrate and tetrabutylammonium hexafluorophosphate (all Aldrich), K2CO3, MgSO4, acetic acid (Lachner) and 4′-bromo-2,2′:6′2′′-terpyridine (TCI) were used as received. Hexane (Lachner) was stored over a molecular sieve, tetrahydrofuran (Aldrich) was distilled from LiAlH4 before use, toluene (Lachner) was distilled from sodium/benzophenone before use, methanol (Aldrich) was bubbled with argon before use, and diethylether, dichloromethane, chloroform (Lachner) and acetonitrile (ACN) were used as obtained.Measurements
1H and 13C NMR spectra were recorded on a Varian UNITYINOVA 400 or a Varian SYSTEM 300 instrument in d8-THF, d2-CD2Cl2, d-CDCl3, d6-DMSO or d4-CD3OD and referenced to the solvent signal: 7.25 ppm (d-CDCl3), 5.32 ppm (d2-CD2Cl2), 3.58 ppm (d8-THF), 2.50 ppm (d6-DMSO) or 3.31 ppm (d4-CD3OD) for 1H and 77.0 ppm (d-CDCl3), 53.84 ppm (d2-CD2Cl2), 67.57 ppm (d8-THF) or 49.15 ppm (d4-CD3OD) for 13C spectra. Coupling constants, J (in Hz), were obtained by the first-order analysis. Infrared spectra were recorded on a Thermo Nicolet 7600 FTIR spectrometer equipped with a Spectra Tech InspectIR Plus microscopic accessory using KBr-diluted samples and the diffuse reflectance technique (DRIFT) (128 or more scans at a resolution of 4 cm−1). Raman spectra of solid samples were recorded on a DXR Raman microscope (Thermo Scientific) using excitations across the whole visible region (λex = 445, 532, 633 and 780 nm) and the usual laser power at the sample of 0.1–0.4 mW. UV/vis spectra were recorded on a Shimadzu UV-2401PC instrument or a SPECORD instrument in methanol or CHCl3/ACN (1/1, v/v); solid samples were coated on the surface of a quartz cuvette. Photoluminescence spectra were recorded on a Fluorolog 3-22 Jobin Yvon Spex instrument, using a four-window quartz cuvette (1 cm) for solutions and using quartz glass for films. The emission spectra were recorded with the excitation wavelength, λex, matching the absorption maximum of the measured compound. Quantum yields, λF, of photoluminescence were measured using the integration sphere Quanta-φ F-3029. Fluorescence decay was monitored with a FluoroHub single photon counting controller on a Fluorolog 3-22 Jobin Yvon Spex instrument using excitation at λex = 378 nm for solutions and λex = 472 nm for films. Viscometric measurements were performed on a Microviscometer Lovis 2000 M/Me (Anton Paar). SEC records were obtained using a Spectra Physics Analytical HPLC pump P1000 with two SEC columns: Polymer Labs (Bristol, USA) Mixed-D and Mixed-E. The system was equipped with a Thermo UV6000 DAD detector. 0.05 M tetrabutylammonium hexafluorophosphate in CHCl3/ACN (1/1, v/v, CHROMASOLV, Riedel-deHaen) was used as an eluent (0.7 mL min−1).:2). Orange powder (0.12 g, 17%). 