
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCationic rhenium complexes ligated with N-heterocyclic carbenes – an overview
Claudia
Hille
and
Fritz E.
Kühn
*
Chair of Inorganic Chemistry/Molecular Catalysis, Department of Chemistry, Catalysis Research Center, Technische Universität München, Lichtenbergstr. 4, D-85747 Garching bei München, Germany. E-mail: fritz.kuehn@ch.tum.de; Fax: +49 89 289 13473; Tel: +49 89 289 13096
First published on 4th November 2015
Abstract
This review provides an overview of the currently known cationic rhenium NHC complexes. Synthesis, structures and properties are described. The title compounds are potential candidates for both catalytic and medical applications. Besides the variety of ancillary ligands, which are in some cases easily substituted, functionalization can be carried out in the side chain or at the backbone of the carbene ligand as well as – in the case of biscarbene ligands – at the bridging moiety. Cationic Re NHC complexes are promising precursors for radiopharmaceuticals and diagnostics – not only because of the possibility to radiolabel the metal (steps in this direction have been made and described already) – but rather the opportunity to link the complexes to biomolecules via the different possibilities provided by the ligands. The development of OLEDs based on luminescent Re(I) carbene complexes renders another potential application.
1. Introduction
In recent years several overview articles and reviews concerning the metal rhenium and NHCs – including their applications in catalysis and radiochemistry – as well as group 7 metal–NHC complexes have been published.1The profound interest in this research area is due to the fact that rhenium is cheaper than most of the noble metals which are commonly used in catalysis, and it can bear a variety of different ligand systems1h and has two isotopes, which can be easily produced – even directly in hospitals – which are therefore suitable for radiotherapeutic purposes.2 Rhenium further provides an almost unrivalled variety of oxidation states that are accessible under “non-exotic” reaction conditions.3
1.1 Pharmaceuticals
The reactor-produced radionuclide 186Re with a half-life of 89.2 h and moderate emission energy (Emax/averageβ = 1.1/0.36 MeV; Eγ = 137 keV) can be received in analogy to 188Re (Emax/averageβ = 2.1/0.8 MeV; Eγ = 155 keV), which is produced by using a 188W/188Re generator system in the form of Na[188ReO4].2a,b Although both isotopes are able to transfer energy to cancer tissue and can be identified by single photon emission computed tomography imaging, 188Re features a more convenient half-life time (t1/2 = 16.9 h).1d,2c,4The application of radioactive rhenium complexes is variegated – not only for treatment of cancer (bone, liver, ovarian, breast etc.) but also for inflammable joint diseases (e.g. arthritis).5 For nuclear medicine therapies like radiation synovectomy 186/188Re sulfur colloids have been applied and 188Re–tin colloids are already in clinical trials.5c,s–u188Re-HEDP therapy for bone pain palliation proved to be effective5e and Lipiodol with radiolabelled rhenium is in clinical trials for the metabolic radiotherapy of heptacellular carcinoma.5d,f For this most common type of liver cancer several complexes with a 186/188rhenium(V) oxo scaffold are known.5h–k The design of radio labeled antibodies with rhenium is popular with respect to radio immunotherapy.5a,l,m,6 Labeling of MAG3 peptide conjugates with rhenium oxo cores as the chelate site – for instance 186Re-MAG3-HBP is expected to be a useful radiopharmaceutical for the palliation of metastatic bone pain5n or 186/188Re(CO)3-A7 showed high uptake in tumor-bearing mice.5p Besides the conventional cancer therapy, Re(I)(CO)3 derivatives showing anticancer activity are promising candidates as organometallic therapeutic and diagnostic agents.7
1.2 Catalysis
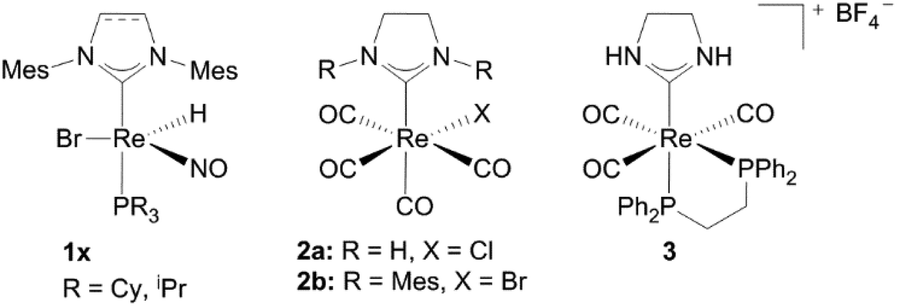
Although rhenium complexes are known in different oxidation states (−III to +VII) and with a variety of ligand systems, particularly rhenium oxo and carbonyl complexes are established in catalysis.1h,8 Besides the oxygen atom transfer reaction – for instance deoxygenation reactions as well as dehydration or deoxydehydration reactions can be catalyzed by Re(VII) complexes,8a,i rhenium carbonyl complexes can act as catalysts in quite a number of different reactions e.g. hydroarylations, transfer-hydrogenation reactions and formation of carbon–carbon or carbon–heteroatom bonds, which were summarized by Kuninobu and Takai in 2011.1h,8k,lNHC complexes with almost every metal in the periodic table are known as homogeneous as well as heterogeneous catalysts.9 In addition to the “classical” catalysis in organic solvents the field of catalysis in aqueous media or water became more interesting in the past few years.1g,10 However, only two groups have reported on rhenium carbene complexes as catalysts so far.11 Rhenium(I) hydride complexes 1x catalyze the dehydro coupling of Me2NH·BH3 and ammonia borane NH3·BH3 as well as the transfer hydrogenation of several olefins, where Me2NH·BH3 donates hydrogen to produce the corresponding alkanes.11a Liu et al. tested the catalytic activity of several rhenium carbonyl complexes in the insertion reaction of terminal alkynes into β keto esters. Complexes 2, 3 and [(NHC)Re2(CO)9] (NHC = 1,3-bis(hydro)-4,5-dihydroimidazol-2-ylidene) are able to catalyze the formation of ethyl(2E)-2-methyl-5-oxo-3-phenyl-2-hexenoate but need the support of photoirradiation and show only moderate activities (Fig. 1).11b
