
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegioselectivity in C–H activation: reagent control in cyclometallation of 2-(1-naphthyl)-pyridine†
Mikhail
Kondrashov
,
David
Provost
and
Ola F.
Wendt
*
Centre for Analysis and Synthesis, Department of Chemistry, Lund University, P. O. Box 124, S-221 00 Lund, Sweden. E-mail: Ola.Wendt@chem.lu.se
First published on 18th November 2015
Abstract
2-(1-Naphthyl)-pyridine (1) possesses sp2 C–H bonds in both the γ- and δ-positions and is therefore a suitable substrate for studying the cyclometallation selectivity with different reagents and conditions. Such selectivity studies are reported. Based on deuterium-exchange experiments it is concluded that cycloruthenation with RuCl2(p-cymene) dimer is reversible with kinetic and thermodynamic preference for γ-substitution. Electrophilic cycloborylation, on the other hand, shows unusual δ-substitution. The previously published cyclopalladation and cycloauration of the substrate was studied in detail and was shown to be irreversible; they proceed under kinetic control and give γ- and δ-substitution for palladium and gold, respectively.
Introduction
Although major advances in the area of C–H activation have been made in the last decades, selectivity remains a critical issue. Ligand-directed C–H activation has been a successful approach towards selectivity and here, typically, the position γ to the directing atom is ultimately functionalised,1 since five-membered metallacycles are normally preferred, especially for palladium. This is also the selectivity found in the majority of ortho-metallation reactions of aromatic substrates, where it is dictated by the geometric accessibility. However, certain procedures allowing other selectivities are known. To override the γ-selectivity, the most common approach is to make the γ-position of the substrate hindered2 or unavailable for substitution,3 and only in a few cases an inherent, preferential substitution of the β-position has been observed.4One of the substrates used in the screening of novel catalytic protocols, 2-(1-naphthyl)-pyridine (1), possesses sp2 C–H bonds in both the γ- and δ-positions. This is unusual, and 1 is therefore an excellent substrate to use as a model for investigating the regioselectivity ligand-directed C–H activation with different reagents and conditions. As expected, ligand-directed substitution reactions performed with 1 almost always resulted in γ-substitutions,5 the only exception being the work by Pan and Chen and co-workers5r in which the selectivity remains unclear, since different substitution patterns are reported in the main article and the supporting information, cf.Scheme 1.
 | ||
| Scheme 1 Selected examples of metal-catalysed substitutions of 2-(1-naphthyl)-pyridine. | ||
In a recent study we could show that 1 can be cyclometallated with palladium or gold precursors and while cyclopalladation occurred at the expected γ-position (as previously indicated in catalysis),5a the cycloauration took place with exclusive δ-selectivity and the so formed gold complex could be functionalised with N-bromo succinimide, cf. Scheme 2.6
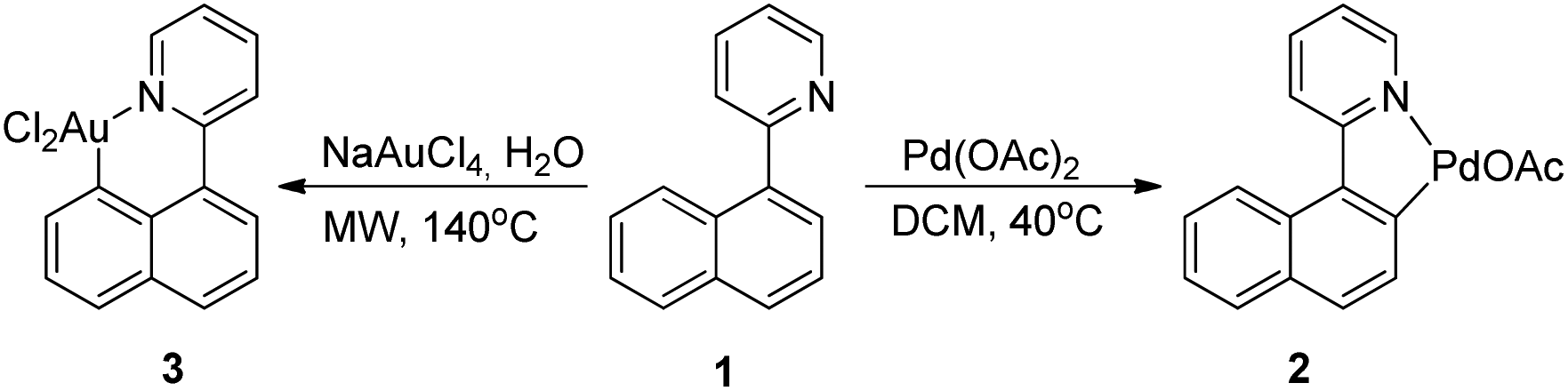 | ||
| Scheme 2 Cyclometallation of 1 with Au and Pd precursors. | ||
This unusual orthogonal regioselectivity could open up for development of catalytic reactions giving isomeric products based on a choice of the metal catalyst. Therefore, we decided to investigate the reactivity of 1 further, and here we report its reactivity with other standard cyclometallating agents, viz. ruthenium and boron precursors, and we also report further studies with all reagents to unravel kinetic and thermodynamic preferences for the different reactivities.