1H NMR (400 MHz, d-CDCl3) δ ppm 8.78–8.76 (m, 4H, A6), 8.68 (s, 4H, B3), 8.65 (m, 4H, A3), 7.89 (td, J1 = 7.7, J2 = 1.7, 4H, A4), 7.66 (s, 2H, C4), 7.38 (ddd, J1 = 7.5, J2 = 4.7, J3 = 1.3, 4H, A5), 7.21 (d, J = 3.9, 2H, D3 or D4), 7.17 (d, J = 3.9, D4 or D3), 2.87 (t, J = 3.9, 4H, Hex1), 1.82–1.72 (m, 4H, Hex2), 1.50–1.36 (m, 12H, Hex3–Hex5), 0.97–0.89 (m, 4H, Hex6). 13C NMR (101 MHz, d-CDCl3) δ ppm 156.05, 149.14, 142.96, 140.96, 139.17, 137.10, 136.87, 135.09, 132.42, 128.91, 126.85, 124.17, 123.92, 121.35, 116.59, 31.69, 30.57, 29.73, 29.73, 22.64, 14.13. IR (DRIFT), cm−1 3063 (m), 3013 (w), 2957 (m), 2925 (s), 2852 (m), 1974 (w), 1955 (w), 1724 (m), 1599 (s), 1583 (s), 1567 (s), 1544 (m), 1468 (s), 1458 (s), 1434 (m), 1403 (s), 1386 (m), 1361 (m), 1340 (w), 1298 (w), 1265 (m), 1254 (m), 1210 (m), 1195 (w), 1177 (w), 1146 (w), 1124 (m), 1093 (m), 1070 (m), 1037 (m), 1021 (m), 990 (m), 970 (w), 918 (w), 897 (w), 879 (m), 848 (m), 839 (m), 789 (s), 776 (s), 742 (s), 728 (m), 715 (w), 693 (w), 682 (m), 663 (m), 658 (m), 642 (m), 631 (m), 622 (m), 585 (w), 566 (w), 524 (w), 501 (m), 461 (w), 403 (m). HRMS found m/z: 961.32106 [M+H]+, C58H33N6S4 requires: 961.32090.
:1) was added and the reaction mixture was heated at 90 °C for 24 hours. After cooling to room temperature the reaction mixture was extracted with dichloromethane, the organic phase was dried with MgSO4, filtered off and evaporated. The crude product was purified by precipitation from concentrated THF solution by using hexane. Orange powder (3.4 g, 97%). 1H NMR (400 MHz, d-CDCl3) δ ppm 7.19 (d, J = 5.2, 2H, C4 or C5), 7.12 (d, J = 3.8, 2H, D3 or D4), 7.01 (d, J = 3.8, 2H, D4 or D3), 6.95 (d, J = 5.2, 2H, C5 or C4), 6.81–6.83 (m, 8H, –Ph), 3.90 (t, J = 6.6, 4H, Hex6), 3.76 (s, 6H, –OCH3), 2.81 (t, J = 7.9, 4H, Hex1), 1.65–1.81 (m, 8H, Hex2 + Hex5), 1.41–1.51 (m, 8H, Hex3 + Hex4). 13C NMR (101 MHz, d-CDCl3) δ ppm 160.43, 153.65, 153.24, 150.09, 144.13, 139.65, 135.24, 130.39, 126.60, 123.86, 115.41, 114.60, 69.10, 68.54, 30.53, 29.31, 29.18, 27.14, 25.87. IR (DRIFT), cm−1 3096 (m), 3070 (m), 3056 (m), 3042 (w), 3001 (m), 2937 (s), 2928 (s), 2855 (s), 2834 (m), 2067 (w), 1857 (w), 1750 (m), 1615 (w), 1591 (m), 1539 (w), 1508 (s), 1495 (m), 1467 (m), 1456 (m), 1442 (m), 1422 (m), 1389 (m), 1378 (w), 1347 (w), 1322 (m), 1308 (m), 1294 (m), 1231 (s), 1193 (s), 1176 (m), 1154 (w), 1146 (w), 1108 (m), 1090 (m), 1071 (s), 1056 (m), 1038 (s), 1005 (w), 989 (s), 958 (w), 940 (m), 929 (m), 919 (m), 906 (w), 889 (w), 876 (m), 855 (m), 826 (s), 805 (s), 792 (s), 757 (m), 742 (s), 722 (m), 699 (m), 687 (m), 669 (m), 654 (m), 638 (w), 631 (w), 599 (w), 586 (w), 566 (w), 521 (s), 506 (m), 472 (w), 462 (m), 426 (w). HRMS found m/z: 765.21732 [M+Na]+, C42H46O4NaS4 requires: 765.21711.