Although the metal rhenium is known since 1925, significant amounts were accessible only since the 1950s.12 Synthesis routes of a broad variety of rhenium complexes are well established since then and some general reviews were published, this review focuses on the synthesis and properties of cationic rhenium NHC complexes – particularly with regard to their potential application in medicine.
2. Cationic rhenium mono-NHC complexes
2.1 Re(I) complexes bearing saturated mono-NHC ligands
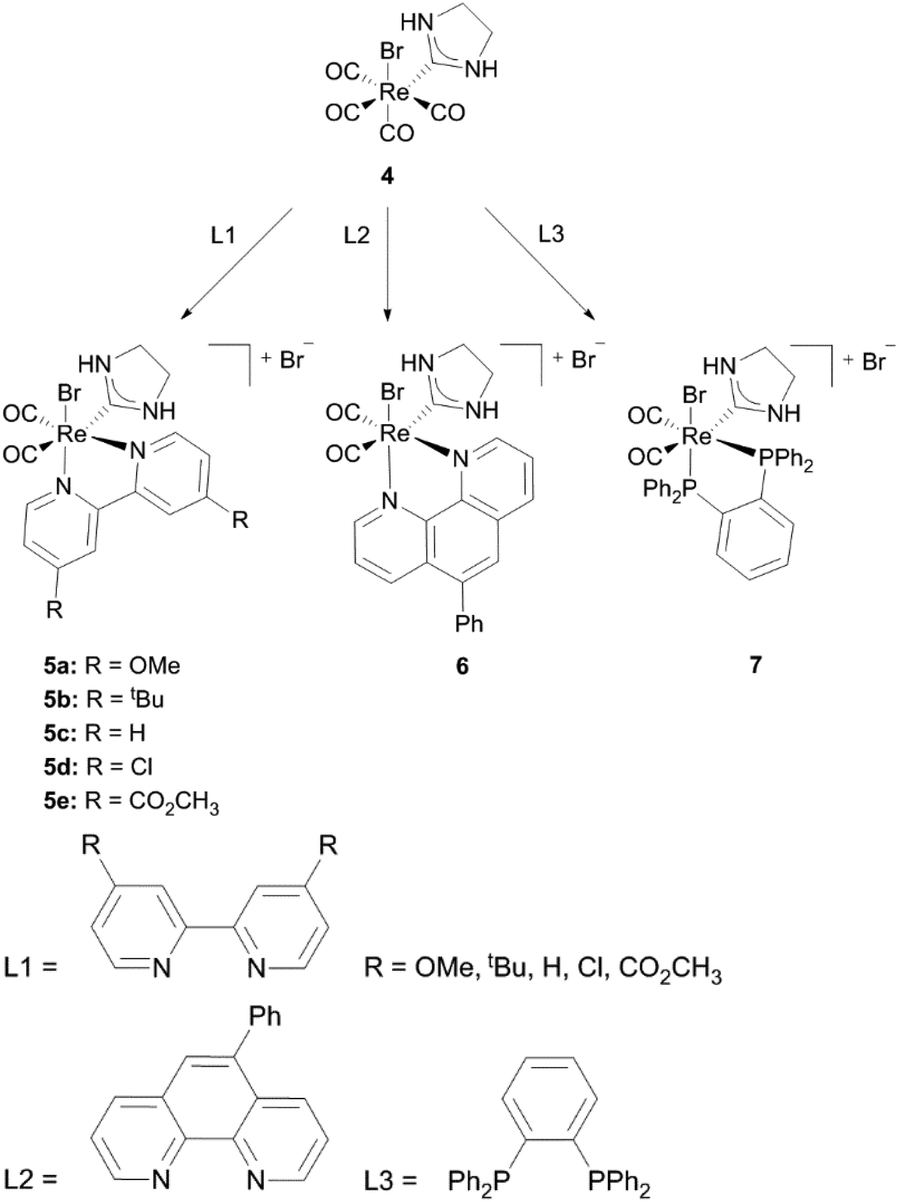
The Re(CO)3 fragment, which can be found in the majority of cationic Re(I) NHC complexes, has attracted attention concerning luminescence for potential application in light emitting diodes (LEDs), organic light emitting diodes (OLEDs) and biological labelling markers.1a,13The first cationic Re(I) NHC complexes were isolated by Che and coworkers.14 These compounds contain bidentate diimine or phosphine ligands and could be obtained by the reaction of the corresponding diimines (L1, L2) or chelating phosphines (L3) with the literature known complex 4.15 All complexes (5–7) are assumed to display a pseudo-octahedral geometry, although only the molecular structures of 5c, 5e and 7 have been determined by X-ray analysis. Based on molecular orbital calculations and photophysical studies it is found out that changing the electron-donating/accepting ability of the bidentate diimine ligand via modification of the electron withdrawing character of the R group leads to reasonable emission lifetimes, quantum yields and well defined redox potentials. The metal to ligand charge transfer (MLCT) excited stage energy is strongly influenced by the R group of complexes 5 and can only be altered by changing the polarity of the solvent at room temperature. However a change to intra-ligand (IL) charge transfer occurs at 77 K in the case of the Phphen ligand. At this temperature all complexes were found to be emitting, nevertheless only complexes 5 and 6 maintained this behaviour at room temperature.14
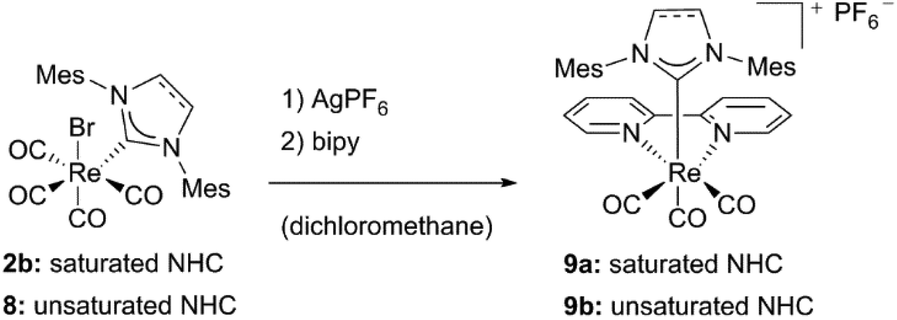
Related to complex 5c, bipyridine NHC Re(I) complexes bearing mesityl substituted ligands could be isolated by the same group.16 In order to achieve substitution with bipyridine, addition of a silver salt has been necessary. Liu et al. ascribe this to the steric bulkiness of the ligands, which may exacerbate the replacement of the CO ligands. Both the saturated and unsaturated compounds 9a and 9b have been characterized by NMR, IR and mass spectrometry, but no crystal structures are reported (Schemes 1 and 2).16
 | ||
| Scheme 1 Luminescent cationic Re(I) monocarbene complexes synthesized by Che and coworkers.14 | ||
 | ||
| Scheme 2 Synthesis of bipyridine NHC Re(I) complexes by Liu and coworkers.16 | ||
A cationic macrocycle substituted Re(I) complex with a coordinated PCC ligand was achieved via template (controlled) synthesis in 2007.17 Starting from Re(I) template 2a and a fluorinated diphosphine (1,2-bis(di(o-fluorophenyl)phosphine)benzene) a bidentate phosphine complex 10a could be formed selectively in acetonitrile. Efforts to carry out this synthesis according to the literature-known conditions14 with similar diphosphine resulted in mixtures of complexes 10a, 11 and 12. This formation of mixtures was ascribed to the simultaneously activation of NH and CF, respectively the insolubility of the products in solvents of low polarity (Scheme 3).18
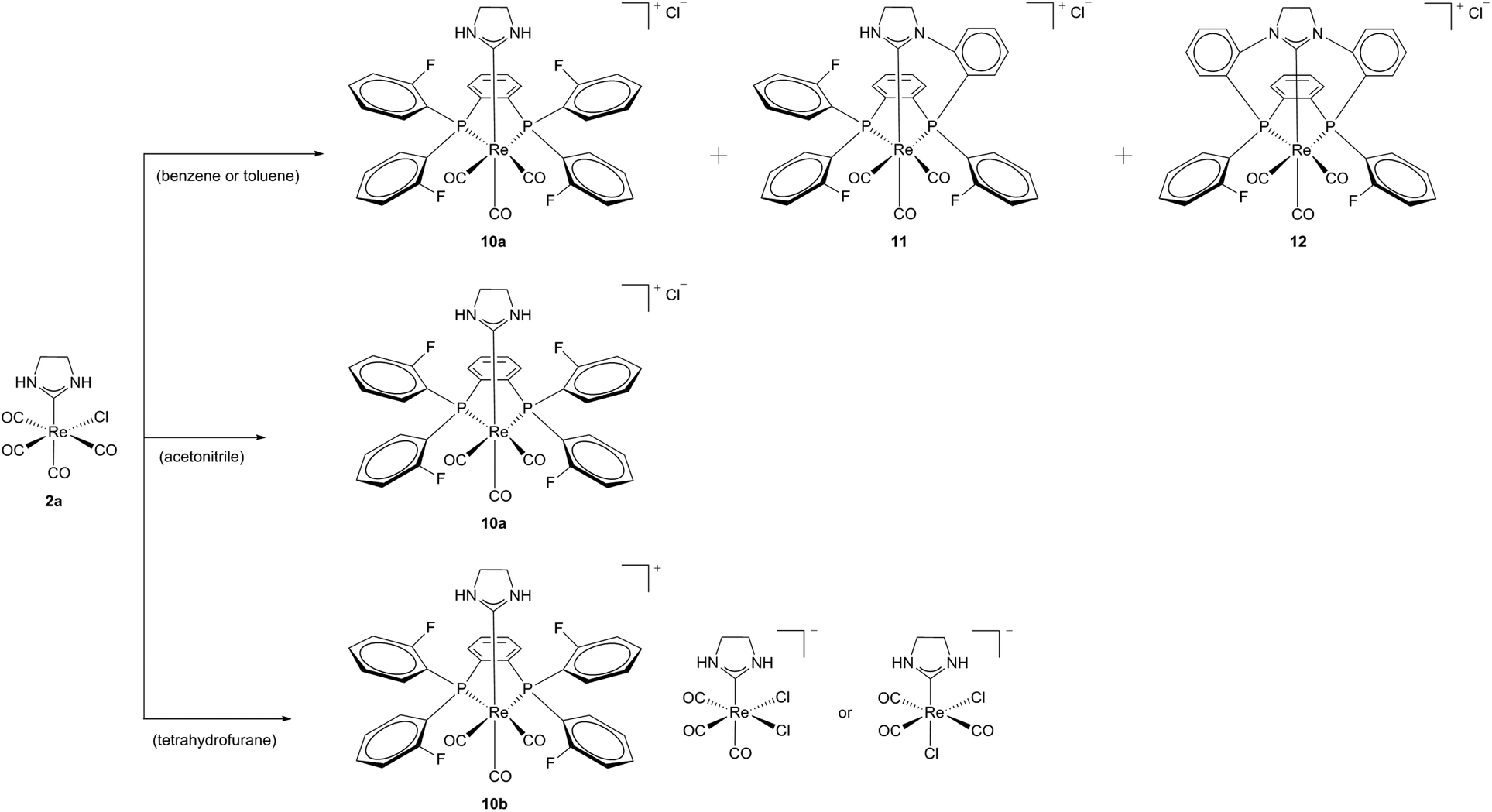 | ||
| Scheme 3 Products of the reaction of 2a with a fluorinated diphosphine in various solvents.18 | ||
The air and water stable macrocyclic [11]ane-P2CNHC Re(I) complex 12 was obtained via the template controlled ring formation reaction via the isolatable neutral intermediate 13.17,18 Deprotonation of 10a and a SN2Ar type attack of the deprotonated nitrogen atoms at the C–F bond of the o-fluorenyl diphosphine connects it to the carbene ligand, resulting in a macrocyclic and polydentate NHC complex 12 (Scheme 4).17–19
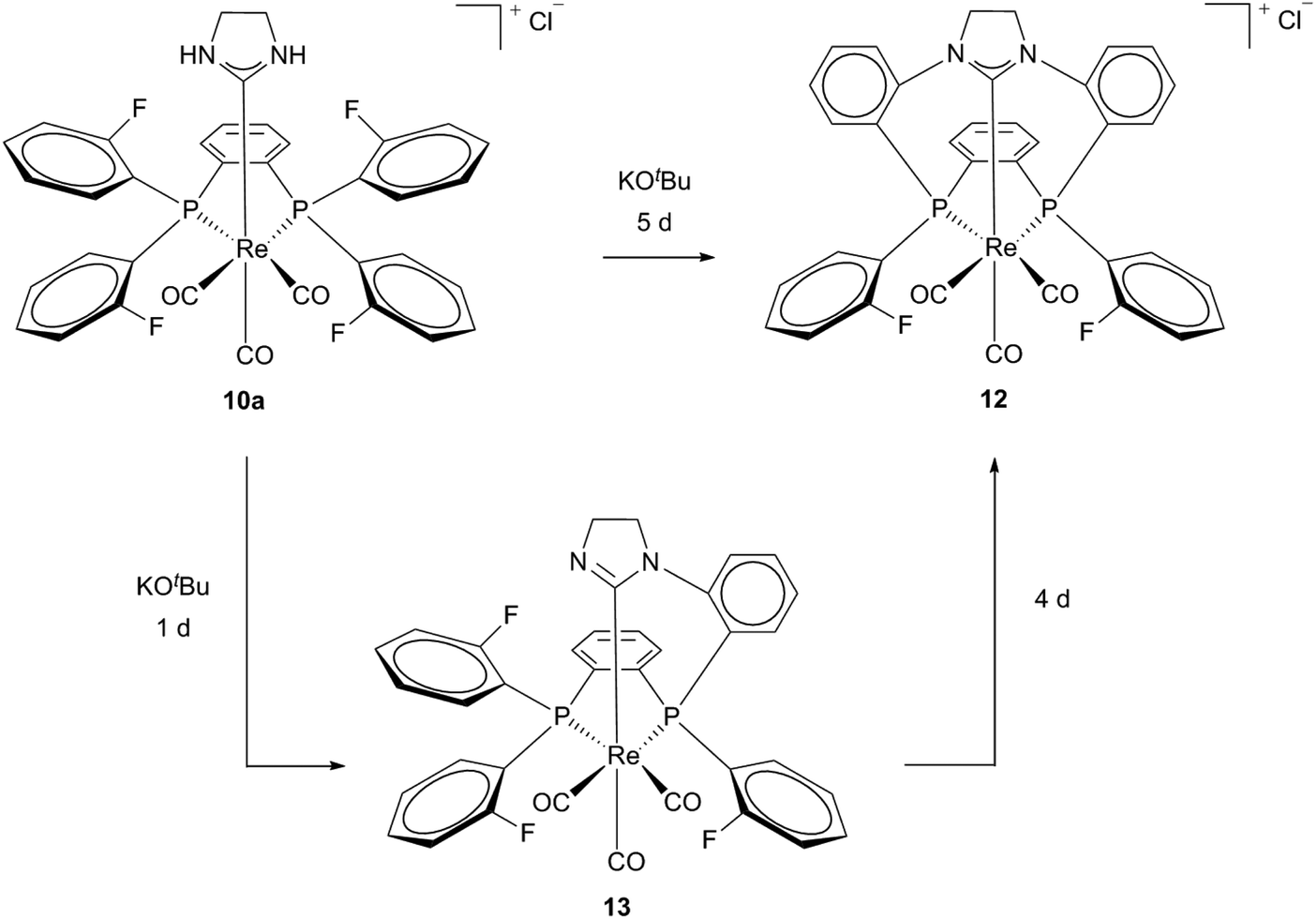 | ||
| Scheme 4 Generation of macrocyclic polydentate Re(I) NHC complex 12via neutral intermediate 13.17–19. | ||
In addition, cationic Re(I) macrocycles with a mixed NHC/phosphine donor set (14, 15) could be obtained by the same group. Using the same method of template synthesis ethylene bridged diphosphines and NHC ligands could be facially coordinated to the metal. No intermediates have been reported (Scheme 5).20
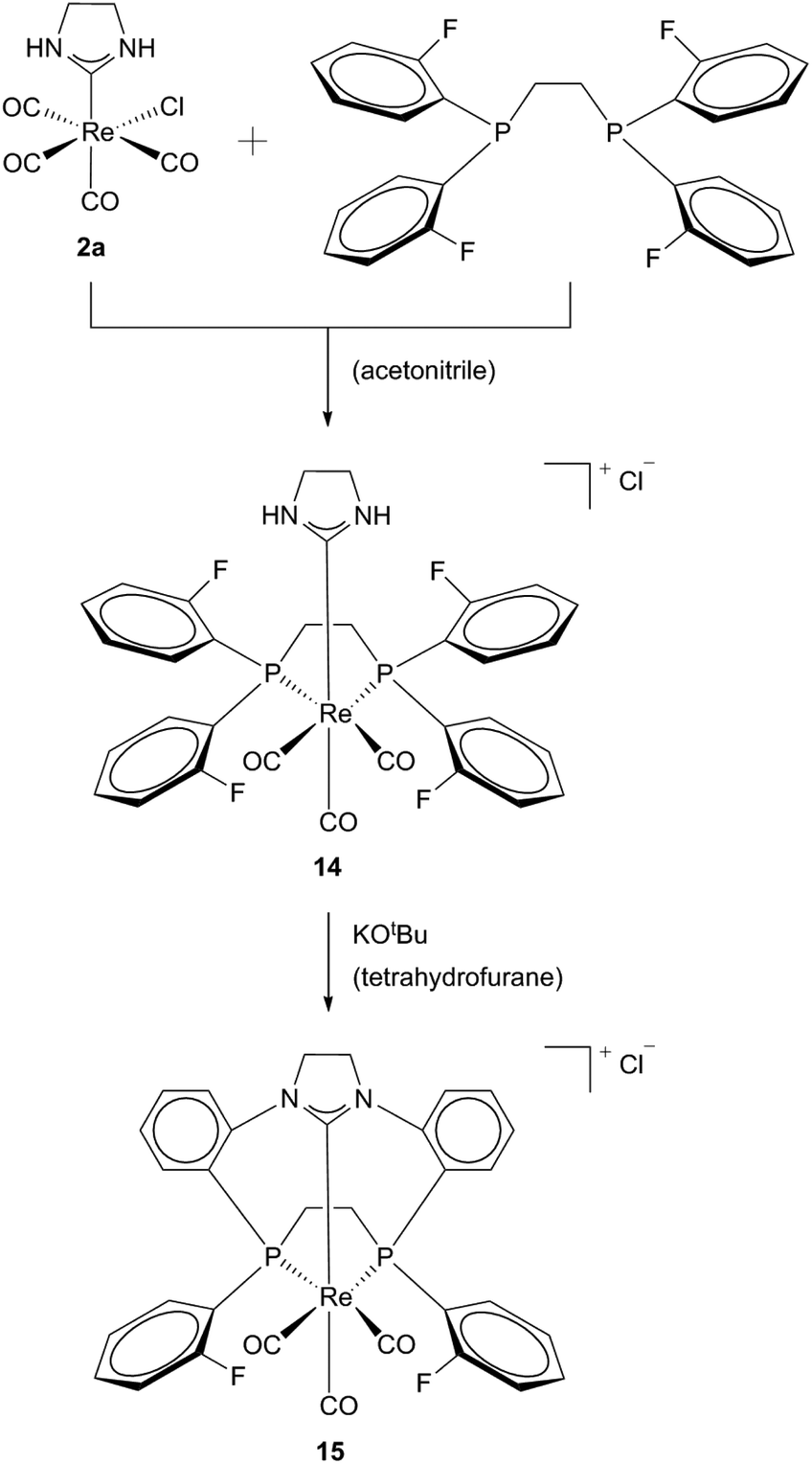 | ||
| Scheme 5 Synthesis of Re(I)-[11]-ane-P2CNHC macrocycles by Hahn et al.20 | ||
As a comparatively easy method, to generate carbene complexes of different metals, is known since a couple of years.1i Using functionalized isocyanides, which contain – beside the isocyanide moiety – a nucleophilic part, leads via intramolecular 1,2 addition usually to imidazolidin-2-ylidenes.17,21
Hahn and coworkers established a cationic tris-NHC–Re(I) by template controlled cyclization of a β-azido functionalized isocyanide.22 As the metal template they used complex 17,15 which contains besides strong π-acceptor CO-ligands two strong σ-donor NHCs and one bromide ligand, so that the abstraction of the halide followed by attaching 2-azidoethyl isocyanide at the ‘free coordination site’ yields complex 18. IR spectroscopy revealed that the functionalized isocyanide is activated for a nucleophilic attack upon coordination to the rhenium core. As a result the formation of tris(NHC) complex 19 could be achieved by reduction and of the azido function and ensuing intramolecular cyclization (Scheme 6).22
 | ||
| Scheme 6 Synthesis of Re(I) monocarbene complexes via cyclization reactions.22 | ||
