
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Coordination chemistry and applications of versatile 4,5-diazafluorene derivatives
Vincent T.
Annibale†
and
Datong
Song
*
Davenport Chemical Research Laboratories, Department of Chemistry, University of Toronto, 80 St. George Street, Toronto, Ontario, Canada M5S 3H6. E-mail: dsong@chem.utoronto.ca; Fax: +1 416 978 7013; Tel: +1 416 978 7014
First published on 1st December 2015
Abstract
This perspective review will examine the coordination chemistry and applications of metal complexes of 4,5-diazafluorene derivatives. The versatile derivatives of 4,5-diazafluorene can serve multiple roles, and display a number of coordination modes. The ambidentate derivatives with multiple coordination sites can allow for the syntheses of coordination polymers, multimetallic, and macrocyclic complexes. In addition, certain 4,5-diazafluorene derivatives can serve as spectator ligands to support reactivity at the metal centre, or as reactive actor ligands engaging in atypical reactivity patterns. The applications of metal complexes of 4,5-diazafluorene derivatives in catalysis, photochemistry and photophysics, as well as in bioinorganic chemistry are also surveyed.
 Vincent Annibale | Vincent Annibale received his Hon. B.Sc. degree in 2008 from the University of Toronto at Mississauga under the supervision of Prof. Ulrich Fekl, and his Ph.D. degree from the University of Toronto in 2014 under the tutelage of Prof. Datong Song. He is currently an NSERC postdoctoral fellow in the group of Prof. Michael Fryzuk at the University of British Columbia. He is a recipient of several awards including NSERC postgraduate and postdoctoral scholarships, and Ontario Graduate scholarships among others. His current research interests include multidentate ligand designs for cooperative small molecule activation and catalysis. |
 Datong Song | Datong Song received his B.Sc. degree at Nankai University in 2000 and Ph.D. degree at Queen's University in 2003. After a one-year postdoctoral research at the University of Toronto, he did a postdoctoral research at MIT supported by an NSERC Postdoctoral Fellowship. In 2006, he started his independent career at the University of Toronto, where he is now an Associate Professor of Chemistry. His broad research interests include ligand design, synthetic inorganic and organometallic chemistry, small molecule activations, luminescent materials, and catalysis. |
I. Introduction
4,5-Diazafluorene (dafH) was first reported in the late 1970s,1,2 and synthesized in two steps from 1,10-phenanthroline (phen). The first step is an oxidative ring contraction of phen with permanganate in basic aqueous media giving 4,5-diazafluoren-9-one (dafo),3 and the second step is a Wolff–Kishner reduction of dafo with hydrazine monohydrate at high temperature (Scheme 1).4,5 The contraction of the middle ring of phen increases the distance between the two N-donors, i.e., 2.72 Å for phen6 and 3.05 Å for dafo7 and dafH.8 In coordination chemistry, the dafH ligand has been conventionally viewed as a 2,2′-bipyridine (bpy) derivative with a methylene linker tethering the two pyridine rings together (Fig. 1). The methylene group effectively ties back the two pyridine rings, resulting in a longer N–N distance in dafH (2.82(3) Å) compared to bpy (2.63(4) Å) in complexes. Consequentially, the overlap between the nitrogen lone pairs and the metal orbitals is less effective for dafH compared to bpy (Fig. 1).9–13 The dafo ligand has an even longer N–N distance (2.96(7) Å) since C9 is sp2 hybridized in dafo.14 Although dafH derivatives were considered merely as bpy analogues in the late 1970s, they have recently gained more attention in many areas. Ono15 and Wong16 previously reviewed the synthesis and coordination chemistry of several dafH derivatives. The purpose of this perspective review is to highlight the versatile nature of dafH derivatives as ligands, and also to detail some of the recent advances made using this ligand family.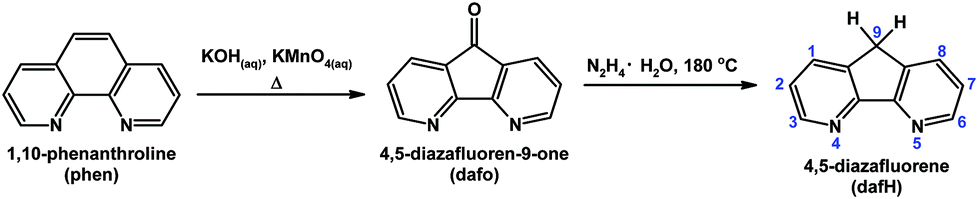 | ||
| Scheme 1 Synthesis of dafH from 1,10-phenanthroline with numbering scheme of dafH ligand is shown.4,5 | ||
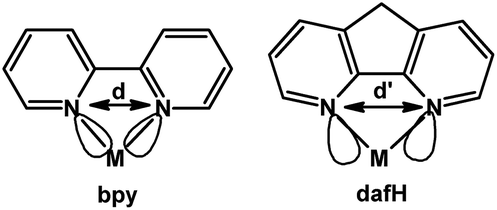 | ||
| Fig. 1 Comparison between bpy and dafH.17 | ||
II. Fundamental coordination chemistry of dafH derivatives
II.1. Coordination chemistry of the parent dafH and daf− ligands
Although being conventionally viewed as a bpy derivative, the dafH ligand can also be viewed as two pyridine rings fused onto a central cyclopentadiene (CpH) ring in a syn fashion. The methylene group of dafH is acidic, analogously the pKa of CpH is 18, and that of fluorene is 22.6 in DMSO.18 The methylene linker of dafH may be deprotonated to form the monoanionic 4,5-diazafluorenide (daf−) (Scheme 2A).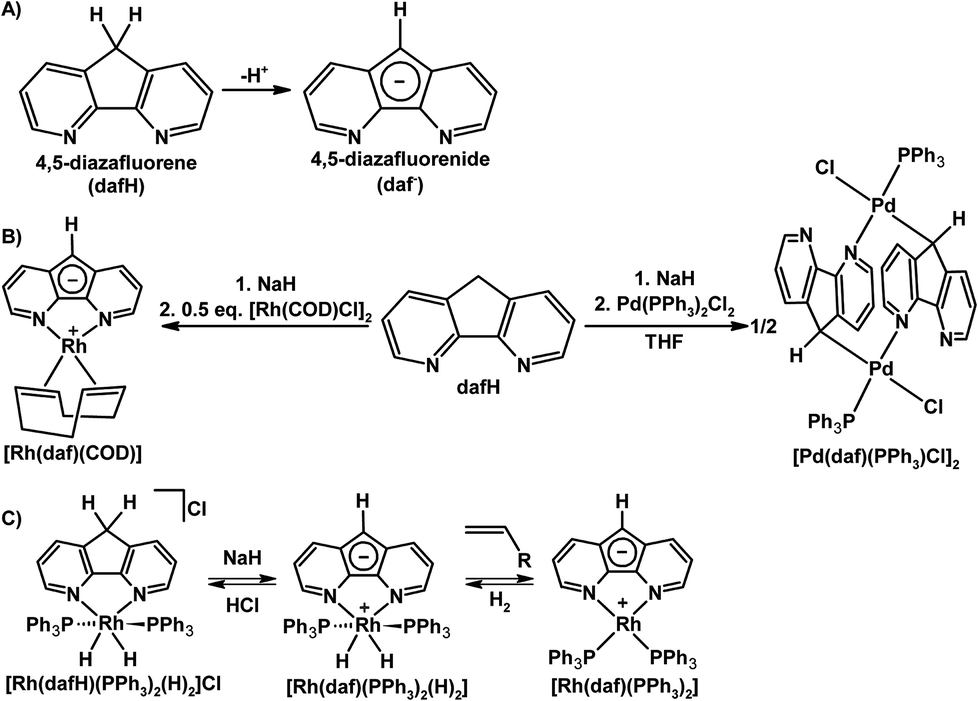 | ||
| Scheme 2 (A) Deprotonation of dafH to give daf−.19 (B) Synthesis of [Rh(daf)(COD)] and [Pd(daf)(PPh3)Cl]2.19 (C) Interconversion between Rh 4,5-diazafluorene complexes.20 | ||
Ambidentate ligands containing multiple potential coordination sites of different nature can be used to construct linkage isomers, homo- and heteromultimetallic complexes, and complex molecular architectures through coordination-driven self-assembly. Several 4,5-diazafluorene derivatives possess multiple coordination sites, especially those derivatives that are functionalized at the C9-position discussed later. The daf− ligand is potentially an ambidentate ligand with two types of metal binding sites, an N,N-chelate site, and the C-donors of the central cyclopentadienyl-like moiety. However in most examples daf− binds metals with its nitrogen donors,19–24 without utilizing the C-donors. In addition the daf− ligand is able to form zwitterionic complexes when only two N-donors are bound to the metal centre with a −1 charge localized onto the ligand backbone. A recurring theme in this review is the reactivity of the ligand backbone in a zwitterionic setup.
Our group's initial investigations into the coordination chemistry of the 4,5-diazafluorenide (daf−) ligand were reported in 2008.19 The reaction of Na[daf] with [Pd(PPh3)2Cl2] gave a dinuclear Pd complex [Pd(daf)(PPh3)Cl]2, in this exceptional case the daf− ligand was bound to Pd through one of the N-donors and the anionic C-donor of the ligand backbone in an η1(σ)-fashion (Scheme 2B).19 A zwitterionic Rh(I) complex [Rh(daf)(COD)] can also be straightforwardly prepared by reacting Na[daf] with 0.5 equiv. of [Rh(COD)Cl]2 (Scheme 2B).
An extension of the work with Rh 4,5-diazafluorene derivatives involved the syntheses of complexes with PPh3 as an auxiliary ligand (see Scheme 2C).20 The Rh(I) complex [Rh(daf)(PPh3)2] can be synthesized from the reaction of Na[daf] and Wilkinson's catalyst. Complex [Rh(daf)(PPh3)2] reacts with H2 to give a Rh(III) dihydride complex [Rh(daf)(PPh3)2(H)2], which reacts with HCl to give [Rh(dafH)(PPh3)2(H)2]Cl (Scheme 2C). Alternatively, [Rh(dafH)(PPh3)2(H)2]Cl can also be synthesized by reacting dafH with Wilkinson's catalyst under a H2 atmosphere, and subsequent deprotonation with NaH gives [Rh(daf)(PPh3)2(H)2] cleanly.
Parts of Scheme 3 highlight the ambidentate nature of daf− in its coordination chemistry with a {RuCp*}+ fragment.25 The addition of neutral dafH to 0.25 equiv. of [RuCp*(μ3-Cl)]4 starting material gave [RuCp*(dafH)(Cl)] where the dafH ligand coordinates through the nitrogen chelate (Scheme 3).25 However the addition of Na[daf] to 0.25 equiv. of [RuCp*(μ3-Cl)]4 gave a monomeric sandwich complex [RuCp*(daf)] where the daf− ligand coordinates through the central cyclopentadienyl-like ring in an η5-fashion leaving the nitrogen chelate vacant (Scheme 3).25 Surprisingly when the [RuCp*(μ3-Cl)]4 starting material is treated with K[daf], or if the coordinated 4,5-diazafluorene ligand of [RuCp*Cl(dafH)] is deprotonated with K[HBEt3] the self-assembly of a tetraruthenamacrocycle [RuCp*(daf)]4 occurred where the daf− ligand coordinated through both the N-donors and the C-donor of the ligand backbone in an η1(σ)-fashion (Scheme 3).25 The different coordination behaviours of daf− in [RuCp*(daf)]n (n = 1 or 4), a monomeric sandwich complex and a tetraruthenamacrocyle, demonstrate its ambidentate nature.
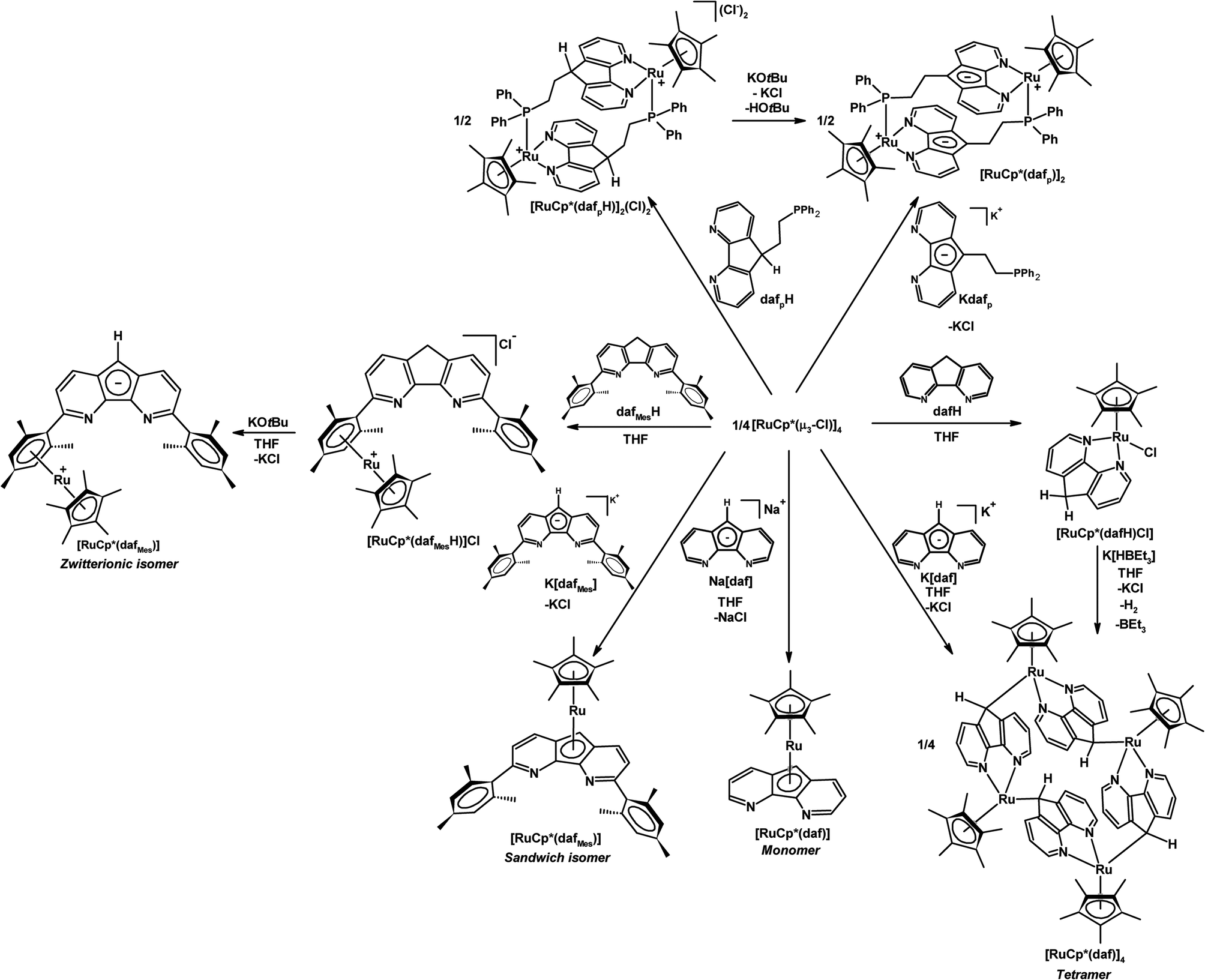 | ||
| Scheme 3 Coordination chemistry of ambidentate 4,5-diazafluorene derivatives with a RuCp* synthon.25 | ||
We have used 4,5-diazafluorenide derivatives to synthesize heterobimetallic complexes.26,27 When the salt [Na(dme)3][Pt(daf)(Ph)2] is treated with [Cu(IPr)Cl], a Pt(II)–Cu(I) heterobimetallic complex [(IPr)Cu(daf)Pt(Ph)2] was obtained where C9 of daf− is coordinated to the {Cu(IPr)}+ fragment in an η1(π) fashion (Scheme 4).26 Surprisingly when the metallation sequence is reversed and the [Cu(daf)(IPr)] complex is treated with [Pt(Ph)2(μ-SMe2]n (n = 2 or 3), the exact same Pt(II)–Cu(I) heterobimetallic complex (as opposed to an isomer) was obtained.26 In the process of generating [(IPr)Cu(daf)Pt(Ph)2] from [Cu(daf)(IPr)] the {Cu(IPr)}+ fragment was replaced by the {Pt(Ph)2} unit at the N,N-chelate site and migrated to the backbone C-donor. The built-in self-correction featured in the synthesis of the [(IPr)Cu(daf)Pt(Ph)2] system allowed for a one pot synthesis to be successfully carried out (Scheme 4).