Experimental section
General
All manipulations were conducted under ambient conditions, unless noted. Solvents and chemicals were purchased from commercial suppliers and used as received. Compounds 1 and 3 were synthesised according to the literature.6a NMR spectra were acquired on a Bruker Avance 400 FT-NMR spectrometer (1H: 400.1 MHz) or a Varian Unity INOVA 500 spectrometer (1H: 499.76 MHz). Residual solvent peaks were used as an internal reference. Elemental analyses were performed by H. Kolbe Microanalytisches Laboratorium, Mülheim an der Ruhr, Germany. Microwave experiments were performed on Biotage Initiator 2.5 microwave synthesiser with automatic power adjustment using “high absorption” setting.1H NMR (400 MHz, CDCl3) δ 9.36 (d, J = 6.7 Hz, 1H), 8.34 (vt, J = 8.2 Hz, 2H), 8.25 (d, J = 8.4 Hz, 1H), 7.85 (dd, J = 8.1, 1.4 Hz, 1H), 7.71 (ddd, J = 8.3, 7.3, 1.7 Hz, 1H), 7.59 (d, J = 8.2 Hz, 1H), 7.47 (ddd, J = 8.5, 6.8, 1.4 Hz, 1H), 7.33 (ddd, J = 7.9, 6.8, 1.1 Hz, 1H), 7.05 (ddd, J = 7.2, 5.7, 1.3 Hz, 1H), 5.58 (dd, J = 11.1, 6.0 Hz, 2H), 5.27 (d, J = 6.0 Hz, 1H), 5.04 (d, J = 4.9 Hz, 1H), 2.40 (hept, J = 7.0 Hz, 1H), 2.07 (s, 3H), 0.96 (d, J = 6.8 Hz, 3H), 0.89 (d, J = 7.2 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 188.0, 165.3, 155.4, 138.4, 136.9, 136.5, 132.1, 131.1, 129.6, 128.4, 126.4, 123.2, 123.1, 121.9, 120.2, 101.6, 101.5, 91.6, 89.8, 84.9, 82.3, 31.0, 22.6, 22.0, 19.0. HR-MS ES+ (H2O–CH3CN): 440.0951, calculated for C25H24NRu ([M − Cl]+) 440.0952.
Compound 1-d2 was isolated in the following way: the reaction mixture above was partitioned between 5% HCl and dichloromethane. The aqueous phase was separated and neutralised with aqueous potassium carbonate and extracted with diethyl ether. The new organic phase was dried over potassium carbonate and evaporated. This yielded 26 mg (90%) of a colourless oil. Alternatively, the deuteration can be performed with lower loading of ruthenium catalyst (0.05 equiv.) and at lower temperature (80 °C) if the heating is extended to 3 days. A higher percent of deuteration can be achieved if the isolated product is subjected to the same procedure again.