:2) to obtain the product as an orange solid (0.18 g, 32%). 1H NMR (400 MHz, d-CDCl3) δ ppm 8.75–8.78 (m, 4H, A6), 8.68–8.65 (m, 8H, B3 + A3), 7.89 (td, J1 = 8.0, J2 = 1.8, 4H, A4), 7.66 (s, 2H, C4), 7.38 (ddd, J1 = 7.6, J2 = 4.9, J2 = 1.5, 4H, A5), 7.19 (d, J = 3.8, 2H, D3 or D4), 7.14 (d, J = 4.10, 2H, D4 or D3), 6.79–6.86 (m, 8H, –Ph), 3.93 (t, J = 6.3, 4H, Hex6), 3.75 (s, 6H, –OCH3), 2.88 (t, J = 7.9, 4H, Hex1), 1.76–1.86 (m, 8H, Hex2 + Hex5), 1.52–1.59 (m, 8H, Hex3 + Hex4). 13C NMR (101 MHz, d-CDCl3) δ ppm 156.07, 156.03, 153.65, 153.27, 149.14, 142.91, 140.72, 139.27, 137.10, 136.86, 135.00, 132.48, 128.84, 126.93, 124.22, 123.92, 121.33, 116.53, 115.44, 114.59, 68.56, 55.72, 30.47, 29.57, 29.34, 29.32, 25.94. IR (DRIFT), cm−1 3060 (m), 3050 (m), 3011 (w), 2930 (s), 2858 (m), 1601 (s), 1584 (s), 1568 (s), 1545 (m), 1508 (s), 1466 (s), 1441 (m), 1401 (s), 1384 (w), 1334 (w), 1303 (w), 1289 (w), 1263 (m), 1231 (s), 1181 (m), 1146 (w), 1123 (m), 1105 (m), 1095 (m), 1071 (m), 1039 (m), 1019 (m), 990 (m), 882 (m), 831 (m), 823 (m), 787 (s), 774 (m), 740 (m), 726 (m), 706 (w), 670 (w), 660 (m), 630 (m), 622 (m), 567 (w), 525 (w), 498 (w), 468 (w), 425 (w), 400 (m). HRMS found m/z: 1205.39458 [M+H]+, C72H65O4N6S4 requires: 1205.39446.
:1) to obtain the pure product (0.62 g, 48%). 1H NMR (300 MHz, d-CDCl3) δ ppm 7.29 (d, J = 5.4, 2H, D4 or D5), 6.96 (d, J = 5.1, 2H, D5 or D4), 6.82 (s, 8H, –Ph), 3.84 (t, J = 6.5, 4H, Hex6), 3.77 (s, 6H, –OCH3), 2.52 (t, J = 7.7, 4H, Hex1), 1.74–1.67 (m, 4H, Hex5), 1.63–1.56 (m, 4H, Hex2), 1.44–1.29 (m, 8H, Hex3 + Hex4). 13C NMR (101 MHz, d-CDCl3) δ ppm 153.32, 152.93, 141.80, 128.43, 128.19, 125.0, 115.08, 114.27, 68.21, 55.43, 30.29, 28.94, 28.77, 28.34, 25.49. IR (DRIFT), cm−1 3107 (m), 3064 (m), 3046 (m), 3011 (m), 2932 (s), 2854 (s), 2836 (m), 1510 (s), 1476 (m), 1466 (m), 1442 (m), 1395 (m), 1379 (w), 1349 (w), 1335 (w), 1291 (s), 1269 (m), 1239 (s), 1179 (m), 1154 (w), 1130 (w), 1109 (m), 1090 (m), 1073 (w), 1038 (s), 1016 (m), 1001 (m), 943 (w), 932 (m), 915 (m), 894 (m), 884 (m), 826 (s), 789 (w), 767 (w), 742 (s), 732 (m), 717 (s), 695 (m), 686 (m), 663 (m), 629 (m), 602 (w), 571 (w), 531 (m), 521 (m), 509 (m), 490 (w), 436 (w), 422 (w). HRMS found m/z: 579.25984 [M+H]+, C34H43O4S2 requires: 579.25973.