2.2 Re(I) complexes bearing unsaturated mono-NHC-ligands
Formation of cationic agostic and solvent stabilized 16 electron complexes via halide abstraction was achieved by Whittlesey et al.23 By reacting neutral mono-Re NHC precursors (20, 23) with NaBAr4F salts, complexes 21 and 24 are generated. Attempts to isolate agostic product 21 failed leading to an undefined rhenium carbonyl species and the additionally appearing tetracarbonyl compound [Re(IiPr2Me2)2(CO)4]BAr4F (22). Directly bubbling of CO into the reaction mixture of 20 and NaBAr4F leads to selective formation of an 18 electron complex cis-22. Addition of acetonitrile to the reaction mixture instead of CO results in the formation of [Re(IiPr2Me2)2(CO)3(MeCN)]BAr4F. These observations suggest that even less weakly coordinating ligands are able to displace the agostic interactions in complex 21. As a less electrophilic bis-NHC species 21 is stabilized via the agnostic bond of one NHC, monocarbene complex 24, which contains a less nucleophilic ligand, and binds a less strongly coordinating solvent molecule to become stable. Preparation of this complex is achieved by reacting the neutral triflate rhenium precursor 23 with the sodium salt of the non-coordinating anion BAr4F in dichloromethane. After halide abstraction subsequent monodentate binding of the solvent takes place, whereas in the presence of CO the carbonylated cationic rhenium complex 25 is formed (Schemes 7 and 8).23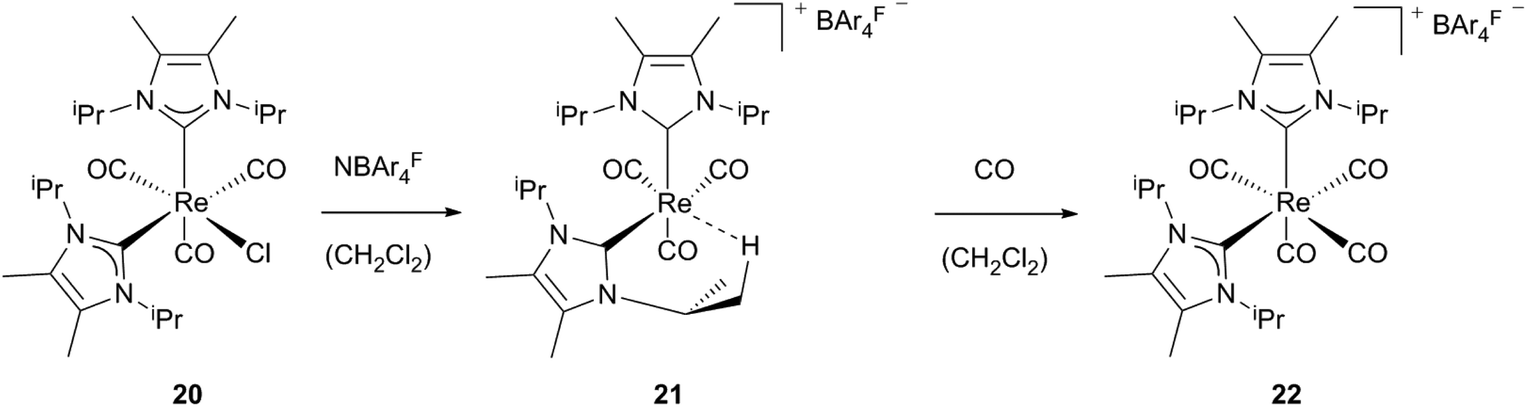 | ||
| Scheme 7 Generation of cationic Re(I) compounds bearing two unsaturated NHCs.23 | ||
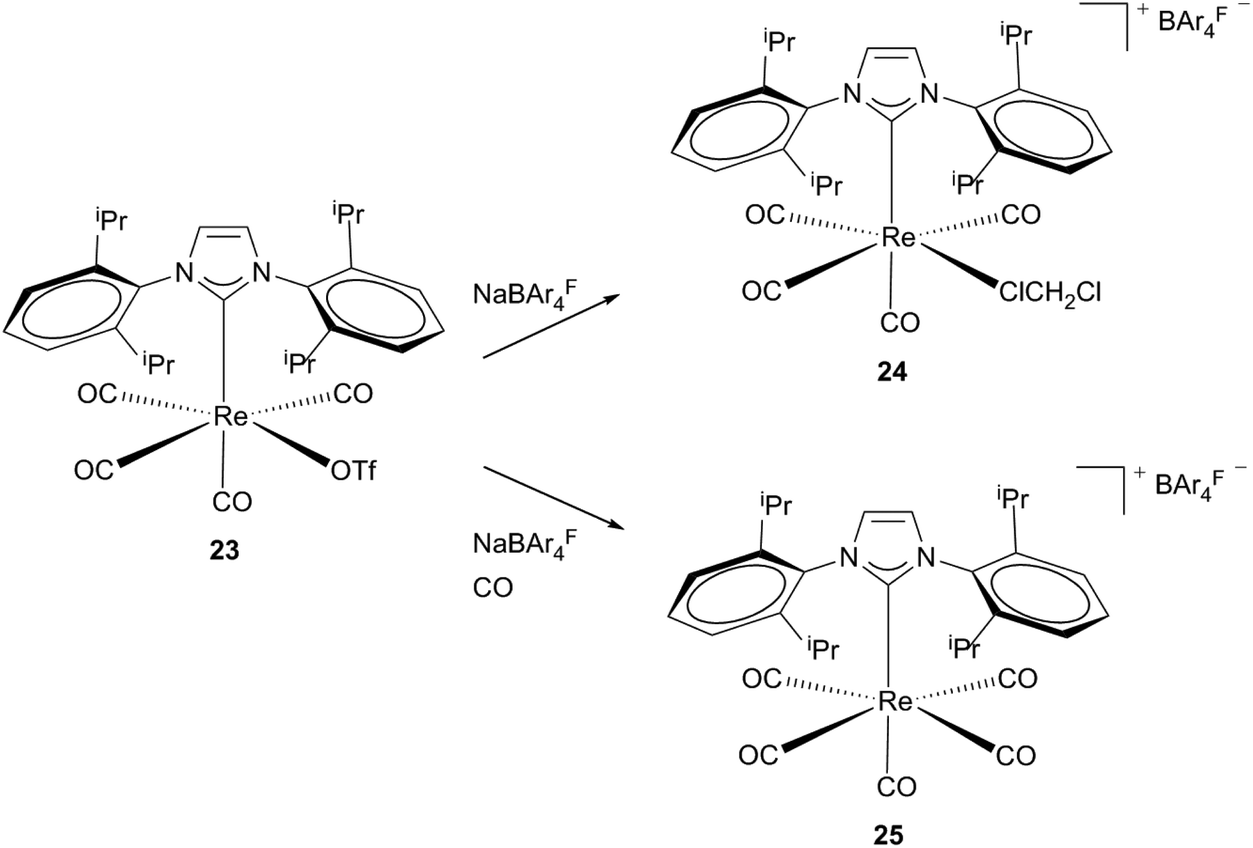 | ||
| Scheme 8 Pathways by Whittlesey et al. for cationic unsaturated Re(I) monocarbene complexes.23 | ||
Another literature known synthetic route to NHC complexes with different metals is the transformation of coordinated imidazoles to NHC ligands.24
Pérez, Riera, López and coworkers described the formation of cationic rhenium(I) carbonyl complexes containing both NHC and N-alkylimidazole ligands as a pseudo-tautomerisation.24a,c,e,25 Starting from a highly stable cationic N-alkylimidazole containing fac-Re[(CO)3] complex 26, one of the imidazole ligands is deprotonated by a strong base resulting in a stable neutral but not isolable intermediate 27, which features an imidazole-2-yl ligand. The non-substituted nitrogen atom at the NHC of the highly reactive compound 27 can be protonated or methylated easily by electrophiles. Using methyltrifluoro-methanesulfonic acid affords the cationic complex 29 and trifluoromethanesulfonic acid leads to 28, which contains a NH–NHC ligand, respectively (Scheme 9).24a–c
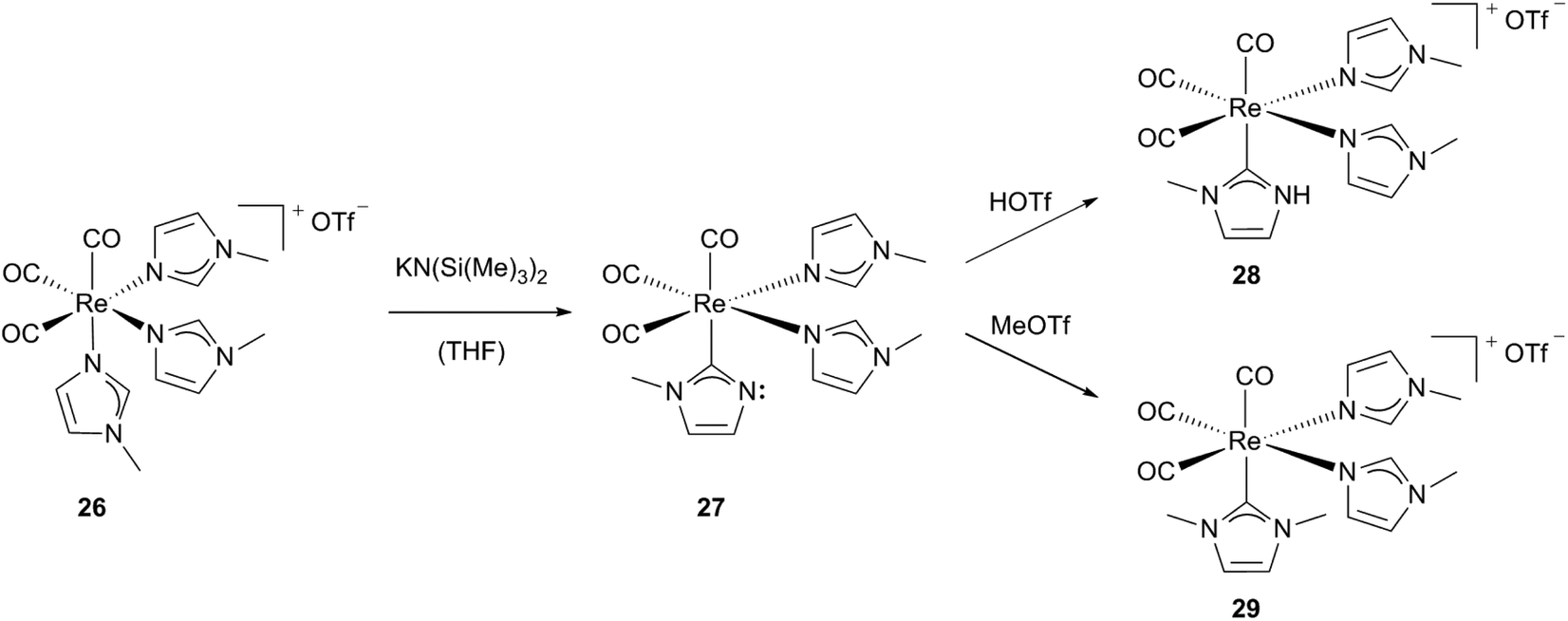 | ||
| Scheme 9 Synthesis of Re(I) carbonyl complexes bearing one NHC and two N-alkylimidazole ligands.24a,b | ||
In addition, the group discovered that besides the nature of the imidazole ligands, the ancillary substituents at the metal influence the resulting product.26
Starting from [Re(OTf)(CO)3(N-MeIm)2] (30) the triflate ligand was substituted, supported by NaBAr′4 to obtain complex 31a.24a The subsequent change of the coordination mode of one imidazole ligand by deprotonation with KN(SiMe3)2 led to the imidazole-2-yl complex 32. From the latter, the cationic Re–NHC complex 33 could be generated via methylation at the non-coordinated nitrogen atom. The influence of the substituent on the phosphane ligand becomes apparent when complexes 31b and 31c, which can be synthesized analogously to complex 31a, were treated under similar conditions as described for complex 31a. In the case of PMe3 (in complex 31a) a cationic Re(I)–NHC complex 33 is created whereas arylphosphane ligands lead to binuclear Re(I)–Re(I) complexes (35) bearing abnormal NHC ligands obtained via double activation of the N-methylimidazole (Scheme 10).
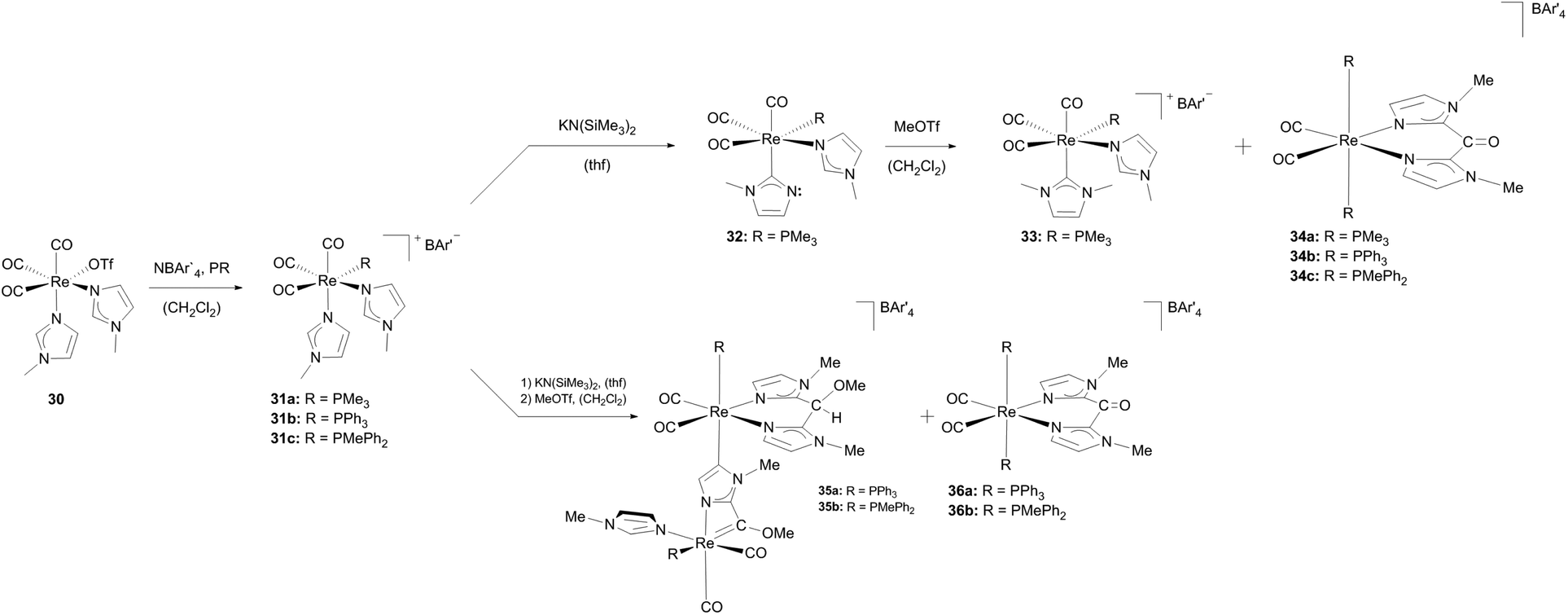 | ||
| Scheme 10 Formation of different cationic Re(I) NHC complexes influenced by ancillary ligands at the metal core.24a,26 | ||
A methoxycarbene ligand is generated by the formation of a four-membered cycle including one rhenium atom and methylation of the oxygen atom of the cis-carbon-monoxide ligand. The bridging and build-up of the abnormal NHC moiety is achieved via deprotonation of the backbone of the N-methylimidazole and coordination to a second rhenium atom. This leads to a formal intramolecular ligand coupling resulting in an N,N-bidentate ligand coordinated at rhenium-2. cis/trans-Rhenium complexes featuring N,N-bidentate chelate ligands (36) formed as side products here.27
Various benzoxazol-2-ylidene-substituted Re(I) phenanthroline complexes can be obtained starting from acetonitrile containing rhenium carbonyl complexes (37, 42x).28 As previously reported, N,O-heterogenocarbene complexes can be built via intramolecular cyclization, if the isocyanide C atom is not deactivated by strong (d → p)π-back-bonding from the metal core.1i,29 Contrary to similar rhenium complexes published by Hahn et al.,29a the strong π-accepting carbonyl ligands coordinated to the rhenium precursor support the formation of the reported luminescent rhenium complexes 38, 39 and 43a–c (Scheme 11).
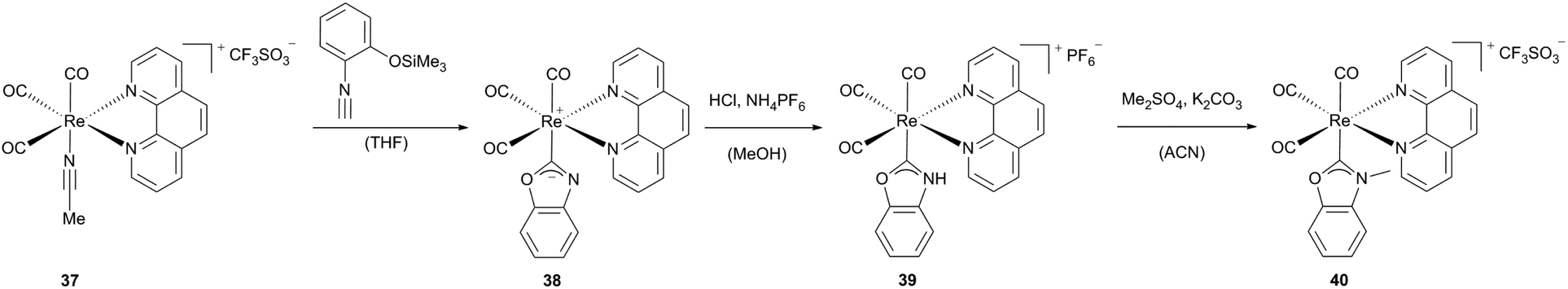 | ||
| Scheme 11 Synthesis of luminescent Re(CO)3 phenanthroline complexes.28 | ||
Complex 39, which may be considered as the N-protonated form of the neutral rhenium complex 38 bearing an anionic N-deprotonated carbene ligand, could be generated via acidification and subsequent metathesis. This complex as well as the methylated equivalent 40 adopts a distorted octahedral geometry with a facial arrangement of the three carbonyl ligands.