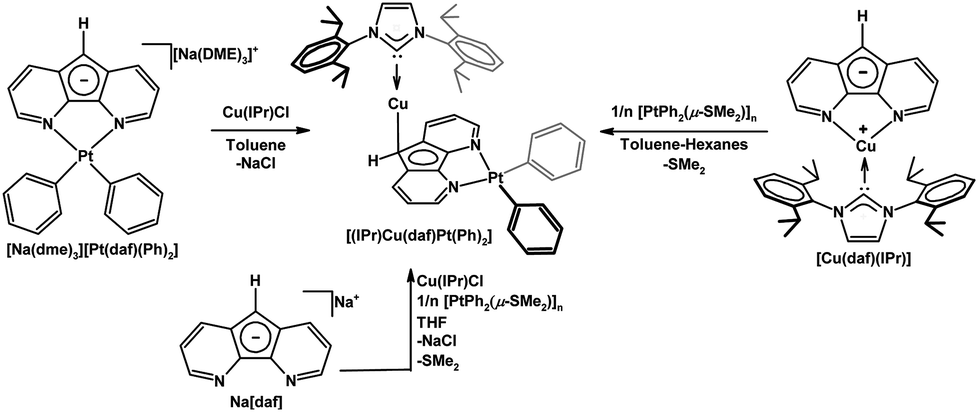 | ||
| Scheme 4 Three synthetic routes to [(IPr)Cu(daf)Pt(Ph)2].26 | ||
II.2. Coordination chemistry of the aryl substituted dafMesH and dafMes− ligands
A 3,6-dimesityl substituted 4,5-diazafluorene ligand (dafMesH) has been synthesized, which can also be deprotonated to give dafMes−.25 These bulkier 4,5-diazafluorene derivatives are also ambidentate. The dafMes− ligand has an N,N-chelate site and the C-donors of the central cyclopentadienyl moiety; in addition, there is also the possibility of the mesityl substituent to participate in bonding. The coordination chemistry of neutral dafMesH and monoanionic dafMes− ligands toward the {RuCp*}+ fragment was examined (Scheme 3). When K[dafMes] was added to 0.25 equiv. of [RuCp*(μ3-Cl)]4, a sandwich complex [RuCp*(dafMes)] formed where the nitrogen chelate is vacant and the dafMes− ligand is coordinated through the central C5-ring in an η5-fashion (Scheme 3).25 However upon adding the neutral dafMesH ligand to 0.25 equiv. [RuCp*(μ3-Cl)]4, [RuCp*(dafMesH)]Cl was obtained. Since the mesityl groups prevent the N-donors from coordinating to the {RuCp*}+ fragment, the {RuCp*}+ fragment coordinates to the arene (Scheme 3).25 After deprotonation of [RuCp*(dafMesH)]Cl with KOtBu, a zwitterionic linkage isomer of [RuCp*(dafMes)] formed, where the Ru(II) remains coordinated to the arene (Scheme 3).25 Interestingly two different linkage isomers of [RuCp*(dafMes)], a sandwich complex and a zwitterionic complex, were formed depending on the reaction sequence.A variety of heterobimetallic complexes can also be synthesized where the central cyclopentadienyl ring of 4,5-diazafluorenide is coordinated to one metal centre and the nitrogen chelate is coordinated to a second metal centre, and the ligand displays either η5,κ2-[N,N] or η5,κ1-N coordination modes.27 For example, [PtPh2(daf)]− and [PtPh2(dafMes)]− can be metallated successfully with the [RuCp*(μ3-Cl)]4 to obtain Ru(II)–Pt(II) heterobimetallic complexes [RuCp*(daf)Pt(Ph)2] and [RuCp*(dafMes)Pt(Ph)2] (Scheme 5). These Ru(II)–Pt(II) heterobimetallic complexes can also be synthesized from the Ru sandwich complexes, [RuCp*(daf)] and [RuCp*(dafMes)] (Scheme 5).27 The [RuCp*(dafMes)] sandwich isomer with a vacant N,N-chelate can also serve as a metalloligand for the complexation with metal halides such as CuCl, FeCl2(THF)1.5 and CoCl2(THF)1.5 (Scheme 5).27
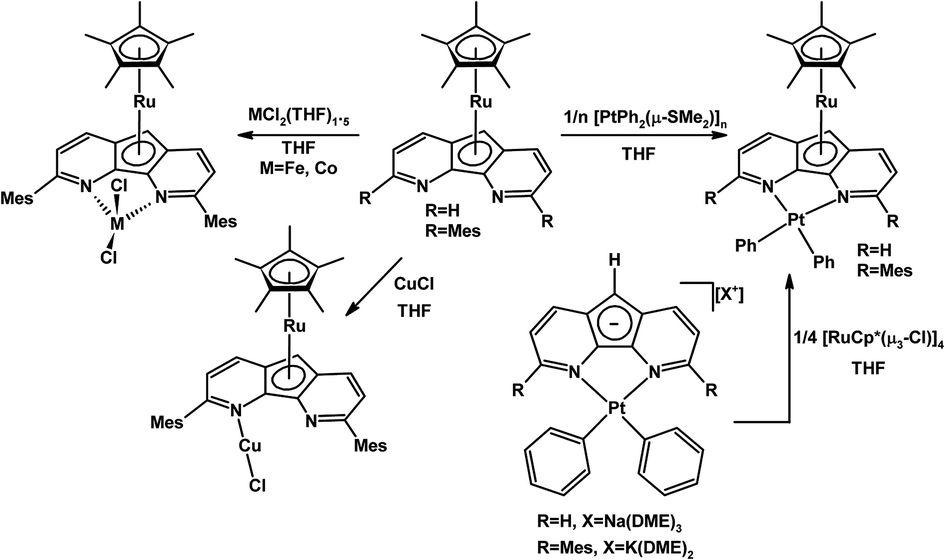 | ||
| Scheme 5 Synthesis of heterodinuclear Ru(II)–M complexes where the daf− or dafMes− ligand displayed η5,κ2-[N,N] or η5,κ1-N coordination modes.27 | ||
II.3. Coordination chemistry of the phosphine donor functionalized dafpH and dafp− ligands
Our group also installed a phosphine arm at the C9-position of 4,5-diazafluorene to give the dafpH ligand, which can also be deprotonated to form the dafp− ligand. Both dafpH and dafp− have been used to assemble head-to-tail macrocycles with {RuCp*}+ (Scheme 3).25 We further demonstrated the transfer of dafp− from [Cu(IPr)(dafp)]n (n = 1 or 2) to either Rh(I) or Au(I) resulted in macrocyclic complexes (Scheme 6).28 There are a few benefits of constructing these macrocyclic Rh(I) and Au(I) complexes through ligand transfer from a {Cu(IPr)}+ fragment, compared to the conventional synthesis by directly reacting the dafp− salt with metal chloride starting materials. The benefits include improved yields, shortened reaction time, and simplified isolation of the product as the soluble [Cu(IPr)Cl] byproduct can easily be removed by filtration. In addition, dafpH and dafp− are also ambidentate ligands with phosphine and N-donor coordination sites, while dafp− can also anchor a metal in the P,C-coordination site. The dafp− ligand displayed several coordination modes, where the diazafluorenyl moiety coordinates through one or both N-donors and the P-donor with or without the participation of the C-donor of the cyclopentadienyl-like moiety.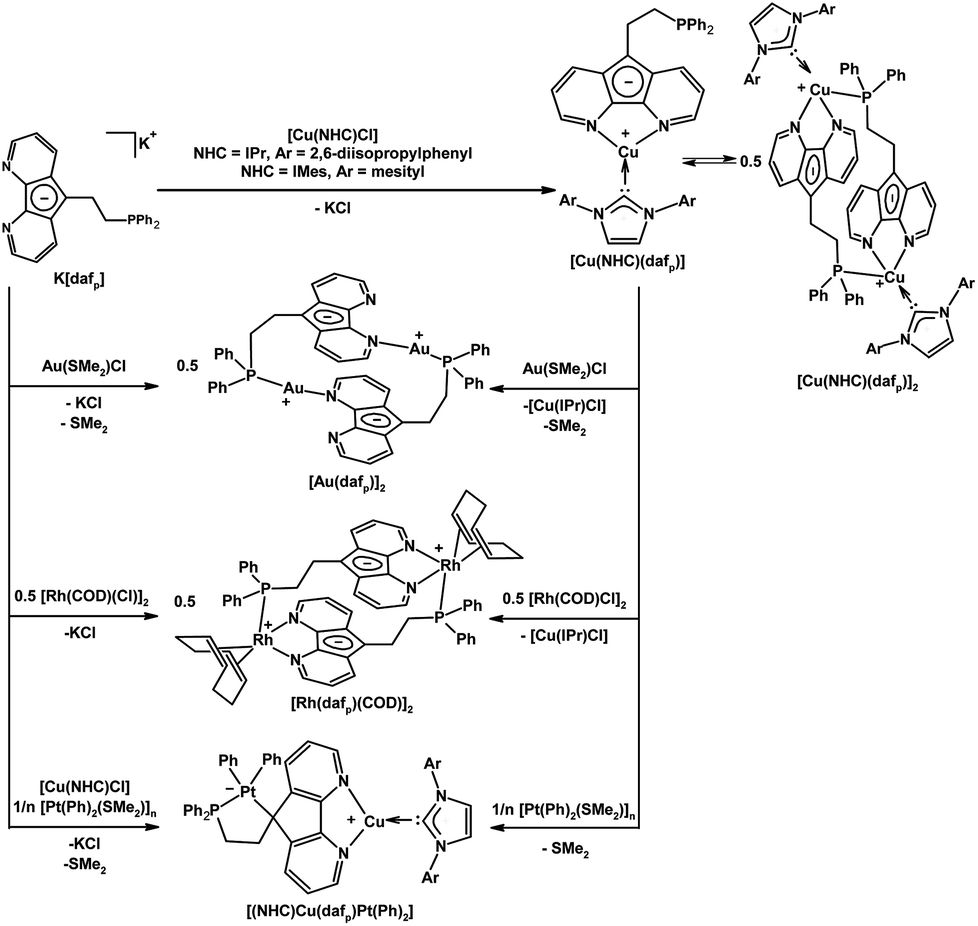 | ||
| Scheme 6 Synthesis of [Cu(NHC)(dafp)] from K[dafp], and the synthesis of macrocyclic complexes [Au(dafp)]2, [Rh(dafp)(COD)]2 through salt metathesis, and dafp− ligand transfer from a Cu(NHC) complex.28 In addition the selective syntheses of Pt(II)–Cu(I) heterobimetallic complexes is shown.26 | ||
We also have demonstrated that in the heterodinuclear complex [(NHC)Cu(dafp)Pt(Ph)2] the tethered phosphine of dafp− helps anchor the Pt(II) center onto the carbon site, and the Cu(I) center is bound to the N,N-chelate site (Scheme 6).26 The synthesis of these PtII–CuI heterobimetallics is highly regioselective, and can also be performed in one-pot (Scheme 6).26
II.4. Coordination chemistry of sulfur donor functionalized 4,5-diazafluorene derivatives
Baudron, Hosseini, and co-workers have created ambidentate ligands with sulfur donors and demonstrated their use in the stepwise synthesis of multimetallic complexes and coordination polymers.29–32 Both 4,5-diazafluorenyl-9-dithiolene (L1) ligands30,32 and 4,5-diazafluorenyl-9-dithiolate (L2)29,31,32 are generated in situ by the removal of propionitrile S-protecting groups using NR4OH (Scheme 7). Both ambidentate dianionic ligands provide different chelates, a dithiolate or dithiolene chelate and the 4,5-diazafluorene moiety. The homoleptic mononuclear complexes of the generic form [M(L1)2](NnBu4)2 (where M = Ni(II) or Hg(II)),30 and [M(L2)2](NEt4)2 (M = Ni(II), Pd(II), Zn(II), or Hg(II))31 can be synthesized (Scheme 7). The homoleptic complexes all have a central metal which is coordinated by the dithiolate or dithiolene moiety of the ligands, leaving the 4,5-diazafluorene chelate sites vacant. These mononuclear complexes form one-dimensional coordination polymers in the presence of Na+ ions (Scheme 7).30,31 The heterotrimetallic complex [(cyclenNi)2Pd(L2)2](BF4)2 can be prepared where the {(cyclen)Ni}2+ fragment is coordinated to the 4,5-diazafluorenyl moiety via the N,N-chelate (Scheme 7).31 The 4,5-diazafluorenyl-9-dithiolate ligand can serve as a bridging ligand through the two anionic S-donors to assemble an octanuclear Cu complex [Cu8(L2)6]4− where the eight Cu centres occupy the vertices of a cube and on the periphery of the complex are the potentially chelating 4,5-diazafluorenyl moieties (Fig. 2).29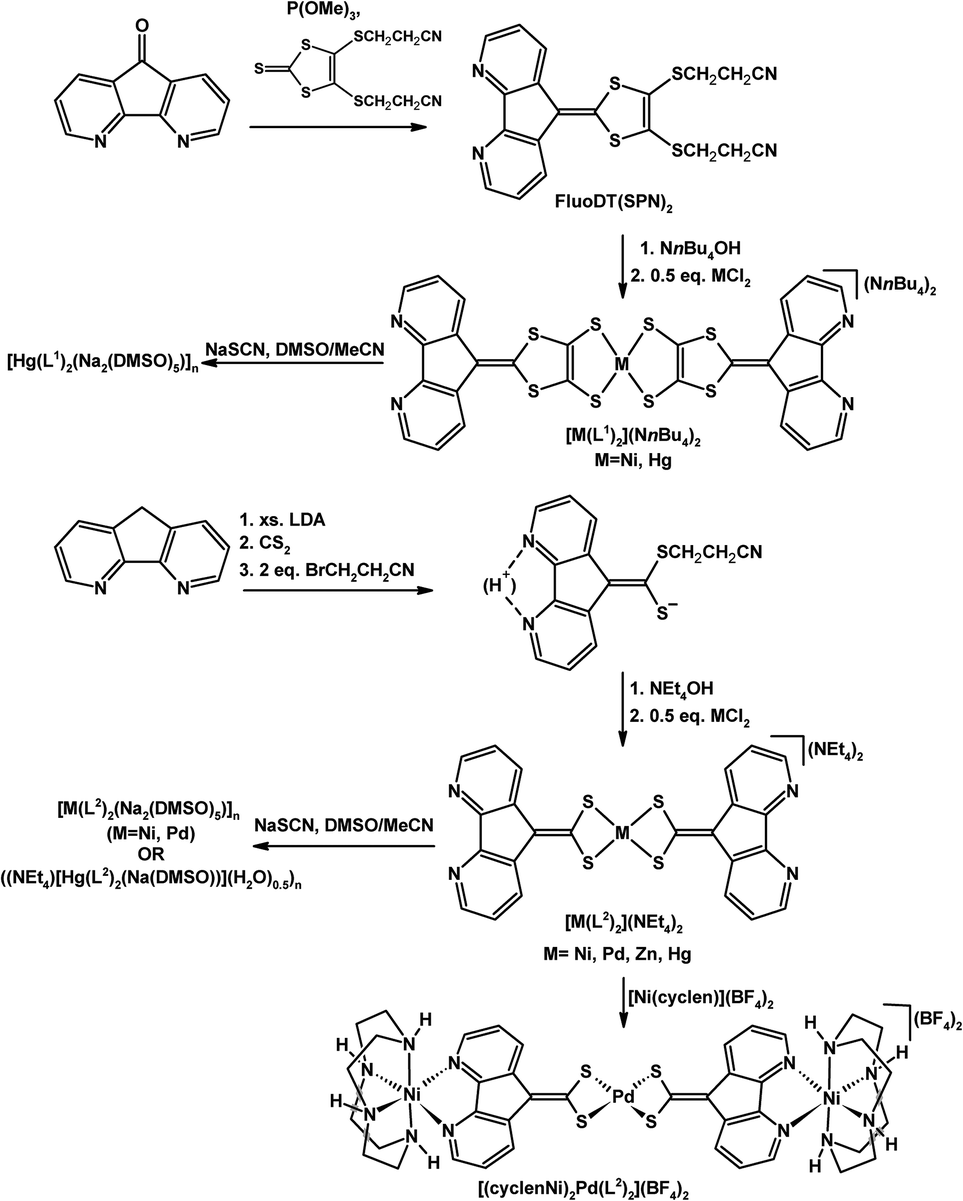 | ||
| Scheme 7 Synthesis of homoleptic complexes [M(L1)2](NnBu4)2 and [M(L2)2](NEt4)2 and subsequent synthesis of a heterotrimetallic complex [(cyclenNi)2Pd(L2)2](BF4)2, and Na+ containing coordination polymers.30,31 | ||
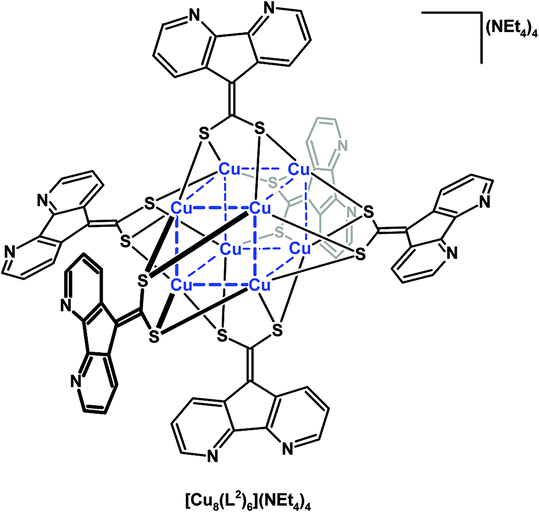 | ||
| Fig. 2 Octanuclear Cu cluster, [Cu8(L2)6](NEt4)4, composed of (L2) 4,5-diazafluorenyl-9-dithiolate ligands.29 | ||
Heteronuclear metallamacrocyles can be constructed from L1 and L2 ligands (Scheme 8).32 The dianionic ligands are first metallated with (dppp)M1Cl2 (where M1 = Pd, Pt, and dppp = 1,2-bis(diphenylphosphino)propane) which bears a diphosphine capping ligand allowing for a discrete neutral species of the general form [(dppp)M1(L1)] or [(dppp)M1(L2)] to be isolated, where both L1 and L2 ligands coordinate to M1 through the S,S-chelate, leaving the 4,5-diazafluorenyl moiety available for a second metal (Scheme 8).32 The addition of a second group 10 metal halide M2X leads to the formation of head-to-tail metallamacrocycles where the formation of metal–sulfur linkages are involved in the assembly (Scheme 8).32
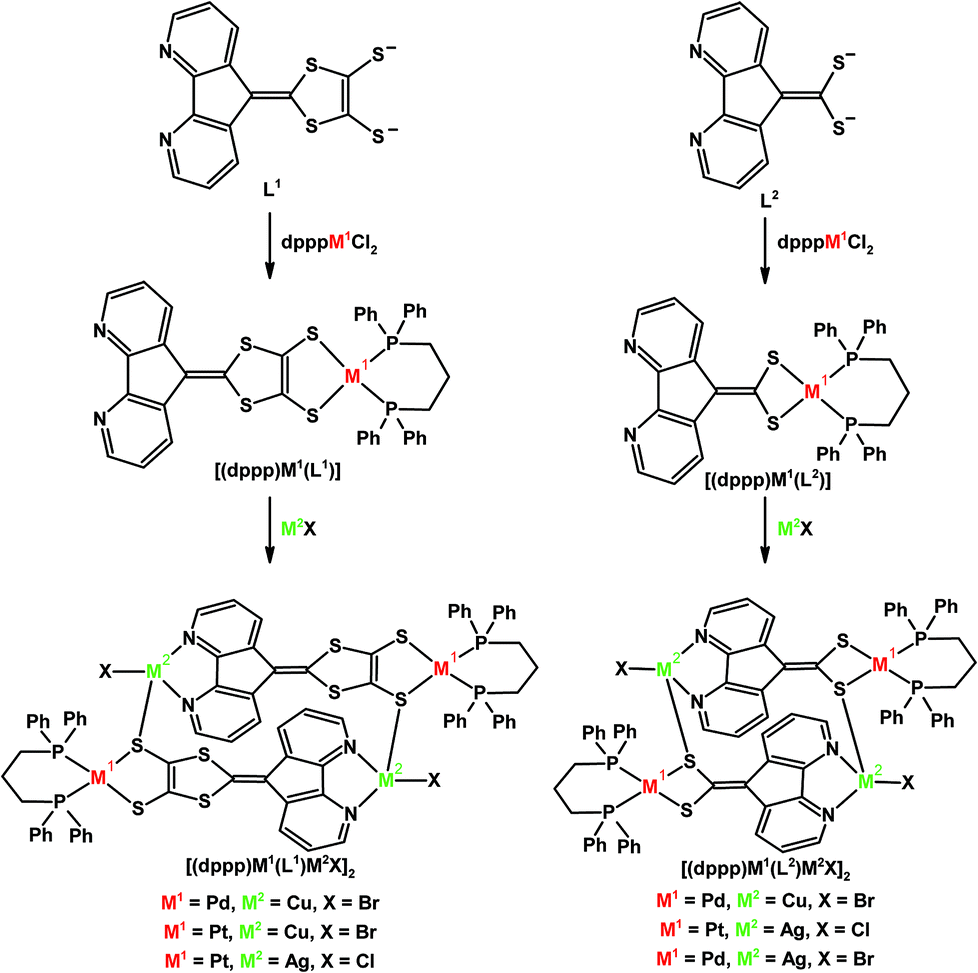 | ||
| Scheme 8 Heteronuclear metallamacrocycles constructed from 4,5-diazafluorenyl-9-dithiolene (L1), and 4,5-diazafluorenyl-9-dithiolate (L2).32 | ||
The heteroleptic Ir(III) complex [Ir(dfppy)2(FluoDT(SPN)2)]+ with the propionitrile-protected 4,5-diazafluorene-9-dithiolene proligand (where dfppy = 2-(2,4-difluorophenyl)pyridine and FluoDT(SPN)2 = 4,5-diaza-9-[4,5-bis(cyanoethylsulfanyl)-1,3-dithiol-2-ylidene]fluorene, see Scheme 7 for the structure of FluoDT(SPN)2) has been used in a cascade reaction in basic media as both the reducing agent for chloroauric acid and the capping agent for the resulting gold nanoparticles.33 In basic media the protecting groups of [Ir(dfppy)2(FluoDT(SPN)2)]+ are cleaved resulting in the 4,5-diazafluorene-9-dithiolene (L1) ligand which is redox-active and able to reduce Au3+ to Au0, and the anionic S-donors allow the Ir(III) capping agent to bind strongly to the surface of the Au0 nanoparticles.33 The coordination chemistry of the bis-thioether analogue 4,5-diaza-9-[4,5-bis(methylthio)-1,3-dithiol-2-ylidene]fluorene (L3) with transition metals was explored.34–40 The solid state structure, the IR spectrum and the solution UV-vis spectrum, and the magnetic properties have been reported for [M(L3)(tpa)](SbF6)2 (where tpa = tris(2-pyridylmethyl)amine). The L3 ligand is labile and readily displaced in coordinating solvents such as acetone, or MeCN.40
II.5. Coordination chemistry of 9-hydroxy-9-alkynyl functionalized dafH derivatives
The coordination chemistry of 9-hydroxy-9-alkynyl-4,5-diazafluorene derivatives which possess several potential sites of coordination has been explored.7,41,42 The 9-hydroxy-9-ethynyl-4,5-diazafluorene ligand can be dehydrated upon binding with a 16-electron coordinatively unsaturated species such as in situ generated [M(diphosphine)2Cl]+ (M = Ru, Os)41 and [Ru(dppe)2Cl](OTf) precursor42 (Scheme 9). The resulting Ru and Os allenylidene complexes feature a vacant N,N-chelate which is a potential site for further coordination. Indeed this N,N-chelate moiety can be coordinated to a second metal centre to form Ru(II)–Ru(II), and Ru(II)–Re(I) multimetallic complexes.42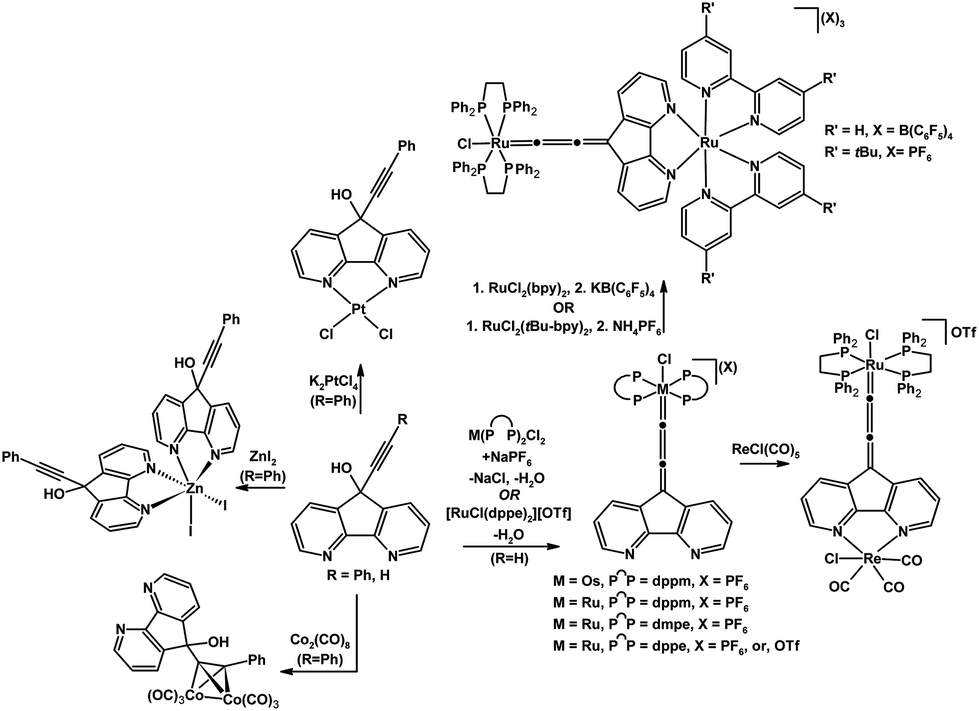 | ||
| Scheme 9 Reactions of 9-hydroxy-9-alkynyl-4,5-diazafluorene.7,41,42 | ||