1H NMR (400 MHz, CDCl3) δ 8.81 (ddd, J = 4.9, 1.8, 1.0 Hz, 1H), 7.92 (dd, J = 7.9, 2.0 Hz, 2H), 7.83 (td, J = 7.7, 1.8 Hz, 1H), 7.65–7.44 (m, 4H), 7.34 (ddd, J = 7.6, 4.9, 1.2 Hz, 1H). ESI-MS m/z 208 ([M + H])+.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of MeOH/HFaq (48%), and the mixture was stirred for 30 minutes. Water and dichloromethane were added, followed by aqueous potassium carbonate. The layers were separated and the organic phase was washed with 10% HCl to eliminate the byproduct 1. Drying over MgSO4 and filtering through a short plug of silica yielded 303 mg (73%) of product as a white powder after evaporation of the solvent. X-ray quality crystals were obtained by vapour-exchange crystallisation using dichloromethane as a solvent and pentane as a counter-solvent.
:1 mixture of MeOH/HFaq (48%), and the mixture was stirred for 30 minutes. Water and dichloromethane were added, followed by aqueous potassium carbonate. The layers were separated and the organic phase was washed with 10% HCl to eliminate the byproduct 1. Drying over MgSO4 and filtering through a short plug of silica yielded 303 mg (73%) of product as a white powder after evaporation of the solvent. X-ray quality crystals were obtained by vapour-exchange crystallisation using dichloromethane as a solvent and pentane as a counter-solvent.
1H NMR (400 MHz, CDCl3) δ 9.06 (dd, J = 6.0, 1.7 Hz, 1H), 8.47 (d, J = 8.5 Hz, 1H), 8.32 (dd, J = 7.5, 1.0 Hz, 1H), 8.20–8.13 (m, 2H), 8.08 (d, J = 8.1 Hz, 1H), 7.91 (dd, J = 8.1, 1.3 Hz, 1H), 7.71 (dd, J = 8.2, 6.8 Hz, 1H), 7.64–7.53 (m, 2H).
13C NMR (101 MHz, CDCl3) δ 152.4, 143.2 (br.t., J = 3.5 Hz), 141.6, 134.3, 132.8, 131.6, 130.9, 128.2, 127.7, 125.7, 125.0, 124.8, 123.3, 122.2. Elemental analysis, calcd for C15H10BF2N: C, 71.19; H, 3.98; N, 5.54; found: C, 71.27; H, 3.93; N 5.49.
:EtOAc 9:1). The mixture was quenched with 3 M NaOH and extracted with 3 × 5 mL of DCM. The organic phase was dried over K2CO3 and evaporated to result in 4.5 mg (93%) of colourless oil.
1H NMR (400 MHz, CDCl3) δ 8.80 (ddd, J = 4.9, 1.8, 0.9 Hz, 1H), 7.92 (d, J = 7.2 Hz 2H), 7.83 (td, J = 7.7, 1.8 Hz, 1H), 7.67–7.43 (m, 5H), 7.34 (ddd, J = 7.6, 4.9, 1.2 Hz, 1H).
Crystallography
XRD-quality crystals of compounds were obtained as described above for 4 and 6. Intensity data were collected with an Oxford Diffraction Excalibur 3 system, using ω-scans and Mo Kα (λ = 0.71073 Å) radiation.10 The data were extracted and integrated using Crysalis RED.11 The structures were solved by direct methods and refined by full-matrix least-squares calculations on F2 using SHELXL12 and SIR-92.13 Molecular graphics were generated using Diamond 3.2i.14 CCDC deposition numbers are 1404269 for 4 and 1404270 for 6.Results and discussion
Selectivity in cycloruthenation
Ruthenium metallacycle 4 was synthesised by cyclometallation of 1 with ruthenium(p-cymene) dichloride dimer as shown in Scheme 3. The 1H COSY NMR spectrum indicates the presence of three chains of neighbouring 1H resonances, containing 4, 4 and 2 resonances each. From this we concluded that the structure of the compound contained a five-membered ring, meaning that the γ-position was functionalised, which is in agreement with previous results from Inoue and co-workers,5d where ruthenium-catalysed arylation takes place in the γ-position. | ||
| Scheme 3 Isolation of metallacycle 4 – a possible intermediate in the catalytic arylation published by Inoue.5d | ||
The structure was unambiguously assigned with the help of X-ray crystallography, cf.Fig. 1. The structure contains moderate distortions: the exocyclic C–C bond and the C–H bond in the peri-position are found to have a dihedral angle (C5–C6–C14–H14) of 14°, which is almost coplanar compared to the 40° observed for similar positions in auracycle 3.6a The ruthenium atom is tilted away from the naphthalene ring, creating a dihedral angle C5–C6–C7–Ru1 of 6°, which is less than the corresponding angle in palladacycle 2.6a The ruthenium atom has a distorted octahedral geometry with a C7–Ru1–N1 angle of 77°, which is substantially lower than expected for an octahedron, but in good agreement with what is found in the square-planar complexes with Au and Pd.