Similar reactions with several phosphines bearing rhenium phenanthroline precursors 41x lead to carbene complexes 43x where both carbonyl ligands are arranged in the cis conformation (Scheme 12).28 Furthermore, the same group reported – based on their previous work28 – in 2014 luminescent N,S-, N,N-, and N,O-NHC rhenium(I) complexes via a ligand substitution reaction.13b
 | ||
| Scheme 12 Generation of benzoxazol-2-ylidene-substituted phenanthroline complexes by Ko and coworkers.28 | ||
Starting from Re(CO)5Br (16) via a neutral bis(N,O-carbene) rhenium precursor 44 several cationic mono carbene rhenium compounds (45x, 46), could be obtained. X-Ray analysis shows that complex 46 bears two unsymmetrical cis carbonyl ligands, one is located trans to the carbene ligand, the other, as well as the isocyanide ligand are trans to the diimine ligand (Scheme 13).
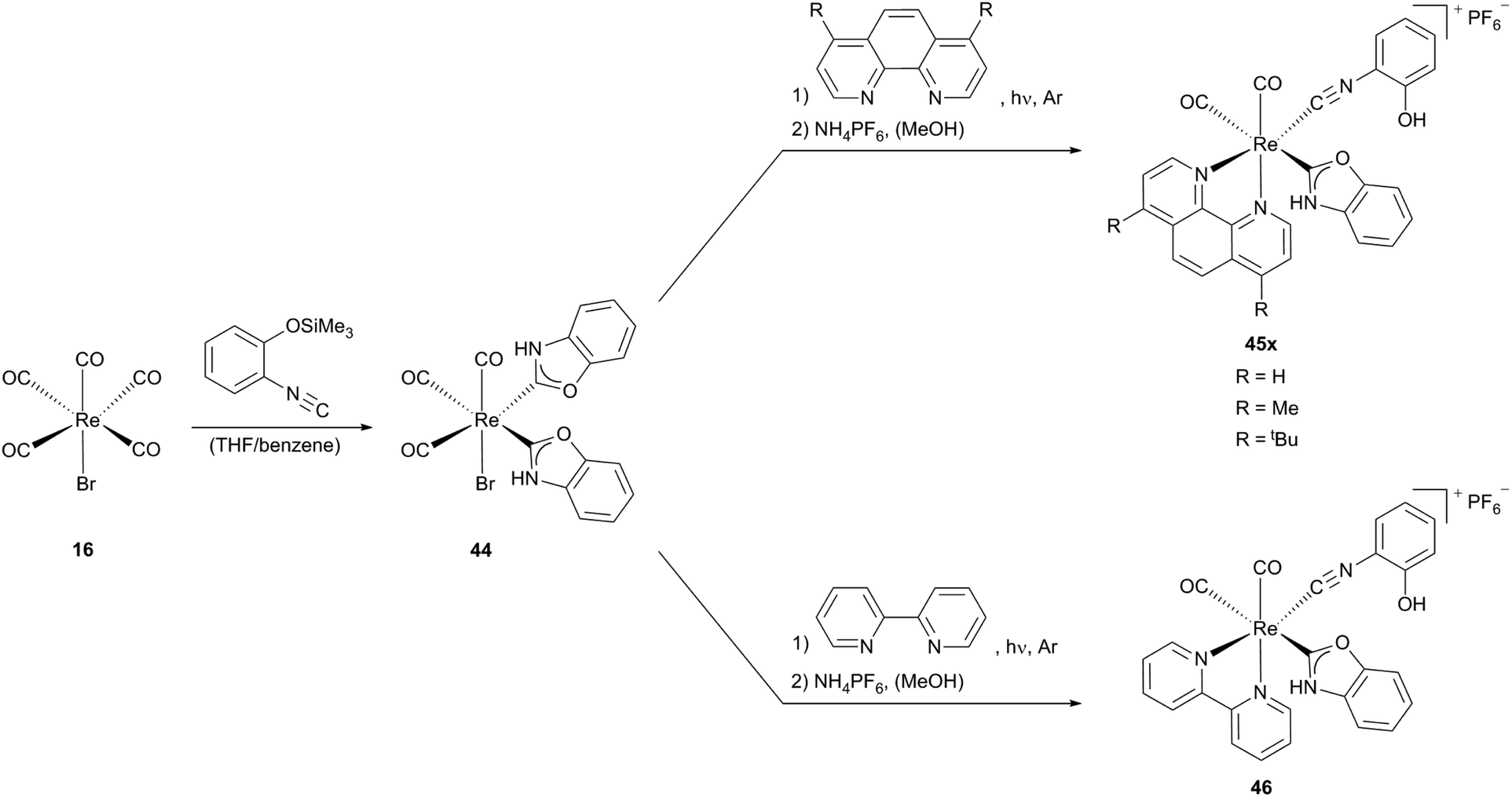 | ||
| Scheme 13 Oxazole–Re(I) complexes containing phenol substituted isocyanides.13b,28 | ||
The opening of one NHC ligand in complex 44 and thus the formation of an isocyanide moiety are favoured by the substitution of the CO ligand, positioned trans to the carbene by a diimine, as a consequence of the weakening of the pπ (carbene)–pπ (O) interaction resulting from enhanced π-(rhenium → carbene)-back-donation.
For complexes 45x and 46 an interconversion between the isocyanide and the N,O-heterocyclic carbene form occurs, while complexes 48x and 49 are generated by the conversion of one coordinated isocyanide ligand to a carbene ligand. The formation of only one carbene ligand – even with a large excess of nucleophiles – results from the different reactivities of the coordinated isocyanide ligands in precursor complex 47. While the isocyanide ligand, standing in the trans position to the carbonyl ligand is susceptible to the nucleophilic attack, the isocyanide C atom positioned trans to the diimine receives a stronger π-back bonding from the metal and hence is more electron rich, consistent with the observations for complexes 45x and 46. Reacting complex 50b with concentrated sulfuric acid leads to the distorted octahedral target NHC complex 51 (Scheme 14).13b
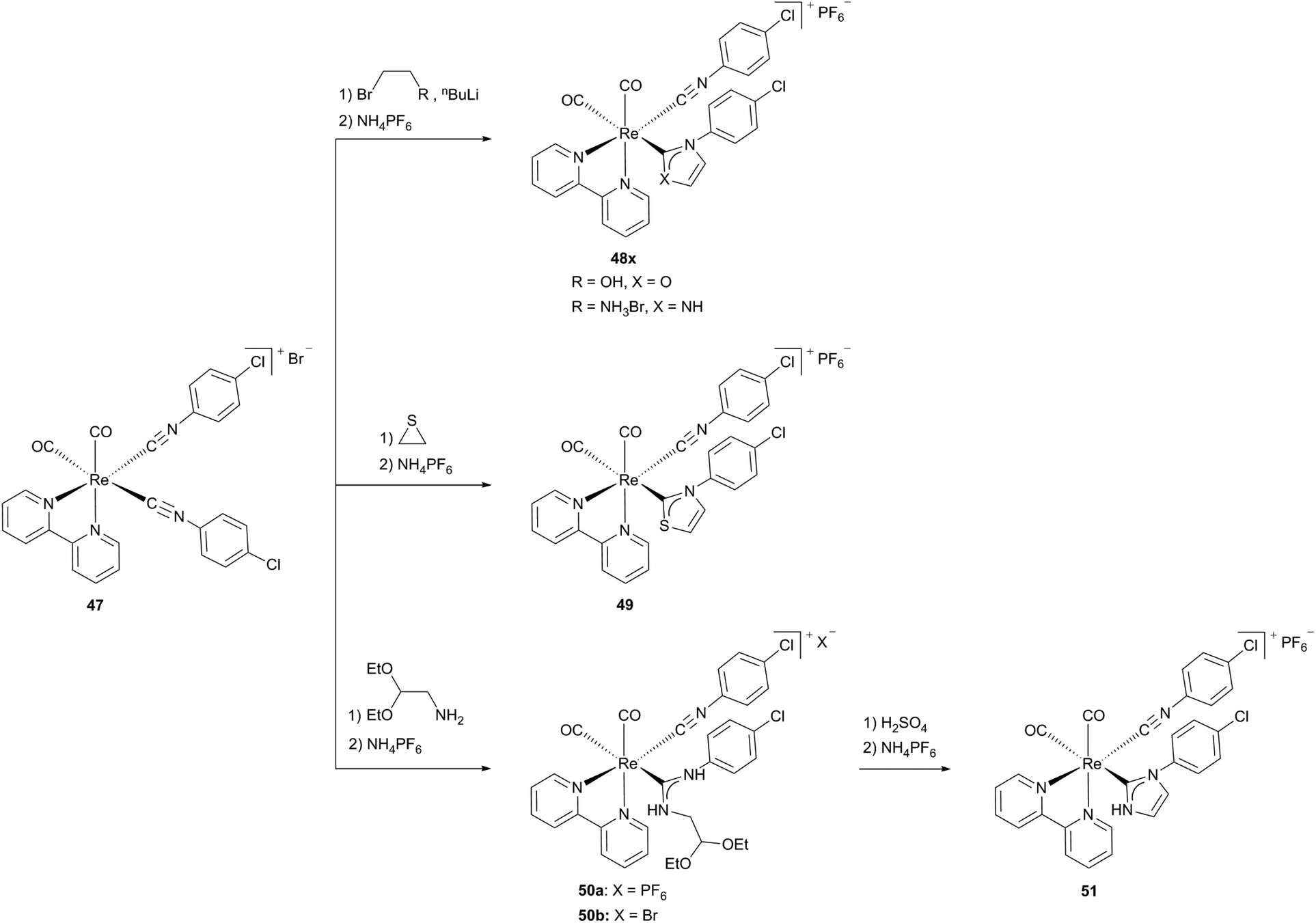 | ||
| Scheme 14 Generation of cationic N,S-, N,N- and N,O-NHC Re(I) diimine complexes.13b | ||
As the first investigations of luminescence of N-heterocyclic rhenium(I) complexes were reported by Che et al.14 (see before), Ko and coworkers studied the photophysical and electrochemical properties of rhenium diimine complexes bearing N,N-, N,S- and N,O-NHC ligands.13b,28
The knowledge about the influence of the relatively strong π-acceptor ability of almost every modification of or at a ligand (complexes 38–40, 43, 45, 46, 48x–51) can be used for the development of further luminescent NHC complexes for various applications (e.g. photocatalysis of CO2 reduction) by tuning the emission properties.13b,28
2.3 Unsaturated Re(V) complexes bearing mono-NHC ligands
Various cationic Re(V) complexes with unsaturated mono-NHC ligands have been prepared.4,30Royo, Romão et al.30a and Abram et al.30b were the first to obtain cationic Re(V) oxo complexes bearing four unsaturated carbene ligands, by reacting strong Lewis basic NHCs with various rhenium precursors.
Dicationic monooxo-complex 53 is the reaction product of [ReOCl3(PPh3)2] with an excess of free carbene 52. The triphenylphosphane- and two of the chloride-ligands are displaced by four monocarbene ligands.30a The green complex 53 is described as moisture-sensitive in solution – traces of water lead to the formation of a dioxo species30b (see below) – and air-stable for a short time as a solid.
By using the same ligand (52) with different rhenium compounds and reaction conditions, monocationic tetrakis(carbene)dioxorhenium complexes 54 and 55 can be formed. The brown solid (54) can be generated starting from [ReO2I(PPh3)2]. [ReO2Me(PhCCPh)] yields yellow crystals (55) featuring an approximate D4 symmetry with perrhenate as an anion and one thf-solvent molecule. Royo, Romão et al. assumed that the formation of complex 55 is caused by adventitious water in the reaction mixture, with some unidentified products additionally found (Scheme 15).30a