:2) to obtain the product as an orange solid (0.28 g, 28%). 1H NMR (400 MHz, d-CDCl3) δ ppm 8.77–8.75 (m, 4H, A6), 8.70 (s, 4H, B3), 8.67 (dt, J1 = 8.0, J2 = 1.1, 4H, A3), 7.89 (td, J1 = 7.8, J2 = 1.8, 4H, A4), 7.71 (d, J = 7.3, 2H, C4), 7.38 (ddd, J1 = 7.4, J2 = 4.8, J3 = 1.4, 4H, A5), 7.24 (d, J = 3.6, 2H, C3), 7.19 (s, 2H, D4), 6.8 (s, 4H, –Ph), 6.79 (s, 4H, –Ph), 3.89 (t, J = 6.5, 4H, Hex6), 3.71 (s, 6H, –OMe), 2.61 (t, J = 7.6, 4H, Hex1), 1.80–1.73 (m, 4H, Hex5), 1.70–1.64 (m, 4H, Hex2), 1.50–1.40 (m, 8H, Hex3 + Hex4). 13C NMR (101 MHz, d-CDCl3) δ ppm 156.12, 155.99, 153.60, 153.26, 149.16, 143.53, 142.98, 140.34, 138.88, 136.86, 136.23, 127.99, 126.64, 125.88, 124.62, 123.92, 121.30, 116.66, 115.38, 114.57, 68.54, 55.66, 30.52, 29.28, 29.06, 28.89, 25.84. IR (DRIFT), cm−1 3064 (m), 3049 (m), 3004 (m), 2936 (s), 2859 (m), 2831 (w), 1599 (m), 1582 (s), 1566 (m), 1552 (m), 1507 (s), 1466 (s), 1446 (m), 1399 (m), 1382 (w), 1362 (w), 1303 (w), 1290 (w), 1265 (m), 1231 (s), 1181 (m), 1152 (w), 1146 (w), 1125 (w), 1107 (m), 1095 (m), 1085 (w), 1065 (m), 1038 (m), 1011 (m), 990 (m), 946 (w), 919 (w), 907 (w), 882 (m), 878 (m), 862 (w), 848 (w), 825 (s), 789 (s), 773 (m), 743 (m), 729 (m), 720 (m), 701 (w), 680 (m), 658 (m), 633 (m), 622 (m), 584 (w), 565 (w), 535 (w), 523 (m), 506 (w), 494 (w), 468 (w), 456 (w), 418 (w), 403 (m). HRMS found m/z: 1205.39479 [M+H]+, C72H65O4N6S4 requires: 1205.39446.
:1) (0.42 g, 30%). 1H NMR (400 MHz, d2-CD2Cl2) δ ppm 7.18 (d, J = 5.1, 2H, E5), 7.02 (s, 2H, F3), 6.95 (d, J = 5.5, 2H, E4), 6.78 (s, 8H, –Ph), 6.76 (s, 8H, –Ph), 3.87–3.81 (m, 8H, Hex6), 3.72 (s, 12H, –OCH3), 2.80 (t, J = 7.8, 4H, Hex1), 2.59 (t, J = 7.8, 4H, Hex1), 1.75–1.61 (m, 16H, Hex2 + Hex5), 1.46–1.39 (m, 16H, Hex3 + Hex4). 13C NMR (101 MHz, d2-CD2Cl2) δ ppm 154.25, 153.85, 143.37, 140.11, 136.57, 131.12, 130.77, 128.76, 128.00, 124.20, 115.82, 115.04, 69.04, 56.13, 32.16, 31.20, 29.84, 29.72, 26.44. IR (DRIFT), cm−1 3105 (w), 3074 (w), 3047 (w), 2997 (w), 2935 (s), 2858 (s), 1512 (s), 1466 (m), 1439 (w), 1389 (w), 1288 (m), 1242 (s), 1215 (s), 1180 (m), 1157 (w), 1111 (m), 1068 (m), 1041 (s), 987 (w), 941 (w), 922 (w), 825 (s), 741 (m), 721 (m), 687 (w), 652 (w), 525 (m). HRMS found m/z: 1155.49656 [M+H]+, C68H83O8S4 requires: 1155.49653.