 | ||
| Fig. 1 Molecular structure of 4 at the 30% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and bond and torsion angles (°) with estimated standard deviations: Ru1–N1 2.088(3), Ru1–C7 2.051(3), C7–Ru1–N1 76.6(1), C5–C6–C7–Ru1 6.2, C5–C6–C14–H14 14.3. | ||
It should be noted that the catalytic reaction published by Inoue and co-workers proceeds under different conditions and they proposed cyclometallation to take place at a Ru(IV) metal centre. In our view, the present results suggest that cyclometallation can also proceed at a Ru(II) centre, and a compound similar to 4 could be an intermediate in the reaction published by Inoue, if oxidation takes place after the cyclometallation, contrary to the original proposal. (Scheme 3).5d
A similar ruthenacycle with a 2-(1-naphthyl)-pyridine ligand was previously synthesised by Wright and co-workers,5ab but it was made via transmetallation from an organomercury compound, rather than in a direct cyclometallation (Scheme 4).
 | ||
| Scheme 4 Synthesis of ruthenacycle by Wright.5ab | ||
In order to gain some mechanistic insight we decided to run a deuterium exchange experiment using catalytic amounts of the ruthenium salt (Scheme 5). Compound 1 was dissolved in acetonitrile-d3 and heated in a J. Young tube with an excess of acetic acid-d4 and 10 mol% of ruthenium(p-cymene) dichloride dimer. Following the disappearance of certain 1H resonances we were able to detect the reaction progress. The deuterium exchange is superstoichiometric indicating that the cycloruthenation process is reversible and catalytic in Ru, which is in agreement with previous mechanistic studies by Dixneuf and Jutand.17 At 70 °C the deuterium exchange occurs in both γ- and δ-positions, but γ-deuteration is significantly faster, as shown by a ca. 5-fold excess of deuterium in the γ-position after 1 h. At elevated temperatures or prolonged reaction times the δ-position can be fully deuterated as well. Thus, the γ-position is both kinetically and thermodynamically preferred.
 | ||
| Scheme 5 Ru-catalysed deuterium exchange in 1. | ||
Interestingly, the substitution ambiguity does not present itself in the arylation reaction. Repeating the reaction published by Inoue and co-workers,5d we did not observe any disubstituted product even at elevated temperatures in the presence of excess of the arylating agent. A probable explanation for this difference between deuterium exchange and the arylation process is that the substitution in the γ-position creates a rotational restriction for the pyridine ring, thus blocking the potential δ-cyclometallation (Scheme 6).
 | ||
| Scheme 6 Attempted bis-arylation of 1. | ||
Selectivity in cycloborylation