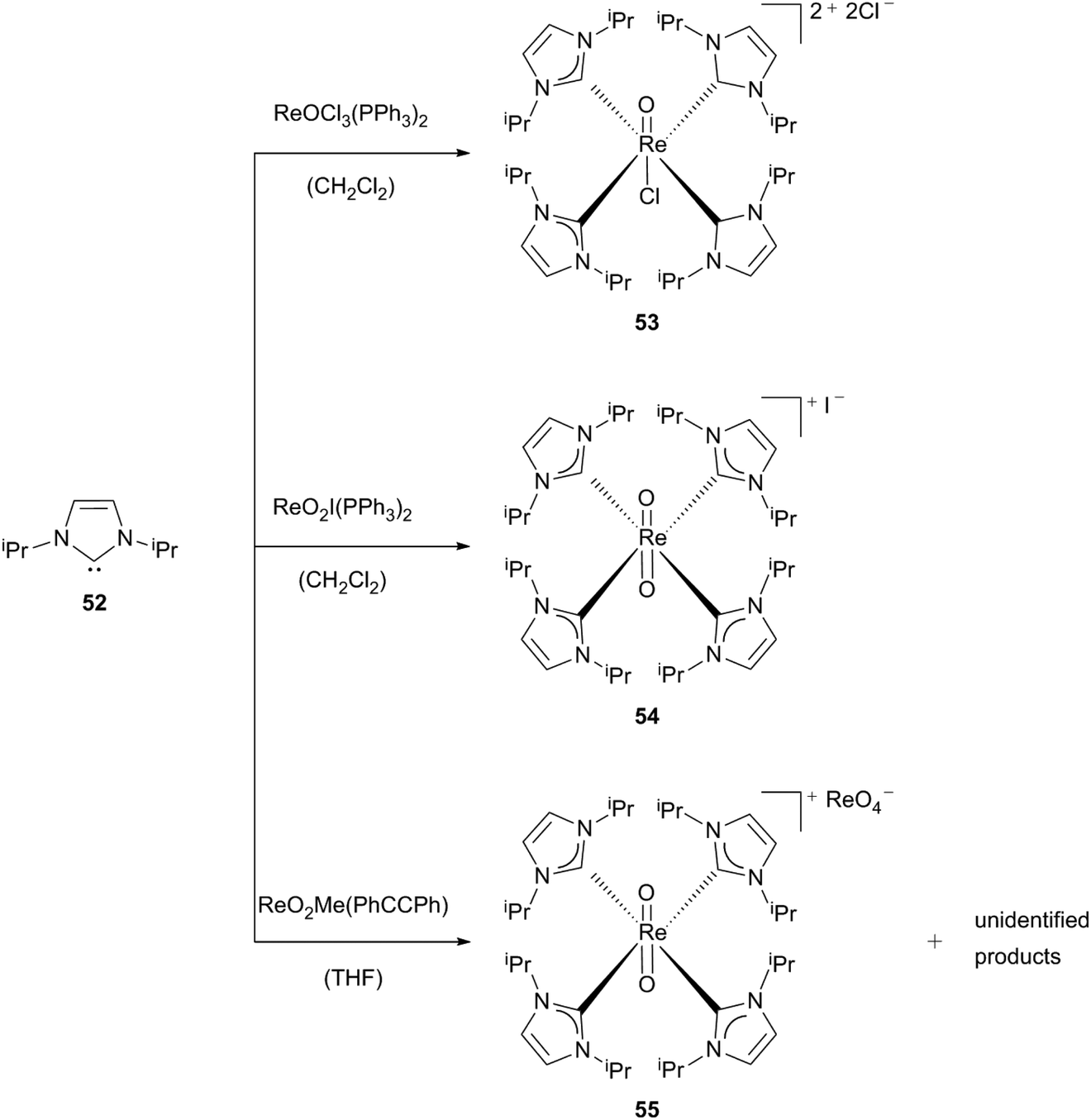 | ||
| Scheme 15 Synthesis of the first cationic Re(V) NHC oxo complexes.30a | ||
Starting from three different rhenium precursors 57–59 Abram et al. reported a cationic dioxorhenium(V) complex bearing four carbene ligands, which are methyl-substituted at the backbone.30b,d
Depending on the reaction conditions hexafluorophosphate (60b) or perrhenate salts (60a) of the complex could be isolated. Performing the reaction of [ReOCl3(PPh3)2] with an excess of the free carbene 56 in air in combination with traces of water in the solvent leads to 60a and HLiPr as a side product, whereas the addition of ammonium hexafluorophosphate yields the stable 60b (Scheme 16).30b
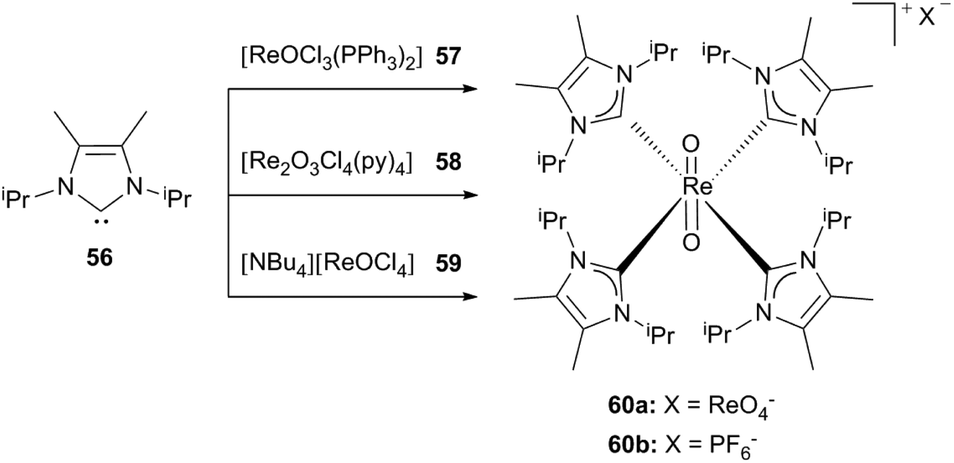 | ||
| Scheme 16 Tetrakis(1,3-diisopropyl-4,5-dimethyl-imidazol-2-ylidene)rhenium(V) dioxo complexes.30b | ||
Using the free carbenes 61/65 under the same reaction conditions as applied for the synthesis of compound 60, the monooxo species 62/68 can be isolated as green powders. By solving complexes 62 and 68 in methanol and adding hexafluorophosphate salts in the presence of LMe (61), an exchange of the chloro ligand for a methoxy ligand takes place whereas for LEt (65) crystals of the composition [ReOCl(LEt)4][PF6]·2KPF6 (68b) are obtained (Scheme 17).30d
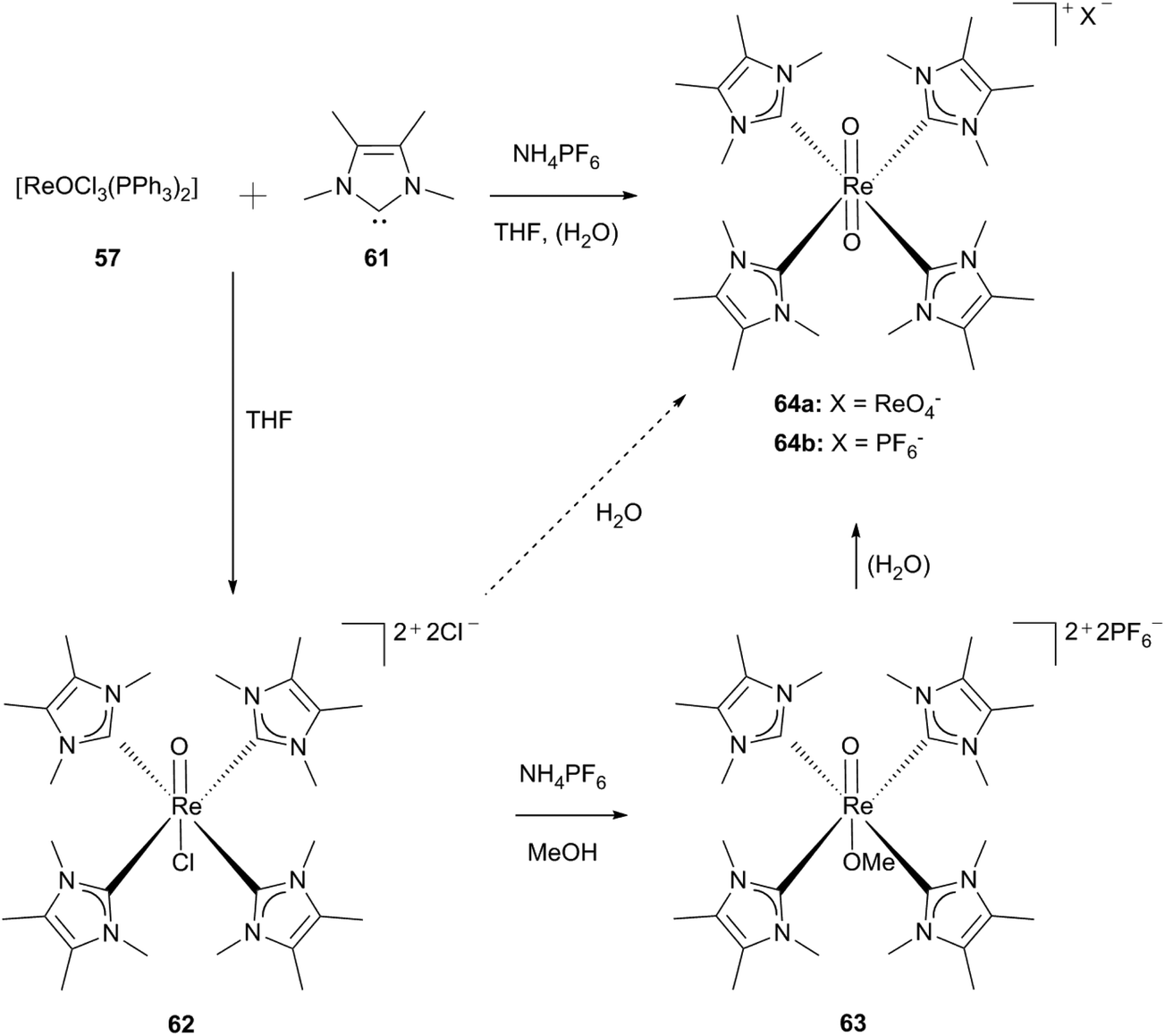 | ||
| Scheme 17 Tetrakis(1,3-dimethyl-4,5-dimethyl-imidazol-2-ylidene)rhenium(V) oxo complexes.30d | ||
Another synthetic approach via NHC transfer reagents for compounds considered for radiopharmaceutical applications was shown for 65. By reacting silver tetrafluoroborate with an excess of free carbene (56, 65) the corresponding silver(I)NHC complexes could be obtained, which then can be used as transmetallation agents. The known precursors for Re(V) carbene complexes, such as [ReOCl3(PPh3)2] and [NBu4][ReOCl4] form the orange-red product 67 with an excess of the silver carbene (66) and addition of (NH4)(CF3SO3) (Scheme 18).30c
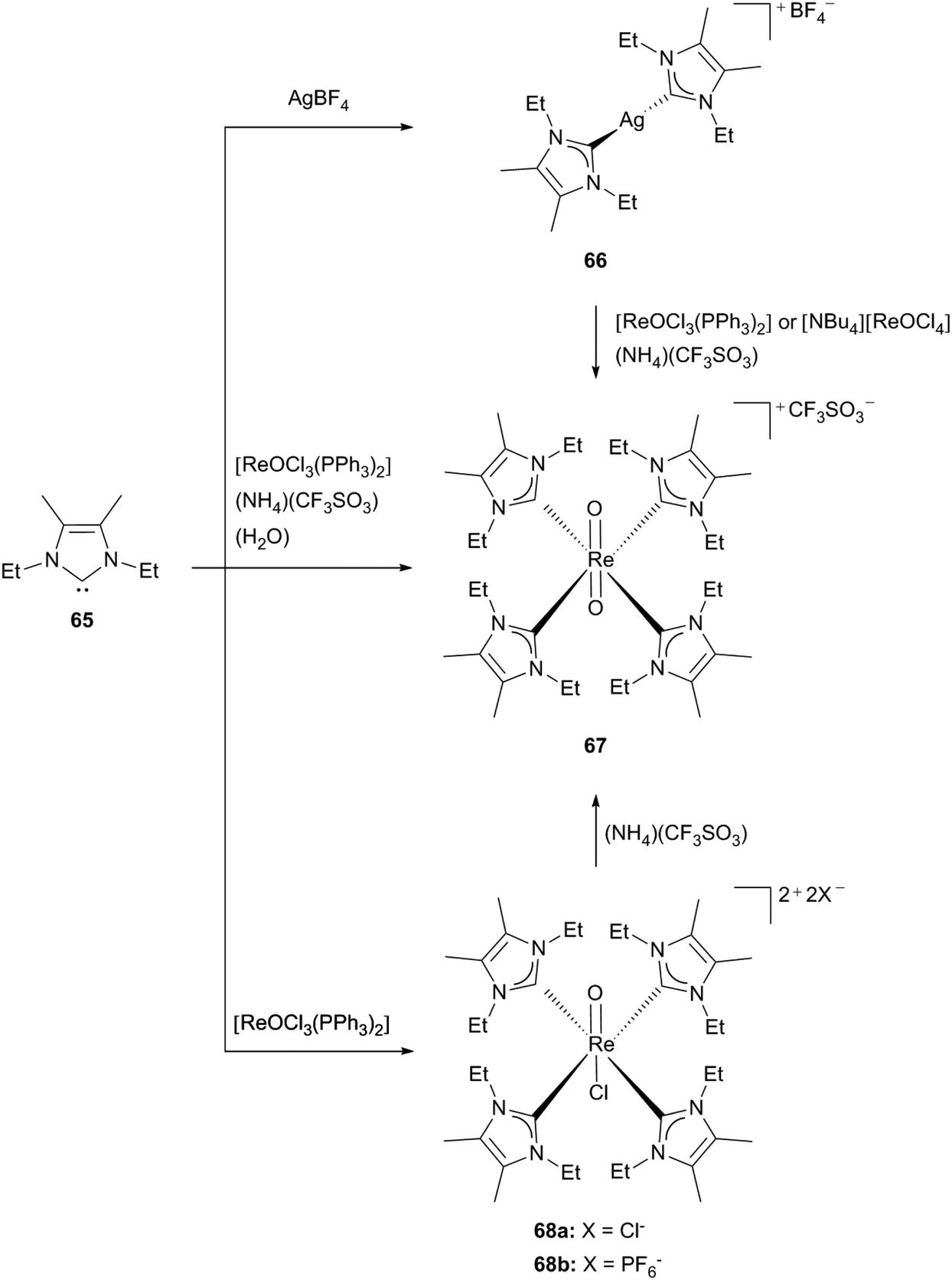 | ||
| Scheme 18 Synthesis of Re(V) NHC complexes starting from 65.30c,d | ||
The dioxo species (60, 64, 67) as well as complexes 62, 63, 66 and 68 feature planar coordination spheres with equatorially bonded NHC ligands.30b–d Although the steric demand of the ligands does not affect the paddle-wheel type ligand arrangement, it influences the stabilities as well as the structures of the complexes depending on the nature of the alkyl substituents at the NHC ligands. Isopropyl substituted imidazolylidene ligands seem to be more basic than the methyl substituted analogues, which signifies a higher nucleophilicity at the carbene C-atom. Consequently dioxo rhenium complexes (60, 64, 67) more stable due to their ability to (better) stabilize the augmented electron density.1k,31
Furthermore nitridorhenium(V) as well as phenyl-imidorhenium(V) complexes with ligands LMe (61) and LEt (65) were published by the same group.30e,f
They used [ReNCl2(PR′2Ph)3] (R′ = Me, Et) as the rhenium precursor to yield 69a,b as stable solids by application of an excess of the corresponding carbenes (61, 65) in dry solvents. Attempts to generate the analogous isopropyl complex have failed so far. Due to the different steric bulkiness of the NHC ligands, caused by the alkyl side chains, hydrolysis induced by moisture is favoured for [ReNCl(61)4]Cl (69a); whereas [ReO2(65)4][ReO4] (70) is formed in hot methanol (Scheme 19).30f
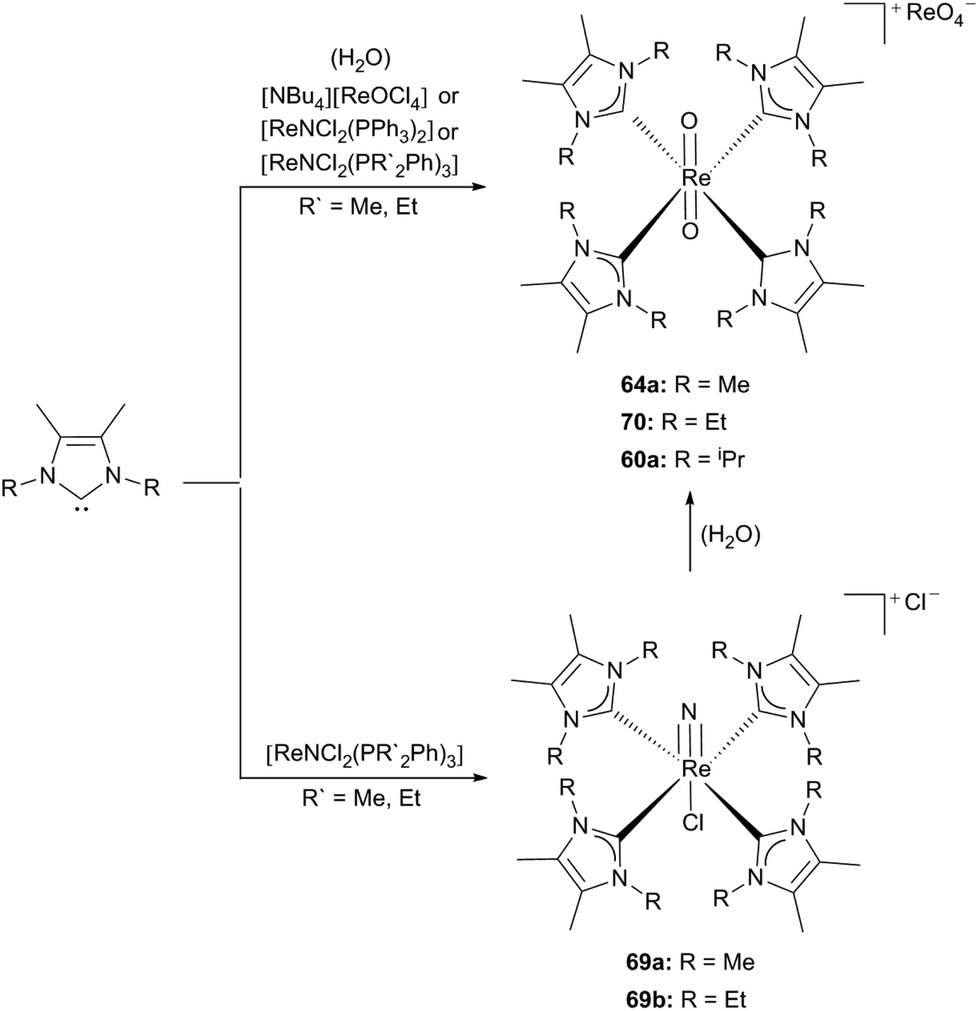 | ||
| Scheme 19 Formation of cationic Re(V) nitride or oxo complexes due to the reaction conditions.30f | ||
Compared to the hydrolysis susceptibility of nitridorhenium carbene complexes (69), using the formally dianionic ligand “PhN2−”, isoelectronic with an oxo ligand and thus able to stabilize rhenium in higher oxidation states, leads to “hydrolysis-inert” phenylimidorhenium(V) carbene complex 71.30e