:2). Orange solid (0.17 g, 33%). 1H NMR (400 MHz, d2-CD2Cl2) δ ppm 8.74 (m, 4H, A6), 8.70 (s, 4H, B3), 8.67 (m, 4H, A3), 7.91 (td, J1 = 7.7, J2 = 2.1, 4H, A4), 7.67 (s, 2H, C4), 7.40–7.37 (m, 4H, A5), 7.19 (s, 2H, D4), 6.80–6.72 (m, 16H, –Ph), 3.90–3.83 (m, 8H, Hex6), 3.69 (s, 6H, –OCH3), 3.66 (s, 6H, –OCH3), 2.89 (t, J = 7.8, 4H, Hex1), 2.66 (t, J = 7.8, 4H, Hex1), 1.80–1.69 (m, 16H, Hex2 + Hex5), 1.45–1.38 (m, 16H, Hex3 + Hex4). 13C NMR (101 MHz, d2-CD2Cl2) δ ppm 156.71, 156.41, 154.23, 153.85, 149.74, 143.72, 143.37, 141.35, 139.47, 137.42, 136.23, 133.28, 129.58, 129.34, 128.46, 124.58, 121.62, 116.88, 115.83, 115.03, 69.06, 56.09, 31.06, 29.92, 29.70, 26.50, 26.42. IR (DRIFT), cm−1 3063 (w), 3009 (w), 2935 (s), 2854 (m), 1601 (m), 1581 (s), 1566 (s), 1508 (s), 1462 (s), 1400 (w), 1288 (w), 1238 (w), 1180 (w), 1149 (w), 1107 (m), 1072 (w), 1038 (s), 991 (w), 883 (w), 825 (m), 795 (m), 744 (m), 679 (w), 656 (m), 633 (w), 621 (w), 525 (w). HRMS found m/z: 1617.65658 [M+H]+, C98H101O8N6S4 requires: 1617.65582.
General procedure for bromination of A-unimers
A unimer was dissolved in dichloromethane (to a concentration of ca. 0.02 M), the solution was then cooled in an ice bath and BBr3 was added (excess). After 4 hours of stirring the cooling bath was removed and the solution was poured into water. The mixture was carefully neutralized with a saturated solution of K2CO3. Then the product was extracted with dichloromethane, dried with MgSO4, filtered off and evaporated to obtain the desired product.General procedure for quaternization of Br-unimers
A unimer was dissolved in toluene (to a concentration of ca. 6.5 mM) and the flask was flushed with argon. Triethylphosphine (PEt3) was added as 1 M solution in THF (ca. 20 eq.) and the reaction was heated to 110 °C for 4 days during which the quaternized product gets precipitated from the solution. After cooling to room temperature the product was filtered and washed with toluene and diethylether. The desired product was dried in vacuo.Acknowledgements
This work was supported by the Czech Science Foundation (P108/12/1143), the Grant Agency of Charles University (project 64213) and the COST Action CM1302 (SIPS) European Network on Smart Inorganic Polymers.References
- E. Figgemeier, V. Aranyos, E. C. Constable, R. W. Handel, C. E. Housecroft, C. Risinger, A. Hagfeldt and E. Mukhtar, Inorg. Chem. Commun., 2004, 7, 117–121 CrossRef CAS.
- C. Houarner, E. Blart, P. Buvat and F. Odobel, Photochem. Photobiol. Sci., 2005, 4, 200–204 CAS.
- C. Houarner-Rassin, E. Blart, P. Buvat and F. Odobel, J. Photochem. Photobiol., A, 2007, 186, 135–142 CrossRef CAS.
- S. Caramori, J. Husson, M. Beley, C. a. Bignozzi, R. Argazzi and P. C. Gros, Chem. – Eur. J., 2010, 16, 2611–2618 CrossRef CAS PubMed.
- K. W. Cheng, C. S. C. Mak, W. K. Chan, A. M. Ching NG and A. B. Djurišič, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1305–1317 CrossRef CAS.
- V. Duprez and F. C. Krebs, Tetrahedron Lett., 2006, 47, 3785–3789 CrossRef CAS.