We were also interested in comparing the selectivity in reactions with transition metals to that of main-group cyclosubstitution reagents. As a test reaction electrophilic cycloborylation with boron tribromide was chosen, a reaction which was previously performed on similar 2-aryl-pyridine substrates by Murakami and co-workers.9 The direct product of the cycloborylation decomposes on a silica gel column, but after an anion exchange with HF, a more stable difluoride could be purified and isolated in good yield (Scheme 7). The COSY spectrum and the X-ray crystallographic study show that the product contains a six-membered ring, and that, hence, the substitution proceeds with δ-selectivity. The molecular structure is shown in Fig. 2. The cycloborylation presumably proceeds via a highly electrophilic borenium cation,15 and, under kinetic control, the nucleophilicity of the different positions determine the selectivity. Hence, the observed selectivity is in agreement with our previous assumption6a that the δ-position, belonging to the less substituted ring is significantly more nucleophilic.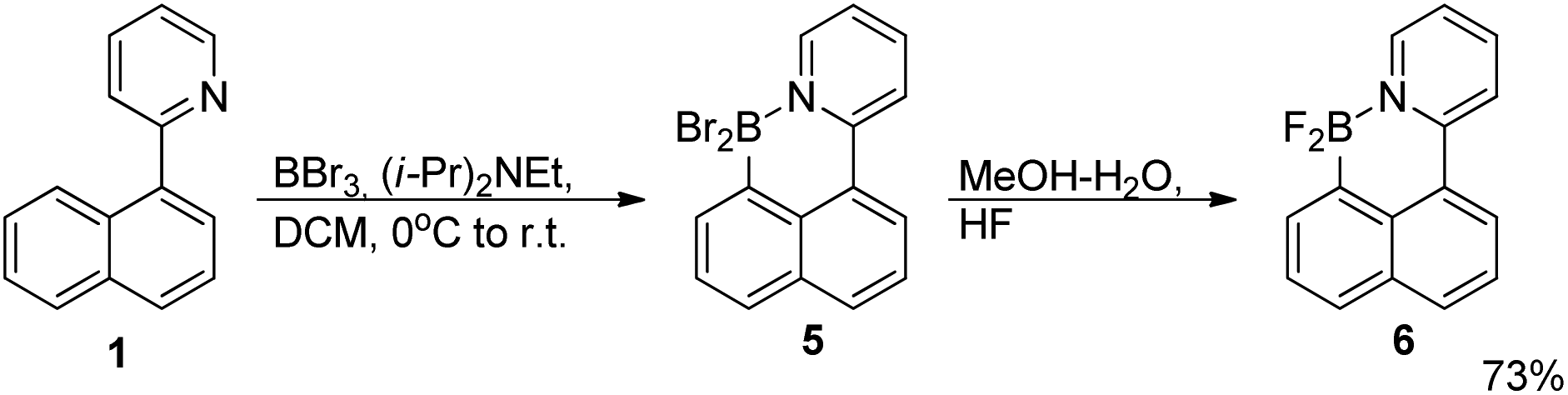 | ||
| Scheme 7 Synthesis of the boracycle 6. | ||
 | ||
| Fig. 2 Molecular structure of 6 at the 30% probability level. Selected bond lengths (Å) and bond and torsion angles (°) with estimated standard deviations: B1–N1 1.604(2), B1–C14 1.576(2), N1–B1–C14 110.3(1), B1–C14–C6–C5 4.7, C5–N1–B1–C14 12.3. | ||
It is important to note that the geometric requirements for the tetrahedrally substituted boron atom are different compared to the square-planar d8-metal complexes. In the six-membered ring the angles around the atom of boron are negligibly different from the expected 109° and the whole structure is almost flat with a dihedral angle between exocyclic C–C and C–B bonds of 5°. It would therefore seem that for boron, six-membered cycle is not only kinetically preferred, but also thermodynamically more stable.
Interestingly, the selectivity of this direct cycloborylation is also orthogonal to the published iridium-,5q rhodium-5ac and palladium-catalysed5z and transition metal-free5z borylations (Scheme 8).
 | ||
| Scheme 8 Published,5q,z,ac protocols for the synthesis of five-membered boracycles of 1. | ||
Acidic hydrolysis of this compound in deuterated acid afforded a δ-monodeuterated 2-(1-naphthyl)-pyridine, isomeric to the product of the low temperature ruthenium-catalysed deuterium exchange (Scheme 9).
 | ||
| Scheme 9 Acid-catalysed hydrolysis of 6. | ||
Unfortunately, our attempts to employ 6 in further electrophilic substitutions failed. While 6 was unreactive in electrophilic bromination and transmetallation to palladium precursors, the Cu-catalysed iodination, which was previously published16 for the similar 2-(phenyl)-pyridine dibromoboracycles unfortunately resulted in predominant formation of free 1, cf. Scheme 10.