When the 9-hydroxy-9-phenylethynyl-4,5-diazafluorene ligand is reacted with K2PtCl4 or ZnI2, the corresponding mononuclear complex forms with the metal bound to the N,N-chelate (Scheme 9).7 However the reaction of 9-hydroxy-9-phenylethynyl-4,5-diazafluorene with Co2(CO)8 led to a (μ-alkyne)hexacarbonyldicobalt complex where the N,N-chelate remains unoccupied (Scheme 9).7
II.6. Coordination chemistry of the dafo ligand
Dafo has been used as a ligand for metals across the periodic table: Mn,43 Co,44,45 Ni,46–48 Cu,44,48–62 Zn,7 Mo,63 Ru,9,64–73 Pd,17,44,74–87 Ag,88–90 Cd,62,90,91 Re,92 Ir,93 Pt,7,17,81 Hg,48 and the lanthanides.50,94–96 In the vast majority of cases dafo behaves as a bidentate chelate ligand, however there are a few examples where dafo adopts either a monodentate κ1-N coordination mode,7,17,50,54,56,88,89 a bis-monodentate bridging coordination mode,17,59,60,79,81,97 or even an η4-cyclopentadienone-like coordination mode in the [Cp*Co(η4-dafo)] complex.45II.7. Coordination polymers containing dafH derivatives
Recently metal–organic frameworks (MOFs) have received substantial attention due to the wide variety of potential architectures arising from different metal–ligand combinations, and the possibility of creating materials with intriguing applications. A variety of MOFs with different architectures were constructed from Zn(II) ions, a variety of aromatic polycarboxylic acid ligands and a 4,5-diazafluoren-9-oxime ligand.98 The polynuclear Zn secondary building units (SBUs) comprising the various MOFs were modulated by the 4,5-diazafluoren-9-oxime ligand, which can coordinate to Zn either in a chelating or monodentate fashion.98Azide containing MOFs can potentially be used as molecule-based magnets. A three-fold interpenetrating MOF of the general formula [(Mn–μ1,3-N3–μ1,1-N3)3(L4)] where L4 is the bis(bidentate) Schiff base ligand 4,5-diazafluoren-9-one azine exhibited spin-canted long-range ferromagnetic ordering.99 The L4 ligand served as the long link in the 3D structure while the chain of Mn–μ1,3-N3–μ1,1-N3 served as the SBU.99 A series of isostructural tetranuclear clusters [M4(dafo)4(N3)2(μ1,1-N3)4(μ1,1,1-N3)2] have been synthesized with azido ligands bridging the four M2+ ions, and the dafo coordinated in a chelating fashion to the metal vertices of the clusters, where the metal can be Co,43,100 Mn,43 Cd,91 or Cu.49
A luminescent Ag(I) one-dimensional polymer chain of the generic formula [Ag2(L5)2(ClO4)2]n where L5 = 4,5-diaza-9,9′-spirobifluorene was structurally characterized.101 Both of the L5 ligands adopt a bis-monodentate bridging mode between the two crystallographically independent Ag(I) centres where the Ag–Ag distance is 2.776(1) Å, and one of ClO4− ligands also bridges two adjacent Ag centres to construct the polymer chain (Fig. 3).101
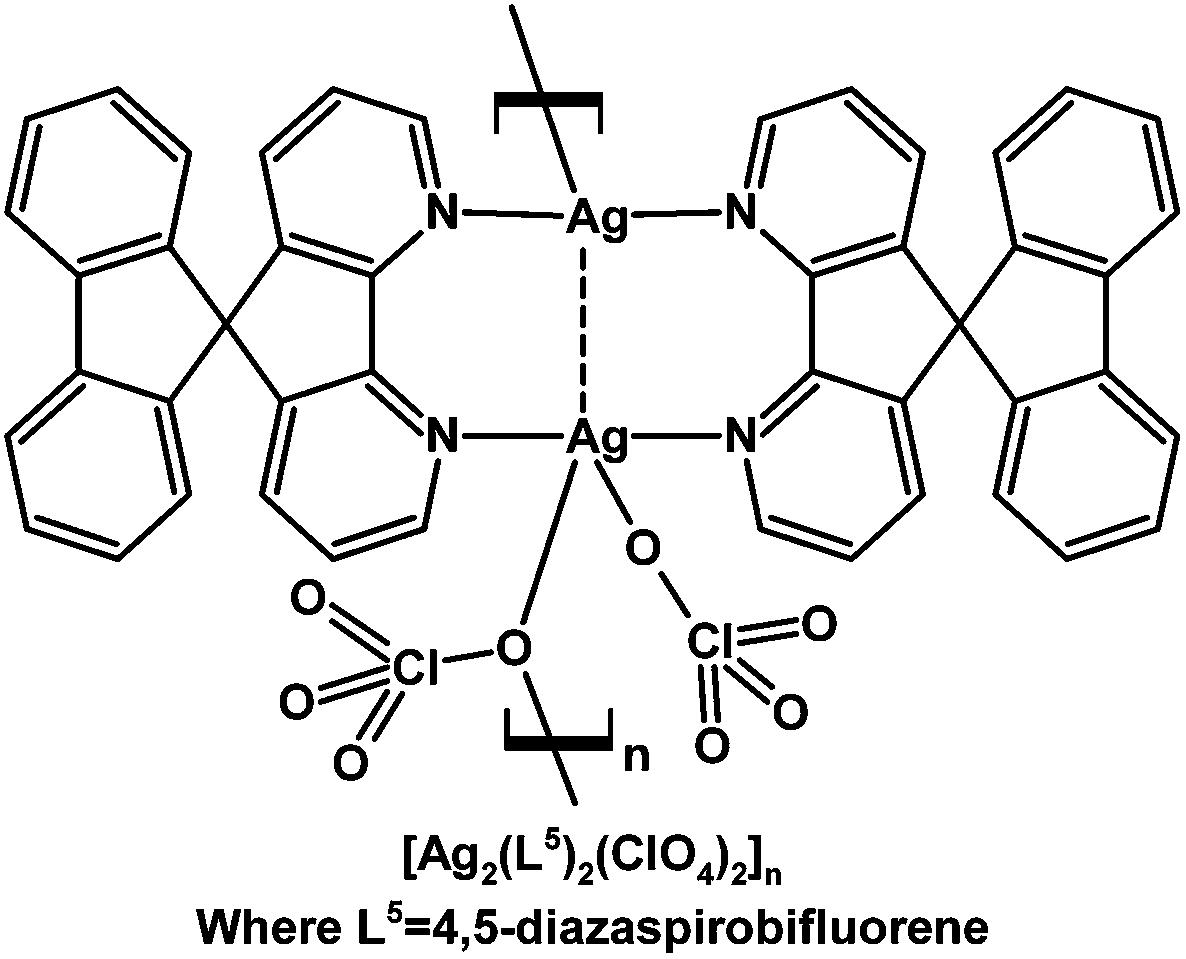 | ||
| Fig. 3 Coordination polymer constructed from Ag(I) and 4,5-diaza-9,9′-spirobifluorene (L5).101 | ||
The fullerene C60 with four peripheral malonate groups can be functionalized with two 4,5-diazafluorene moieties in the trans-1 positions, since 4,5-diazafluorene is planar the N,N-chelates can be situated 180° relative to each other.102 The reaction between AgOTf and this large ditopic bis(4,5-diazafluorene)tetrakis(malonate) substituted fullerene ligand (L6) gives a 1-D coordination polymer [Ag2(L6)(OTf)2]n where each Ag(I) centre is four-coordinate and is bound to the N,N-chelate of one of the 4,5-diazafluorene moieties, an O-donor from the −OTf ligand, and a C-donor from a neighboring C60 cage in an η1-fashion (Fig. 4).102 In the solid state of this fullerene-based coordination polymer the two antiparallel 4,5-diazafluorene moieties from two neighboring L6 ligands engage in face-to-face π–π interactions.102 The choice of Ag(I) precursor is crucial for coordination polymer formation. For example, if AgBF4 is used instead, a non-polymeric salt [((L6)(Ag(toluene)2)](BF4)2·CH2Cl2 forms, where each Ag(I) centre is coordinated to the N,N-chelate of the 4,5-diazafluorene moieties and is bound to one η1-toluene and one η2-toluene.102
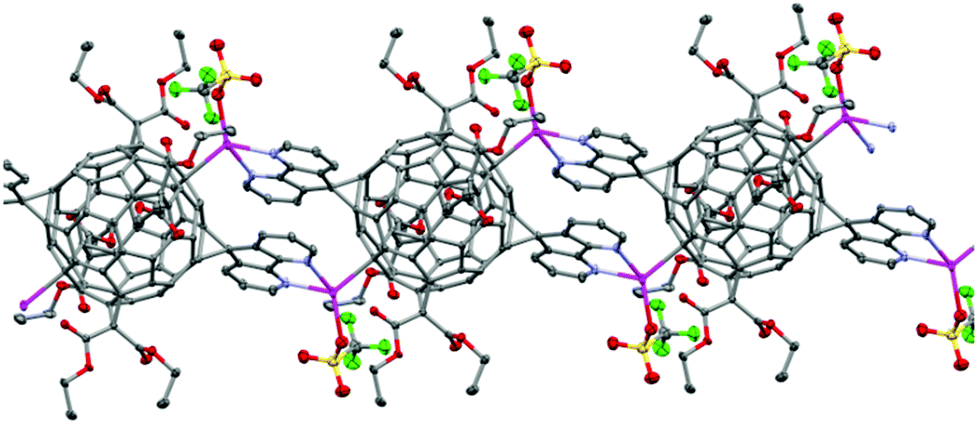 | ||
| Fig. 4 A portion of the solid-state structure of the one-dimensional coordination polymer [((L6)(AgOTf)2)·5(toluene)]n where L6 is a bis(4,5-diazafluorene)tetrakis(malonate) substituted fullerene, toluene solvent molecules and H-atoms omitted for clarity. Ag: pink, S: yellow, F: green, O: red, N: blue, C: grey.102 | ||
A simple coordination polymer of [Cd(dafo)(NCS)2]n can be prepared where each Cd centre adopts a distorted octahedral geometry and the ambidentate NCS− ligands bridge adjacent Cd centres through both the N and S termini.103
Jung, Lee, and co-workers demonstrated the use of a heteroditopic bis(4,5-diazafluorenylimino)dibenzo[18]crown-6 ether based ligand which could be used to synthesize a coordination polymer gel in the presence of Zn2+ and Cs+ ions.104 The Zn2+ ion is bound by two 4,5-diazafluorenylimino moieties with a tetrahedral coordination geometry and one Cs+ ion is sandwiched between two crown ether rings to give a highly cross-linked coordination polymer gel, where the rheological properties and microstructure are strongly dependent on the presence and concentration of Cs+ ions.104
There are several examples in the literature regarding inorganic–organic hybrid polyoxometallates which contain the Keggin-type cluster anions and metal–dafo complex cations (typically Cu, and in a few examples Ag or Cd).50,51,57,59,60,90,97,105 The metal–dafo cation within these hybrid compounds seems to direct the assembly of other supramolecular interactions within the solid state. The coordination geometry of the metal, how the Keggin cluster is coordinated to the cation, and even the number of metals can play a role. For example helical assemblies can form when dinuclear [Cu2(dafo)2(H2O)]2+ cations are used as a hinge-like motif to link Keggin clusters together to form a three-dimensional framework.59,60
III. Applications of 4,5-diazafluorene derivatives
III.1. As actor ligands in reactivity chemistry
Most of inorganic and organometallic chemistry is dominated by metal centred reactivity where the ligand is a spectator, hence the term “spectator ligand”. There are emerging examples of ligand-centred, or metal–ligand cooperative reactivity where reactions with incoming substrates occur at the “actor ligands”. The use of multifunctional actor ligands in small molecule activation and catalysis has gained significant interest in the chemistry community and there are few recent reviews on this topic.106–113 There are several examples where 4,5-diazafluorene derivatives behave as actor ligands, where the majority of the ligand-based reactions occur at the reactive C9.Rillema and co-workers have uncovered divergent chemical behaviour for coordinated dafo ligand and free dafo in reactions with nucleophiles.70,71 Free dafo reacts with 2-aminopyridine in ethanol to give a Schiff-base product (Scheme 10A), however under the same reaction conditions the coordinated dafo ligand of [Ru(bpy)2(dafo)]2+ reacts with 2-aminopyridine to give a ring-opened product possessing a coordinated esterified 2,2′-bipyridine ligand (Scheme 10B).71 In another example no reaction occurs between free dafo and DBU in wet dichloromethane (Scheme 10A), while coordinated dafo in [Ru(bpy)2(dafo)]2+ reacts with DBU to give another ring-opened product (Scheme 10B).70 The spectator metal centre plays a major role in altering the reaction pathways of dafo with nucleophiles; the driving force to form a coordinated 2,2′-bipyridine ligand from a coordinated dafo is the release of coordination-induced ring strain and the formation of shorter, stronger Ru–N bonds.70,71 There are two potential nucleophilic sites in 2-aminopyridine, the amine and the pyridyl ring nitrogen atoms; the spectator metal ion increases the electron density on the carbonyl carbon atom of dafo thus hindering the reaction with the amine nitrogen and the formation of the Schiff-base product.71 Free dafo does not react with ethylene glycol, meanwhile coordinated dafo of [Ru(bpy)n(dafo)m]2+ (n = 1 and m = 2, or, n = 0, and m = 3) reacts to give the corresponding ketal ligand coordinated to Ru(II) which is resistant to hydrolysis with HCl(aq) – a highly unusual feature compared with most organic ketals.72 Siemeling and co-workers synthesized the sandwich complex [Cp*Co(η4-dafo)], where the dafo ligand is coordinated through the π system leaving the [N,N]-chelate vacant.45 The {Cp*Co} fragment coordinates η2 to each of the two six-membered dearomatized rings of dafo; this results in cyclopentadienone-like behaviour where the nucleophilicity of the oxygen atom is increased substantially versus free dafo.45 The nucleophilic, coordinated, cyclopentadienone-like dafo of [Cp*Co(η4-dafo)] reacts with electrophiles such as acetyl chloride to give the O-acylated cobaltocenium species (Scheme 10C); in contrast free dafo does not react with acetyl chloride (Scheme 10A).45
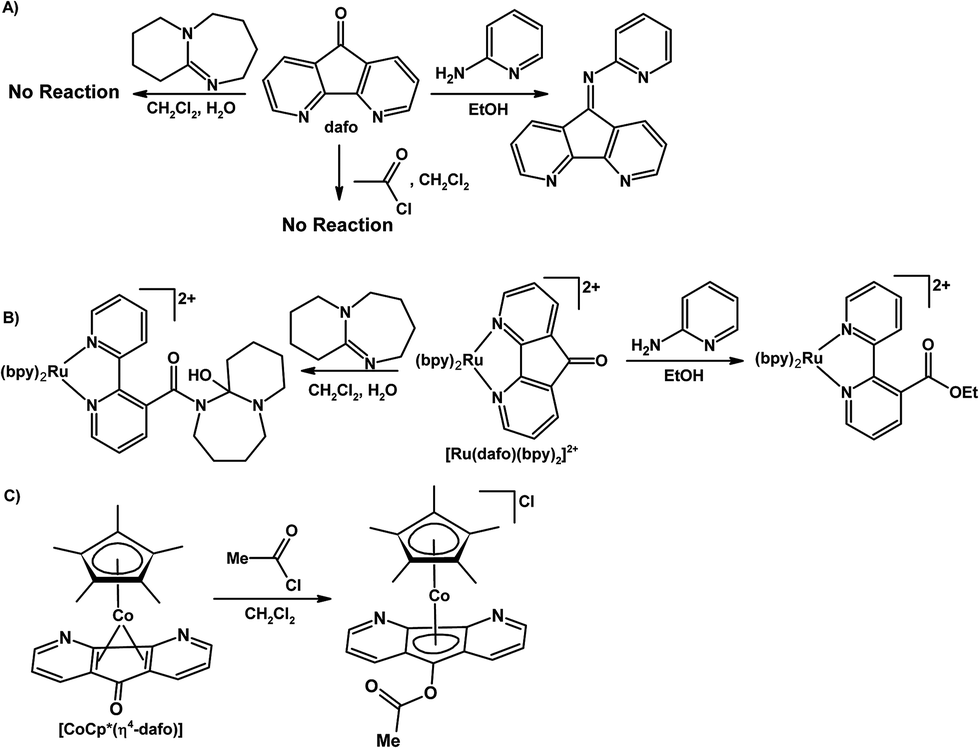 | ||
| Scheme 10 (A) Reactivity of uncoordinated free dafo with 2-aminopyridine, DBU, and acetyl chloride.70,71 (B) Reactivity of [Ru(dafo)(bpy)2]2+ with 2-aminopyridine, and DBU.70,71 (C) Reactivity of [CoCp*(η4-dafo)] with acetyl chloride.45 | ||
Our group discovered a surprising example of ligand-based reactivity: free 4,5-diazafluorene is air-stable but the coordinated 4,5-diazafluorene ligand in [Ru(dafH)(PPh3)2(Cl)2] can selectively undergo an aerobic oxidation reaction giving a coordinated dafo ligand (Scheme 11).66 The selectivity of this ligand-based oxidation reaction is surprising since the typically oxygen-sensitive phosphine ligands are left intact.
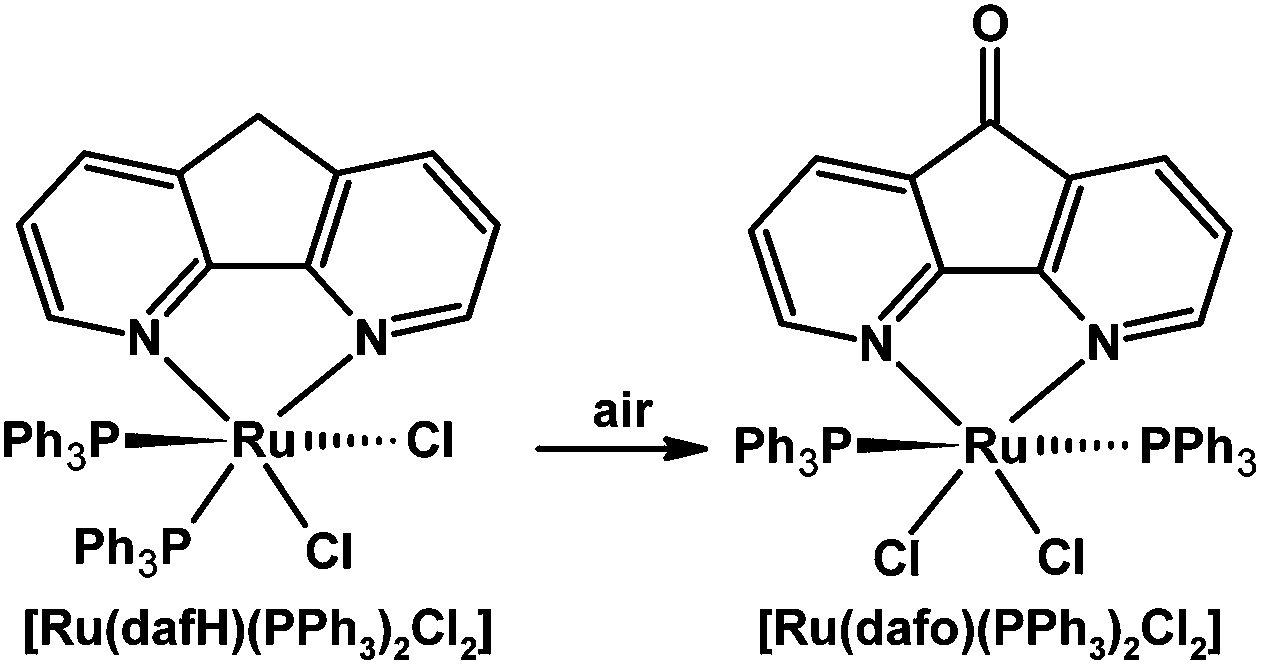 | ||
| Scheme 11 Selective oxidation of a coordinated dafH ligand.66 | ||
Our group also previously demonstrated an interesting example of metal–ligand cooperativity. The Ru(II) 4,5-diazafluorenide complex, [Ru(daf)(PPh3)2(H)(N2)], can be synthesized where the daf− ligand possesses a central negatively charged cyclopentadienyl like moiety that remains uncoordinated and thus has unquenched basicity (Scheme 12).21 [Ru(daf)(PPh3)2(H)(N2)] reversibly splits dihydrogen over a long-range, between the metal centre and the backbone carbanion, at a distance of ∼5 Å, to yield complex [Ru(dafH)(PPh3)2(H)2].21 The π-system of the diazafluorenide ligand is disrupted and restored during the forward and backward reactions, respectively. It is also worth noting that the reversible synthesis of a metal–dinitrogen complex via the metal-hydride route, a route which circumvents the need for harsh reducing agents, is quite unique.114
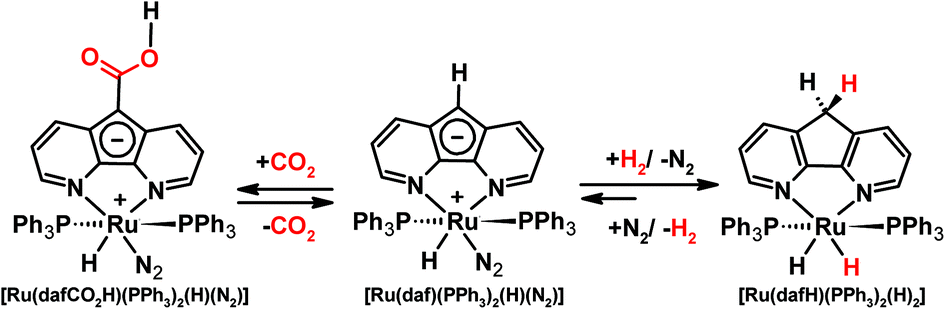 | ||
| Scheme 12 Reactions of [Ru(daf)(PPh3)2(H)(N2)] toward reversible H2 splitting,21 and a reversible formal insertion of CO2 into a daf− C–H bond.115 | ||
The formation of H2 from a coordinated 4,5-diazafluorene ligand giving a coordinated 4,5-diazafluorenyl ligand has been noted by other groups. Mach and co-workers observed the evolution of H2 when 4,5-diazafluorene was added to [Cp*2Ti(Me3SiCCSiMe3)] which gave the paramagnetic [Cp*2Ti(III)(daf)] adduct (Scheme 13A).23 Andersen and co-workers found that the stabilized biradical [Cp*2Yb(dafH)] adduct slowly eliminated H2 to give [Cp*2Yb(daf)]; in addition, the dinuclear Yb complex prepared with the 9,9′-bis-4,5-diaza-9H-fluorene ligand also thermally produced [Cp*2Yb(daf)] (Scheme 13B).24 The mechanism for dihydrogen formation is proposed on the basis of kinetic and labelling experiments to involve the dinuclear Yb complex as an intermediate.24 The oxidation states of Yb were described as being intermediate between +2 and +3 for both [Cp*2Yb(dafH)] and [Cp*2Yb(bpy)] with an equilibrium between the at least two low-lying open-shell singlet states.24 Andersen and co-workers also performed calculations on the 4,5-diazafluorene ligand in an attempt to understand how even though [Cp*2Yb(dafH)] and [Cp*2Yb(bpy)] have multiconfigurational ground states, they differ in reactivity where [Cp*2Yb(dafH)] eliminates H2, and [Cp*2Yb(bpy)] does not.24 The dafH ligand has unpaired spin density distributed in various pπ orbitals on the nitrogen and carbon atoms.24 The LUMO+1 of 2b1 symmetry has unpaired spin density at the 9-position carbon, this orbital is possibly responsible for the chemistry observed.24 The unpaired spin density at the 9-position aids in the cleavage of the C–H allowing the formation of H2 and a C–C bond to give the dinuclear Yb complex.
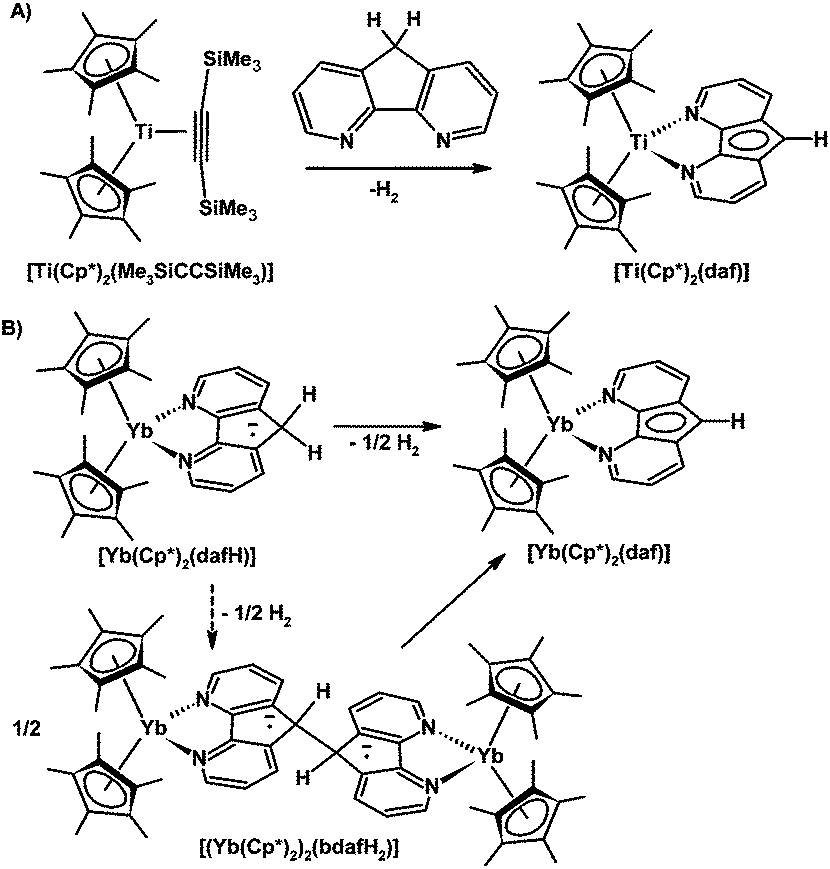 | ||
| Scheme 13 Two studies involving the formation of H2 from a coordinated dafH ligand. (A) Isolation of [Ti(Cp*)2(daf)].23 (B) Synthesis of [Yb(Cp*)2(daf)] from the stabilized biradical, [Yb(Cp*)2(dafH)], or the dinuclear complex, [(Yb(Cp*)2)2(bdafH2)].24 | ||
Our group also investigated the reactivity of the zwitterionic Ru(II) diazafluorenide complex [Ru(daf)(PPh3)2(H)(N2)] toward CO2 and uncovered an interesting example of ligand-based reactivity.115 At room temperature [Ru(daf)(PPh3)2(H)(N2)] selectively and reversibly undergoes a formal insertion of CO2 into a remote ligand C–H bond to generate a monoanionic 4,5-diazafluorenyl-9-carboxylic acid ligand (dafCO2H−) on Ru(II) (Scheme 12).115 The activation of CO2 in our system occurs at the ligand backbone remote from the metal centre where the metal's role is to adjust the nucleophilicity of the ligand-based carbanion, the acidity of the C–H bond involved in proton migration, as well as the strength of the newly formed C–C bond. Given the unusual situation of having an actor ligand and a spectator metal centre, a variety of spectator metal centres were used to tune the reactivity and electronics of the actor daf− ligand for tandem CO2 and C–H activation.22 Since Rh(III) is isoelectronic with Ru(II), the complex [Rh(daf)(PPh3)2(H)2] also reacts with CO2 in an analogous way where CO2 reversibly inserts into the C–H bond of the ligand backbone (Scheme 14).22
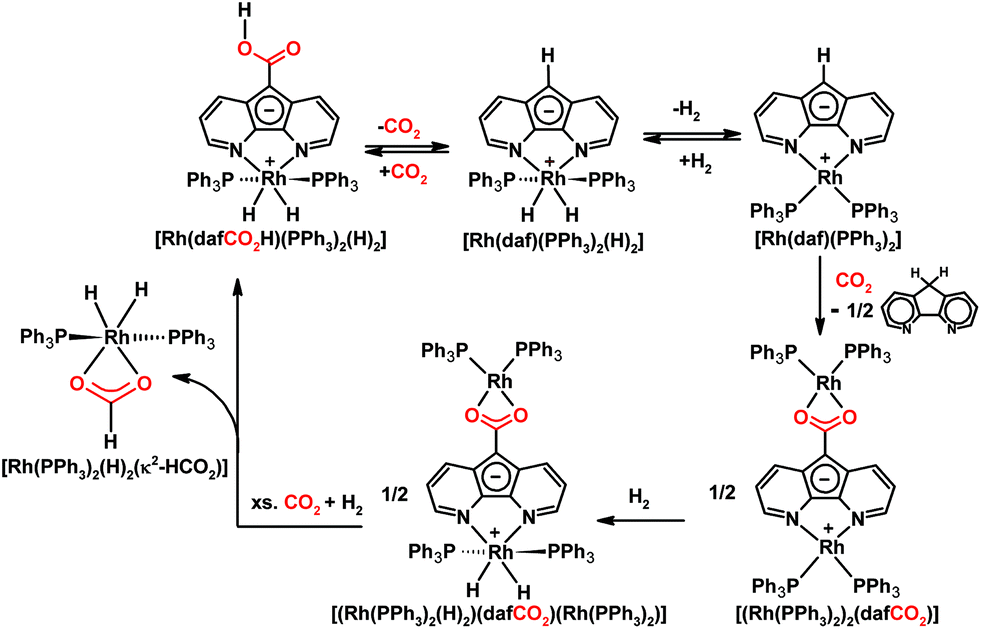 | ||
| Scheme 14 Chemistry of Rh 4,5-diazafluorenyl complexes with CO2 and H2.20,22,116 | ||
In contrast, when the more electron-rich Rh(I) complex [Rh(daf)(PPh3)2] is placed under CO2, a dinuclear Rh(I) complex, [(Rh(PPh3)2)2(dafCO2)], is formed where the two Rh(I) centres are bridged by a dianionic 4,5-diazafluorenyl-9-carboxylate ligand (dafCO22−), along with the formation of free dafH (Scheme 14).22 The result of tuning daf− with a more electron rich metal centre is the increased basicity of the ligand-based carbanion which can deprotonate the carboxylic acid initially formed from CO2 insertion into the ligand C–H bond. This proton transfer gives [Rh(dafH)(PPh3)2]+ and [Rh(dafCO2)(PPh3)2]−; the carboxylate of the latter replaces the dafH ligand of the former to yield the dinuclear product.22 As a result CO2 is trapped by the second metal centre, and also there is no proton on O that can engage in proton transfer necessary for the decarboxylation.22 The dinuclear [(Rh(PPh3)2)2(dafCO2)] complex was reacted with H2 to attempt ligand-based CO2 reduction.116 A series of stepwise stoichiometric reactions with H2, NMR experiments at low temperatures with added PPh3 or CO2, along with 13C-labelling experiments were conducted in an attempt to gain some mechanistic insight.116 Upon the addition of a CO2 and H2 gas mixture to [(Rh(PPh3)2)2(dafCO2)], a mixture of the carboxylated Rh(III) complex [Rh(dafCO2H)(PPh3)2(H)2] and [Rh(PPh3)2(H)2(κ2-HCO2)] results (Scheme 14).116
It is worth noting that the carboxylic acid intermediate in the reaction between [Rh(daf)(PPh3)2] and CO2 could not be isolated or even observed in NMR experiments, presumably because the highly basic carbanion in [Rh(daf)(PPh3)2] effected by the electron rich Rh(I) centre deprotonates the carboxylic acid to trigger the formation of the final product too quickly before the concentration of the carboxylic acid intermediate could build up. In order to observe and isolate both the kinetic and thermodynamic products of CO2 reaction, the slightly less electron donating Cu(I) was used as the spectator metal centre.22 When CO2 is added to [Cu(daf)(IPr)] in toluene the kinetic product [Cu(dafCO2H)(IPr)] precipitates from solution (Scheme 15).22 Since [Cu(dafCO2H)(IPr)] and [Cu(daf)(IPr)] are both soluble in DMSO, the reaction between these two species is readily observable when DMSO is used as the solvent for both the carboxylation of [Cu(daf)(IPr)] with CO2 and the decarboxylation of [Cu(dafCO2H)(IPr)] under an N2 atmosphere. In both cases, a dinuclear Cu(I) complex [(Cu(IPr))2(dafCO2)] and dafH are obtained as thermodynamic products (Scheme 15).22 In both the Cu(I) and Rh(I) cases, the metal 4,5-diazafluorenyl and the metal 4,5-diazafluorenyl-9-carboxylic acid complexes react with each other in solution, in contrast to the less electron rich Ru(II) and Rh(III) systems.