Matching the analogous nitrido complex 69b, the phenylimido ligand is able to generate a conjugated π-system and hence can compensate for the electron density that is donated by the carbene ligands to the rhenium centre. Again, by performing the reaction in dry solvents Abram and coworkers obtained [Re(NPh)X(65)4]2+ (71x; X: Br, Cl), whereas the traces of water in the reaction mixture resulted in the hydroxo derivative (72). Purple crystals of 72c could be obtained via recrystallization in methanol, which is mandatory to get rid of the carbenium salt that is formed as a side product. Coordination of a hydroxo instead of a methoxo ligand can be explained by the steric shielding of the NHC ligands, which allow less free space in the trans position to the imido ligand.30e This behaviour was observed before for rhenium oxo complex 63.30d The fact that only stable phenylimido rhenium complexes with carbene 65 can be prepared to date, whereas efforts to generate analogous complexes with 56/61 lead to dioxo complexes may be explained by the matched shielding of the ethyl substituted carbene caused by its fitting steric demand (Scheme 20).30e
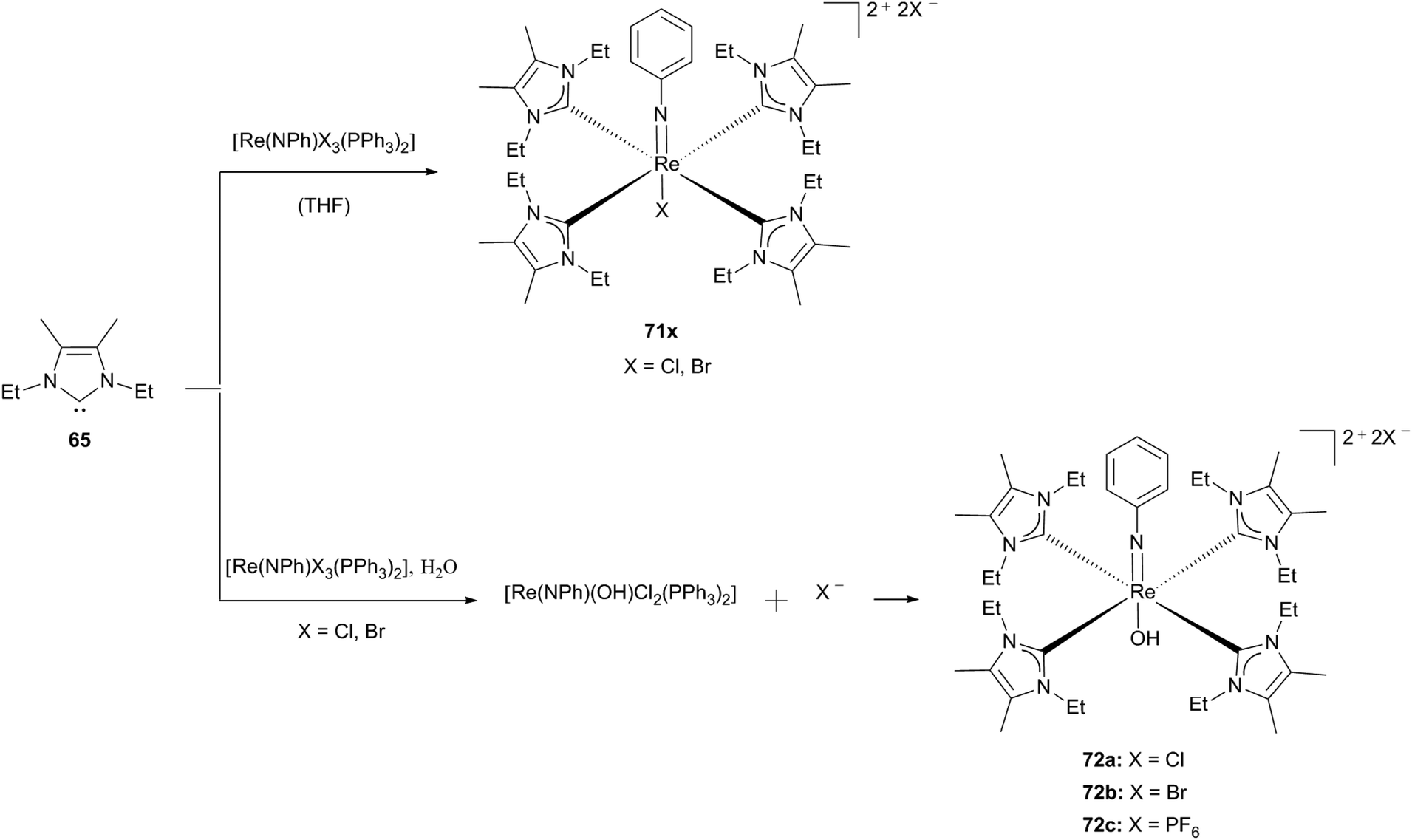 | ||
| Scheme 20 Phenylimidorhenium(V) carbene complexes synthesised by Abram and coworkers.30e | ||
3. Cationic rhenium bis-NHC complexes
3.1 Cationic rhenium(I) bis-NHC complexes
Based on the previous work,32 Herrmann, Kühn and coworkers reported the synthesis of chelated NHC cationic Re(I) carbonyl complexes with weakly coordinating anions (WCA).33The uncharged rhenium precursors fac-bromotricarbonyl(NHC)Re(I) (75x) were synthesized by treatment of [NEt4]2[ReBr3(CO)3] with free carbenes (74x), which were obtained starting from N substituted imidazoles (73x) via the SN2 reaction and subsequent deprotonation with a base.32 These stable bis(NHC) complexes (75x) are able to react with silver WCA salts (AgPF6, Ag[Al(OC(CF3)3)4] in acetonitrile at room temperature to give the corresponding rhenium(I) bis(NHC) acetonitrile WCA complexes (76, 77) (Scheme 21).
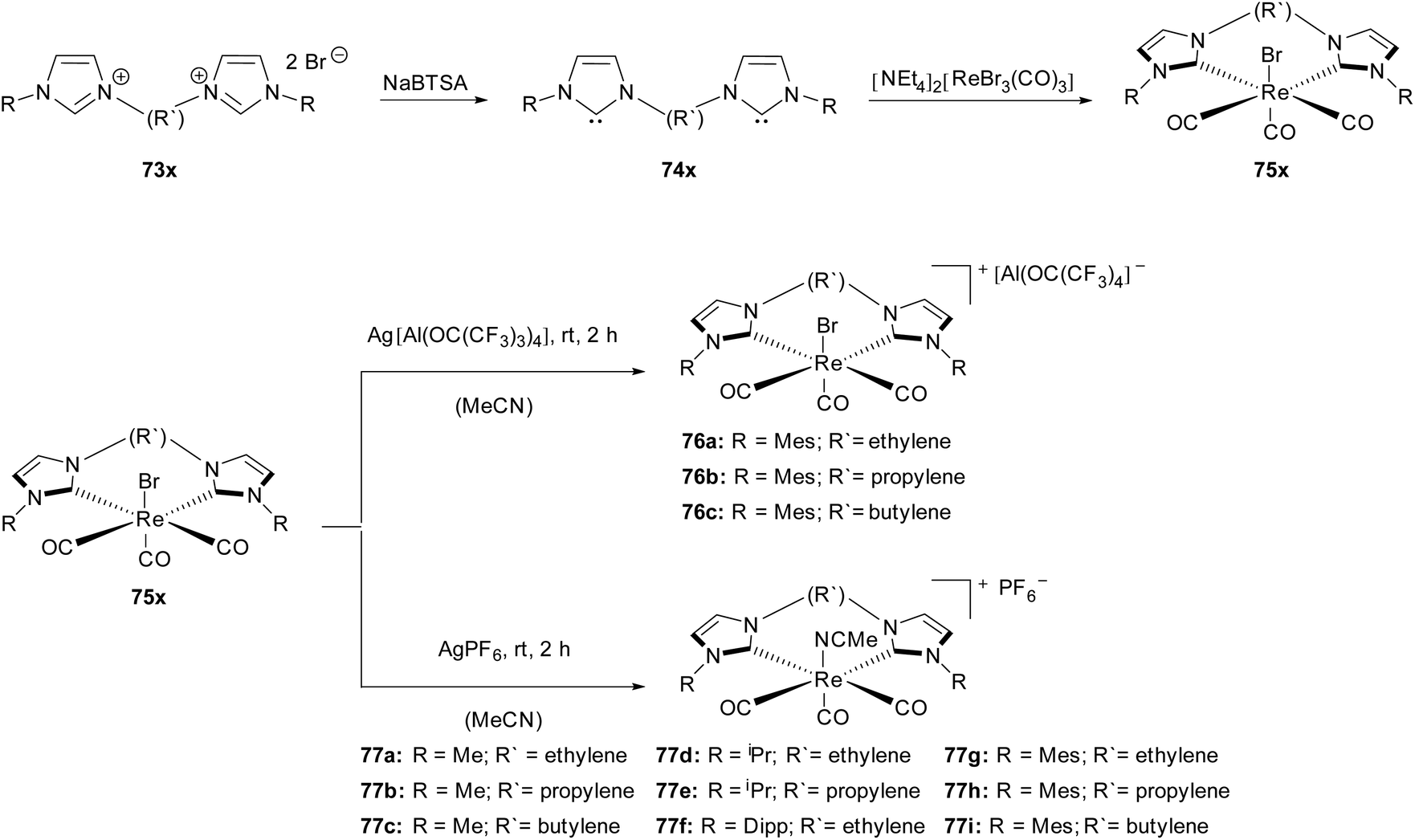 | ||
| Scheme 21 Cationic Re(I)(CO)3bis(NHC)(WCA) by Herrmann, Kühn and coworkers.33 | ||
All obtained complexes were characterized by mass spectrometry and spectroscopic methods. Interestingly, the variation of the bridge length and the substituents as well as the WCA anions did not strongly influence both the yield and the IR resonances. All the Re(CO)3 moieties display a distorted C3v local symmetry. This asymmetry results in a degenerate CO stretching mode, where one of the CO groups differs from the other two. Though, despite the observed little resonance variance no general trend relating to the influence of the different bridge length could be determined.
The distorted octahedral symmetry is confirmed by X-ray structure analysis for complexes 77a, 77b, 77h and 77i. In comparison with their uncharged Re(I) NHC analogues fairly similar bond distances between the metal and the trans-carbonyl group have been found.32 However, the steric demand of the acetonitrile ligand, being different from the bromo ligand, influences the position of the bulky mesitylene ligands independent of the length of the bridging moiety in complexes 77h and 77i. The increasing bridge length between the NHC moieties is, however, responsible for a higher torsion angle between the imidazole rings. In the case of a large WCA ligand as a counterion (76a), only a minor change of the bond length compared to the uncharged derivative appears. All attempts to obtain single crystals of the complexes (77c–f) as well as 76b have failed so far, whereas the crystallisation of 76c has led to the first Re(I) NHC CCC pincer complex.33
With regard to potential application in catalysis or pharmaceutical chemistry the stability of the Re(I) NHC compounds was examined by temperature dependent NMR experiments. The labile acetonitrile ligand can be easily exchanged by DMSO, changing also the geometry of complex 77h. The compound remains stable up to 93 °C for several minutes. This allows for potential use as a homogeneous transition metal catalyst, for e.g. hydroamination reactions.