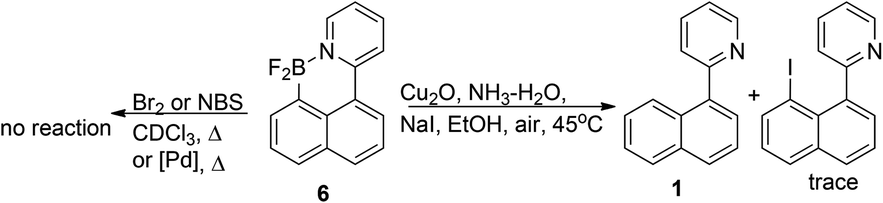 | ||
| Scheme 10 Attempts of electrophilic substitutions of 6. | ||
Deuterium labelling studies for gold and palladium
To gain more insight in the cycloauration and -palladation reactions we performed some labelling studies for these reactions. Heating 1 with catalytic amounts of palladium acetate in the presence of acetic acid-d4 showed no H-D exchange, which indicates that the cyclopalladation process is irreversible and proceeds under kinetic control, which is in agreement with previous studies by Dixneuf and Jutand (Scheme 11).17 | ||
| Scheme 11 Attempted deuterium-exchange in 1 in the presence of palladium acetate. | ||
Unfortunately, a similar experiment could not be performed for the cycloauration procedure, since both 1 and 3 have very poor solubility in water, which is the reaction solvent in this case. Instead we performed a stoichiometric reaction in D2O. The product formed showed no sign of deuteration in the γ-position. Similarly, when we used a bis-deuterated starting material 1-d2 and performed the reaction in H2O, the γ-position remained completely deuterated. This is somewhat surprising since it is known that the cycloauration in the γ-position is possible at these conditions for very similar substrates.18 Thus, for cycloauration the reaction proceeds irreversibly under kinetic control with the δ-position kinetically favourable (Scheme 12).
 | ||
| Scheme 12 Attempt of deuterium-exchange during the synthesis of auracycle 3. | ||
While the mechanisms of the majority of cyclometallations,19 including cyclopalladation and cycloruthenation,17 have been studied to some extent, little is known about the mechanism of cycloauration. In order to shine more light on this mechanism we decided to also study the kinetic isotope effect for this reaction. Hence, we decided to run a reaction between 1 equivalent of sodium tetrachloroaurate, and 1.1 equivalent of each 1 and 1-d2. The observed ratio of the deuterated and non-deuterated metallacyclic products was 40:60, indicating a k.i.e. of 1.5, cf.Scheme 13. This observed value is potentially lower than the real value since the concentration of the non-deuterated starting material is dropping faster than the concentration of the non-deuterated starting material. However, we believe that this effect has very little significance since the total conversion in C–H activation at this conditions is very low. Assuming an electrophilic aromatic substitution-type mechanism, we can conclude that the value is somewhat higher than what is found for standard electrophiles,20 but significantly lower than the value for electrophilic mercuration21 where C–H bond cleavage is proposed to be rate-determining.
 | ||
| Scheme 13 Kinetic isotope effect of the cycloauration. | ||
Conclusions
In summary we have studied the regioselectivity of directed aromatic substitution of 2-(1-naphthyl)-pyridine and shown that the choice of metal precursor dictates the reactivity. Thus, palladium gives an irreversible γ-selective cyclometallation, on one hand, whereas gold gives an irreversible δ-selective cyclometallation. Hence, it is clear that both positions can be kinetically preferred. Cycloruthenation, on the other hand, is reversible and the γ-position is both kinetically and, as expected, thermodynamically preferred. Electrophilic cycloborylation, finally, also resulted in formation of a δ-substituted product. The probable origin of the observed regioselectivity for gold and boron is the higher nucleophilicity of the unsubstituted naphthalene ring, which leads to its higher reactivity towards the electrophilic metal centres. Simultaneously, the cyclopalladation and cycloruthenation proceed via a CMD-type mechanism,22 which poses less restrictions on the nucleophilicity of the reactive carbon atom. Accordingly, the relative thermodynamic stability of five-membered rings is relevant for the case of ruthenium, since cycloruthenation proceeds under equilibrium conditions. These results show the possibility of controlling the selectivity in ligand-directed C–H activation reactions forming a variety of substituted products in a selective manner. Accordingly, 2-(1-naphthyl)-pyridine can be used as a model compound to study the properties and preferences of different new catalytic systems.Acknowledgements
Financial support from the Swedish Research Council, the Knut and Alice Wallenberg Foundation and the Royal Physiographic Society in Lund is gratefully acknowledged.References
- For example see: L. X. Campeau, D. J. Schipper and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 3266 CrossRef CAS PubMed.
- B. V. S. Reddy, L. R. Reddy and E. J. Corey, Org Lett., 2006, 8, 3391 CrossRef CAS PubMed.