- A. Wild, F. Schlütter, G. M. Pavlov, C. Friebe, G. Festag, A. Winter, M. D. Hager, V. Cimrová, U. S. Schubert, V. Cimrova and U. S. Schubert, Macromol. Rapid Commun., 2010, 31, 868–874 CrossRef CAS PubMed.
- R. Siebert, A. Winter, M. Schmitt, J. Popp, U. S. Schubert and B. Dietzek, Macromol. Rapid Commun., 2012, 33, 481–497 CrossRef CAS PubMed.
- P. D. Vellis, J. A. Mikroyannidis, C. Lo and C. Hsu, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7702–7712 CrossRef CAS.
- X. Chen, Q. Zhou, Y. Cheng, Y. Geng, D. Ma, Z. Xie and L. Wang, J. Lumin., 2007, 126, 81–90 CrossRef CAS.
- X. Chen, L. Ma, Y. Cheng, Z. Xie and L. Wang, Polym. Int., 2007, 56, 648–654 CrossRef CAS.
- R. Siebert, A. Winter, B. Dietzek, U. S. Schubert and J. Popp, Macromol. Rapid Commun., 2010, 31, 883–888 CrossRef CAS PubMed.
- S. Etienne and M. Beley, Inorg. Chem. Commun., 2006, 9, 68–71 CrossRef CAS.
- L. D. Carlos, R. a. S. Ferreira, V. De Zea Bermudez and S. J. L. Ribeiro, Adv. Mater., 2009, 21, 509–534 CrossRef CAS PubMed.
- M. Barboiu, Y.-M. Legrand, L. Prodi, M. Montalti, N. Zaccheroni, G. Vaughan, A. van der Lee, E. Petit and J.-M. Lehn, Eur. J. Inorg. Chem., 2009, 2009, 2621–2628 CrossRef.
- S. Encinas, L. Flamigni, F. Barigelletti, E. C. Constable, C. E. Housecroft, E. R. Schofield, E. Figgemeier, D. Fenske, M. Neuburger, J. G. Vos and M. Zehnder, Chem. – Eur. J., 2002, 8, 137–150 CrossRef CAS.
- T. K. Sievers, A. Vergin, H. Möhwald, D. G. Kurth, A. Verging, H. Möhwald and D. G. Kurth, Langmuir, 2007, 23, 12179–12184 CrossRef CAS PubMed.
- G. Schwarz, I. Haßlauer and D. G. Kurth, Adv. Colloid Interface Sci., 2014, 207, 107–120 CrossRef CAS PubMed.
- F. S. Han, M. Higuchi and D. G. Kurth, Adv. Mater., 2007, 19, 3928–3931 CrossRef CAS.
- Y. Y. Chen, Y.-T. Tao and H.-C. Lin, Macromolecules, 2006, 39, 8559–8566 CrossRef CAS.
- A. Winter, C. Friebe, M. Chiper, M. D. Hager and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2009, 3, 4083–4098 CrossRef.
- A. Wild, A. Teichler, C.-L. Ho, X.-Z. Wang, H. Zhan, F. Schlütter, A. Winter, M. D. Hager, W.-Y. Wong and U. S. Schubert, J. Mater. Chem. C, 2013, 1, 1812–1822 RSC.
- R. Siebert, Y. Tian, R. Camacho, A. Winter, A. Wild, A. Krieg, U. S. Schubert, J. Popp, I. G. Scheblykin and B. Dietzek, J. Mater. Chem., 2012, 22, 16041–16050 RSC.
- Y. Y. Chen and H. C. Lin, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 3243–3255 CrossRef CAS.
- R. Dobrawa, M. Lysetska, P. Ballester, M. Grüne and F. Würthner, Macromolecules, 2005, 38, 1315–1325 CrossRef CAS.
- C. Ringenbach, A. De Nicola and R. Ziessel, J. Org. Chem., 2003, 68, 4708–4719 CrossRef CAS PubMed.
- S. C. Yu, C. C. Kwok, W. K. Chan and C. M. Che, Adv. Mater., 2003, 15, 1643–1647 CrossRef CAS.
- P. Bláhová, J. Zedník, I. Šloufová, J. Vohlídal and J. Svoboda, Soft Matter, 2014, 12, 214–229 CrossRef.
- P. Štenclová-Bláhová, J. Svoboda, I. Šloufová and J. Vohlídal, Phys. Chem. Chem. Phys., 2015, 17, 13743–13756 RSC.
- J. Lehn, Prog. Polym. Sci., 2005, 30, 814–831 CrossRef CAS.
- J. W. Steed and J. L. Atwood, Supramolecular chemistry, John Wiley & Sons, Ltd, 2nd edn, 2009 Search PubMed.
- A. Ciferri, Macromol. Rapid Commun., 2002, 23, 511–529 CrossRef CAS.
- J. Svoboda, P. Stenclova, F. Uhlík, J. Zedník and J. Vohlídal, Tetrahedron, 2011, 67, 75–79 CrossRef CAS.
- D. Bondarev, J. Zedník, I. Šloufová, A. Sharf, M. Procházka, J. Pfleger and J. Vohlídal, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3073–3081 CrossRef CAS.
- C. G. Bochet, C. Piguet and A. F. Williams, Helv. Chim. Acta, 1993, 76, 372–384 CrossRef CAS.
- D. Rais, M. Menšík, P. Štenclová-Bláhová, J. Svoboda, J. Vohlídal and J. Pfleger, J. Phys. Chem. A, 2015, 119, 6203–6214 CrossRef CAS PubMed.
- I. Šloufová, B. Vlčková, M. Procházka, J. Svoboda and J. Vohlídal, J. Raman Spectrosc., 2014, 45, 338–348 CrossRef.
- S. Kazim, J. Pfleger, K. Halašová, M. Procházka, D. Bondarev and J. Vohlídal, Eur. Phys. J.: Appl. Phys., 2011, 55, 23905 CrossRef.
- P. S. Braterman, J.-I. Song and R. D. Peacock, Inorg. Chem., 1992, 31, 555–559 CrossRef CAS.
- F. Schlütter, A. Wild, A. Winter, M. D. Hager, A. Baumgaertel, C. Friebe and U. S. Schubert, Macromolecules, 2010, 43, 2759–2771 CrossRef.
- T. Vitvarová, J. Zedník, M. Bláha, J. Vohlídal and J. Svoboda, Eur. J. Inorg. Chem., 2012, 2012, 3866–3874 CrossRef.
- V. Stepanenko, M. Stocker, P. Müller, M. Büchner and F. Würthner, J. Mater. Chem., 2009, 19, 6816–6826 RSC.
- R. A. Dobrawa, Synthesis and Characterization of Terpyridine-based Fluorescent Coordination Polymers, Bayerischer Julius-Maximilians-Universität, Würzburg, 2004 Search PubMed.
- J. N. Demas and B. A. DeGraft, Topics in Fluorescence Spectroscopy: Volume 4: Probe Design and Chemical Sensing, 1994 Search PubMed.
- R. Englman and J. Jortner, Mol. Phys., 1970, 18, 145–164 CrossRef CAS.
- R. Dobrawa and F. Würthner, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4981–4995 CrossRef CAS.
- U. S. Schubert, H. Hofmeier and G. R. Newkome, Modern Terpyridine Chemistry, Wiley-VCH Verlag GmbH, Weinheim, 2006 Search PubMed.
- R. Hogg and R. G. Wilkins, J. Chem. Soc., 1962, 341–350 RSC.
- R. H. Holyer, C. D. Hubbard, S. F. A. Kettle and R. G. Wilkins, Inorg. Chem., 1966, 5, 622–625 CrossRef CAS.
- J. R. Winkler, C. Creutz and N. Sutin, J. Am. Chem. Soc., 1987, 109, 3470–3471 CrossRef CAS.
- J. Burgess, S. Radulović and F. Sánchez, Transition Met. Chem., 1987, 12, 529–536 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5dt04133c |
| This journal is © The Royal Society of Chemistry 2016 |