Catalytic applications in the hydroamination of 4-pentyn-1-amine as examined by Müller, Yan et al.34 have failed so far, so that the focus should be laid on other catalytic reactions or on modification of the complex. Replacing one of the strongly binding NHC ligands – in combination with an WCA – for example pyridine containing Re(I) carbonyl complexes as reported by Stagni, Massi, Brown et al.35 and Yang et al.36 – could create efficient catalysts.37
3.2 Cationic rhenium(V) bis-NHC complexes
The group of Hor reported the first Re(V) complexes in which the trans oxo-hydroxo core is stabilized by two chelating N-heterocyclic dicarbene ligands.30g The air and moisture stable complexes 79 were obtained via transmetalation from a silver(I) carbene precursor 78![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 38 with the commercially available complex ReOCl3(PPh3)2 (57) in good yields. For 79a/b the characterization of the complexes was carried out via NMR, MS, EA, FTIR and single crystal XRD. All three cationic mononuclear complexes (79) occur in an octahedral geometry with the two NHC ligands orthogonal to the trans oxo-rhenium-hydroxo axis. The asymmetric units ([ReO(OH)(LR)2 ]2+) (79) contain in all cases two disordered anions in the ratios of PF6−:ReO4− = 0.7:0.3 (79a); 0.74:0.26 and 0.66:0.34 (79b); 0.65:0.35 (79c). The presence of an oxo-rhenium-hydroxo motif is explained as the most likely one. On the one hand the observed length of the rhenium oxo bonds do not fit in the typically literature known range of rhenium oxo bonds in a stable [O
38 with the commercially available complex ReOCl3(PPh3)2 (57) in good yields. For 79a/b the characterization of the complexes was carried out via NMR, MS, EA, FTIR and single crystal XRD. All three cationic mononuclear complexes (79) occur in an octahedral geometry with the two NHC ligands orthogonal to the trans oxo-rhenium-hydroxo axis. The asymmetric units ([ReO(OH)(LR)2 ]2+) (79) contain in all cases two disordered anions in the ratios of PF6−:ReO4− = 0.7:0.3 (79a); 0.74:0.26 and 0.66:0.34 (79b); 0.65:0.35 (79c). The presence of an oxo-rhenium-hydroxo motif is explained as the most likely one. On the one hand the observed length of the rhenium oxo bonds do not fit in the typically literature known range of rhenium oxo bonds in a stable [O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ReO]2+ rhenium dioxo core with monodentate NHC ligands,30a,b,d on the other hand the di-hydroxy rhenium motif is excluded due to incongruence regarding charge neutrality and the lower probability of the occurring of a trication (Scheme 22).30g
ReO]2+ rhenium dioxo core with monodentate NHC ligands,30a,b,d on the other hand the di-hydroxy rhenium motif is excluded due to incongruence regarding charge neutrality and the lower probability of the occurring of a trication (Scheme 22).30g
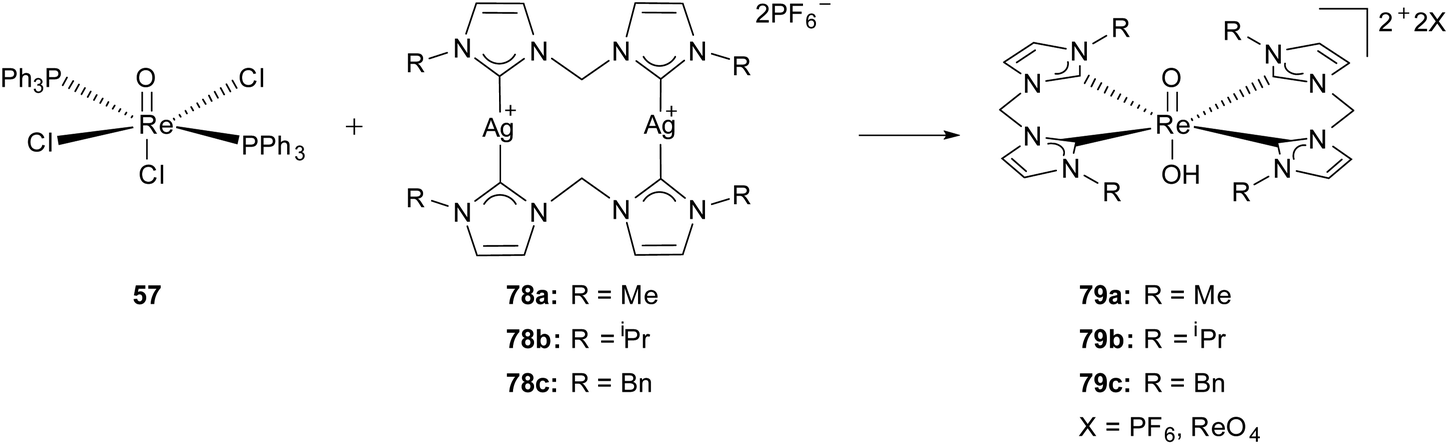 | ||
| Scheme 22 Generation of cationic Re(V) oxo-hydroxo biscarbene complexes by Hor et al.30g | ||
Based on this work, the groups of Reiner and Kühn developed a route towards a radiolabeled 188Re N-heterocyclic carbene complex (188Re-80).4 The cold trans-dioxobis(1,1′-methylene-bis(3,3′-diisopropylimidazolium-2-ylidene))rhenium(V)hexa-fluorophosphate (80) was synthesized following a previously published procedure.4,30g By reacting ReOCl3(PPh3)2 with one equivalent of the corresponding silver carbene (78b) in acetonitrile, the trans-dioxorhenium biscarbene complex (80) is obtained. The synthesis can be carried out under an aerobic atmosphere due to the relative inertness of rhenium(V) NHC complexes with trans-dioxo-rhenium cores towards oxidation, moisture, thermal stress and even nucleophilic attack-caused by the steric shielding of the metal centre owing to the pseudo-octahedral coordination and the closed shell electronic structure (Scheme 23).1f,4,30b,d,f,39
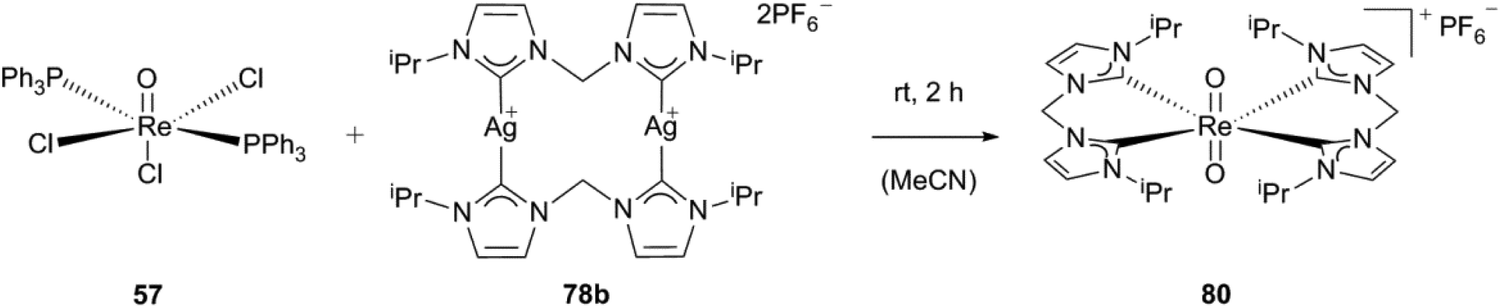 | ||
| Scheme 23 Synthesis of trans-dioxobis(1,1′-methylene-bis(3,3′-diisopropylimidazolium-2-ylidene))rhenium(V) hexafluorophosphate.4 | ||
The synthesis of the generator-produced 188Re–NHC complex (188Re-80) starts with 188ReO4−, which can be produced by using a 188W/188Re-generator. After transformation to the 188ReOCl3(PPh3)2 intermediate (188Re-57), adding cold ReOCl3(PPh3)2, transmetallation via silver carbene (78b) and purification by HPLC, the 188Re-labeled counterpart (188Re-80) can be obtained (Scheme 24).
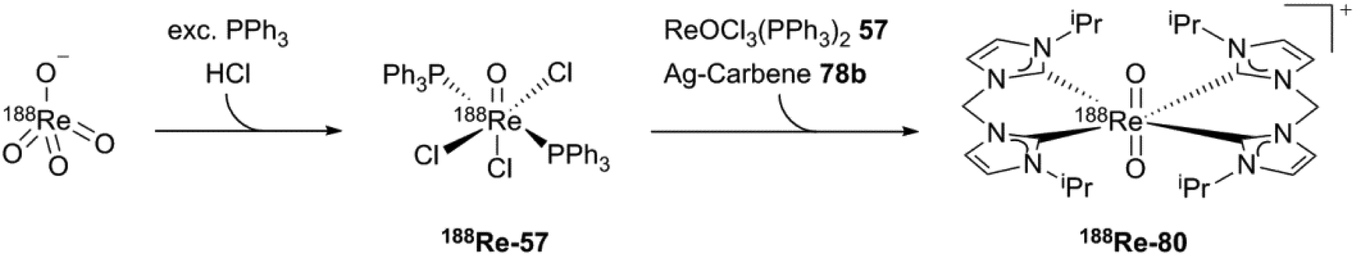 | ||
| Scheme 24 The first generator produced cationic 188Re NHC complex.4 | ||
The carried out stability tests revealed that indeed the unlabelled rhenium complex (80) is stable for 24 h at room temperature in a solution of acetonitrile and water, nevertheless undesirable decomposition occurred by testing the 188Re-labelled counterpart (188Re-80). Under the applied conditions, starting with water at 37 °C in a pH range from 5 to 8, with phosphate buffered saline solutions (37 °C; pH: 5, 6 & 7) as well as bovine serum, which is reasonably similar to the in vivo environment, the decomposition took place more quickly. The authors ascribe this to the bulkiness of the ligands at the rhenium centre and to the presence of potentially coordinating anions in the used environmental media.4
In contrast to the similar water stable 99Tc NHC complex (dioxobis(1,1′-methylene-bis(3,3′-dimethylimidazolium-2-ylidene))technecium(V)) published by Braband and coworkers,40 which differs from the metal only by its less bulky methyl substituents, the bulkiness of the isopropyl ligands and the terminal oxo ligands may promote the de-coordination of the NHC ligand as the primary decomposition pathway in aqueous medium instead of reoxidation to free 188perrhenate. The rapid decomposition in phosphate buffered saline solutions is due to the higher ionic strength of buffered solutions – especially chloride anions can compete with the carbenes at the rhenium – hence imidazolium salts are generated, that do not have the ability to bind to the metal again. Furthermore, biomacromolecules that are abundant in the fetal bovine serum may facilitate the decomposition of the complex (188Re-80) by undergoing nucleophilic reactions with rhenium. The formed adducts of biomacromolecules with the metal leads to the formation of 188ReO4−.
Despite the unfitness of this particular complex for use in radiopharmaceutical applications, due to its reduced stability under physiological conditions, a new approach for carrier-free and carrier-added radiolabeled rhenium NHC complexes has been opened.4 These investigations done by Reiner and Kühn et al.4 illustrate that although several radiopharmaceuticals based on the ReI(CO)3-fragment5o–r,41 as well as Re(V)-complexes5h–j,l are known, only estimations based on steric, kinetic and electronic considerations can be done. Whether Re(I) or Re(V) in combination with different types of ligands are more stable under physiological relevant conditions can only be figured out by testing these compounds.
4. Conclusion and perspectives
In this review, synthetic routes to cationic rhenium NHC complexes and their properties have been summarized. Since the first cationic Re(I) carbene complex was isolated in 199814 a variety of different approaches such as template synthesis or intramolecular cyclization have been reported. Beside the Re(CO)3 core, bearing varying auxiliary ligands rhenium(V) mono- or bis-NHC compounds comprising nitrido, imido and oxo units arouse interest with respect to their potential applications, particularly in medicine. Apart from the direct coordination of a free NHC moiety to the metal, the synthetic approach via transmetalation of stable silver NHC precursors looks quite promising for future clinical applications. With respect to nuclear medicine, creating diagnostic or therapeutic radiopharmaceuticals for instance can be carried out by radiolabelling of rhenium,4 or by linking the complexes bearing potential coupling groups (organoimido, alkylamido or alkylcarboxyl) to biomolecules via the N- or C-terminus of peptides.30e Investigations of high valent Re(V) biscarbene (O, OH) complexes as oxidation catalysts30g or Re(I) complexes bearing NHCs in hydroamination reactions33 or as photocatalysts13b are still ongoing. It also appears to be rather straightforward that in this field further research concerning luminescence, toxicity, air and water stability as well as solubility has to be executed with regard to further development of applications of these complexes.
Acknowledgements
The Margarete Ammon Foundation is acknowledged for a PhD grant to C. H.Notes and references
- (a) R. Visbal and M. C. Gimeno, Chem. Soc. Rev., 2014, 43, 3551–3574 RSC; (b) E. M. Hahn, A. Casini and F. E. Kühn, Coord. Chem. Rev., 2014, 276, 97–111 CrossRef CAS; (c) S. Bellemin-Laponnaz and S. Dagorne, Chem. Rev., 2014, 114, 8747–8774 CrossRef CAS PubMed; (d) S. Jürgens, W. A. Herrmann and F. E. Kühn, J. Organomet. Chem., 2014, 751, 83–89 CrossRef; (e) F. E. Hahn, ChemCatChem, 2013, 5, 419–430 CrossRef CAS; (f) S. J. Hock, L.-A. Schaper, W. A. Herrmann and F. E. Kuhn, Chem. Soc. Rev., 2013, 42, 5073–5089 RSC; (g) L.-A. Schaper, S. J. Hock, W. A. Herrmann and F. E. Kühn, Angew. Chem., Int. Ed., 2013, 52, 270–289 CrossRef CAS PubMed; (h) Y. Kuninobu and K. Takai, Chem. Rev., 2011, 111, 1938–1953 CrossRef CAS PubMed; (i) F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122–3172 CrossRef CAS PubMed; (j) K. R. Jain and W. A. H. a. F. E. Kühn, Curr. Org. Chem., 2008, 12, 1468–1478 CrossRef CAS; (k) H. Braband, T. I. Kückmann and U. Abram, J. Organomet. Chem., 2005, 690, 5421–5429 CrossRef CAS; (l) R. Schibli and A. Schubiger, Eur. J. Nucl. Med. Mol. Imaging, 2002, 29, 1529–1542 CrossRef CAS PubMed; (m) U. Abram and R. Alberto, J. Braz. Chem. Soc., 2006, 17, 1486–1500 CrossRef CAS.
- (a) F. F. Knapp, Cancer Biother. Radiopharm., 1998, 13, 337–349 CrossRef CAS PubMed; (b) T. Mindt, H. Struthers, E. Garcia-Garayoa, D. Desbouis and R. Schibli, CHIMIA, 2007, 61, 725–731 CrossRef CAS; (c) G. R. Morais, A. Paulo and I. Santos, Organometallics, 2012, 31, 5693–5714 CrossRef CAS.
- (a) W. A. Herrmann, Angew. Chem., Int. Ed. Engl., 1988, 27, 1297–1313 CrossRef; (b) J. R. Dilworth and S. J. Parrott, Chem. Soc. Rev., 1998, 27, 43–55 RSC; (c) G. Gasser, I. Ott and N. Metzler-Nolte, J. Med. Chem., 2011, 54, 3–25 CrossRef CAS PubMed.
- T. Wagner, B. M. Zeglis, S. Groveman, C. Hille, A. Pöthig, L. C. Francesconi, W. A. Herrmann, F. E. Kühn and T. Reiner, J. Labelled Compd. Radiopharm., 2014, 57, 441–447 CrossRef CAS PubMed.
- (a) E. Torres-García, G. Ferro-Flores, C. Arteaga de Murphy, L. Correa-González and P. A. Pichardo-Romero, Arch. Med. Res., 2008, 39, 100–109 CrossRef PubMed; (b) L. A. C. Torres, A. Marco, J. F. Batista, A. Casaco, G. Lopez, I. García, A. Perera, Y. Peña, A. Hernández, Y. Sanchez, S. Romero, R. Leyva, A. Prats and R. Fernandez, Nucl. Med. Commun., 2008, 29, 66–75 CrossRef CAS PubMed; (c) K. L. Shin, C. Jung, H. J. Choi, J. M. Jeong, M. Son, Y. J. Lee, E. B. Lee, S. H. Hong and Y. W. Song, Nucl. Med. Commun., 2007, 28, 239–244 CrossRef CAS PubMed; (d) P. Bernal, J.-L. Raoul, G. Vidmar, E. Sereegotov, F. X. Sundram, A. Kumar, J. M. Jeong, P. Pusuwan, C. Divgi, P. Zanzonico, J. Stare, J. Buscombe, C. T. T. Minh, M. M. Saw, S. Chen, R. Ogbac and A. K. Padhy, Int. J. Radiat., Oncol. Biol. Phys., 2007, 69, 1448–1455 CrossRef CAS PubMed; (e) R. H. Knut Liepe, J. Kropp, T. Grüning, R. Runge, R. Koch, F. F. Knapp Jr. and W.-G. Franke, Cancer Biother. Radiopharm., 2000, 15, 261–266 CrossRef PubMed; (f) V. A. Nicolas Lepareur, N. Moiret and E. Garin, Int. J. Mol. Imaging, 2012, 2012, 1–9 CrossRef PubMed; (g) H. R. Maxon, E. A. Deutsch, S. R. Thomas, K. Libson, S. J. Lukes, C. C. Williams and S. Ali, Radiology, 1988, 166, 501–507 CrossRef CAS PubMed; (h) J. R. Singh, K. Reghebi, C. R. Lazarus, S. E. M. Clarke, A. P. Callahan, F. F. Knapp Jr. and P. J. Blower, Nucl. Med. Commun., 1993, 14, 197–203 CrossRef CAS PubMed; (i) B. G. Ande Bao, R. Klipper, G. Negrete and W. T. Phillips, J. Nucl. Med., 2003, 44, 1992–1999 Search PubMed; (j) J. M. Jeong, Y. J. Kim, Y. S. Lee, J. I. Ko, M. Son, D. S. Lee, J.-K. Chung, J. H. Park and M. C. Lee, Nucl. Med. Biol., 2001, 28, 197–204 CrossRef CAS PubMed; (k) S. Seifert, T. Heinrich, C. Jentschel, C. Smuda, R. Bergmann and H.-J. Pietzsch, Bioconjugate Chem., 2006, 17, 1601–1606 CrossRef CAS PubMed; (l) M. G. Gerard, W. M. Visser, J. D. M. Herscheid, G. B. Snow and G. van Dongen, J. Nucl. Med., 1993, 34 Search PubMed; (m) S. Guhlke, A. Schaffland, P. O. Zamora, J. Sartor, D. Diekmann, H. Bender, F. F. Knapp and H. J. Biersack, Nucl. Med. Biol., 1998, 25, 621–631 CrossRef CAS PubMed; (n) K. Ogawa, T. Mukai, Y. Arano, M. Ono, H. Hanaoka, S. Ishino, K. Hashimoto, H. Nishimura and H. Saji, Bioconjugate Chem., 2005, 16, 751–757 CrossRef CAS PubMed; (o) K.-T. Chen, T.-W. Lee and J.-M. Lo, Nucl. Med. Biol., 2009, 36, 355–361 CrossRef CAS PubMed; (p) K. Ogawa, H. Kawashima, S. Kinuya, K. Shiba, M. Onoguchi, H. Kimura, K. Hashimoto, A. Odani and H. Saji, Ann. Nucl. Med., 2009, 23, 843–848 CrossRef PubMed; (q) D. Satpati, A. Korde, K. Kothari, H. D. Sarma, M. Venkatesh and S. Banerjee, Cancer Biother. Radiopharm., 2008, 23, 741–748 CrossRef CAS PubMed; (r) J. H. Yu, O. Urs, J. Xia, S. Li, Mo Dong, D. Yin and Y. Wang, Nucl. Med. Commun., 2005, 26, 453–458 CrossRef CAS PubMed; (s) P. P. Venkatesan, S. Shortkroff, M. R. Zalutsky and C. B. Sledge, Int. J. Radiat. Appl. Instrum., Part B, 1990, 17, 357–362 CrossRef CAS; (t) S.-J. Wang, W.-Y. Lin, B.-T. Hsieh, L.-H. Shen, Z.-T. Tsai, G. Tinge and F. Knapp Jr., Eur. J. Nucl. Med., 1995, 22, 505–507 CrossRef CAS PubMed; (u) J. M. Jeong, Y. J. Lee, Y. J. Kim, Y. S. Chang, D. S. Lee, J.-K. Chung, Y. W. Song and M. C. Lee, Appl. Radiat. Isot., 2000, 52, 851–855 CrossRef CAS PubMed; (v) N. Viola-Villegas, A. E. Rabideau, J. Cesnavicious, J. Zubieta and R. P. Doyle, ChemMedChem, 2008, 3, 1387–1394 CrossRef CAS PubMed; (w) T.-Y. Luo, I. C. Tang, Y.-L. Wu, K.-L. Hsu, S.-W. Liu, H.-C. Kung, P.-S. Lai and W.-J. Lin, Nucl. Med. Biol., 2009, 36, 81–88 CrossRef CAS PubMed.