- For example see: B.-J. Li, S.-L. Tian, Z. Fang and Z.-J. Shi, Angew. Chem., Int. Ed., 2008, 47, 1115 CrossRef CAS PubMed.
- A. McNally, B. Haffemayer, B. S. L. Collins and M. J. Gaunt, Nature, 2014, 510, 129 CrossRef CAS PubMed.
- (a) D. Kalyani, A. R. Dick, W. Q. Anani and M. S. Sanford, Tetrahedron, 2006, 62, 11483 CrossRef CAS; (b) F. Kakiuchi, T. Kochi, H. Mutsutani, N. Kobayashi, S. Urano, M. Sato, S. Nishiyama and T. Tanabe, J. Am. Chem. Soc., 2009, 131, 11310 CrossRef CAS PubMed; (c) N. Chatani, Y. Ie, F. Kakiuchi and S. Murai, J. Org. Chem., 1997, 62, 2604 CrossRef CAS PubMed; (d) S. Oi, S. Fukita, N. Hirata, N. Watanuki, S. Miyano and Y. Inoue, Org. Lett., 2001, 3, 2579 CrossRef CAS PubMed; (e) X. Jia, D. Yang, S. Zhang and J. Cheng, Org. Lett., 2009, 11, 4716 CrossRef CAS PubMed; (f) G. Zhang, G. Lv, C. Pan, J. Cheng and F. Chen, Synlett, 2011, 2991 CAS; (g) S. Oi, S. Fukita and Y. Inoue, Chem. Commun., 1998, 22, 2439 RSC; (h) C. Xu and Q. Shen, Org. Lett., 2014, 16, 2046 CrossRef CAS PubMed; (i) P. Patel and S. Chang, Org. Lett., 2014, 16, 3328 CrossRef CAS PubMed; (j) Q. Wu, C. Du, Y. Huang, X. Liu, Z. Long, F. Song and J. You, Chem. Sci., 2015, 6, 288 RSC; (k) C.-Z. Luo, P. Gandeepan, J. Jayakumar, K. Parthasarathy, Y.-W. Chang and C.-H. Cheng, Chem. – Eur. J., 2013, 19, 14181 CrossRef CAS PubMed; (l) M. Chaitanya, D. Yadagiri and P. Anbarasan, Org. Lett., 2013, 15, 4960 CrossRef CAS PubMed; (m) J. Qi, L. Huang, Z. Wang and H. Jiang, Org. Biomol. Chem., 2013, 11, 8009 RSC; (n) K. Cheng, B. Yao, J. Zhao and Y. Zhang, Org. Lett., 2008, 10, 5309 CrossRef CAS PubMed; (o) X. Wang, L. Truesdale and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 3648 CrossRef CAS PubMed; (p) W.-N. Sit, C.-W. Chan and W.-Y. Yu, Molecules, 2013, 18, 4403 CrossRef CAS PubMed; (q) A. Ros, B. Estepa, R. Lopez-Rodriguez, E. Alvarez, R. Fernandez and J. M. Lassaletta, Angew. Chem., Int. Ed., 2011, 50, 11724 CrossRef CAS PubMed; (r) L. Li, P. Yu, J. Cheng, F. Chen and C. Pan, Chem. Lett., 2012, 41, 600 CrossRef CAS; (s) J. Jin, Q. Wen, P. Lu and Y. Wang, Chem. Commun., 2012, 48, 9933 RSC; (t) R.-J. Tang, C.-P. Luo, L. Yang and C.-J. Li, Adv. Synth. Catal., 2013, 355, 869 CrossRef CAS; (u) M. Iwasaki, M. Iyanaga, Y. Tsuchiya, Y. Nishimura, W. Li, Z. Li and Y. Nishihara, Chem. – Eur. J., 2014, 20, 2459 CrossRef CAS PubMed; (v) Q. Xiao, X. Meng, M. Kanai and Y. Kuninobu, Angew. Chem., Int. Ed., 2014, 53, 3168 CrossRef CAS PubMed; (w) J. Kim and S. Chang, J. Am. Chem. Soc., 2010, 132, 10272 CrossRef CAS PubMed; (x) J. A. Fernandez-Salas, S. Manzini, L. Piola, A. M. Z. Slawin and S. P. Nolan, Chem. Commun., 2014, 50, 6782 RSC; (y) S. A. Denisov, Y. Cudre, P. Verwilst, G. Jonusauskas, M. Marin-Suarez, J. F. Fernandez-Sanchez, E. Baranoff and N. D. McClenaghan, Inorg. Chem., 2014, 53, 2677 CrossRef CAS PubMed; (z) Y. Kuninobu, T. Iwanaga, T. Omura and K. Takai, Angew. Chem., Int. Ed., 2013, 52, 4431 CrossRef CAS PubMed; (a a) Z.-H. Guan, Z.-H. Ren, Y.-M. Liang, S. M. Spinella, S. Yu and X. Zhang, J. Am. Chem. Soc., 2009, 131, 729 CrossRef CAS PubMed; (a b) A. M. Clark, C. E. F. Rickard, W. R. Roper and L. J. Wright, J. Organomet. Chem., 2000, 598, 262 CrossRef; (a c) E. C. Keske, B. D. Moore, O. V. Zenkina, R. Wang, G. Schatte and C. M. Crudden, Chem. Commun., 2014, 50, 9883 RSC.
- (a) M. Kondrashov, S. Raman and O. F. Wendt, Chem. Commun., 2015, 51, 911 RSC; (b) For related halofunctionalisations of organometallic intermediates, see for example: T. Wang, S. Shi, M. Rudolph and A. S. K. Hashmi, Adv. Synth. Catal., 2014, 356, 2337 CrossRef CAS.
- B. Li, T. Roisnel, C. Darcel and P. H. Dixneuf, Dalton Trans., 2012, 41, 10934 RSC.
- The compound 1-d2 contains approximately 7% of monodeuterated analogue. The residual 1H is equivalently distributed between two positions. Overall this contamination has a negligible effect on the calculated k.i.e. value.
- N. Ishida, T. Moriya, T. Goya and M. Murakami, J. Org. Chem., 2010, 75, 8709 CrossRef CAS PubMed.
- Crysalis CCD, Oxford Diffraction Ltd., Abingdon, Oxfordshire, UK, 2005 Search PubMed.
- H. Putz and K. Brandenburg, Crystal Impact – GbR, Kreuzherrenstr. 102, 53227 Bonn, Germany Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- A. Altomare, G. Cascarano, C. Giacovazzo, A. Guagliardi, M. C. Burla and M. Polidori, J. Appl. Crystallogr., 1994, 27, 435 Search PubMed.
- Crysalis RED, Oxford Diffraction Ltd, Abingdon, Oxfordshire, UK, 2005 Search PubMed.
- A borenium cation was the proposed intermediate in the original publication by Murakami,9 but also directly observed in a similar non-directed borylation in the following article: A. Del Grosso, P. J. Singleton, C. A. Muryn and M. J. Ingleson, Angew. Chem., Int. Ed., 2011, 50, 2102 CrossRef CAS PubMed.
- L. Niu, H. Yang, D. Yang and H. Fu, Adv. Synth. Catal., 2012, 354, 2211 CrossRef CAS.
- I. Fabre, N. von Wolff, G. Le Duc, E. F. Flegeau, C. Bruneau, P. H. Dixneuf and A. Jutand, Chem. – Eur. J., 2013, 19, 7595 CrossRef CAS PubMed.
- A. P. Shaw, M. Tilset, R. H. Heyn and S. J. Jakobsen, Coord. Chem., 2011, 64, 38 CrossRef CAS.
- For example see reviews: (a) Y. Boutadla, D. L. Davies, S. A. Macgregor and A. I. Poblador-Bahamonde, Dalton Trans., 2009, 5820 RSC; (b) D. Balcells, E. Clot and O. Eisenstein, Chem. Rev., 2010, 110, 749 CrossRef CAS PubMed.
- E. V. Anslyn and D. A. Dougherty, Modern Physical Organic Chemistry, University Science Books, Sausalito California, 2006, p. 608, ISBN 978-1-891389-31-3 Search PubMed.
- A. J. Kresge and J. F. Brennan, Proc. Chem. Soc., 1963, 215 CAS.
- For the role of carboxylate ligands enabling the CMD mechanism of cyclometallation see, for example, L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray data in cif format. CCDC 1404269 and 1404270. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt04068j |
| This journal is © The Royal Society of Chemistry 2016 |