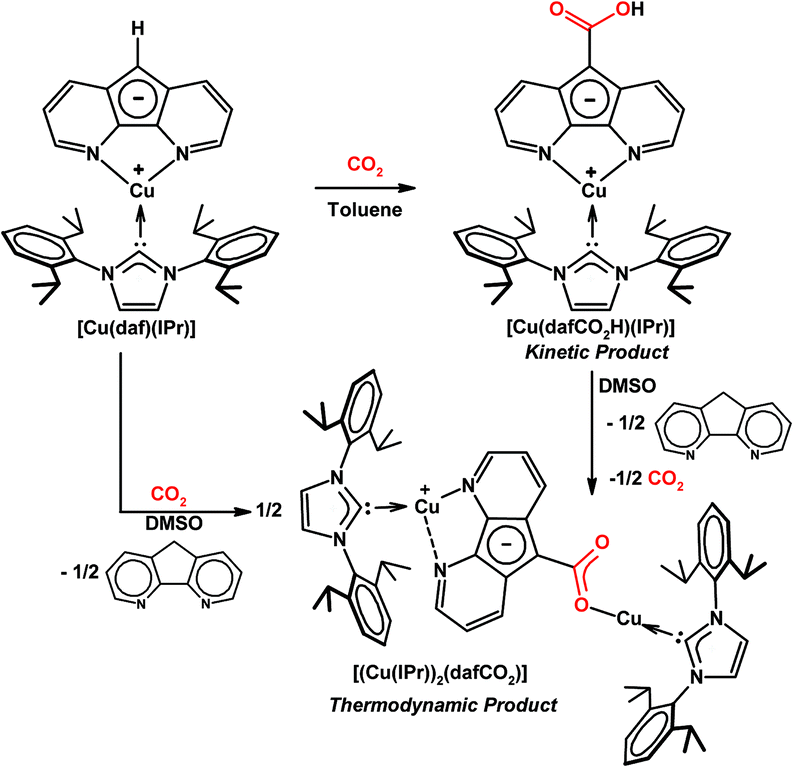 | ||
| Scheme 15 Reaction of [Cu(daf)(IPr)] with CO2 which gives [Cu(dafCO2H)(IPr)] as the kinetic product and a mixture of [(Cu(IPr))2(dafCO2)] and dafH as thermodynamic products.22 | ||
III.2. 4,5-Diazafluorene derivatives as ligands in catalysis
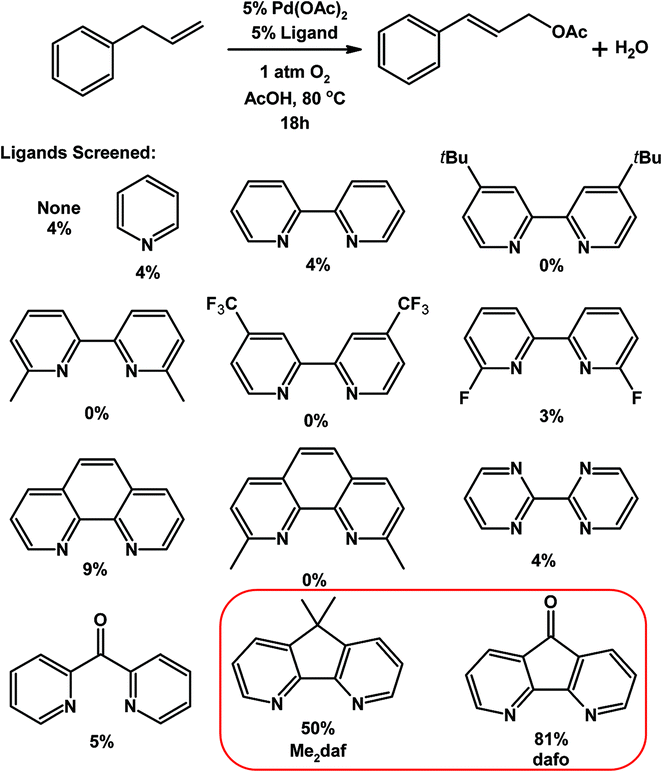
Nitrogen-donor ligands have been used extensively in oxidative aerobic organic transformations, especially given their robustness versus traditional phosphine ligands under oxidizing reaction conditions. Stahl and co-workers initially explored the use of 4,5-diazafluorene derivatives in oxidative organic reactions where O2 is the oxidant.76 A variety of nitrogen-donor ligands were screened in the Pd-catalyzed aerobic allylic acetoxylation of allylbenzene; most of the ligands screened gave low yields of the cinnamyl acetate product (Table 1).74 Conversely, when 9,9-dimethyl-4,5-diazafluorene (Me2daf) was used as the ligand, a 50% yield of cinnamyl acetate was obtained, the yield was further improved to 81% when dafo was used as the ligand.74 The structures of 4,5-diazafluorene derivatives seem to have a large impact on the catalytic results obtained relative to the other nitrogen-donor ligands tested. Both the ability of dafo to withdraw electron density through π back-bonding and the unique ligand bite angle may play a role in catalysis. Recently Stahl reported the mechanistic investigations where they suggest that the dafo ligand promotes the C–O reductive elimination, but further studies are needed.77 Stahl and co-workers have also demonstrated the use of 4,5-diazafluorene derivatives as ancillary ligands in the aerobic Pd-catalyzed cross-coupling of indoles with benzene.75 The regioselectivity for arylation at the C2- vs. the C3-position of the indole compound was dramatically affected by the identity of 4,5-diazafluorene derivative and the anionic ligand used (Scheme 16).75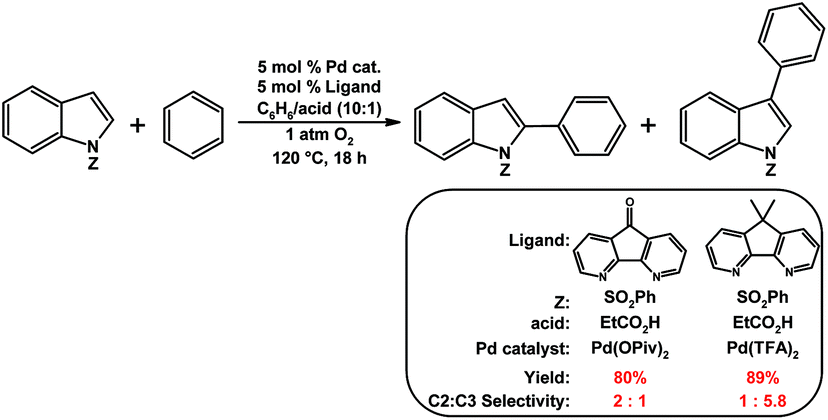 | ||
| Scheme 16 Pd-catalyzed aerobic coupling of indoles with benzene using 4,5-diazafluorene derivatives as ligands.75 | ||
|
Stahl,78 along with Zhao and Huang,80 independently and simultaneously reported the use of the dafo ligand in Pd-catalyzed aerobic dehydrogenation to form α,β-unsaturated aldehydes, ketones, esters, and azobenzenes (Scheme 17). Typically enones and other α,β-unsaturated carbonyl compounds are prepared in stepwise protocols. The aerobic Pd-dafo catalyzed reaction is a much more efficient alternative.
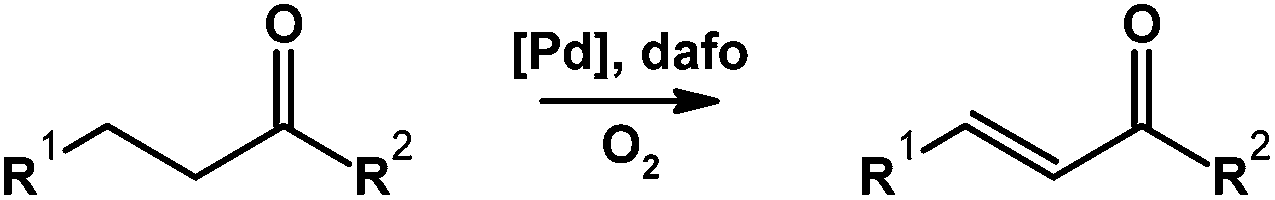 | ||
| Scheme 17 Pd-dafo-catalyzed aerobic dehydrogenation to form double bonds.78,80 | ||
Recently it has been shown that dafo is an effective ligand for the Pd-catalyzed aerobic dehydrogenative Heck reaction to couple furans and thiophenes with cinnamic acid and stilbene derivatives.85 In addition a variety of alkenes could be coupled with ferrocene using a Pd dafo catalyst in an aerobic dehydrogenative Heck reaction (Scheme 18).83 A combination of kinetics, competition and ESI-MS (to characterize catalytic intermediates) experiments suggest that dafo plays a role at each stage of the catalytic cycle,85i.e., the dafo influences C–H bond activation, insertion of alkenes, the stereo-selective step, and the regeneration of the catalyst with O2.85
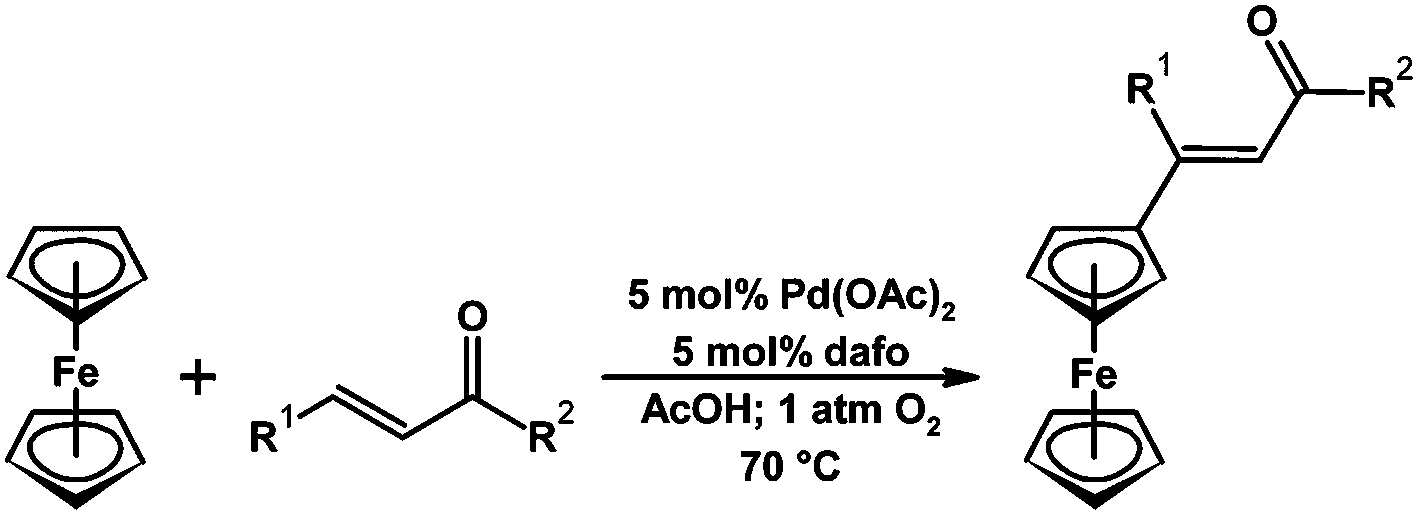 | ||
| Scheme 18 Pd-dafo-catalyzed aerobic Heck coupling reaction of an alkene with ferrocene.83 | ||
Elsevier and co-workers have looked at the influence of various nitrogen chelates on Pd catalyzed C–C bond formation reactions.79 Zerovalent mono and binuclear palladium and platinum bis(quinone) complexes of dafH and dafo have been prepared, where the 4,5-diazafluorene derivative either acts as a chelate, monodentate, or bridging ligand.17,81,82 In the reaction of cinnamyl chloride with benzyl Grignard, the regioselectivity when palladium complexes of 4,5-diazafluorene derivatives are used is extremely high for substitution at the less substituted allylic carbon with less than 5% of the homo-coupling product, a sharp contrast to when phosphine complexes are used.79
[Rh(daf)(COD)] can catalyze the hydrogenation of olefins, however it is not as fast a catalyst as [Rh(PPh3)3Cl] or [Ru(PPh3)3Cl2], and does not hydrogenate internal olefins.19 Wilkinson's catalyst [Rh(PPh3)3Cl] dissociates in solution and produces hydride species under H2. In contrast, no hydride was observed when a solution of [Rh(daf)(COD)] was exposed to H2.
[Rh(dafH)(PPh3)2(H)2]Cl was also found to be a selective olefin hydrogenation catalyst, and can even hydrogenate internal olefins and substrates with pyridyl or carbonyl groups.20 The chloride counterion appears to play a role in catalysis, i.e., if the counterion is replaced with triflate the complex is inactive towards olefin hydrogenation. Exogenous chloride anions however appear to slow down the catalysis.
Recently it was shown that 1,5-dihydro-2H-cyclopenta[2,1-b:3,4-b′]dipyridin-2-one (a 4,5-diazafluorene derivative with keto–enol tautomerism), L7, can be coordinated to Ru(II) to give [Ru(L7)(bpy)2](PF6)2.117 [Ru(L7)(bpy)2](PF6)2 was used as a photosensitizer in dye-sensitized solar cells (DSSCs) and as a catalyst for the transfer hydrogenation of ketones.117
The complex [MoO2Cl2(dafo)] was quite an active and selective olefin epoxidation catalyst when compared with other polypyridine ligands.63 Of the polypyridine ligands tested the best catalysts seemed to have moderate donating capability and little steric hindrance with respect to the {MoO2Cl2} fragment.63
Huang and co-workers reported the use of dafo as a ligand in organic transformations mediated by a gold catalyst and secondary amine that work synergistically (Scheme 19).118,119 Tri- and tetrasubstituted allenes can be synthesized in a α-vinylidenation or an α-vinylidenation/γ-vinylidenation cascade from aldehydes and silyl-EBX.118 The trisubstituted allenyl aldehyde product produced in this gold–amine system is sensitive toward O2 to yield ynone products, the result of oxidative C–C bond cleavage (Scheme 19).119
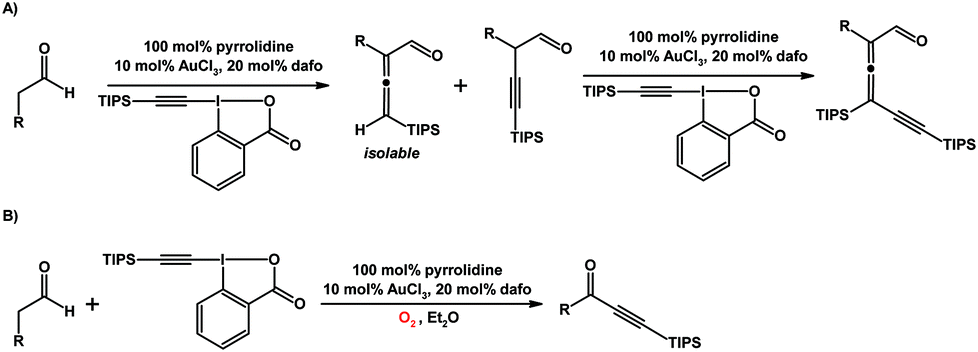 | ||
| Scheme 19 (A) α-Vinylidenation of aldehydes to give trisubstituted allenes, or an α-vinylidenation/γ-vinylidenation cascade to give tetrasubstituted allenes.118 (B) Aerobic C–C bond cleavage reaction with aldehydes which yields an ynone product. Both reactions are catalyzed by an Au–dafo/amine catalyst system.119 | ||
Stahl and coworkers have demonstrated Pd catalyzed aerobic intramolecular aryl C–H amination to give carbazole derivatives as products (Scheme 20).86 The 1,4-dioxane solvent decomposes in the presence of O2 to form in situ an alkyl peroxide that promotes the efficient Pd catalyzed aryl C–H amination.86 The neutral dafo ligand was found to be the most efficient at promoting carbazole synthesis starting from the N-benzenesulfonyl-2-aminobiphenyl test substrate giving an 80% yield, Me2daf can also promote the reaction giving a 33% yield.86
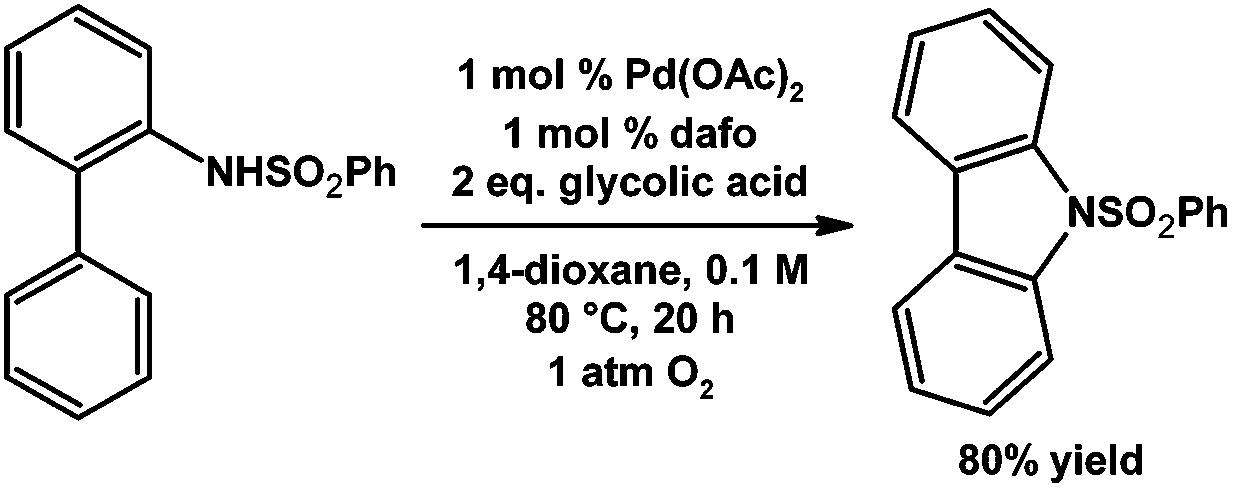 | ||
| Scheme 20 Pd catalyzed aerobic aryl C–H amination.86 | ||
One interesting catalytic application of 4,5-diazafluorene derivatives is the introduction of a catalytically active metal to the surface of nanoparticles to create nanocatalysts. Silica-coated magnetite nanoparticles (SMNPs) were silanated with 3-aminopropyltriethoxysilane, the exposed –NH2 group was reacted with dafo to give the imine that was subsequently reduced to the amine (Scheme 21A).120 The 4,5-diazafluorene-functionalized SMNPs were coordinated with Pd(II) to give a Pd-4,5-diazafluorenyl SMNP catalyst for C(sp2)–C(sp2) cross-coupling reactions (Scheme 21B).120 An elegant feature of this heterogeneous SMNP nanocatalyst is that it can be separated from the reaction mixture by using an external magnet, and reused successfully in subsequent cross-coupling reactions. Using a similar protocol silica nanospheres were functionalized with a 4,5-diazafluorene-imine group, and subsequently reacted with ferric chloride.121 The silica nanosphere-based iron nanocatalyst was used to perform the one-pot coupling reaction of a terminal alkyne, dichloromethane, and amines to give propargylamines.121 In addition, the silica nanosphere-based iron catalyst could be recycled at least seven times without any appreciable loss of catalytic activity.121
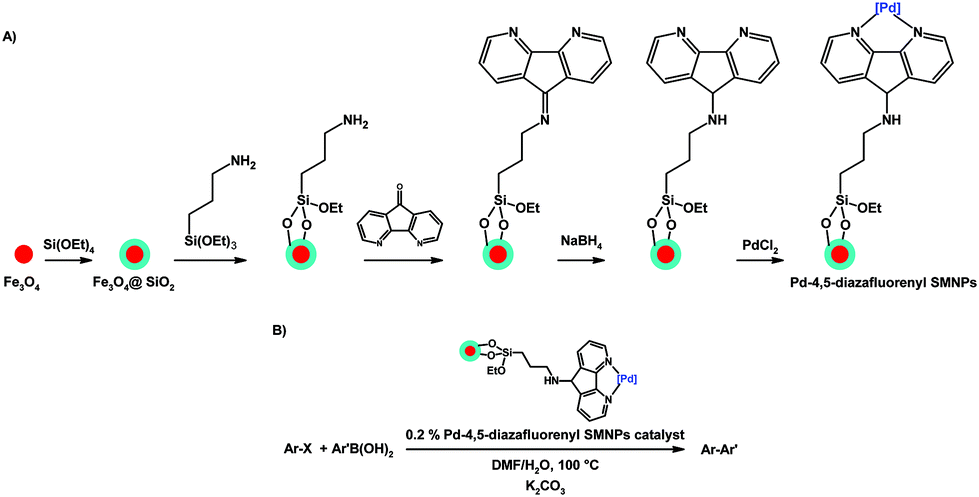 | ||
| Scheme 21 (A) Synthesis of magnetically-separable Pd-4,5-diazafluorenyl SMNP nanocatalysts, and (B) their use as catalysts in cross-coupling reactions.120 | ||
III.3. Photophysical and photochemical applications
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1)).9,10 The most substantial difference is that the emission quantum yield of [Ru(bpy)3]2+ in water (φ(298 K) = 0.042, τ(298 K) = 630 ns) is ∼50 fold larger than that of [Ru(dafH)(bpy)2]2+ (φ(298 K) = 8 × 10−4) and the lifetime of [Ru(dafH)(bpy)2]2+ was so short that it could not be determined.9,10 The dafH ligand with a longer N–N distance was found to be lower than bpy in the spectrochemical series which translated into an energetic lowering of ligand field excited states and a dramatic decrease in emission intensity on approaching room temperature.9,10
:1)).9,10 The most substantial difference is that the emission quantum yield of [Ru(bpy)3]2+ in water (φ(298 K) = 0.042, τ(298 K) = 630 ns) is ∼50 fold larger than that of [Ru(dafH)(bpy)2]2+ (φ(298 K) = 8 × 10−4) and the lifetime of [Ru(dafH)(bpy)2]2+ was so short that it could not be determined.9,10 The dafH ligand with a longer N–N distance was found to be lower than bpy in the spectrochemical series which translated into an energetic lowering of ligand field excited states and a dramatic decrease in emission intensity on approaching room temperature.9,10
A series of multinuclear Ru(II) complexes supported by multinucleating ligands possessing two or more chelating 4,5-diazafluorenyl moieties have been explored.122–138 The spacers between the coordinating 4,5-diazafluorenyl moieties have been varied substantially to allow for the metal–metal separation, and the degree of intramolecular charge transfer to be finely tuned. In some cases disappointingly the Ru(II) complexes had extremely short excited state lifetimes at room temperature on the order of ∼0.03 ns (e.g., Ru complex of the ditopic ligand with an adamantyl spacer).122
One of the major challenges in the field of DSSCs is to identify molecules that can efficiently harvest sunlight when incorporated into mesoscopic semiconductor thin films such as mesoporous TiO2. Ru(II) complexes of 4,5-diazafluorene derivatives have been investigated as sensitizers for TiO2, especially for their ability to inject electrons into TiO2 from their excited states. Following adsorption of [Ru(dafo)(bpy)2]2+ onto TiO2 it was noted by Heuer, Meyer and coworkers that the dafo ligand engaged in a ring-opening reaction resulting in a coordinated 3-(CO2H)-2,2′-bipyridine ligand with the carboxylate group anchoring the complex to TiO2.65 The control experiment of independently anchoring the analogous Ru(II) complex of 3-(CO2H)-2,2′-bipyridine gave comparable results; however, the excited-state injection yield for the [Ru(dafo)(bpy)2]2+/TiO2 system was consistently lower.65 Ru(II)-dafo complexes with either one or two 4,4′-dicarboxy-2,2′-bipyridine (dcbpy) ligands to anchor the complex onto TiO2 have also been explored.69,139 The thermal stability of [Ru(dafo)(dcbpy)(SCN)2] at high temperatures has also been investigated where the final products are Ru and RuS2; again the ring-opening of dafo seems to play a role in complex decomposition.64 Another ligand design tested to anchor Ru(II) polypyridine complexes to TiO2 was an ambidentate ligand (4,5-diazafluoren-9-ylidene)malonic acid (dfm), which has a 4,5-diazafluorenyl chelate with an olefin bridge to two carboxylic acid groups.140 The metal-to-ligand charge transfer (MLCT) excited state lifetime of [Ru(dfm)(bpy)2]2+ was extremely short in solution, yet the interfacial electron transfer to TiO2 was efficient (ϕinj = 0.70 ± 0.05).140 Some Ru photosensitizers with record high extinction coefficients possess 4,5-diazafluorenylidene ligands substituted with a 1,3-dithiole group (ε ≥ 40000 M−1 cm−1 @ 470 nm), displaying rapid and efficient charge injection to TiO2.141,142 In addition, Ru(II) complexes of 4,5-diazafluorene functionalized at the 9-position with carbazole groups,143 aryl groups,144 or bis(thiophene)pyrrolyl groups,139 have all been used to create DSSCs based on TiO2.
Cationic Ir(III) complexes of the form [Ir(Ln)(dfppz)2](PF6) (n = 5, 8, 9, 10, 11) with cyclometallated 1-(2,4-difluorophenyl)pyrazole (dfppz) have been prepared, where the parent 4,5-diaza-9,9′-spirobifluorene ligand (L5) is substituted at the 3,6-positions with one (L8) or two (L9) pendant phenyl groups (see Fig. 5 for labelling scheme).148 Interestingly these pendent phenyl groups can engage in π–π stacking interactions with the dfppz ligand(s) within complexes; these interactions may limit the elongation of the Ir–ligand bonds in the excited state protecting the metal centre.148
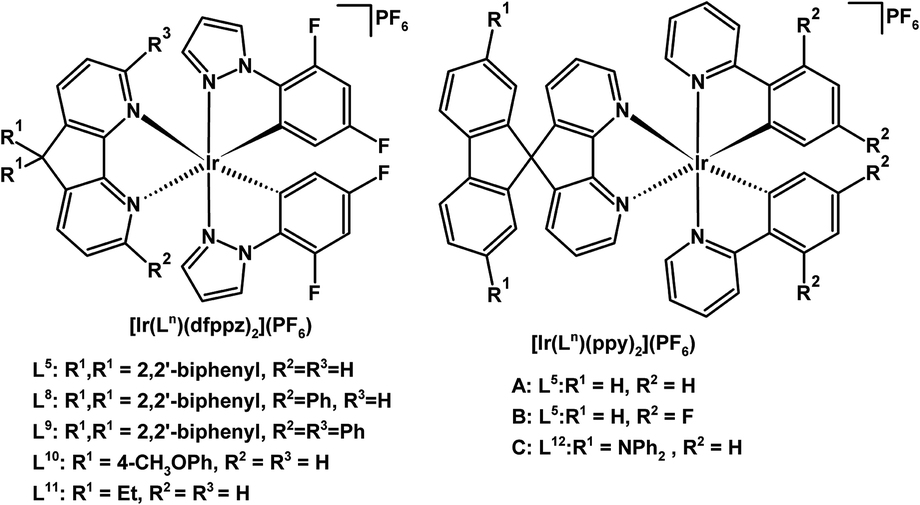 | ||
| Fig. 5 Luminescent cationic Ir(III) complexes of 4,5-diazafluorene derivatives for use in solid-state light-emitting electrochemical cells.145–148 | ||
A white-light emitting LEC was created using a mixture of red-, green- and blue-emitting cationic Ir(III) complexes, where the green and blue emitters have 4,5-diazafluorene derivatives disubstituted at the C9-position with gem-diaryl (L10) or gem-dialkyl (L11) groups (Fig. 5).146 Substitution at the C9-position was found to be crucial for the synthesis of Ir(III) complexes with high photoluminescence quantum yields.146 Likewise, Ir(III) complexes [Ir(L5)(ppy)2](PF6) (where ppy is a cyclometallated 2-phenylpyridine derivative) A and B (Fig. 5) show high electroluminescence efficiencies: 7.1%, 22.6 lm W−1 for orange A, and 7.1%, 26.2 lm W−1 for green B,145 owing to the spiro structural feature of L5, which limits self-quenching phenomena without greatly perturbing the energy gaps of the compounds.
A conceptually interesting approach toward incorporating three functions into a single molecule, a luminescent chromophore with hole and electron transport capability was explored by the Wong group. In luminescent Ir(III) complex C (Fig. 5), the ligand L12 has an electron-transporting 4,5-diazafluorene site, and hole-transporting –NPh2 groups.147 Unfortunately the LEC device performance using this tri-functional Ir(III) complex was rather low. There is no 3MLCT contribution to the lowest transition for the complex in the triplet state.147
Luminescent lanthanide complexes exhibit very sharp emission bands. The design and tuning of sensitizing ligands that allow for efficient ligand-to-metal energy transfer is an area of particular interest. Luminescent lanthanide tris(β-diketonate) complexes of various 4,5-diazafluorene derivatives such as dafo,94–96,149 9,9-diaryl substituted 4,5-diazafluorene,150 4,5-diazafluoren-9-imine derivatives,151 and 4,5-diaza-9,9′-spirobifluorene152 have been synthesized and characterized. Several of these lanthanide complexes exhibit NIR luminescence94–96 photo- and electroluminescence,150,152 and even triboluminescence.149
Green phosphorescent Re(I) complexes of various 4,5-diazafluorene derivatives have been investigated for their performance in OLEDs.153–156 In comparing [Re(N^N)(CO)3Br] type complexes of dafH and a 9,9-di(ethoxyphenyl) substituted 4,5-diazafluorene derivative, both complexes give approximately the same emission wavelength, however the complex with bulky groups at the C9 position alleviates to a large extent the self-quenching at high doping concentrations.153 A dinuclear Re(I) complex of 9,9′-bi-4,5-diazafluorenyl can serve as a highly efficient green phosphorescent emitter in OLEDs with a maximum luminance of 2026 cd m−2 and a peak current efficiency of 8.2 cd A−1.154 Non-doped devices containing [(L5)Re(CO)3Br] had outstanding performance with a further improved peak luminance of 8531 cd m−2 and maximum current efficiency of 16.8 cd A−1.156
The phosphine Cu(I) complexes of 3,3′-methylen-4,4′-diphenyl-2,2′-biquinoline (mdpbq) exhibited red phosphorescence (Fig. 6).157 Complexes of the extremely rigid mdpbq ligand showed decent photoluminescence quantum yields in 20 wt% poly(methylmethacrylate) (PMMA) films, 0.56 and 0.43 for [Cu(mdpbq)(PPh3)2](BF4) [Cu(mdpbq)(DPEphos)](BF4), respectively.157 The OLEDs doped with these Cu(I) complexes gave a current efficiency up to 6.4 cd A−1 for a multi-layer device.157
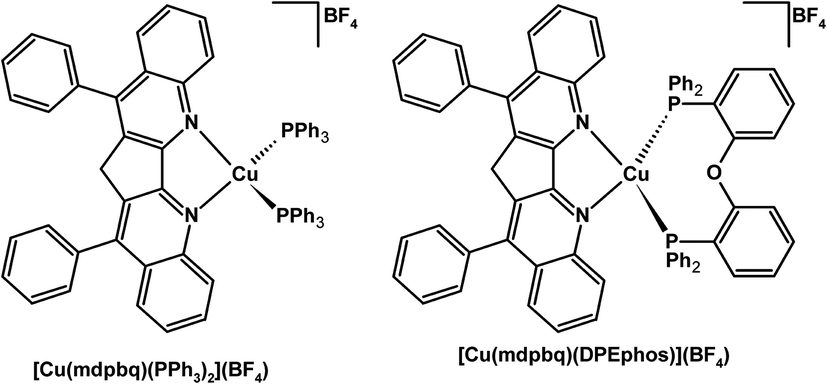 | ||
| Fig. 6 Structures of red phosphorescent Cu(I) complexes.157 | ||
Ag–dafo complexes with either carborane-based diphosphines, or simple classical diphosphine ligands of the general formula [Ag(dafo)(P–P)]OTf are luminescent. The emissions seem independent of the phosphine ligand or the coordination environment of the Ag+ ion.158 The free dafo emits at 537 nm (τ = 5 ns), while the luminescent Ag–dafo complexes emit across the blue to orange region of the spectrum with lifetimes also in the ns range, similar to free dafo.158 The dafo ligand centred transitions seem to be responsible for the luminescence behaviour of [Ag(dafo)(P–P)]OTf and [Ag(dafo)(PPh3)(OTf)] complexes, where the electron density at the Ag centre tuned by the different phosphine ligands modified the emission energy.158
Ir complexes of the form [Ir(L^L)(ppy)2]n+ where the bidentate L^L is either dafH, 9-fulleriden-4,5-diazafluorene, or cyclometallated 9-fulleriden-4-azafluorene can be used as harmonophores with second-order NLO properties.159 The introduction of the fullerene moiety weakens the interaction between the cationic Ir(III) complex and the anion, which also leads to an overall increase in the NLO response with large and negative μβ1.907 values (−600 to −2190 × 10−48 esu).159 For the charge neutral complex with cyclometallated 9-fulleriden-4-azafluorene ligand the μβ1.907 value is lower than the cationic complexes.159 In addition the second-order NLO properties of Ru(II) complexes of the forms [Ru(N^N)(PPh3)2Cl2] and [Ru(N^N)(CO)2Cl2] where the N^N chelate is either dafH or 9-fulleriden-4,5-diazafluorene, have also been examined.160 The greater absolute values of μβ1.907 for complexes with the fullerene substituted ligand compared to those with unsubstituted dafH suggest the importance of a highly polarizable C60 group.160
Ru(II) bipyridine complexes of 4,5-diazafluorene-9-imine derivatives with long alkyl chains off of the imine moiety have been incorporated as surfactants into Langmuir–Blodgett thin films and have activities for second order harmonic generation that are 2.6 to 3.6 times greater than that of the organic standard (E)-N-methyl-4-(2-(4-octadecyloxyphenyl)ethenyl)-pyridinium iodide.161 Similarly [Re(N^N)(CO)3Cl] complexes of similar 4,5-diazafluorene-9-imine derivatives can also be formed into stable Langmuir–Blodgett thin films, though a lower than expected measurement for the second-order harmonics NLO signal was observed.162
Zn(II) complexes of the highly conjugated push–pull 4,5-diazafluoren-9-ylidene ligand family terminated with either an N,N-dibutylamino or an azulenyl moiety exhibit amongst the highest reported μβ1.907 value for a Zn(II) complex.163 With respect to the free ligands, coordination to Lewis acidic Zn(II) enhances the μβ1.907 value presumably by red-shifting the ILCT transition.163
Duan, Bai, and coworkers reported [Ru(L13)(bpy)2]2+ (where L13 is 4,5-diazafluoren-9-one-2,4-dinitrophenylhydrazone) as a selective chromo- and fluorogenic dual responding fluoride sensor.166 Not only does the presence of F− enhance the luminescence intensity but also triggers a dramatic color change from yellow to magenta. The Ru(II) complex could even be adsorbed onto paper to allow for the preparation of colorimetric testing strips for F− concentration in water. Interestingly, even when the concentration of F− is as low as 1 mg L−1, the color change is visible to the naked eye.
III.4. Bioinorganic chemistry of 4,5-diazafluorene derivatives
Recently Ru(II) complexes [Ru(L14)(bpy)2](PF6)2 (where L14 is a 4,5-diazafluoren-9-imine ligand with various N-aryl groups) have been found to be effective topical antibiotics against the bacteria Staphylococcus aureus which is resistant to the antibiotic methicillin.167 In particular the derivative with a –OC7H15 group attached to the N-aryl group, which is non-toxic toward human skin keratinocyte cells, exhibits strong microbicidal and bacterial growth inhibitory effects.167 One possible mechanism, for how these Ru(II) complexes are active against S. aureus, is through the generation of reactive oxygen species.167Two crystal structures of the bis-dafo Ag(I) nitrate complexes were obtained where the nitrate ion is either bound to the metal centre in the case of [Ag(dafo)2(NO3)],168 or not in the case of [Ag(dafo)2](NO3)·H2O.88,168 These complexes exhibited broad spectrum antibacterial properties against six different clinically resistant strains of diabetic foot bacteria, and had a significantly lower minimum inhibitory concentration (MIC) compared with currently available commercial antibiotics.168 Initial studies also showed that the silver complexes could be loaded into hydrogels for possible incorporation into wound dressings.168 The in vitro linear dichroism studies also showed that both dafo and the silver complexes bind to calf thymus DNA, however the complexes showed significantly stronger binding to DNA compared with the free ligand.168
The free 9-diazo-4,5-diazafluorene can be photochemically activated to release dinitrogen as a byproduct and generate a triplet carbene.169 Matrix photolysis experiments of complex [Cu(9-diazo-4,5-diazafluorene)2(NO3)2] (Fig. 7) indicate the formation of a Cu(I)–L˙+ species (S = 1/2), where the radical is primarily localized at the C9-position.169 Presumably the Cu(II) center is reduced by the triplet carbene formed via photolysis. In solution the nitrate ligands are labile, and other chemical or biological substrates such as DNA can coordinate to Cu. Consequently, [Cu(9-diazo-4,5-diazafluorene)2(NO3)2] cleaves DNA upon irradiation with visible light under anaerobic conditions; in contrast, the free 9-diazo-4,5-diazafluorene ligand is significantly less effective for the photocleavage of DNA.169–171
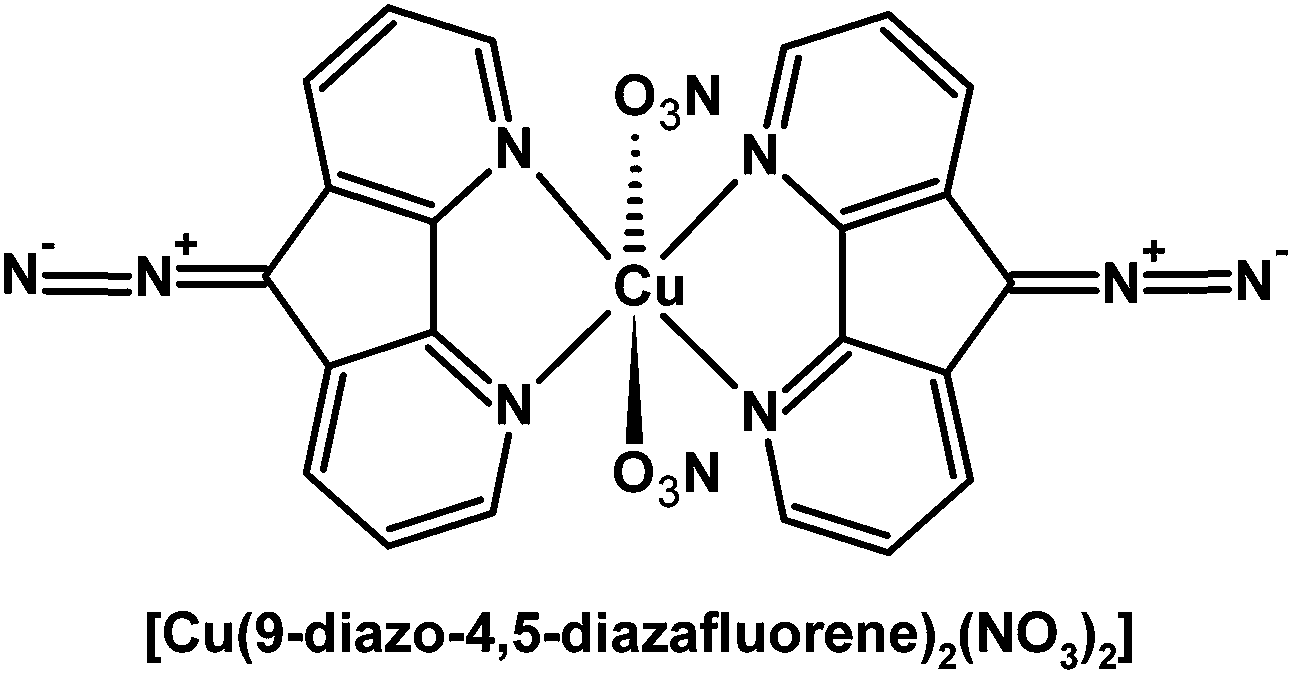 | ||
| Fig. 7 Structure of [Cu(9-diazo-4,5-diazafluorene)2(NO3)2].169,170 | ||
IV. Conclusions
Since the first report on the synthesis of dafH, the chemistry of dafH derivatives as ligands has evolved into a vibrant field. From the fundamental chemistry point of view, the parent compounds dafH and dafo can be derivatized in many ways to achieve the desired functions. In particular, the functionalization at the C9 position has been in the spotlight for generating new ligand series. Many of the ambidentate derivatives have been utilized to construct coordination polymers, self-assembled macrocycles, and heteromultimetallic complexes with great specificity. The reactive nature of the 9-position of dafH, daf− and dafo ligands gave rise to the actor-ligand behavior in the corresponding metal complexes, which distinguish dafH derivatives from bpy. From an applications point of view, dafH derivatives have found use in many areas. Their use as ancillary ligands in catalysis has been fruitful, especially in transformations performed under oxidizing conditions. The photochemistry and physics of metal complexes of 4,5-diazafluorene derivatives have been studied as emitters in solid state lighting, photosensitizers in DSSCs, and harmonophores in non-linear optics. Biological studies of 4,5-diazafluorene metal complexes have included DNA binding and cleavage, and antimicrobial properties. Further research may lead to new types of fundamental reactivities as well as applications.References
- K. Kloc, J. Mlochowski and Z. Szulc, J. Prakt. Chem., 1977, 319, 959–967 CrossRef CAS.
- K. Kloc, Z. Szulc and J. Mlochowski, Heterocycles, 1978, 9, 849–852 CrossRef CAS.
- I. F. Eckhard and L. A. Summers, Aust. J. Chem., 1973, 26, 2727–2728 CrossRef CAS.
- R. P. Thummel, F. Lefoulon and R. Mahadevan, J. Org. Chem., 1985, 50, 3824–3828 CrossRef CAS.
- M. J. Plater, S. Kemp and E. Lattmann, J. Chem. Soc., Perkin Trans. 1, 2000, 971–979 RSC.
- S. Nishigaki, H. Yoshioka and K. Nakatsu, Acta Crystallogr., Sect. B: Struct. Sci., 1978, 34, 875–879 CrossRef.
- L. Maguire, C. M. Seward, S. Baljak, T. Reumann, Y. Ortin, E. Banide, K. Nikitin, H. Müller-Bunz and M. J. McGlinchey, Eur. J. Inorg. Chem., 2009, 3250–3258 CrossRef CAS.
- M. Riklin, A. von Zelewsky, A. Bashall, M. McPartlin, A. Baysal, J. A. Connor and J. D. Wallis, Helv. Chim. Acta, 1999, 82, 1666–1680 CrossRef CAS.
- L. J. Henderson, F. R. Fronczek and W. R. Cherry, J. Am. Chem. Soc., 1984, 106, 5876–5879 CrossRef CAS.
- W. M. Wacholtz, R. A. Auerbach, R. H. Schmehl, M. Ollino and W. R. Cherry, Inorg. Chem., 1985, 24, 1758–1760 CrossRef CAS.
- R. P. Thummel, F. Lefoulon and J. D. Korp, Inorg. Chem., 1987, 26, 2370–2376 CrossRef CAS.
- T. C. Strekas, H. D. Gafney, S. A. Tysoe, R. P. Thummel and F. Lefoulon, Inorg. Chem., 1989, 28, 2964–2967 CrossRef CAS.
- K. Maruszewski and J. R. Kincaid, Inorg. Chem., 1995, 34, 2002–2006 CrossRef CAS.
- D. Aguilà, E. Escribano, S. Speed, D. Talancón, L. Yermán and S. Alvarez, Dalton Trans., 2009, 6610–6625 RSC.
- K. Ono and K. Saito, Heterocycles, 2008, 75, 2381–2413 CrossRef CAS.
- W. Wong, Coord. Chem. Rev., 2005, 249, 971–997 CrossRef CAS.
- R. A. Klein, P. Witte, R. van Belzen, J. Fraanje, K. Goubitz, M. Numan, H. Schenk, J. M. Ernsting and C. J. Elsevier, Eur. J. Inorg. Chem., 1998, 319–330 CrossRef CAS.
- F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456–463 CrossRef CAS.
- H. Jiang and D. Song, Organometallics, 2008, 27, 3587–3592 CrossRef CAS.
- H. Jiang, E. Stepowska and D. Song, Eur. J. Inorg. Chem., 2009, 2083–2089 CrossRef CAS.
- E. Stepowska, H. Jiang and D. Song, Chem. Commun., 2010, 46, 556–558 RSC.
- V. T. Annibale, D. A. Dalessandro and D. Song, J. Am. Chem. Soc., 2013, 135, 16175–16183 CrossRef CAS PubMed.
- P. T. Witte, R. Klein, H. Kooijman, A. L. Spek, M. Polášek, V. Varga and K. Mach, J. Organomet. Chem., 1996, 519, 195–204 CrossRef CAS.
- G. Nocton, C. H. Booth, L. Maron and R. A. Andersen, Organometallics, 2013, 32, 1150–1158 CrossRef CAS.
- V. T. Annibale, R. Batcup, T. Bai, S. J. Hughes and D. Song, Organometallics, 2013, 32, 6511–6521 CrossRef CAS.
- R. Batcup, F. S. N. Chiu, V. T. Annibale, J. U. Huh, R. Tan and D. Song, Dalton Trans., 2013, 42, 16343–16350 RSC.
- R. Batcup, V. T. Annibale and D. Song, Dalton Trans., 2014, 43, 8951–8958 RSC.
- R. Tan, F. S. N. Chiu, A. Hadzovic and D. Song, Organometallics, 2012, 31, 2184–2192 CrossRef CAS.
- S. A. Baudron, M. W. Hosseini and N. Kyritsakas, New J. Chem., 2006, 30, 1083–1086 RSC.
- S. Baudron and M. Hosseini, Inorg. Chem., 2006, 45, 5260–5262 CrossRef CAS PubMed.
- S. A. Baudron, M. W. Hosseini, N. Kyritsakas and M. Kurmoo, Dalton Trans., 2007, 1129–1139 RSC.
- S. A. Baudron and M. W. Hosseini, Chem. Commun., 2008, 4558–4560 RSC.
- G. Nasr, A. Guerlin, F. Dumur, S. A. Baudron, E. Dumas, F. Miomandre, G. Clavier, M. Sliwa and C. R. Mayer, J. Am. Chem. Soc., 2011, 133, 6501–6504 CrossRef CAS PubMed.
- K. Sako, M. Kusakabe and H. Tatemitsu, Mol. Cryst. Liq. Cryst. Sci. Technol., Sect. A, 1996, 285, 101–106 CrossRef CAS.
- Q. Zhu, J. Dai, D. Jia, L. Cao and H. Lin, Eur. J. Inorg. Chem., 2004, 4789–4794 CAS.
- Q. Zhu, J. Dai, D. Jia, L. Cao and H. Lin, Polyhedron, 2004, 23, 2259–2264 CrossRef CAS.
- Q. Zhu, Y. Zhang, R. Dai, G. Bian, D. Jia, J. Zhang and L. Guo, Chem. Lett., 2003, 32, 762–763 CrossRef CAS.
- Q. Zhu, W. Lu, Y. Zhang, G. Bian, J. Gu, X. Lin and J. Dai, Eur. J. Inorg. Chem., 2008, 230–238 CrossRef CAS.
- Q. Zhu, L. Yu, Y. Qin, L. Huo, M. Shao and J. Dai, CrystEngComm, 2011, 13, 2521–2528 RSC.
- T. Culpitt, I. A. Guzei, L. C. Spencer, A. Simonson, J. S. Miller, M. R. Wimmer and K. J. Nelson, Inorg. Chim. Acta, 2015, 427, 162–167 CrossRef CAS.
- M. P. Cifuentes, M. G. Humphrey, G. A. Koutsantonis, N. A. Lengkeek, S. Petrie, V. Sanford, P. A. Schauer, B. W. Skelton, R. Stranger and A. H. White, Organometallics, 2008, 27, 1716–1726 CrossRef CAS.
- O. Pélerin, C. Olivier, T. Roisnel, D. Touchard and S. Rigaut, J. Organomet. Chem., 2008, 693, 2153–2158 CrossRef.
- J. Zhao, R. Zhao, Q. Yang, B. Hu, F. Liu and X. Bu, Dalton Trans., 2013, 42, 14509–14515 RSC.
- O. Jung and C. G. Pierpont, Inorg. Chem., 1994, 33, 2227–2235 CrossRef CAS.
- U. Siemeling, I. Scheppelmann, B. Neumann, H. G. Stammler and W. W. Schoeller, Organometallics, 2004, 23, 626–628 CrossRef CAS.
- D. Li and H. Zeng, Appl. Organomet. Chem., 2013, 27, 89–97 CrossRef CAS.
- D. Li and Z. Tao, J. Coord. Chem., 2013, 66, 4237–4254 CrossRef CAS.
- B. Machura, M. Wolff, J. Palion, A. Świtlicka, I. Nawrot and K. Michalik, Struct. Chem., 2011, 22, 1053–1064 CrossRef CAS.
- B. K. Babu, A. R. Biju, S. Sunkari, M. V. Rajasekharan and J. P. Tuchagues, Eur. J. Inorg. Chem., 2013, 1444–1450 CrossRef CAS.
- X. Chen, D. Wang, Q. Luo and R. Wang, J. Coord. Chem., 2009, 62, 3895–3904 CrossRef CAS.
- F. Dong, L. Che, L. Li and F. Luo, Transit. Met. Chem., 2011, 36, 125–130 CrossRef CAS.
- P. Kulkarni, S. Padhye and E. Sinn, Inorg. Chim. Acta, 2001, 321, 193–199 CrossRef CAS.
- P. Kulkarni, S. Padhye, E. Sinn, C. E. Anson and A. K. Powell, Inorg. Chim. Acta, 2002, 332, 167–175 CrossRef CAS.
- D. Li, J. Tian, W. Gu, X. Liu, H. Zeng and S. Yan, J. Inorg. Biochem., 2011, 105, 894–901 CrossRef CAS PubMed.
- G. Li, N. Liu, S. Liu and S. Zhang, Electrochim. Acta, 2008, 53, 2870–2876 CrossRef CAS.
- B. Li, B. Li, X. Zhu and Y. Zhang, Inorg. Chem. Commun., 2003, 6, 1304–1306 CrossRef CAS.
- J. Meng, X. Wang, E. Wang, H. Fu and Y. Zhong, Transit. Met. Chem., 2009, 34, 361–366 CrossRef CAS.
- S. Menon and M. V. Rajasekharan, Inorg. Chem., 1997, 36, 4983–4987 CrossRef CAS.
- H. Pang, J. Sha, J. Peng, A. Tian, C. Zhang, P. Zhang, Y. Chen, M. Zhu and Y. Wang, Inorg. Chem. Commun., 2009, 12, 735–738 CrossRef CAS.
- H. Pang, C. Zhang, J. Peng, Y. Wang, J. Sha, A. Tian, P. Zhang, Y. Chen, M. Zhu and Z. Su, Eur. J. Inorg. Chem., 2009, 5175–5180 CrossRef CAS.
- M. N. Patel, B. S. Bhatt and P. A. Dosi, Chem. Biodiversity, 2012, 9, 2810–2824 CAS.
- S. Shao, Y. Zhu, K. Ma, H. Zhao and Y. Qiu, J. Coord. Chem., 2013, 66, 2702–2711 CrossRef CAS.
- A. Günyar and F. E. Kühn, J. Mol. Catal. A: Chem., 2010, 319, 108–113 CrossRef.
- F. M. Emen, K. Ocakoglu and N. Kulcu, J. Therm. Anal. Calorim., 2012, 110, 799–805 CrossRef CAS.
- W. B. Heuer, H. Xia, M. Abrahamsson, Z. Zhou, S. Ardo, A. A. Narducci Sarjeant and G. J. Meyer, Inorg. Chem., 2010, 49, 7726–7734 CrossRef CAS PubMed.
- H. Jiang, E. Stepowska and D. Song, Dalton Trans., 2008, 5879–5881 RSC.
- K. Ocakoglu, E. Harputlu, P. Guloglu and S. Erten-Ela, Inorg. Chem. Commun., 2012, 24, 118–124 CrossRef CAS.
- A. M. Pyle, J. P. Rehmann, R. Meshoyrer, C. V. Kumar, N. J. Turro and J. K. Barton, J. Am. Chem. Soc., 1989, 111, 3051–3058 CrossRef CAS.
- W. K. Seok, A. K. Gupta, S. Roh, W. Lee and S. Han, Bull. Korean Chem. Soc., 2007, 28, 1311–1316 CrossRef CAS.
- Y. Wang and D. P. Rillema, Inorg. Chem. Commun., 1998, 1, 27–29 CrossRef CAS.
- Y. Wang and D. P. Rillema, Tetrahedron Lett., 1997, 38, 6627–6630 CrossRef CAS.
- Y. Wang, W. Perez, G. Y. Zheng and D. P. Rillema, Inorg. Chem., 1998, 37, 2051–2059 CrossRef CAS.
- G. Yang, L. Ji, X. Zhou and Z. Zhou, Transit. Met. Chem., 1998, 23, 273–276 CrossRef CAS.
- A. N. Campbell, P. B. White, I. A. Guzei and S. S. Stahl, J. Am. Chem. Soc., 2010, 132, 15116–15119 CrossRef CAS PubMed.
- A. N. Campbell, E. B. Meyer and S. S. Stahl, Chem. Commun., 2011, 47, 10257–10259 RSC.
- A. N. Campbell and S. S. Stahl, Acc. Chem. Res., 2012, 45, 851–863 CrossRef CAS PubMed.
- T. Diao and S. S. Stahl, Polyhedron, 2014, 84, 96–102 CrossRef CAS PubMed.
- T. Diao, T. J. Wadzinski and S. S. Stahl, Chem. Sci., 2012, 3, 887–891 RSC.
- C. J. Elsevier, Coord. Chem. Rev., 1999, 185–6, 809–822 CrossRef.
- W. Gao, Z. He, Y. Qian, J. Zhao and Y. Huang, Chem. Sci., 2012, 3, 883–886 RSC.
- R. A. Klein, C. J. Elsevier and F. Hartl, Organometallics, 1997, 16, 1284–1291 CrossRef CAS.
- R. A. Klein, R. van Belzen, K. Vrieze, C. J. Elsevier, R. P. Thummel, J. Fraanje and K. Goubitz, Collect. Czech. Chem. Commun., 1997, 62, 238–256 CrossRef CAS.
- M. Piotrowicz and J. Zakrzewski, Organometallics, 2013, 32, 106–109 CrossRef.
- J. W. Sprengers, M. de Greef, M. A. Duin and C. J. Elsevier, Eur. J. Inorg. Chem., 2003, 3811–3819 CrossRef CAS.
- A. Vasseur, C. Laugel, D. Harakat, J. Muzart and J. Le Bras, Eur. J. Org. Chem., 2015, 944–948 CrossRef CAS.
- A. B. Weinstein and S. S. Stahl, Catal. Sci. Technol., 2014, 4, 4301–4307 CAS.
- Y. Xia, Z. Miao, F. Wang, H. Yao, M. Cui, Y. Ma, Z. Qi and Y. Sun, J. Organomet. Chem., 2015, 779, 81–85 CrossRef CAS.
- A. R. Biju and M. V. Rajasekharan, Polyhedron, 2008, 27, 2065–2068 CrossRef CAS.
- A. A. Massoud, Y. M. Gohar, V. Langer, P. Lincoln, F. R. Svensson, J. Jänis, S. T. Gårdebjer, M. Haukka, F. Jonsson, E. Aneheim, P. Löwenhielm, M. A. M. Abu-Youssefe and L. R. Öhrström, New J. Chem., 2011, 35, 640–648 RSC.
- J. Meng, X. Wang, Y. Ma, E. Wang and X. Xu, J. Coord. Chem., 2008, 61, 2853–2860 CrossRef CAS.
- B. Machura, I. Nawrot and K. Michalik, Polyhedron, 2012, 31, 548–557 CrossRef CAS.
- B. Machura, M. Wolff and A. Świtlicka, Inorg. Chem. Commun., 2011, 14, 17–21 CrossRef CAS.
- B. Tong, F. Wu, Q. Mei and Q. Zhang, Z. Naturforsch., 2010, 65, 511–515 CAS.
- S. Dang, J. Yu, X. Wang, Z. Guo, L. Sun, R. Deng, J. Feng, W. Fan and H. Zhang, J. Photochem. Photobiol., A, 2010, 214, 152–160 CrossRef CAS.
- S. Dang, J. Yu, X. Wang, L. Sun, R. Deng, J. Feng, W. Fan and H. Zhang, J. Lumin., 2011, 131, 1857–1863 CrossRef CAS.
- S. Dang, J. Yu, J. Yu, X. Wang, L. Sun, J. Feng, W. Fan and H. Zhang, Mater. Lett., 2011, 65, 1642–1644 CrossRef CAS.
- A. Tian, Z. Han, J. Peng, J. Ying, J. Sha, B. Dong, J. Zhai and H. Liu, Inorg. Chim. Acta, 2008, 361, 1332–1338 CrossRef CAS.
- K. Shao, Y. Zhao, Y. Lan, X. Wang, Z. Su and R. Wang, CrystEngComm, 2011, 13, 889–896 RSC.
- C. Liu, S. Gao, D. Zhang, Y. Huang, R. Xiong, Z. Liu, F. Jiang and D. Zhu, Angew. Chem., Int. Ed., 2004, 43, 990–994 CrossRef CAS PubMed.
- J. Cano, F. A. Mautner, C. Berger, R. C. Fischer and R. Vicente, Polyhedron, 2013, 50, 240–245 CrossRef CAS.
- C. Wang, C. Yang, S. Tseng, S. Lin, T. Wu, M. Fuh, G. Lee, K. Wong, R. Chen, Y. Cheng and P. Chou, Inorg. Chem., 2004, 43, 4781–4783 CrossRef CAS PubMed.
- P. Peng, F. Li, F. L. Bowles, V. S. P. K. Neti, A. J. Metta-Magana, M. M. Olmstead, A. L. Balch and L. Echegoyen, Chem. Commun., 2013, 49, 3209–3211 RSC.
- A. Pal, B. Biswas, M. Mitra, C. S. Purohit, C. Lin and R. Ghosh, J. Chem. Sci., 2014, 126, 717–725 CrossRef CAS.
- E. Noh, S. Park, S. Kang, J. Y. Lee and J. H. Jung, Chem. – Eur. J., 2013, 19, 2620–2627 CrossRef CAS PubMed.
- S. Wang, Z. Sun, C. Zhang, L. Ni, C. Wang, Y. Gao, L. Lv, J. Chang and W. Hao, Inorg. Chem. Commun., 2014, 41, 47–50 CrossRef CAS.
- B. Askevold, H. W. Roesky and S. Schneider, ChemCatChem, 2012, 4, 307–320 CrossRef CAS.
- S. Schneider, J. Meiners and B. Askevold, Eur. J. Inorg. Chem., 2012, 412–429 CrossRef CAS.
- C. Gunanathan and D. Milstein, Acc. Chem. Res., 2011, 44, 588–602 CrossRef CAS PubMed.
- C. Gunanathan and D. Milstein, Top. Organomet. Chem., 2011, 37, 55–84 CrossRef CAS.
- J. I. van der Vlugt, Eur. J. Inorg. Chem., 2012, 363–375 CrossRef CAS.
- J. I. van der Vlugt and J. N. H. Reek, Angew. Chem., Int. Ed., 2009, 48, 8832–8846 CrossRef CAS PubMed.
- R. H. Crabtree, New J. Chem., 2011, 35, 18–23 RSC.
- V. T. Annibale and D. Song, RSC Adv., 2013, 3, 11432–11449 RSC.
- J. Ballmann, R. F. Munhá and M. D. Fryzuk, Chem. Commun., 2010, 46, 1013–1025 RSC.
- V. T. Annibale and D. Song, Chem. Commun., 2012, 48, 5416–5418 RSC.
- V. T. Annibale and D. Song, Organometallics, 2014, 33, 2776–2783 CrossRef CAS.
- A. Baysal, M. Aydemir, F. Durap, S. Özkar, L. T. Yildirim and Y. S. Ocak, Polyhedron, 2015, 89, 55–61 CrossRef CAS.
- Z. Wang, X. Li and Y. Huang, Angew. Chem., Int. Ed., 2013, 52, 14219–14223 CrossRef CAS PubMed.
- Z. Wang, L. Li and Y. Huang, J. Am. Chem. Soc., 2014, 136, 12233–12236 CrossRef CAS PubMed.
- M. A. Zolfigol, T. Azadbakht, V. Khakyzadeh, R. Nejatyami and D. M. Perrin, RSC Adv., 2014, 4, 40036–40042 RSC.
- R. K. Sharma, S. Sharma and G. Gaba, RSC Adv., 2014, 4, 49198–49211 RSC.
- L. De Cola, V. Balzani, F. Barigelletti, L. Flamigni, P. Belser and S. Bernhard, Recl. Trav. Chim. Pays-Bas, 1995, 114, 534–541 CrossRef CAS.
- R. N. Warrener, A. B. B. Ferreira, A. C. Schultz, D. N. Butler, F. R. Keene and L. S. Kelso, Angew. Chem., Int. Ed. Engl., 1996, 35, 2485–2487 CrossRef CAS.
- R. N. Warrener, A. C. Schultz, M. A. Houghton and D. N. Butler, Tetrahedron, 1997, 53, 3991–4012 CrossRef CAS.
- Y. Wang, W. J. Perez, G. Y. Zheng, D. P. Rillema and C. L. Huber, Inorg. Chem., 1998, 37, 2227–2234 CrossRef CAS PubMed.
- F. Cheng, C. He, H. Yin and N. Tang, Transit. Met. Chem., 2013, 38, 259–265 CrossRef CAS.
- F. Cheng, C. He, H. Yin, N. Tang and N. Hou, Z. Anorg. Allg. Chem., 2013, 639, 1284–1290 CrossRef CAS.
- F. Cheng, J. Chen, F. Wang, N. Tang and L. Chen, J. Coord. Chem., 2012, 65, 205–217 CrossRef CAS.
- F. Cheng, J. Chen, C. Sun, N. Tang and L. Chen, Z. Anorg. Allg. Chem., 2011, 637, 160–166 CrossRef CAS.
- F. Cheng, J. Chen, F. Wang, N. Tang and L. Chen, Transit. Met. Chem., 2011, 36, 573–578 CrossRef CAS.
- F. Cheng, N. Tang, J. Chen, F. Wang and L. Chen, Z. Naturforsch. B, 2011, 66, 923–929 CrossRef CAS.
- F. Cheng, N. Tang, J. Chen, L. Chen, L. Jia and G. Chen, Inorg. Chem. Commun., 2010, 13, 258–261 CrossRef CAS.
- F. Cheng, L. Chen, G. Bo and N. Tang, Inorg. Chem. Commun., 2009, 12, 227–230 CrossRef CAS.
- F. Cheng, L. Chen, J. Zhang, J. Wang and N. Tang, Inorg. Chem. Commun., 2009, 12, 728–730 CrossRef CAS.
- F. Cheng and N. Tang, Inorg. Chem. Commun., 2008, 11, 939–942 CrossRef CAS.
- F. Cheng and N. Tang, Inorg. Chem. Commun., 2008, 11, 243–245 CrossRef CAS.
- F. Cheng, N. Tang and L. Chen, Z. Anorg. Allg. Chem., 2008, 634, 1608–1612 CrossRef CAS.
- F. Cheng, C. He, L. Yao, F. Wang and N. Tang, J. Coord. Chem., 2015, 68, 704–716 CrossRef CAS.
- K. Ocakoglu, S. Sogut, H. Sarica, P. Guloglu and S. Erten-Ela, Synth. Met., 2013, 174, 24–32 CrossRef CAS.
- W. B. Heuer, H. Xia, W. Ward, Z. Zhou, W. H. Pearson, M. A. Siegler, A. A. Narducci Sarjeant, M. Abrahamsson and G. J. Meyer, Inorg. Chem., 2012, 51, 3981–3988 CrossRef CAS PubMed.
- A. Staniszewski, W. B. Heuer and G. J. Meyer, Inorg. Chem., 2008, 47, 7062–7064 CrossRef CAS PubMed.
- M. Abrahamsson, J. H. J. Hedberg, H. Becker, A. Staniszewski, W. H. Pearson, W. B. Heuer and G. J. Meyer, ChemPhysChem, 2014, 15, 1154–1163 CrossRef CAS PubMed.
- R. Sivakumar, A. Manivel, M. Meléndrez, J. Martínez-Oyanedel, M. Bunster, C. Vergara and P. Manidurai, Polyhedron, 2015, 87, 135–140 CrossRef CAS.
- K. Ono, H. Tanaka, M. Shiozawa, T. Motohiro, S. Kunikane and K. Saito, Chem. Lett., 2007, 36, 892–893 CrossRef CAS.
- H. Su, F. Fang, T. Hwu, H. Hsieh, H. Chen, G. Lee, S. Peng, K. Wong and C. Wu, Adv. Funct. Mater., 2007, 17, 1019–1027 CrossRef CAS.
- H. Su, H. Chen, F. Fang, C. Liu, C. Wu, K. Wong, Y. Liu and S. Peng, J. Am. Chem. Soc., 2008, 130, 3413–3419 CrossRef CAS PubMed.
- H. Chen, K. Wong, Y. Liu, Y. Wang, Y. Cheng, M. Chung, P. Chou and H. Su, J. Mater. Chem., 2011, 21, 768–774 RSC.
- H. Chen, W. Hung, S. Chen, T. Wang, S. Lin, S. Chou, C. Liao, H. Su, H. Pan, P. Chou, Y. Liu and K. Wong, Inorg. Chem., 2012, 51, 12114–12121 CrossRef CAS PubMed.
- X. Chen, X. Zhu, Y. Xu, S. S. S. Raj, S. Öztürk, H. Fun, J. Ma and X. You, J. Mater. Chem., 1999, 9, 2919–2922 RSC.
- Z. Liu, F. Wen and W. Li, Thin Solid Films, 2005, 478, 265–270 CrossRef CAS.
- K. Z. Wang, L. H. Gao, C. H. Huang, G. Q. Yao, X. S. Zhao, X. H. Xia, J. M. Xu and T. K. Li, Solid State Commun., 1996, 98, 1075–1079 CrossRef CAS.
- S. Wang, J. Zhang, Y. Hou, C. Du and Y. Wu, J. Mater. Chem., 2011, 21, 7559–7561 RSC.
- X. Li, D. Zhang, W. Li, B. Chu, L. Han, T. Li, Z. Su, J. Zhu, S. Wu, Y. Chen, P. Lei, Z. Hu and Z. Zhang, Synth. Met., 2009, 159, 1340–1344 CrossRef CAS.
- X. Li, D. Zhang, W. Li, B. Chu, L. Han, T. Li, Z. Su, J. Zhu, Y. Chen, Z. Hu, P. Lei and Z. Zhang, Opt. Mater., 2009, 31, 1173–1176 CrossRef CAS.
- X. Li, D. Zhang, H. Chi, G. Xiao, Y. Dong, S. Wu, Z. Su, Z. Zhang, P. Lei, Z. Hu and W. Li, Appl. Phys. Lett., 2010, 97, 263303 CrossRef.
- X. Li, H. Chi, G. Lu, G. Xiao, Y. Dong, D. Zhang, Z. Zhang and Z. Hu, Org. Electron., 2012, 13, 3138–3144 CrossRef CAS.
- Q. Zhang, J. Ding, Y. Cheng, L. Wang, Z. Xie, X. Jing and F. Wang, Adv. Funct. Mater., 2007, 17, 2983–2990 CrossRef CAS.
- O. Crespo, M. C. Gimeno, A. Laguna, R. Marriott, J. M. Sáez-Rocher and M. D. Villacampa, Dalton Trans., 2014, 43, 12214–12220 RSC.
- C. Dragonetti, A. Valore, A. Colombo, S. Righetto, G. Rampinini, F. Colombo, L. Rocchigiani and A. Macchioni, Inorg. Chim. Acta, 2012, 382, 72–78 CrossRef CAS.
- A. Valore, M. Balordi, A. Colombo, C. Dragonetti, S. Righetto, D. Roberto, R. Ugo, T. Benincori, G. Rampinini, F. Sannicolò and F. Demartin, Dalton Trans., 2010, 39, 10314–10318 RSC.
- B. W. Chu and V. W. Yam, Inorg. Chem., 2001, 40, 3324–3329 CrossRef CAS PubMed.
- V. W. W. Yam, K. Wang, C. Wang, Y. Yang and K. Cheung, Organometallics, 1998, 17, 2440–2446 CrossRef CAS.
- A. Colombo, C. Dragonetti, S. Righetto, D. Roberto, A. Valore, T. Benincori, F. Colombo and F. Sannicolò, J. Mater. Chem., 2012, 22, 19761–19766 RSC.
- K. Sako, T. Kakehi, S. Nakano, H. Oku, X. F. Shen, T. Iwanaga, M. Yoshikawa, K. Sugahara, S. Toyota, H. Takemura, T. Shinmyozu, M. Shiotsuka and H. Tatemitsu, Tetrahedron Lett., 2014, 55, 749–752 CrossRef CAS.
- I. Erden, A. Erdoğmuş, N. Demirhan and U. Avciata, Transit. Met. Chem., 2008, 33, 439–442 CrossRef CAS.
- Z. Lin, Y. Zhao, C. Duan, B. Zhang and Z. Bai, Dalton Trans., 2006, 3678–3684 RSC.
- P. Lam, G. Lu, K. Hon, K. Lee, C. Ho, X. Wang, J. C. O. Tang, K. Lam, R. S. M. Wong, S. H. L. Kok, Z. Bian, H. Li, K. Lee, R. Gambari, C. Chui and W. Wong, Dalton Trans., 2014, 43, 3949–3957 RSC.
- A. A. Massoud, Y. M. Gohar, V. Langer, P. Lincoln, F. R. Svensson, J. Janis, S. T. Gardebjer, M. Haukka, F. Jonsson, E. Aneheim, P. Lowenhielm, M. A. M. Abu-Youssefe and L. R. Ohrstrom, New J. Chem., 2011, 35, 640–648 RSC.
- B. J. Kraft, H. J. Eppley, J. C. Huffman and J. M. Zaleski, J. Am. Chem. Soc., 2002, 124, 272–280 CrossRef CAS PubMed.
- H. J. Eppley, S. M. Sato, A. D. Ellington and J. M. Zaleski, Chem. Commun., 1999, 2405–2406 RSC.
- H. J. Eppley, V. J. Isada, S. M. Lato, J. C. Huffman, A. D. Ellington and J. M. Zaleski, J. Inorg. Biochem., 1999, 74, 124–124 Search PubMed.
Footnote |
| † Current Address: Department of Chemistry, The University of British Columbia, 2036 Main Mall, Vancouver, British Columbia, Canada V6T 1Z1. |
| This journal is © The Royal Society of Chemistry 2016 |