- P. V. Paul, L. Beaumier, J.-L. Vanderheyden, W. D. Burgua, L. L. Kunz, A. R. Fritzberg, P. G. Abrams and A. C. Morgan Jr., Cancer Res., 1991, 51, 676–681 Search PubMed.
- (a) K. Wähler, A. Ludewig, P. Szabo, K. Harms and E. Meggers, Eur. J. Inorg. Chem., 2014, 2014, 807–811 CrossRef PubMed; (b) A. Leonidova, V. Pierroz, R. Rubbiani, Y. Lan, A. G. Schmitz, A. Kaech, R. K. O. Sigel, S. Ferrari and G. Gasser, Chem. Sci., 2014, 5, 4044–4056 RSC; (c) A. Leonidova, V. Pierroz, R. Rubbiani, J. Heier, S. Ferrari and G. Gasser, Dalton Trans., 2014, 43, 4287–4294 RSC; (d) A. Kastl, S. Dieckmann, K. Wähler, T. Völker, L. Kastl, A. L. Merkel, A. Vultur, B. Shannan, K. Harms, M. Ocker, W. J. Parak, M. Herlyn and E. Meggers, ChemMedChem, 2013, 8, 924–927 CrossRef CAS PubMed; (e) T. Joshi and G. Gasser, Synlett, 2015, 275–284 CrossRef CAS.
- (a) T. J. Korstanje, J. T. B. H. Jastrzebski and R. J. M. Klein Gebbink, Chem. – Eur. J., 2013, 19, 13224–13234 CrossRef CAS PubMed; (b) Y. Wang, L. Zhang, Y. Yang, P. Zhang, Z. Du and C. Wang, J. Am. Chem. Soc., 2013, 135, 18048–18051 CrossRef CAS PubMed; (c) D. Xia, Y. Wang, Z. Du, Q.-Y. Zheng and C. Wang, Org. Lett., 2012, 14, 588–591 CrossRef CAS PubMed; (d) M. Shiramizu and F. D. Toste, Angew. Chem., Int. Ed., 2012, 51, 8082–8086 CrossRef CAS PubMed; (e) I. Ahmad, G. Chapman and K. M. Nicholas, Organometallics, 2011, 30, 2810–2818 CrossRef CAS; (f) S. C. A. Sousa and A. C. Fernandes, Tetrahedron Lett., 2011, 52, 6960–6962 CrossRef CAS; (g) E. Arceo, J. A. Ellman and R. G. Bergman, J. Am. Chem. Soc., 2010, 132, 11408–11409 CrossRef CAS PubMed; (h) S. Vkuturi, G. Chapman, I. Ahmad and K. M. Nicholas, Inorg. Chem., 2010, 49, 4744–4746 CrossRef CAS PubMed; (i) A. T. Herrmann, T. Saito, C. E. Stivala, J. Tom and A. Zakarian, J. Am. Chem. Soc., 2010, 132, 5962–5963 CrossRef CAS PubMed; (j) J. E. Ziegler, M. J. Zdilla, A. J. Evans and M. M. Abu-Omar, Inorg. Chem., 2009, 48, 9998–10000 CrossRef CAS PubMed; (k) Y. Jiang and H. Berke, Chem. Commun., 2007, 3571–3573, 10.1039/B708913A; (l) Y. Kuninobu, K. Kikuchi, Y. Tokunaga, Y. Nishina and K. Takai, Tetrahedron, 2008, 64, 5974–5981 CrossRef CAS.
- (a) O. Schuster, L. Yang, H. G. Raubenheimer and M. Albrecht, Chem. Rev., 2009, 109, 3445–3478 CrossRef CAS PubMed; (b) C. Samojłowicz, M. Bieniek and K. Grela, Chem. Rev., 2009, 109, 3708–3742 CrossRef PubMed; (c) E. A. B. Kantchev, C. J. O'Brien and M. G. Organ, Angew. Chem., Int. Ed., 2007, 46, 2768–2813 CrossRef CAS PubMed; (d) W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290–1309 CrossRef CAS.
- (a) E. Levin, E. Ivry, C. E. Diesendruck and N. G. Lemcoff, Chem. Rev., 2015, 115(11), 4607–4692 CrossRef CAS PubMed; (b) H. D. Velazquez and F. Verpoort, Chem. Soc. Rev., 2012, 41, 7032–7060 RSC.
- (a) Y. Jiang, O. Blacque, T. Fox, C. M. Frech and H. Berke, Organometallics, 2009, 28, 5493–5504 CrossRef CAS; (b) T.-C. Su, Y.-H. Liu, S.-M. Peng and S.-T. Liu, Eur. J. Inorg. Chem., 2013, 2013, 2362–2367 CrossRef CAS.
- C. C. R. F. E. Kühn and W. A. Herrmann, Organometallic Complexes of Rhenium, 2003 Search PubMed.
- (a) J. G. Vaughan, B. L. Reid, P. J. Wright, S. Ramchandani, B. W. Skelton, P. Raiteri, S. Muzzioli, D. H. Brown, S. Stagni and M. Massi, Inorg. Chem., 2014, 53, 3629–3641 CrossRef CAS PubMed; (b) C.-O. Ng, S.-M. Yiu and C.-C. Ko, Inorg. Chem., 2014, 53, 3022–3031 CrossRef CAS PubMed; (c) V. Fernandez-Moreira, F. L. Thorp-Greenwood and M. P. Coogan, Chem. Commun., 2010, 46, 186–202 RSC; (d) P. D. Rachel, C. Evans and C. J. Winscom, Coord. Chem. Rev., 2006, 250, 2093–2126 CrossRef; (e) L. Sacksteder, M. Lee, J. N. Demas and B. A. DeGraff, J. Am. Chem. Soc., 1993, 115, 8230–8238 CrossRef CAS.
- W.-M. Xue, M. C.-W. Chan, Z.-M. Su, K.-K. Cheung, S.-T. Liu and C.-M. Che, Organometallics, 1998, 17, 1622–1630 CrossRef CAS.
- C.-Y. Liu, D.-Y. Chen, G.-H. Lee, S.-M. Peng and S.-T. Liu, Organometallics, 1996, 15, 1055–1061 CrossRef CAS.
- C.-H. Chen, Y.-H. Liu, S.-M. Peng, J.-T. Chen and S.-T. Liu, Dalton Trans., 2012, 41, 2747–2754 RSC.
- O. Kaufhold, A. Stasch, P. G. Edwards and F. E. Hahn, Chem. Commun., 2007, 1822–1824, 10.1039/b617033a.
- O. Kaufhold, A. Stasch, T. Pape, A. Hepp, P. G. Edwards, P. D. Newman and F. E. Hahn, J. Am. Chem. Soc., 2008, 131, 306–317 CrossRef PubMed.
- P. G. Edwards and F. E. Hahn, Dalton Trans., 2011, 40, 10278–10288 RSC.
- V. Blase, T. Pape and F. E. Hahn, J. Organomet. Chem., 2011, 696, 3337–3342 CrossRef CAS.
- F. E. Hahn, V. Langenhahn and T. Pape, Chem. Commun., 2005, 5390–5392, 10.1039/B510996E.
- A. Flores-Figueroa, O. Kaufhold, K.-O. Feldmann and F. E. Hahn, Dalton Trans., 2009, 9334–9342, 10.1039/b915033a.
- T. A. Martin, C. E. Ellul, M. F. Mahon, M. E. Warren, D. Allan and M. K. Whittlesey, Organometallics, 2011, 30, 2200–2211 CrossRef CAS.
- (a) M. A. Huertos, J. Pérez, L. Riera, J. Díaz and R. López, Angew. Chem., Int. Ed., 2010, 49, 6409–6412 CrossRef CAS PubMed; (b) M. A. Huertos, J. Pérez, L. Riera and A. Menéndez-Velázquez, J. Am. Chem. Soc., 2008, 130, 13530–13531 CrossRef CAS PubMed; (c) M. A. Huertos, J. Pérez, L. Riera, J. Díaz and R. López, Chem. – Eur. J., 2010, 16, 8495–8507 CrossRef CAS PubMed; (d) J. Ruiz and B. F. Perandones, J. Am. Chem. Soc., 2007, 129, 9298–9299 CrossRef CAS PubMed; (e) V. Miranda-Soto, D. B. Grotjahn, A. G. DiPasquale and A. L. Rheingold, J. Am. Chem. Soc., 2008, 130, 13200–13201 CrossRef CAS PubMed; (f) J. Ruiz, A. Berros, B. F. Perandones and M. Vivanco, Dalton Trans., 2009, 6999–7007, 10.1039/B906450H; (g) K. Araki, S. Kuwata and T. Ikariya, Organometallics, 2008, 27, 2176–2178 CrossRef CAS.
- M. A. Huertos, J. Pérez, L. Riera, J. Díaz and R. López, Angew. Chem., Int. Ed., 2010, 122, 6553–6556 CrossRef.
- M. A. Huertos, J. Pérez and L. Riera, Chem. – Eur. J., 2012, 18, 9530–9533 CrossRef CAS PubMed.
- M. A. Huertos, J. Pérez and L. Riera, Chem. – Eur. J., 2012, 18, 9530–9533 CrossRef CAS PubMed.
- C.-C. Ko, C.-O. Ng and S.-M. Yiu, Organometallics, 2012, 31, 7074–7084 CrossRef CAS.
- (a) F. E. Hahn and L. Imhof, Organometallics, 1997, 16, 763–769 CrossRef CAS; (b) M. Tamm and F. Ekkehardt Hahn, Coord. Chem. Rev., 1999, 182, 175–209 CrossRef.
- (a) B. Royo, E. Herdtweck and C. C. Romão, Eur. J. Inorg. Chem., 2004, 2004, 3305–3309 CrossRef; (b) H. Braband, T. I. Zahn and U. Abram, Inorg. Chem., 2003, 42, 6160–6162 CrossRef CAS PubMed; (c) E. Oehlke, T. Kückmann and U. Abram, Z. Anorg. Allg. Chem., 2007, 633, 830–834 CrossRef CAS; (d) T. I. Kückmann and U. Abram, Inorg. Chem., 2004, 43, 7068–7074 CrossRef PubMed; (e) H. Braband, D. Przyrembel and U. Abram, Z. Anorg. Allg. Chem., 2006, 632, 779–785 CrossRef; (f) H. Braband, E. Oehlke and U. Abram, Z. Anorg. Allg. Chem., 2006, 632, 1051–1056 CrossRef CAS; (g) R. Lum, H. Zhang, W. Zhang, S.-Q. Bai, J. Zhao and T. S. A. Hor, Dalton Trans., 2013, 42, 871–873 RSC.
- A. M. Magill, K. J. Cavell and B. F. Yates, J. Am. Chem. Soc., 2004, 126, 8717–8724 CrossRef CAS PubMed.
- (a) D. Canella, S. J. Hock, O. Hiltner, E. Herdtweck, W. A. Herrmann and F. E. Kühn, Dalton Trans., 2012, 41, 2110–2121 RSC; (b) O. Hiltner, F. J. Boch, L. Brewitz, P. Härter, M. Drees, E. Herdtweck, W. A. Herrmann and F. E. Kühn, Eur. J. Inorg. Chem., 2010, 2010, 5284–5293 CrossRef.
- S. J. Hock, L.-A. Schaper, A. Pöthig, M. Drees, E. Herdtweck, O. Hiltner, W. A. Herrmann and F. E. Kühn, Dalton Trans., 2014, 43, 2259–2271 RSC.
- (a) T. E. Müller, M. Grosche, E. Herdtweck, A.-K. Pleier, E. Walter and Y.-K. Yan, Organometallics, 1999, 19, 170–183 CrossRef; (b) L. L. Ouh, T. E. Müller and Y. K. Yan, J. Organomet. Chem., 2005, 690, 3774–3782 CrossRef CAS.
- L. A. Casson, S. Muzzioli, P. Raiteri, B. W. Skelton, S. Stagni, M. Massi and D. H. Brown, Dalton Trans., 2011, 40, 11960–11967 RSC.
- H. Zhu, Z. Yang, N. Li, X.-J. Wang, F. Wang, H. Su, Q. Xie, Y. Zhang, Y.-X. Ma and B.-H. Lin, J. Organomet. Chem., 2012, 716, 95–102 CrossRef CAS.
- S. J. Hock, PhD Thesis, Technical University of Munich, 2013.
- C. A. Quezada, J. C. Garrison, M. J. Panzner, C. A. Tessier and W. J. Youngs, Organometallics, 2004, 23, 4846–4848 CrossRef CAS.
- I. Demachy and Y. Jean, Inorg. Chem., 1996, 35, 5027–5031 CrossRef CAS PubMed.
- M. Benz, B. Spingler, R. Alberto and H. Braband, J. Am. Chem. Soc., 2013, 135, 17566–17572 CrossRef CAS PubMed.
- C. Kluba and T. Mindt, Molecules, 2013, 18, 3206 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |