
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Critical design of heterogeneous catalysts for biomass valorization: current thrust and emerging prospects
Sudipta
De
a,
Saikat
Dutta
b and
Basudeb
Saha
*b
aDepartment of Chemical and Biomolecular Engineering, National University of Singapore, 4 Engineering Drive 4, 117585, Singapore
bCatalysis Center for Energy Innovation, ISE Laboratory, University of Delaware, 221 Academy Street, Newark, Delaware, USA. E-mail: bsaha@udel.ecu
First published on 26th August 2016
Abstract
Catalysis in the heterogeneous phase plays a crucial role in the valorization of biorenewable substrates with controlled reactivity, efficient mechanical process separation, greater recyclability and minimization of environmental effects. In this minireview, we have critically analyzed the design aspects of catalysts with multifunctional properties in making biomass conversion processes more productive and sustainable. The catalytic systems have been categorized into subgroups such as metal oxides and phosphates, supported metals, functionalized porous materials, acidic and ion-exchange resins, zeolites and carbonaceous materials to discuss their structural features and active sites towards intrinsic reactivity and selectivity in the transformation of biomass intermediates via hydrolysis, dehydration, hydrodeoxygenation and oxidation pathways. Recent advances in photocatalytic materials for the oxidation of biomass-derived small molecules (such as sugars, alcohols, carboxylic acids) and lignin model compounds and the role of different heteroatoms in improving photo-electronic properties are discussed. The synergistic effect of metal nanoparticulate sites and acid–base supports of bimetallic catalytic systems in performing multistep cascade processes in one-pot are analyzed.
1. Introduction
Biomass is considered as the most abundant renewable resource with a worldwide production of 170 billion metric tons per year by photosynthesis.1 Calculation shows that ∼1.3 billion tons of lignocellulosic biomass can produce ∼33% of U.S. transportation fuel and 25% of all organic chemicals per year.2 The carbohydrate content of lignocellulosic biomass is typically 75%, which can be converted into soluble sugars directly by employing acid hydrolysis processes. The obtained sugars can then be converted into ethanol, advanced high octane biofuels or fuel additives, various chemicals, foods, and medicines using chemical and biochemical technologies.3 Biomass is typically composed of 40–50% cellulose, 16–33% hemicelluloses, 15–30% lignin, and small amounts of other components.Over the past two decades, several catalytic processes have been developed for the efficient conversion of biomass resources into biofuels and value-added chemicals.4–8 Homogeneous catalytic systems generally offer relatively high activity and selectivity, but these catalysts have some disadvantages in separation, recyclability and disposal. Heterogeneous catalytic systems, on the other hand, offer mighty advantages in terms of easy separation, reusability and de-corrosion with the ability to provide practical conveniences in a continuous system. Another merit of heterogeneous catalysts is their stability to severe reaction environments; some can withstand temperatures as high as 1300 °C and pressure up to 350 bar.9
The first step in biomass processing involves depolymerization of biomass constituents into soluble oligosaccharides, which is often carried out in the presence of a mineral acid. This step, known as lignocellulose pretreatment, is required to reduce cellulose crystallinity by disrupting H-bonding networks, to increase material porosity and to remove lignin. The pretreatment step significantly improves the yield of oligosaccharides and reduces the operating costs of downstream units. However due to severe disadvantages of mineral acids, alternative pathways have been established such as ball-milling,10 solubilization/precipitation in ionic liquids11 and liquid acidic/alkaline solutions,12,13 non-thermal atmospheric plasma14 and steam explosion. After the pretreatment step, soluble oligosaccharides are converted to monosaccharides through the hydrolysis process. Subsequently, monosaccharides, e.g., glucose and xylose, are converted to platform molecules using combined Brønsted (B)–Lewis (L) acid catalytic systems.15–17 The BL acid catalyzed isomerization and dehydration reactions for sugar conversion have been significantly advanced over the past few years with the appropriate design of various types of solid catalysts having a large pore size and strong acid density.7,18 These types of solid catalysts usually possess a high surface area because the reactions take place on the surface of the materials. Thus, it is desirable to maximize the surface area to achieve better performance. The acidity of these materials can be tuned by adopting different synthesis protocols. However, the key challenge with heterogeneous catalysis is to identify the active catalytic sites suitable for a particular type of transformation. In particular, biomass contains a large number of oxygen functional groups that are subject to chemical transformations; thus, the conversion process requires a new paradigm of how to design heterogeneous catalysts that can transform the said functional groups selectively. The adsorption behavior of the functional groups on the catalyst surface has to be extensively understood in order to design an appropriate catalyst for a particular transformation.
Zeolites are excellent solid catalysts which have exhibited excellent catalytic performance in the petrochemical industry over many decades largely due to the presence of uniform microporous networks (0.25–1 nm).19 However, these catalysts pose a challenge in biomass processing because of severe diffusion limitations that arise from the presence of sole micropores. Therefore, a search for materials containing larger pores has stimulated the development of ordered mesoporous materials such as M41S20 and SBA.21 In addition, hierarchically porous materials exhibiting multimodal porosity have been developed. The latter materials with micro-, meso-, and/or macroporous structures have superior mass/heat transfer rates and enhanced accessibility to active sites.22–25 In addition to such features as high surface area and porosity, the presence of large numbers of active sites is an added advantage. However, many of the currently available porous materials fail to meet these criteria, and therefore, it is of special importance to gain a better understanding of the combined effects of pore hierarchy and active sites on catalytic performance to design new catalysts.
This review article presents an overview of recent advances in catalyst design, aiming to improve the efficiency of biomass depolymerization and upgrade to chemicals and fuels. The intrinsic reactivity and selectivity of the catalytic systems are assessed to correlate with the structure and active sites of the catalysts. Particular emphasis has been given to establishing the relationship between catalytic activities and various intrinsic properties including the effect of pore size, surface area, and composition of the solid and Brønsted/Lewis acid density ratio. Photocatalytic materials for the photo-oxidation of biomass-derived molecules and the roles of different heteroatoms in improving photo-electronic properties are discussed at length. The synergistic effect between metal nanoparticle structures and acid–base supports of bimetallic catalysts in performing multistep cascade processes in one pot is analyzed. Scheme 1 illustrates an overview of the fundamental reactions in the biomass transformation process and the different functionalities of heterogeneous catalysts involved therein.
 | ||
| Scheme 1 Key functionalities of heterogeneous catalysts required for fundamental reactions involved in biomass conversion. | ||
2. Metal oxide- and phosphate-based catalysts
Metal oxides are often used as solid catalysts in biomass transformation chemistry. Metal oxides with high specific surface area and large pore size are of special interest because the reactants can come into proximity with the active sites inside the metal oxide pores. Among different metal oxides, titania (TiO2) and zirconia (ZrO2) based materials have attracted special attention due to their good acid–base properties. Only some particular phases of these metal oxides are catalytically active for the conversion of glucose and fructose. For example, a report on the effects of TiO2 (anatase or rutile phase) and ZrO2 (monoclinic/tetragonal mixture of ZrO2) for glucose and fructose conversions in hot-compressed water at 200 °C26 reveals that anatase TiO2, with both acidic and basic sites, can promote high glucose conversion with an excellent HMF yield, while rutile TiO2 is found inactive. A basic monoclinic/tetragonal mixture of ZrO2 (m/c-ZrO2) showed enhanced activity for the reaction. In the fructose study, both anatase TiO2 and m/c-ZrO2 enhanced the conversion of fructose. However, in the case of glucose, anatase TiO2 offers a better HMF yield than m/c-ZrO2. Collective observation helps to find out the appropriate nature of the catalysts suitable for the different steps of the entire transformation. The isomerization of glucose into fructose is actually enhanced by the basic sites of the catalysts, whereas the formation of HMF is enhanced by the acidic sites of the catalysts. It is an established experimental fact that solid base ZrO2 could promote the isomerization of glucose to 1,2-enediol, which is considered to be the decisive step for generating HMF.27 Based on this fact, Liu et al. utilized a catalytic system consisting of solid base ZrO2 together with solid acid SO42−/TiO2–SiO2, which offers a maximum 48% HMF yield from starch hydrolyzate under optimal conditions and at a solid acid-to-solid base ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4.28 Our group reported multiple templating pathways for the preparation of self-assembled mesoporous TiO2 materials.29–31 Calcination at high temperatures provided uniform size- and shape-controlled TiO2 nanoparticles with significantly high surface area. Carboxylic acid containing molecules (such as amino acid) were mainly used as a template which imparted a considerable amount of acidity inside the pores after removal of the templates. The presence of dual Brønsted and Lewis acid sites promoted the conversion of monosaccharides (fructose, glucose, mannose, lactose and galactose) and disaccharides (sucrose, maltose and cellobiose) into HMF in moderate to high yield. Mesoporous catalysts containing dual Brønsted acid (–HSO3, –HPO3) and Lewis acid (TiO2, Sn, Ti) sites effectively converted monosaccharides and oligosaccharides to HMF biphasic solvent systems.32,33
:4.28 Our group reported multiple templating pathways for the preparation of self-assembled mesoporous TiO2 materials.29–31 Calcination at high temperatures provided uniform size- and shape-controlled TiO2 nanoparticles with significantly high surface area. Carboxylic acid containing molecules (such as amino acid) were mainly used as a template which imparted a considerable amount of acidity inside the pores after removal of the templates. The presence of dual Brønsted and Lewis acid sites promoted the conversion of monosaccharides (fructose, glucose, mannose, lactose and galactose) and disaccharides (sucrose, maltose and cellobiose) into HMF in moderate to high yield. Mesoporous catalysts containing dual Brønsted acid (–HSO3, –HPO3) and Lewis acid (TiO2, Sn, Ti) sites effectively converted monosaccharides and oligosaccharides to HMF biphasic solvent systems.32,33
The major factors that affect catalysts concerning hydrolysis–dehydration chemistry are acidity and surface area. Mesoporous Nb–W oxide shows a higher turnover frequency (TOF) for the hydrolysis of sucrose than other solid acids (i.e. Amberlyst-15, Nb2O5).34 The added W-oxide phase shows an extra benefit over pure phase Nb2O5 in terms of reactivity. The acid strength increased gradually with the addition of W, offering the highest reaction rate with mesoporous Nb3W7 oxide. Fig. 1 shows that the tungsten-containing samples have both Brønsted acid and Lewis acid sites whereas Nb oxide has negligible Brønsted acid sites. However, the amount of Brønsted acidity is much lower than that of Lewis acidity even at an optimum Nb/W ratio. The high catalytic performance of Nb3W7 oxide was attributed to the high surface area, mesoporous structure and multiple strong acid sites (Brønsted and Lewis acid sites). It is shown that the number of active acid sites increased with an increase in surface area. The mesoporous Nb3W7 oxide exhibited a two-fold higher reaction rate and turnover frequency than that of bulk Nb3W7 oxide, indicating that the mesoporous structure enhanced the reaction rate owing to the accessible acid sites. Furthermore, the ratio of Brønsted and Lewis acid sites could be easily tuned by varying the Nb/W ratio. However, the Nb–W oxide catalyst had a lower activity for cellobiose hydrolysis due to the low number of Brønsted acid sites.
 | ||
| Fig. 1 FTIR spectra for pyridine adsorbed by mesoporous (a) Nb, (b) Nb7W3, (c) Nb5W5, and (d) Nb3W7 oxides (B = Brønsted acid site; L = Lewis acid site). Assignments: 1590 cm−1 (strong Lewis acid site), 1532 cm−1 (strong Brønsted acid site), 1485 cm−1 (very strong Brønsted acid site or strong Lewis acid site), 1440 cm−1 (very strong Lewis acid site). Adapted from ref. 34. Copyright 2010, with permission from Wiley-VCH.34 | ||
A similar type of mixed oxide phase catalyst, HNbMoO6, exhibited a much higher activity, producing glucose at twice the rate of Amberlyst-15 (Scheme 2 and Fig. 2).35 The improved catalytic activity of this layered structured material could be related to its strong acidity, water-tolerance and intercalation ability. In the previous literature, it was observed that HNbMoO6 was able to intercalate water, alcohol and ketone at room temperature, resulting in a high activity for acid-catalyzed reactions such as Friedel–Crafts alkylation and acetalization.36 The hydrated layered HNbMoO6 materials contained a strong acidic interlayer gallery that accommodated the insertion of saccharides (containing many hydroxyl groups) and successfully intercalated them to achieve increased solubility. In some cases, reactivity does not solely depend on these factors. For instance, zirconium phosphate (ZrP) solid acids with varied surface areas obtained from different preparation methods exhibit similar catalytic properties for the dehydration of fructose.37
 | ||
| Scheme 2 Hydrolysis of cellulose to glucose with layered HNbMoO6 catalyst. Adapted from ref. 35. Copyright 2008, with permission from the Royal Society of Chemistry.35 | ||
 | ||
| Fig. 2 Different hydrolysis rates of sucrose with different solid acids. Adapted from ref. 35. Copyright 2008, with permission from the Royal Society of Chemistry.35 | ||
Metal phosphate-based catalysts are more effective than simple metal oxides in the one-pot conversion of cellulose and carbohydrates into platform chemicals where both isomerization and dehydration steps are required. Due to the presence of both Brønsted and Lewis acidity, phosphate-based systems containing different metal components (such as Ti, V, Zr, Nb, Sn etc.) have achieved high yields of HMF and levulinic acid.32,38–42 To make an effective metal phosphate catalyst, the optimum ratio of Brønsted and Lewis acidity needs to be maintained. It was observed that excessive Brønsted acid sites had a detrimental effect on the isomerization process, whereas excessive Lewis acid sites led to the formation of by-products (e.g., humins) and decreased the selectivity to HMF.42 The introduction of a second metal (such as Ru, Pt etc.) into the dual acidic phosphates can perform the hydrogenation or oxidation of platform molecules, which extends the process in a cascade manner. Such bifunctional systems have been used in the one-pot conversion of cellulose and carbohydrates into different valuable products including hydrocarbons, isosorbide, 2,5-furandicarboxylic acid (FDCA), 2,5-diformylfuran (DFF) etc.43–45
Alongside the thermal catalytic transformation of bio-based feedstocks, photocatalytic transformation has evolved as an alternative technique to derive platform chemicals.46 In principle, biomass substrates contain a large number of oxygen functionalities, e.g., alcohol groups which can be oxidized using a photocatalytic process to obtain various useful chemicals. These functional groups can serve as sacrificial agents (electron donors) for photocatalytic self-oxidation to produce ketones, aldehydes, and/or carboxylic acids.47 Thermodynamically, alcohols with a singlet electronic configuration are unable to react directly with dioxygen, which has a triplet electronic configuration.48 Thus, metal catalysts are necessary to induce the formation of singlet oxygen species.
Heterogeneous photochemistry continues to be an important component of modern chemistry in the 21st century. Growing knowledge on mechanisms, the development of new technologies for storage and conversion of solar energy, environmental detoxification of liquid and gaseous ecosystems, and the photochemical production of new materials have transformed this field as a new avenue in sustainable chemistry research.46 While photochemistry has been applied in organic transformation significantly for the past three decades, its application in biomass conversion has not been explored extensively. Heterogeneous photocatalysts can perform several chemical transformations at room temperature and atmospheric pressure using sunlight as a renewable source of energy.
Photocatalysis is one of the best sustainable routes that use mild reaction conditions for the selective transformation of biomass-derived platform molecules into valuable chemicals.49 Selective photo-oxidation of biomass-derived sugars provides a wide range of high value chemicals such as succinic, 2,5-furandicarboxylic, 3-hydroxypropionic, gluconic, glucaric and levulinic acid, 3-hydroxybutyrolactone etc. (Table 1).50 Titania-based catalysts are of special interest for excellent photocatalytic activity. The strong oxidative potential of the positive holes in titania oxidizes water to form hydroxyl radicals that can oxidize organic substrates directly. Titania materials with improved electronic properties have been synthesized through sol–gel processes by varying the precursor and/or the ageing conditions (magnetic stirring, ultrasound, microwave or reflux) and used for the liquid-phase selective photo-oxidation of bio-derived alcohols to the corresponding aldehydes.46
| Entry | Model biomass-based molecules | Photocatalysts | Products | Ref. |
|---|---|---|---|---|
| 1 | Methanol | Anatase TiO2 | Methyl formate | 58 |
| 2 | Malic acid | TiO2–guanidine–(Ni, Co)–Fe2O4 | Formic acid | 54 |
| 3 | Acetic, propionic, n-butanoic and n-pentanoic acid | Pt/TiO2 (rutile) | Corresponding hydrocarbons | 59 |
| 4 | Glucose | Bare-TiO2 and supported TiO2 on zeolite Y | Glucaric and gluconic acid | 55 and 56 |
| 5 | Glucose | Fe–TiO2 and Cr–TiO2 supported on zeolite Y | Glucaric and gluconic acid | 60 and 61 |
| 6 | Lignocellulose | Commercial TiO2 | Sugars | 62 |
| 7 | Kraft lignin | Ta2O5–IrO2 thin film | Vanillin and vanillic acid | 63 |
Photocatalytic oxidation depends on the type of alcohol groups. Primary alcohols generally show higher reactivity with high selectivity. The proposed mechanism of this reaction is initiated with the interaction of a surface hole (of catalyst) with the hydroxyl group of the alcohol, forming metal-oxo species with proton removal (Scheme 3).51 Alcohols with long carbon chains and branching are the most suitable substrates that make the proton removal step easier. Furthermore, a higher number of adjacent hydrogen atoms is also a key factor for the simpler removal of proton to achieve an improved conversion rate.
 | ||
| Scheme 3 Photocatalytic oxidation of 1-phenyl ethanol to acetophenone over TiO2 (□ = surface hole).51 | ||
The anatase form of TiO2 with a band gap of 3.2 eV is reported to be the best for photo-activity and photo-stability in most reaction media. Practically, TiO2 photo-activation takes place in the range of 300–388 nm. However, the main problem is the inadequate absorption of solar energy at this wavelength (only ca. 5% of the solar light energy can be absorbed by TiO2). The most useful strategy to overcome this problem is the doping of TiO2 with metallic52 (e.g., Cr, Fe, Zn etc.) or non-metallic53 (e.g., N, C, S etc.) elements. Doping with these elements renders the recombination of electron–hole pairs in TiO2 thereby improving the photocatalytic efficiency of TiO2 under visible light and/or sunlight. For example, the selective conversion of maleic acid to formic acid is conducted in an aqueous medium under visible light irradiation using a magnetically separable TiO2–guanidine–(Ni, Co)–Fe2O4 nanocomposite (Table 1, entry 2).54 Using a similar analogy, acetic, propionic, butanoic and n-pentanoic acid could be decarboxylated to hydrocarbons in the absence of oxygen. As a result of the modification with guanidine, the band gap of the metal oxide semiconductor is remarkably decreased and the catalyst exhibits activity under visible light and sunlight irradiation. This simple process opens up a promising selective synthesis method for the production of shorter chain acids or hydrocarbons from higher aliphatic carboxylic acids.
Nanosized TiO2 materials have also offered good photo-oxidation activity for the oxidation of glucose to organic carboxylic acids, namely glucaric (GUA) and gluconic (GA) acid55 with high selectivity. The efficiency of the TiO2 photocatalyst depends on the synthesis methods and the solvent system. For example, TiO2 synthesized by an ultrasound-modified sol–gel method exhibited superior activities over other titania materials. Other TiO2 materials prepared by the solvent refluxing method showed a lower band gap (3.03 eV) with a higher absorption threshold (409 nm). The selectivity of the organic products also depends on the nature of the solvent systems. For example, a change in solvent composition from 10%H2O/90%CH3CN to 50%H2O/50%CH3CN resulted in improved product selectivity from 39% to 71%. The selectivity of the reaction products towards carboxylic acids also improved by modifying the TiO2 catalyst with a zeolite type Y (SiO2:Al2O3 = 80) support.56 The high efficiency of the latter catalyst is due to the charge density of Y type zeolites, which tend to adsorb cationic substrates and repulse anions.57 Due to this electronic feature, the carboxylic acid products are repulsed by the zeolite, thereby facilitating the selective photo-oxidation of glucose. The selectivity can be further improved by doping the supported TiO2 materials with transition metal ions like Fe and Cr. For example, Fe–TiO2 systems showed 94% selectivity after 20 min of illumination.
Numerous efforts have been given to develop strategies for the depolymerization of cellulose and hemicellulose to produce high-value chemicals and fuels. However, understanding of the complex chemical structure and recalcitrant nature of lignin remains elusive.64 Recently, a few promising catalytic lignin depolymerization methods have been developed; however, many of these involve chemical oxidation pathways using homogeneous transition-metal catalysts at elevated temperatures (>80 °C).65–71 These processes are usually energy-intensive and uncontrollable (in terms of selectivity) and less suitable for applications. Therefore, there is a need to develop a room temperature lignin depolymerization method that could offer the selective cleavage of specific linkages within the biopolymer.
Selective oxidation of alcohols to the corresponding carbonyl compounds (especially aldehydes) is one of the important organic transformations as carbonyl compounds are widely used in the food, beverage, and pharmaceutical industries and as precursors in chemical industries.72,73 Among the selective oxidation processes of alcohols, the conversion of aromatic alcohols to aldehydes is of particular interest because the aromatic alcohols represent model lignin compounds obtained from the depolymerization of real lignin.2 Since benzyl alcohol is the key structural unit of lignin, major focus has been given to its reactivity and selective transformation strategies. Another advantage is that benzyl alcohol is the simplest aromatic alcohol and therefore presents fewer analytical challenges relative to the complicated lignin models.
Recently, photo-assisted transformation of benzyl alcohol to benzaldehyde has been the center of focus because this could replace the industrial synthesis route via benzyl chloride hydrolysis derived from toluene chlorination or toluene oxidation. Metal oxide based catalysts such as TiO2,74–76 Nb2O5,77 and ZnO (ref. 78) have been well explored for alcohol oxidation under UV irradiation. Improved activity even in visible light has been achieved by the incorporation of various noble metals including Au,79 Pd,80 and Pt (ref. 81 and 82) on different photoactive catalyst supports. It has been shown that the presence of Brønsted acids in the catalysts can accelerate the aerobic photo-oxidation of alcohols. For example, a small quantity of SiO2 can enhance the activity when incorporated into TiO2via acid pretreatment.83 The incorporated SiO2 increases the proton density that effectively promotes the decomposition of the formed surface peroxide–Ti species, leading to catalyst surface site regeneration. As a result, an about two-fold improvement in conversion was achieved when a small quantity of SiO2 was incorporated into TiO2 after the pre-treatment with acid to afford more Brønsted acid sites.
The mechanism of the photo-oxidation of aromatic alcohols under visible light irradiation was studied over anatase TiO2 photocatalyst in the presence of oxygen.84 The success of the photo-oxidation process relies on the possibility of absorbing visible light by the aromatic alcohol to form surface complex species with TiO2. The formation of such species was confirmed by studying the reaction with fluorinated TiO2 that showed a dramatic decrease in activity. Fig. 3 summarizes the selective photocatalytic oxidation of benzyl alcohol in the presence of oxygen and titania wherein the surface complexes are formed by close interaction of the alcohol groups of the aromatic ring with the surface functionalities. The photo-produced holes abstract hydrogen from the –CH2OH groups of the photo-excited species, which leads to the oxidation of the alcohol to aldehyde. At the same time the photo-induced electrons are trapped by oxygen in the reacting medium. O2 plays a significant role as an electron scavenger and possibly leads to the re-generation of surface hydroxyl groups on TiO2. The mechanism reveals that formation of the surface complex is the crucial step that initiates the photo-oxidation process, where surface hydroxyl groups are important for their formation. Therefore, it can be expected that materials with the ability to form surface complexes in the presence of O2 will undergo visible light induced photo-oxidation in the same manner.
 | ||
| Fig. 3 Proposed reaction mechanism for the photocatalytic selective oxidation of benzyl alcohol under visible light irradiation.84 | ||
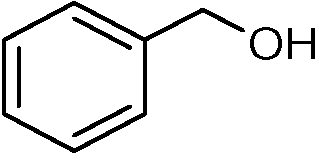
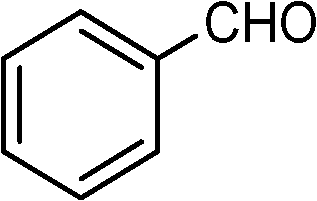
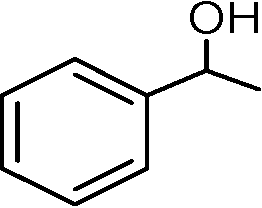
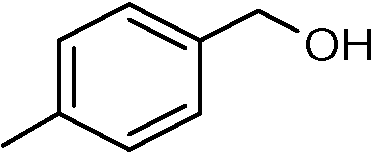
Nanosized noble metals such as Au, Pd and Pt in combination with a semiconductor can take advantage of plasmonic effects to achieve the photocatalytic selective production of several aromatic aldehydes from their corresponding aromatic alcohols (Table 2, entries 2–4).85–87 A similar advantage of the plasmonic effect of Pt can be extended to glucose conversion to aromatic aldehyde as shown in Fig. 4.









 | ||
| Fig. 4 Pt/TiO2 extends the plasmonic effect for the conversion of glucose. | ||
Plasmonic photocatalysts composed of gold nanoparticles supported on the anatase–rutile interphase (Evonik P-25) show a plasmon activation phenomenon where the conduction electrons of Au particles are activated by visible light followed by consecutive electron transfer at the Au/rutile/anatase interphase contact. After the removal of electrons, the positively charged Au particles facilitate the oxidation process with the reduction of oxygen. Metal core–semiconductor shell nanohybrids are also efficient visible-light-driven photocatalysts for the selective oxidation of benzyl alcohol to benzaldehyde. Pt and Pd serve as the cores of the hybrid systems and CeO2 as the shells. Electrons produced under visible light irradiation are trapped by the metal cores and adsorbed benzyl alcohol interacts with the holes to form the corresponding radical cation (Fig. 5). The dioxygen or superoxide radical reacts with these radical cations to form aldehydes.
 | ||
| Fig. 5 (A) TEM image of as-synthesized multi-Pd-core–CeO2-shell nanoparticles. (B) A plausible mechanism for the selective oxidation of aromatic alcohols to aldehydes in the presence of multi-Pd-core–CeO2-shell photocatalyst under visible light irradiation. Adapted from ref. 87. Copyright 2012, with permission from the Royal Society of Chemistry.87 | ||
Polyoxometalate (POM) materials can be used as heterogeneous photocatalysts when supported on amorphous mesoporous molecular sieves, titania, and silica. Heteropolyoxometalates of the type [S2M18O62]4+ (M = W, Mo) are highly active for the photo-oxidation of aromatic alcohols under sunlight and UV/vis light in acetonitrile.88 Near-UV/vis irradiation of a POM solution results in an oxygen-to-metal charge-transfer excited state that has strong oxidation ability, which is responsible for the oxidation of organic substrates.
3. Supported metal catalysts
Hydrodeoxygenation (HDO) is considered as the most effective way to upgrade bio-oils into hydrocarbon fuels. The key challenge of the HDO process is to achieve a high degree of oxygen removal with minimum hydrogen consumption and for this, proper and careful design of catalysts is required. Several catalytic systems have been reported for HDO with various advantages and disadvantages where precious noble metal catalysts (e.g., Pd, Pt, Re, Rh, and Ru) have shown remarkable activities over non-precious transition metal catalysts (e.g., Fe, Ni and Cu).90 However, in terms of process cost, transition metals are more advantageous than noble metals and sometimes exhibit comparable HDO efficiency.One of the major challenges in upgrading biomass-derived platform chemicals is that these chemicals generally contain various functionalities. Taking into account a highly useful platform molecule, 5-hydroxymethylfurfural (HMF), which contains four active functional groups and often undergoes unwanted transformations in the HDO process leading to the formation of several by-products with low DMF yield.
A proper choice of a bimetallic catalytic system can selectively convert HMF to DMF with high yield. In bimetallic systems, the incorporation of a second metal creates a number of possibilities of modifying the surface structure and composition of metal catalysts. The properties of bimetallic catalysts significantly differ from those of their monometallic analogs due to geometric and electronic effects between the two metals.91 The major advantage of the bimetallic systems is the tunable catalytic activity by varying the size and composition. PtCo nanoparticulate (NP) catalyst is an example of this type, which selectively hydrogenates carbonyl groups of substrates in the presence of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds.92 Similarly, PtCo NPs encapsulated in hollow carbon spheres93 are reported to be effective for HDO of HMF at 120 °C to obtain 2,5-bis(hydroxymethyl)furan as the main product in 70% yield. A reaction at higher temperature (160 °C) produced DMF in high yield (96%). Under comparable reaction conditions, activated carbon-supported platinum (Pt/AC) and graphitized carbon-supported platinum (Pt/GC) catalysts achieved only 9% and 56% DMF yields, respectively. This comparison portrays a clear role of the second metal (here Co) in the higher activity of PtCo. Co is believed to alter the electronic environment of PtCo. Recently, Luo et al. revealed the mechanism for the selective HDO of HMF over PtCo nanocrystal catalyst, which demonstrated that the reaction was sequential on both Pt and PtCo alloys, with DMF formed as an intermediate product.94 The reaction of DMF could be greatly suppressed on the alloys, such that a Pt3Co2 catalyst can achieve DMF yields as high as 98%. The Co oxide surface monolayer interacted weakly with the furan ring to prevent side reactions, including overhydrogenation and ring opening, while providing sites for effective HDO to the desired product, DMF. The same group extended the application to other base metals (such as Ni, Cu, and Zn) along with Pt and studied the composition dependent HDO activity of the catalysts.95 Ebitani et al. reported a carbon supported PdAu bimetallic catalyst (PdxAuy/C) for the selective hydrogenation of HMF to DMF.96 The bimetallic PdxAuy/C catalyst exhibited significantly higher activity as compared to monometallic Pd/C and Au/C catalysts due to the existence of a charge transfer phenomenon from Pd to Au atoms as proved by XPS and X-ray absorption near-edge structure (XANES) analyses. A remarkable synergy between Pd and Ir Pd–Ir/SiO2 catalyst has been reported for the hydrogenation of furfural and HMF in aqueous medium.97 It is shown that Ir improved the TOF of the bimetallic catalyst for CO hydrogenation by promoting CO adsorption on the surface and thereby stronger interaction of the surface Pd sites with the furan ring of the substrate.
C bonds.92 Similarly, PtCo NPs encapsulated in hollow carbon spheres93 are reported to be effective for HDO of HMF at 120 °C to obtain 2,5-bis(hydroxymethyl)furan as the main product in 70% yield. A reaction at higher temperature (160 °C) produced DMF in high yield (96%). Under comparable reaction conditions, activated carbon-supported platinum (Pt/AC) and graphitized carbon-supported platinum (Pt/GC) catalysts achieved only 9% and 56% DMF yields, respectively. This comparison portrays a clear role of the second metal (here Co) in the higher activity of PtCo. Co is believed to alter the electronic environment of PtCo. Recently, Luo et al. revealed the mechanism for the selective HDO of HMF over PtCo nanocrystal catalyst, which demonstrated that the reaction was sequential on both Pt and PtCo alloys, with DMF formed as an intermediate product.94 The reaction of DMF could be greatly suppressed on the alloys, such that a Pt3Co2 catalyst can achieve DMF yields as high as 98%. The Co oxide surface monolayer interacted weakly with the furan ring to prevent side reactions, including overhydrogenation and ring opening, while providing sites for effective HDO to the desired product, DMF. The same group extended the application to other base metals (such as Ni, Cu, and Zn) along with Pt and studied the composition dependent HDO activity of the catalysts.95 Ebitani et al. reported a carbon supported PdAu bimetallic catalyst (PdxAuy/C) for the selective hydrogenation of HMF to DMF.96 The bimetallic PdxAuy/C catalyst exhibited significantly higher activity as compared to monometallic Pd/C and Au/C catalysts due to the existence of a charge transfer phenomenon from Pd to Au atoms as proved by XPS and X-ray absorption near-edge structure (XANES) analyses. A remarkable synergy between Pd and Ir Pd–Ir/SiO2 catalyst has been reported for the hydrogenation of furfural and HMF in aqueous medium.97 It is shown that Ir improved the TOF of the bimetallic catalyst for CO hydrogenation by promoting CO adsorption on the surface and thereby stronger interaction of the surface Pd sites with the furan ring of the substrate.
Apart from the noble metal-based catalysts, transition metal-based catalysts have also shown high DMF selectivity.98–100 Bimetallic systems based on the combination of first row transition metals such as Fe, Co, Ni, Cu and Zn were recently reported to produce DMF yields as high as 99%. The carbon nanotube-supported bimetallic Ni–Fe (Ni–Fe/CNT) catalyst was investigated for the selective hydrogenation and hydrogenolysis of HMF.98 Two different metal components presented different behaviors when used individually. The Fe/CNT catalyst showed low catalytic activity at both low and high temperatures, which confirmed the poor hydrogenation property of Fe. Meanwhile, the monometallic Ni/CNT catalyst showed high conversion but poor selectivity under the same conditions. The combination of Ni and Fe in an appropriate atomic ratio of Ni/Fe (2.0) significantly increased the selectivity to DMF, which could be attributed to the formation of Ni–Fe alloy species that selectively cleaved the C–O bond.
The beneficial effect of Fe in Ni–Fe/SiO2 bimetallic catalyst for the conversion of furfural has been reported by comparing the activity of Ni–Fe/SiO2 with Ni/SiO2.101 The Ni/SiO2 catalyzed reaction produced furfuryl alcohol and furan as the primary products via hydrogenation and decarbonylation of furfural. In contrast, the Ni–Fe/SiO2 catalyzed reaction produced 2-methylfuran (2-MF) as the major product. The presence of Fe in Ni–Fe/SiO2 suppressed the decarbonylation activity of Ni while promoting CO hydrogenation and C–O hydrogenolysis. DFT analysis suggests that the selectivity difference of the two catalysts is due to the difference in stability of η2-(C, O) surface species (Scheme 4). A stronger interaction between O (from the carbonyl group) and the oxyphilic Fe atoms makes the η2-(C, O) species more stable on the Ni–Fe/SiO2 surface than those on pure monometallic Ni/SiO2, resulting in ready hydrogenation of furfural to furfuryl alcohol and subsequent hydrogenolysis to 2-MF.
 | ||
| Scheme 4 Various intermediate species on the catalyst surface during the conversion of furfural.101 | ||
The bimetallic Zn/Pd/C system is another good example of the synergistic effect applied in the selective HDO of lignin compounds.102 The β-O-4 linkages in dimeric model lignin and synthetic lignin polymers were successfully cleaved by this system, producing HDO products in high yield without destroying the aromaticity. Controlled experiments suggested that the single use of Pd/C or Zn2+ was unable to promote HDO, indicating a synergistic effect between Pd/C and Zn2+ as represented in a mechanistic approach (Fig. 6); however, the chances of Pd–Zn alloy formation were excluded from an X-ray absorption spectroscopy (EXAFS) study. The same catalyst was effective for HDO of HMF, producing 85% DMF at 150 °C.103
 | ||
| Fig. 6 Proposed pathways for HDO of β-O-4 lignin substrate over Zn/Pd/C catalyst. Pathway (a) involves substrate binding to Zn2+ sites and hydrogen spillover from Pd sites. Pathway (b) represents desorption of Zn ions into solution and activation of substrate via binding; it is the Zn bound substrate that reacts with Pd sites on the catalyst's surface.102 | ||
Catalytic transfer hydrogenation (CTH) is a fascinating way to upgrade furfurals into liquid fuels where the solvent is a hydrogen donor. Recently, the application of a bifunctional Ru/RuOx/C catalyst has been reported for the hydrogenation of furfural and HMF in a series of alcohols.104–106 The metallic Ru sites serve mainly to produce hydrogen from the donor and the Lewis acid RuOx sites to carry out hydrogenolysis. Mechanistic investigation reveals that the hydrogenation of the carbonyl group of furfural to furfuryl alcohol proceeds through a Lewis acid-mediated intermolecular hydride transfer and the hydrogenolysis of furfuryl alcohol occurs mainly via ring-activation involving both metal and Lewis acid sites.
Sugar alcohols (such as sorbitol and mannitol) are valuable chemicals that can be derived from the hydrogenation of sugars. Supported metal catalysts have been widely used in biomass conversion due to their excellent hydrogenation activities. Though the hydrolysis of cellulose to glucose is rarely catalyzed solely by supported metal catalysts, this can be done by a combined system of a metal and an acid catalyst through a tandem process of hydrolysis and hydrogenation. Since the first production of hexitols by Pt/Al2O3 catalyst reported by Fukuoka and Dhepe in 2006,107 there have been numerous reports on the production of hexitols from cellulose by using supported catalysts such as Pt/carbon black,108 Ru/CNTs,109 Pt/MCM-48,110 Rh (or Ir)-Ni/MC,111 Pt-Ni/ZSM-5,112 Ni/CNF (ref. 113) etc. The Nakagawa and Tomishige groups used a bimetallic Ir-ReOx/SiO2 catalyst in combination with an acidic catalyst (HZSM-5) or a mineral acid component (H2SO4 or HCl).114,115 Mechano-catalytically depolymerized cellulose was converted to hexanols over the catalyst in a biphasic reaction system (n-decane + H2O). The organic phase (n-decane) played a crucial role in the product distribution. The catalyst possessed remarkable hydrogenolysis activity for the conversion of hexanols to n-hexane, while n-decane effectively extracted hexanols to suppress degradation of the hydrogenolysis product.
Oxidation is another important process in biomass transformation. Both 2,5-furandicarboxylic acid (FDCA) or 2,5-diformylfuran (DFF) are polyester building-block chemicals of commercial importance that can be obtained from the selective oxidation of HMF. Among the supported metal catalysts, gold is the most effective for the aerobic oxidation of HMF as demonstrated through a Au/CeO2 catalyzed oxidation of HMF into an ester of FDCA, dimethylfuroate, in basic solution, which is readily soluble in most solvents without the presence of a base.116 An excellent yield (99 mol%) of the ester was obtained due to the presence of nanometric CeO2 support acting as an oxygen pump by releasing and adsorbing O2 through a Ce+4/Ce+3 redox process. HMF oxidation to FDCA has also been reported in acidic solution using Au/CeO2, Au/TiO2 and Fe-POP (porous organic polymer) catalysts.117,118
It has been shown that the basicity of the support material of the catalyst plays an important role in HMF oxidation. Because of the basic property of the support, hydrotalcite-supported gold nanoparticles (Au/HT) are reported to be effective for liquid phase oxidation of HMF in water at 95 °C under O2 at atmospheric pressure.119 In addition to basicity, the active metal sites of the catalyst are also important. For example, the Au/HT catalyst exhibits higher activity than Au/MgO, even though MgO is more basic than HT. The Au/HT catalyst, containing highly dispersed and completely reduced Au metal on HT with a particle size distribution of 3.2 nm, achieved 81% FDCA yield in air at atmospheric pressure and at high HMF/metal ratios (150 and 200) with high turnover numbers. In contrast, unreduced Au/HT (without calcination) provided a negligible FDCA yield.
Apart from oxide-based catalyst supports, a new type of mesoporous covalent triazine frameworks (CTFs) with varying pore size, specific surface area, and N content has been recently reported.120 Ru clusters dispersed on CTF supports showed excellent activity for aerobic oxidation of HMF with high selectivity in FDCA. The activity of the CTF supported catalysts was comparably higher than those of activated carbon, γ-Al2O3, hydrotalcite, or MgO supported analogues.
An in situ process for the oxidation of HMF to FDCA has been reported by Kröger et al., where silicone-encapsulated methyl isobutyl ketone (MIBK)-swollen PtBi catalyst was used in a two-phase system consisting of water and MIBK.121 The oxidation process was studied in a membrane reactor and in the presence of an oxidation catalyst by varying the diameter of PtBi/C-silicone beads and the ratios of the acid catalyst (for fructose into HMF) and oxidation catalyst (Fig. 7). In the first step, acid catalyzed dehydration of fructose was carried out with Lewatit SPC 108, a microporous cationic ion exchanger in H+ form. In the second step, in situ formed HMF was oxidized by an MIBK-modified PtBi/C catalyst encapsulated in silicone beads. DFF was formed as the major product in MIBK. The authors claimed that water was needed as a co-substrate for the oxidation of the aldehyde group to form FDCA as the end product. Another report described the use of cobalt acetylacetonate encapsulated in a sol–gel silica matrix for the direct catalytic conversion of fructose to FDCA with 99% selectivity at 72% fructose conversion.122 In all cases, a cooperative effect was observed by the combination of a solid acid and a metal encapsulated in the matrix, leading to an improved conversion and selectivity compared to that of the same reaction performed in two consecutive steps.
 | ||
| Fig. 7 (A) Membrane reactor: (step 1) HMF formation in the water phase, (step 2) diffusion of HMF in the MIBK phase, (step 3) HMF oxidation. (B) Batch reactor for direct transformation of fructose into FDCA: (step 1) acid-catalyzed dehydration of fructose to HMF, (step 2) diffusion of HMF into the silicone network and oxidation into FDCA.121 | ||
Due to the recalcitrant nature of biomass, its conversion often requires harsh reaction conditions and therefore the catalysts must be stable enough at high temperature and pressure. This can be achieved by choosing a suitable support where the active phase can be well dispersed with strong interaction with the support. We have seen that acidic or basic supports can greatly promote some selective reactions due to their tunable functional properties. For example, acidic zeolites promote the dehydration of carbohydrates due to the presence of Brønsted acidity, while basic hydrotalcites are considered as excellent supports for oxidation processes such as the oxidation of HMF to FDCA. Sometimes, the catalyst supports can play an important role in the spillover of H2 or O2 molecules to achieve higher rates of reduction or oxidation, as described for the HDO of lignin by Zn/Pd/C.102
4. Functionalized porous materials
In the biomass process chemistry, product selectivity is one of the main issues. High boiling solvents like dimethylsulfoxide (DMSO) or N,N-dimethylformamide (DMF) can be used as promoters for better dissolution of carbohydrates and cellulose for higher selectivity to HMF. However, the isolation of the product is very complicated due to their high boiling points. Acid functionalization over silica materials could be a good option to eliminate the need for soluble reaction promoters as well as mineral acids. Bifunctional silicas (Taa-SBA-15) containing a propylsulfonic acid catalyst and a thio-ether group as promoter could be prepared either by post-synthetic grafting or by co-condensation (Fig. 8).123 The co-condensation method was more effective in incorporating robust functional groups into mesoporous silica which are more stable than the non-porous silica modified by post-synthetic grafting. This bifunctional catalyst achieved 74% selectivity for HMF at 66% fructose conversion.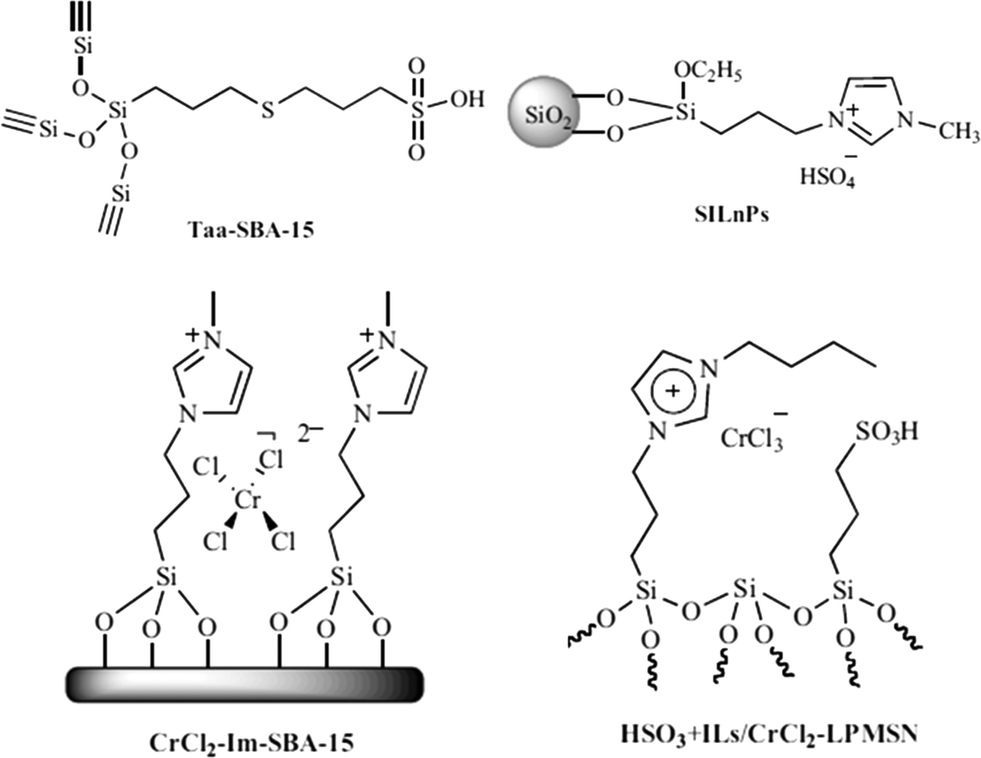 | ||
| Fig. 8 Functionalized silica materials used in acid dehydration of sugars. | ||
Similar issues are found with ionic liquids (ILs) as catalysts, co-solvents or promoters. ILs are effective for acid catalyzed carbohydrate dehydration; however, they have serious drawbacks such as high viscosity, tough separation process, corrosiveness and toxicity. To address these problems, Sidhpuria et al. prepared a series of supported ionic liquid nanoparticles (SILnPs) with particle size ranging from 293 ± 2 to 610 ± 11 nm by immobilization of ionic liquid 1-(triethoxysilyl-propyl)-3-methyl-imidazolium hydrogen sulfate (ILHSO4) on the surface of silica nanoparticles.124 The catalysts showed improved activity over other zeolites and acidic ion exchange resin catalysts, leading to >99% fructose conversion and 63% HMF yield. The main advantage of the catalysts is their easy separation via centrifugation and high recyclability over seven times.
Silica functionalization by creating a local reaction environment by immobilizing Cr on mesoporous SBA-15 resembled the coordination and chemical properties of the ionic liquid medium125 In spite of the unique reactivity of CrCl2 for the dehydration of glucose in 1-ethyl-3-methylimidazolium chloride ([EMIm]Cl) (70% HMF yield),126,127 it exhibited poor selectivity in water. This might be caused by the formation of stable, coordinatively saturated [Cr(H2O)6]2+ complexes.128 CrCl2-Im-SBA-15 (2.98 wt% Cr) offered a maximum HMF selectivity of 70% at a glucose conversion of 50% in the 2-BuOH/MIBK (7:3) extraction phase. A bifunctional solid catalyst (HSO3 + ILs/CrCl2-LPMSN) based on mesoporous silica nanoparticles functionalized with both sulfonic acid (–SO3H) and CrCl2 immobilized by ILs (ref. 129) achieved a high HMF yield (73%) with nearly 100% fructose conversion in DMSO.
Commercially available sulfonated resins have high acidity but may not be suitable as solid acids for liquid-phase reactions due to their very low surface areas of <1 m2 g−1. Micelle-templated silica materials generally possess very high surface areas in the range of 400–1000 m2 g−1 and high pore volumes in addition to narrow pore size distributions (20–100 Å).130 Sulfonic acid functionalized mesoporous silicas can be potentially applied in liquid phase dehydration reactions.
Previously, we stated that the co-condensation method is more effective than post-synthetic modification to incorporate robust functional groups into mesoporous silica. However, for achieving higher product selectivity, post-synthetic modification is more effective. Mesoporous MCM-41 silica catalyst anchored with sulfonic acid groups (SAGs) via post-synthetic modification was studied in the conversion of xylose.131 The catalyst displayed a fairly high selectivity for furfural (ca. 82% in DMSO or water/toluene mixture) at high xylose conversion (>90%). On the other hand, a hybrid-SO3H material, prepared via the co-condensation of (3-mercaptopropyl)trimethoxysilane with bis(trimethoxysilylethyl) benzene, showed a comparatively lower selectivity for furfural. The reason could be the presence of the large unidimensional mesopores in MCM-41-SO3H, which promoted the reaction of xylose to furfural by allowing fast diffusion of furfural out of the catalyst once formed, thereby protecting furfural and avoiding degradation. The hybrid-SO3H catalyst, on the other hand, contains more hydrophobic surface that could enhance the adsorption of furfural at the surface, where it reacted with intermediates on the accessible acid sites, increasing the extent of reactions responsible for furfural loss.
Porous coordination polymers (PCPs) have a large variety of pore surfaces and pore structures that can be decorated with various functional groups. For example, MIL-101, composed of a chromium oxide cluster and a terephthalate ligand, can be functionalized with acidic –SO3H groups (Scheme 5).132 The catalyst possesses a high BET surface area of 1915 m2 g−1 and offers a key advantage that it restricts the hydrolysis of cellulose into xylose, glucose and cellobiose sugars. The mild acidity of the catalyst prevents the formation of side-products such as HMF or levulinic acid which are normally found in the hydrolysis of cellulose under strong acidic conditions. The crystalline structure and water insolubility of cellulose prevents it from going deep inside the pores of MIL-101 but its contact with the acidic sites results in chain scission.
 | ||
| Scheme 5 Schematic representation of the structure of MIL-101-SO3H. Adapted from ref. 132. Copyright 2008, with permission from Wiley-VCH.132 | ||
Similar to porous organic polymers (POPs), functionalized POP materials have been recently explored in biomass conversion.117,133–136 Acidic –SO3H groups grafted on POPs are shown to be highly effective in many acid catalyzed reactions including esterification/transesterification and dehydration of bio-based feedstocks. For example, –SO3H functionalized pyrene-based microporous organic polymers exhibited excellent catalytic activity for the synthesis of biodiesels via esterification/transesterification of long chain fatty acids/esters at room temperature.133 The catalytic activities are directly related to the acidity of these materials, which could be easily regulated by the degree of sulfonation. Another work reported POP materials based on polytriphenylamine that efficiently converted carbohydrates and biomass to HMF. The key factors that controlled the activity of the materials are their very high surface area (1437 m2 g−1) and surface acidity (4.30 mmol g−1).
It is often seen that solid catalysts are difficult to separate from the reaction mixture during the hydrolysis of cellulose or other raw biomass. Highly efficient catalysts can degrade cellulose into soluble sugars in high yields, but the unconverted lignin components remain as a solid residue. Sometimes, humins are formed in large amounts which are difficult to separate from the recovered solid catalysts. To address the problem, Fu et al. developed a magnetic solid acid catalyst based on mesoporous silica (SBA-15), which can be successfully separated from the reaction system by using an external magnetic force.137Scheme 6 represents the preparation method of the sulfonated magnetic acid catalyst. A surfactant-templated sol–gel method was reported in which Fe3O4 magnetic nanoparticles were dispersed in the block copolymer P123 for the co-condensation of tetraethoxysilane (TEOS). After assembly, 3-(mercaptopropyl)trimethoxysilane (MPTMS) was added to introduce mercapto groups. Finally, the mercapto groups were oxidized by H2O2 into sulfonic acid groups inside the pores of the mesoporous silica.
 | ||
| Scheme 6 Preparation of magnetic acid catalyst Fe3O4-SBA-SO3H.137 | ||
Activity measurement experiments revealed that the magnetic solid acid (Fe3O4-SBA-SO3H) showed better performance compared to sulfuric acid in the hydrolysis of cellobiose. Furthermore, the catalyst provided 50% glucose yield in the hydrolysis of ionic liquid pretreated amorphous cellulose. When the catalyst was used for the hydrolysis of microcrystalline cellulose, the yield of glucose decreased to 26%, which is still better than those of other solid acid catalysts (Fig. 9). Unique structural features, e.g., uniform channels in the magnetic solid acid containing concentrated acid sites, allow the reactants to easily enter and interact with these acid sites. Cellulose chains undergo hydrolysis reaction by the protons diffused from these channels of SBA-15. The recycled catalyst was washed with 1 M H2SO4 and water/ethanol and dried at 80 °C for the next use without deactivation.
 | ||
| Fig. 9 Hydrolysis of macrocrystalline cellulose by different solid acids (1.5 g of catalyst, 1.5 g of cellulose, 15 ml of H2O at 150 °C for 3 h). Reproduced from ref. 137. Copyright 2011, with permission from the Royal Society of Chemistry (2011).137 | ||
Scheme 7 shows a strategy of deriving a chemo or enzymatic cascading protocol for the multi-step production of desired products like DMF or FDCA from cellulose where a metal nanoparticle supported by porous functionalized supports or desired enzymes immobilized on a support will play a major role. For example, a multifunctional support material can host a metal nanoparticle and chemical cascading may offer a one-pot conversion from cellulose to the desired products.
 | ||
| Scheme 7 Different functional groups for different transformations. | ||
5. Acidic and ion-exchange resins
Cation-exchange resins are used commercially as solid acid catalysts in many organic reactions, such as esterification,138 alkylation,139 hydration/dehydration,140,141 and condensation reaction.142 Strong acid sulfonated copolymer resin was thought to be unusable above 130 °C. Early in the 1960s, Hartler et al. studied cellulose hydrolysis using acidic resins, which were ineffective in the hydrolysis of glycosidic bonds.143 Qi et al. found that an ion-exchange resin (Dowex 50wx8) could work well above 130 °C or even 150 °C in a microwave.141 The use of a cation exchange resin catalyst together with an acetone–water reaction medium for fructose dehydration resulted in 73% HMF yield at 95% fructose conversion at 150 °C. Li et al. reported different acidic ion-exchange resins showing a visible effect on the dehydration of fructose to HMF.144 Three types of ion-exchange resins (macroporous strong-acid resin such as D072 and D001-cc, macroporous weak-acid resin such as 001×1, 001×7 and 001×14.5, and gel strong-acid resin such as D152) were used among which the macroporous strong acidic resin exhibited better catalytic activity due to its larger surface area, stronger acidity and the presence of a large number of macropores allowing the trapping of fructose molecules into the inner sites of the macroporous strong-acid resin.Amberlyst is another kind of strong acidic heterogeneous catalyst which has been used in various acid catalyzed reactions including hydrolysis. It is a styrene-divinylbenzene polymer based acid that has sulfonic groups (–SO3H). The macroporous structure of this resin allows small molecules to enter into the pores and interact with more active acid sites. Schüth et al. reported the hydrolysis of microcrystalline cellulose and α-cellulose with Amberlyst 15DRY resin where 1-butyl-3-methylimidazolium chloride ([BMIm]Cl) was used as the solvent for better dissolution of cellulose.145 The activity of 15DRY was compared with that of soluble p-toluenesulfonic acid (p-TSA) which contains similar sulfonic acid groups. The hydrolysis performance with Amberlyst 15DRY showed an induction period of about 1.5 h, while no induction period was observed during the first 1.5 h for p-TSA (Fig. 10). During the initial 1.5 h, the p-TSA catalyzed reaction showed faster kinetics, producing a small amount of sugars. The differential effect of the two systems was investigated by the visual appearance of isolated cellulose suspensions and degree of polymerization (DP) analysis (Fig. 11). DP analysis of cellulose at different recovery times demonstrated that cellulose was effectively depolymerized in the presence of Amberlyst 15DRY, producing 90% cello-oligomer yield after the induction period. Further studies showed that depolymerization is a first-order reaction with respect to catalyst concentration and the induction period strongly depends on the amount of acid used.146 Increasing the catalyst concentration from 0.46 to 6.9 mmol of H3O+ decreased the induction time from 1.5 h to less than 5 min. This observation suggests that the acid concentration played an important role in the hydrolysis reaction, and the use of a larger amount of Amberlyst 15DRY showed a similar performance to that of a soluble acid (p-TSA) with no induction period for the production of glucose.
 | ||
| Fig. 10 (a) Total release of reducing sugars from ▲ α-cellulose (using p-TSA), ■ microcrystalline cellulose, ▼ wood (spruce), ● α-cellulose (using Amberlyst 15DRY) determined by 3,5-dinitrosalicylic acid (DNS) assay. (b) Production of mono- and disaccharides, monitored by HPLC, for the hydrolysis of α-cellulose catalyzed by ▲ p-TSA and by ● Amberlyst 15DRY. Reaction conditions: cellulose (5 g) dissolved in 100 g of [BMIm]Cl, Amberlyst 15DRY (1.00 g, 4.6 mmol H+) or p-TSA (4.6 mmol), water (111 mmol), 373 K. Adapted from ref. 145. Copyright 2008, with permission from Wiley-VCH.145 | ||
 | ||
| Fig. 11 Hydrolysis of microcrystalline cellulose. Appearance of cellulose recovered from [BMIm]Cl by the addition of water after the reaction times indicated. The values between parentheses represent the percentage of isolated cellulose. Adapted from ref. 145. Copyright 2008, with permission from Wiley-VCH.145 | ||
The additional advantage of having binding groups in the resin polymer chain for cellulose hydrolysis has been reported.147 Cellulase-mimetic catalyst (CP–SO3H) supported on chloromethyl polystyrene (CP) resin has such a feature in which –Cl groups acted as the cellulose-binding domain and –SO3H groups acted as the catalytic domain (Scheme 8). Activity studies showed the complete hydrolysis of cellobiose into glucose at 120 °C. The excellent activity of CP–SO3H relied on the adsorption ability of cellobiose through hydrogen binding between the chloride groups on the catalyst and the hydroxyl groups of the sugars. The adsorption of cellobiose on the catalyst surface was preferred over glucose which ensured the hydrolysis of cellobiose to proceed and the desorption of glucose product from the catalyst. The catalyst effectively hydrolysed crystalline cellulose (Avicel), producing up to 93% glucose, while almost no glucose was reported using sulfuric acid under the same reaction conditions. The activation energy of cellulose hydrolysis catalyzed by CP–SO3H was calculated as 83 kJ mol−1 at 373–413 K which is much lower than that of sulfuric acid (170 kJ mol−1) and even less than that of sulfonated active carbon (AC–SO3H) (110 kJ mol−1).148
 | ||
| Scheme 8 Hydrolysis mechanism of a cellulase-mimetic solid catalyst.147 | ||
Similar to Amberlyst-type resins, Nafion (sulfonated tetrafluoroethylene based fluoropolymer-copolymer) is also considered as an effective solid acid for the hydrolysis of cellulose due to its very high acidity with strength comparable to that of pure sulfuric acid.149–152 However, the low surface area (∼0.02 m2 g−1) limits the practical application of unswollen Nafion resin in catalysis. The surface area can effectively be increased by modifying the resin using a porous catalytic support. Harmer et al. reported an interesting method of increasing the activity of Nafion resin by developing a resin/silica nanocomposite using a porous silica network in which Nafion particles were entrapped.153 Among the various types of silica-based porous materials, mesocellular silica foams (MCFs) have unique advantages as catalyst supports due to their well-defined ultra-large mesopores (up to 50 nm in diameter) connected by uniformly sized windows.154 Large molecules can easily diffuse through the pores of MCFs, and the mass transfer of reactants and products can easily be achieved because of its open continuous three-dimensional system.155
Because of these advantages of MCFs and excellent acidic properties of Nafion resin, Shen et al. reported a series of Nafion-resin-supported MCF solid acid catalysts for the catalytic dehydration of fructose.156Scheme 9 represents a possible mechanism that shows the interaction between the reactant and the solid catalyst. As confirmed by diffuse reflectance infrared Fourier transform (DRIFT) analysis, electrostatic effects and hydrogen bonds from the polar silanol-rich surface of the MCF weaken the stability of the hydroxyl group and C–H bonds in fructose, thereby facilitating the dehydration reaction. Once the fructofuranose is formed and moved into the cells of the MCF, the hydroxyl group on the saturated carbon is protonated quickly. Because of the strong interaction with Nafion and silanol groups, loss of the first water molecule becomes more efficient, leading to the formation of an enediol intermediate. After deprotonation, the intermediate continues to lose two water molecules under the influence of electrostatic forces and/or H-bonding on the C–O and C–H bonds to form HMF. The additional advantage of this system is the structural space limitation of MCF that prevents side reactions to form soluble polymers and insoluble humins. Moreover, the catalyst can be recycled easily and regenerated by using an ion-exchange method.
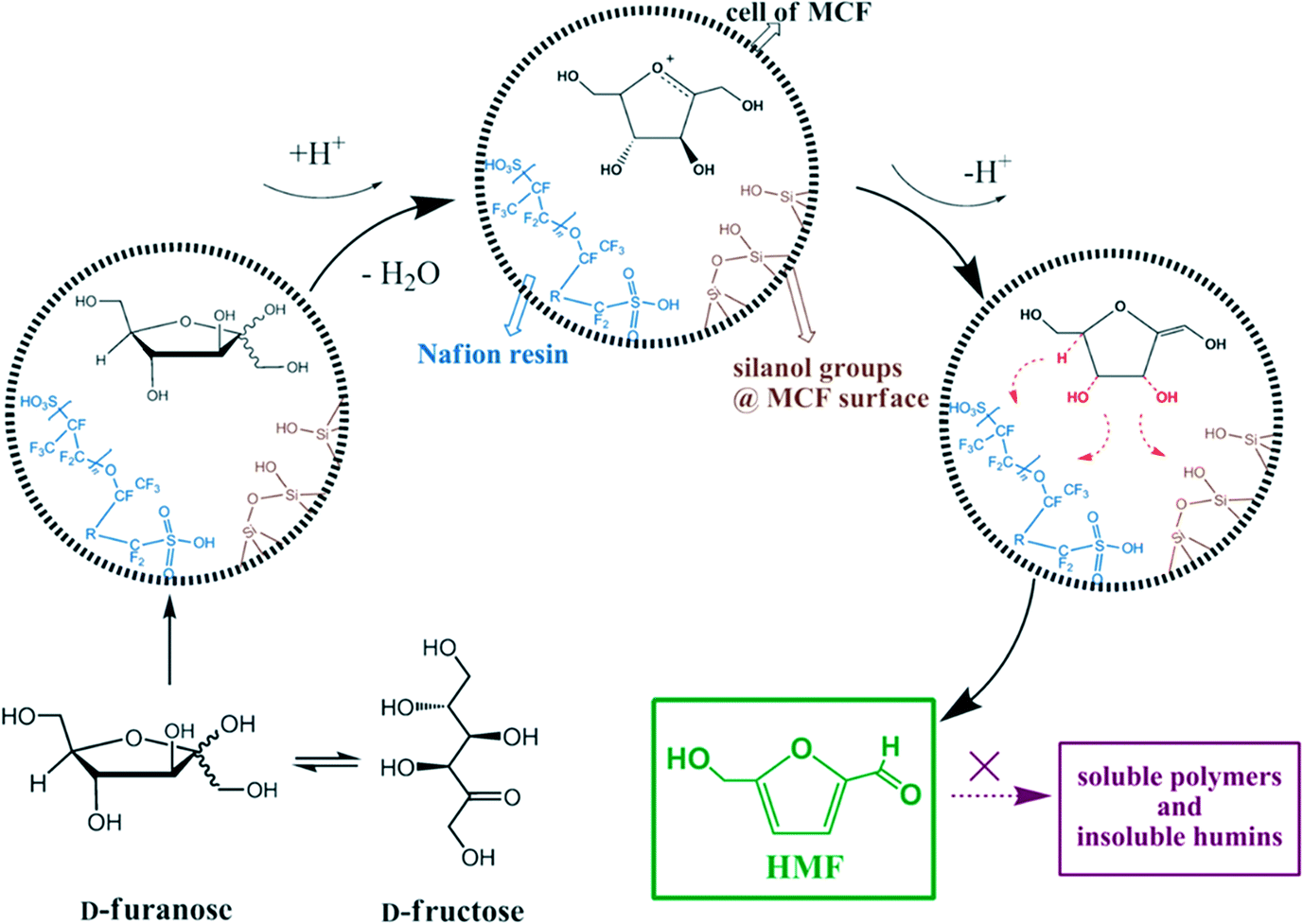 | ||
| Scheme 9 Possible mechanism for the dehydration of D-fructose over Nafion-modified MCF materials. Adapted from ref. 156. Copyright 2013, with permission from Wiley-VCH.156 | ||
6. Zeolites
Zeolites are microporous aluminosilicate minerals that have a porous structure to host a wide variety of cations, such as H+, Na+, K+, and Mg2+. The most important features of zeolite materials are the appropriate Brønsted/Lewis acid ratio, dual meso-/microporosity and excellent mass-transfer ability, which make them good candidates for a wide number of applications including catalysis, adsorption and separation.157–162 The acidity of zeolite depends on the atomic ratio of Si/Al. The amount of Al atoms is proportional to the amount of Brønsted acid sites; the higher the ratio of Al/Si, the higher the acidity of the catalyst.163 Because biomass-derived molecules generally contain carbonyl functionalities, protic polar solvents (especially water) are preferred for their transformations and hence the water-resistant and hydrophobic nature of zeolites is desirable.Transition metal containing zeolites are of special interest due to their Lewis acidity. For example, isolated Ti or Sn species in the framework positions of hydrophobic zeolites, such as high silica zeolites, offer unique Lewis acid sites even in the presence of protic polar solvents (such as water) (Fig. 12).164 Due to their hydrophobic nature, these metallozeolites can be effective for biomass-transformation processes in water. The isomerization of sugars is one of the key steps in biomass transformation in order to derive platform molecules, and as a consequence, metal containing zeolites can serve as potential candidates.
 | ||
| Fig. 12 Metal substituted zeolites with variable Lewis acidic sites. | ||
Davis et al. used Sn-Beta zeolite for the isomerization of glucose to fructose in aqueous medium.165 The advantage of this catalyst is that it is active over a wide temperature range (343–413 K) and acidic medium (pH <2). Isotope labeling experiments proved that the sugar isomerization mechanism using Sn-Beta takes place through a Lewis acid intra-molecular hydride shift from the C-2 to the C-1 position.166 It is also proved that Sn-Beta mediated glucose isomerization follows reaction pathways analogous to those of enzymes, including glucose ring opening in the Lewis acid center, intra-molecular proton shift to form fructose, and finally, ring closure to form the cyclic form of fructose.167 For the isomerization of C5 sugar (e.g., xylose to xylulose), the same pathway is followed by using Sn-Beta.168 Another advantage of Sn-BEA zeolite is the presence of the silanol group adjacent to the active site Sn metal center that can synergistically perform isomerization and epimerization of glucose.169
A significant role of the reaction medium is observed in the transformation of sugars to give different products. A combination of Sn-Beta and borate salts cooperatively catalyzed the epimerization of sugars in aqueous medium, producing mannose as the main product.170 An intermediate complex is formed by the interaction of the borate salt with the initial sugar that reacts on the Lewis acid sites of Sn-Beta by an epimerization pathway through a 1,2-intramolecular carbon shift. This novel methodology has been successfully used to derive mainly the epimer products (mannose, xylose, or ribose) instead of the isomer products (fructose, xylulose, or ribulose) from different monosaccharides (glucose, xylose, or arabinose). The epimerization of glucose is also possible without borate salts, but in this case, the solvent has to be changed from water to methanol.171 On the other hand, in pure aqueous phase, isomerization takes place to produce fructose. The effect of methanol is still unclear, but it is suggested that methanol could interact with framework Sn sites, affecting the intermediate transition state.
In contrast, Ti-Beta has been shown to uniquely catalyze the isomerization of D-glucose in a stereoselective fashion to produce L-sorbose.172 The mechanism follows an intramolecular C5–C1 hydride shift (Fig. 13) for which there are no metalloenzyme or catalytic analogs. In this case, the solvent effect is also very prominent, achieving fructose as the main product in the aqueous medium and sorbose in methanol.
 | ||
| Fig. 13 Stereoselective sugar rearrangements catalyzed by hydrophobic Lewis acid zeolites (Sn-Beta, Ti-Beta) in water and methanol (353–413 K). | ||
Apart from sugar rearrangement, metal containing Lewis acidic zeolites are also able to transform sugars into lactic acid, which is industrially produced mainly through microbial fermentation of glucose and sucrose. Hydrophobic Sn-Beta zeolite has been reported to give a 99 wt% yield of methyl lactate from trioses through isomerization–esterification in methanol.173 The use of hexose sugars as substrates is however very challenging because these monosaccharides undergo several alternative reaction pathways, including dehydration and polymerization reactions, which could affect the yield of the desired product. Direct transformation of common sugars like glucose, fructose or sucrose in alcohols could produce alkyl lactate in high yields (up to 80% or more) using Sn-Beta as the catalyst.174,175 The proposed mechanism involves a retro-aldol fragmentation of hexose followed by the isomerization of the resulting trioses through a 1,2-hydride shift (Fig. 14).
 | ||
| Fig. 14 Reaction pathway for the conversion of fructose to methyl lactate.174 | ||
H-form zeolites are widely used as acid catalysts due to their shape-selective properties in chemical reactions. Although a high Al/Si ratio makes the zeolite more acidic, the hydrolysis of cellulose with different H-form zeolites showed that the zeolites with high Si/Al ratio, such as H-Beta (75) and H-ZSM (45), are more active than those with lower Si/Al such as H-Beta (13) and H-mordenite (10).176 This could be attributed to the highly hydrophobic character of zeolites with high Si/Al ratios.
The shape-selective properties of H-form zeolites can be used in the polymerization reaction to achieve the desired polymer chain. Dusselier et al. recently reported a zeolite-based catalytic process to convert lactic acid into lactide in record yields.177 The new process replaces the current multistep process by avoiding both racemization and side-product formation. The first step of the current process involves the polycondensation of L-lactic acid (L-LA) with water removal to a prepolymer followed by the release of lactide from the chain (Fig. 15, red frame). The second step involves an endothermic transesterification in the presence of a metal salt to give the lactide. The main drawback of this process is the low selectivity (50–60%) to the desired L,L-lactide due to racemization leading to unwanted D,L-lactide and formation of polymeric waste residue. Therefore, the competitive and thermodynamically favored growth of oligomers has to be stopped in order to obtain a high yield of L,L-lactide. This can be done by dimerization of LA to linear dimer (L2A) and its direct cyclization by condensation during water removal (Fig. 15, blue frame). Among the different types of solid acid catalysts (including macroporous resins like Nafion and Amberlyst), H-Beta zeolite gave the highest selectivity for lactide, yielding nearly 79% at full LA conversion.
 | ||
| Fig. 15 Current and proposed chemical process for making lactide and PLA from lactic acid. LnA: linear oligomer of n lactyl units; ROP: ring-opening polymerization. Reproduced from ref. 177. Copyright 2015 with permission from AAAS.177 | ||
Though the lactide selectivity strongly depends on the pore size and acidity of the catalysts, the unique shape-selectivity of zeolite is essential to enhance lactide yield. An acidic mesoporous aluminosilicate, H-Al-MCM-41, with a pore diameter about four times that of zeolite Beta, showed lower selectivity (and activity) towards lactide, emphasizing the zeolite's shape-selective control. In addition, the presence of Brønsted acidity is essential, as zeolite in its non-acidic form (NH4-Beta, Si/Al = 12.5) and a low-aluminum H-Beta (Si/Al = 180) offer poor conversion and very low lactide yield. On the other hand, microporous zeolites like H-Beta produced high lactide selectivity due to pore confinement effects.
It is very important to understand the relationship of activity with the pore structure and acidity of the catalyst. In a recent publication, Yi et al. reported different aluminosilicate materials with different pore diameters and acidity for the dehydration of glycerol into acrolein.178 Hierarchically mesoporous acidic aluminosilicate (ASN-X) and aluminosilicophosphate (ASPN-X, X = Si/Al molar ratio) with identical pore structures but different acid sites were tested and their activities were compared to those of microporous (HZSM-5) and mesoporous (AlMCM-41) aluminosilicate acid catalysts. The unique features of ASN and ASPN materials enabled elucidating how the reaction was governed by the combined action of pore structures and acid sites in the catalysts. Taking into account the acidic properties of the catalysts, HZSM-5 has the highest number of acid sites and the highest B/L ratio and thus can be expected to have the highest activity for the conversion of glycerol. In contrast, HZSM-5 demonstrated a slower initial conversion due to its smaller average pore diameter.
To understand the relationship between acid sites and activity, two catalysts (ASPN-40 and ASPN-60) with identical pore structures were chosen. ASPN-60 has less acid density and a lower B/L ratio than ASPN-40 and thus offered lower activity than ASPN-40. However, the intrinsic activity of a single active site on ASPN-60 was shown to be higher than that on ASPN-40. This can be explained by the surface density of the acid sites. Assuming that the cross-sectional area of glycerol is ca. 0.3 nm2,179 and the average size of a surface hydroxyl group (Brønsted acid site) is ca. 0.1 nm2,180 ASPN-60 has a surface acid density of 0.195 nm−1, which corresponds to ca. 4 acid sites per 20 nm2 of surface area. The acid sites on the surface are sufficiently far apart from each other, and therefore glycerol molecules can interact with all of the acid sites without any severe hindrance (Fig. 16A). The surface acid density of ASPN-40 is 0.524 nm−1, equivalent to ca. 10 acid sites per 20 nm2 of surface area. Compared to ASPN-60, the acid sites on the ASPN-40 surface are in close proximity, and therefore a steric hindrance is apparent during the interaction of glycerol molecules with an individual acid site (Fig. 16B). As a result, an inverse relationship was found between the TOF(0) and the surface density of the acid sites (Fig. 16C).
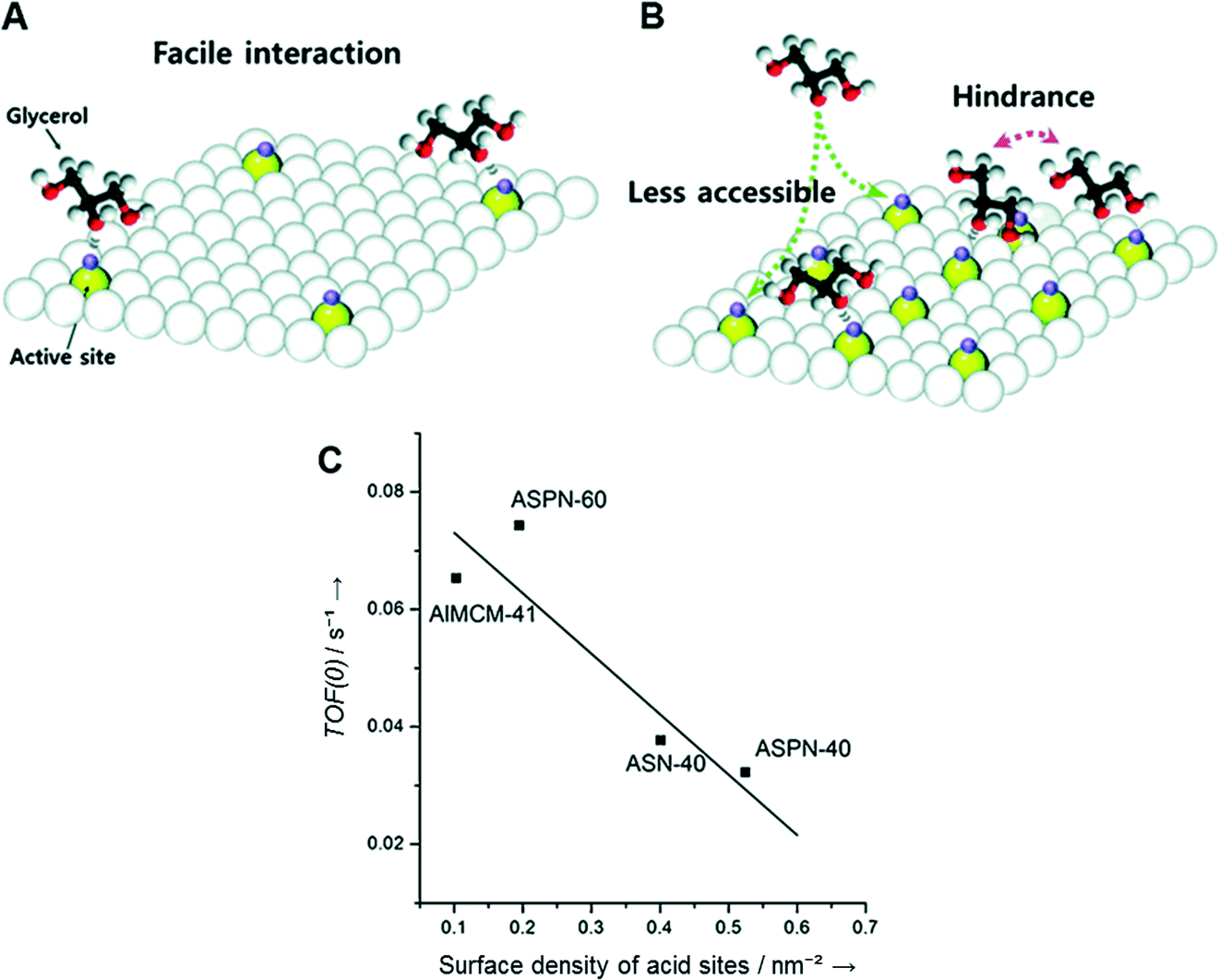 | ||
| Fig. 16 Schematic representation of the effect of surface density of acid sites on the turnover frequency of (A) ASPN-60 and (B) ASPN-40. (C) Correlation between the surface density of acid sites and the turnover frequency at time zero. Reproduced from ref. 178. Copyright 2015, with permission from Wiley-VCH.178 | ||
7. Carbonaceous materials
Carbon-based solid-acid catalysts have gained special attraction due to their hydrophobic nature, which makes the separation process easier and thus reduces the production cost. Among other advantages, carbon supports are resistant to acidic or basic media and are stable at high temperatures (even above 1000 K). Carbon supports are generally amphoteric in nature and exhibit an acid–base character due to the presence of multiple types of oxygenated functional groups, which enhances metal adsorption and catalyst dispersion (Fig. 17). | ||
| Fig. 17 Different oxygen functionalities on activated carbon. | ||
The presence of Brønsted acidic –SO3H groups in the sulfonated carbon catalysts improves the hydrolysis rate of cellulose. It is reported that sucrose-derived sulfonated CMK-3 catalyst hydrolyzed cellulose to glucose with 75% yield at 94% cellulose conversion, which is better than that of any other solid-acid catalysts reported so far.181 The analysis suggests that the activity depends on the acid density, which in turn relies on the sulfonic group concentration and the reaction temperature of the catalyst preparation. The higher preparation temperature (250 °C) of the catalyst enhanced overall acid density as well as the specific surface area. Apart from the sulfonation conditions, the carbon source and the carbonization procedure also significantly influenced the loading of the functional groups on the carbon surface and hence the catalytic activity. For example, CMK-3 showed higher activity in hydrolysis as compared to other carbon-based materials such as acetylene carbon black, MWCNT, cell-carbon, resin-carbon and coconut shell active carbon.
Introduction of hydrophilic functionalities on the catalyst surface increases the interaction with hydroxyl groups of cellulose through H-bonding to destruct the 3-D robust network of cellulose.182,183 An amorphous carbon material containing –SO3H, –COOH, and –OH groups showed efficient hydrolysis of cellulose to saccharides.148,184 A synergetic effect of strong Brønsted acid sites (–SO3H), efficient adsorption of β-1,4-glucan by –OH and –COOH, and facile access to the material bulk are attributed to the high activity of the amorphous carbon. The activation energy of the carbon catalyst in the hydrolysis of cellulose into glucose is 110 kJ mol−1, which is lower than that of H2SO4 (170 kJ mol−1). H-mordenite, Nafion, and Amberlyst-15 catalysts showed non-adsorption of cellobiose in water after the solution was stirred for 24 h, whereas the carbon materials adsorbed 24% of cellobiose in 3 h. This suggests a stronger affinity of carbon materials towards β-1,4 glucan by the surface –OH groups, and hence a higher degree of hydrolysis.185
An interesting phenomenon exhibited by the carbon materials is “hydrogen spillover”. This makes a support material catalytically active as the dissociative absorption of hydrogen atoms migrates from a metal NP surface to the support surface.186 The spillover effect is generally promoted by the electrostatic interactions between oxygen-bearing carboxyl and hydroxyl groups on the carbon surface and metal cations or anions.187 The spillover effect is very important in the hydrogenation reaction where the reaction rate is dependent on the spillover effect of both metal and support surfaces. The cumulative beneficial effect has been successfully applied in the direct conversion of sorbitol from cellulose using Pt NP catalysts loaded on reduced graphene oxide (Pt/RGO).188 The appropriate Pt particle size and the hydrogen spillover effect of the Pt/rGO enabled an excellent conversion (92%) of cellulose into sorbitol.
Biomass itself can serve as a great source of carbonaceous materials with advanced structures and catalytic properties.189 Template-directed synthesis is of particular interest in the synthesis of structurally ordered porous carbon materials. Selection of a suitable template can make porous carbons with controlled architecture and desirable physical and chemical properties. Generally, water-soluble carbohydrates (such as fructose, glucose, sucrose etc.) are used as carbon precursors and in most cases, the structures of the carbon materials mainly depend on the template used. However, carbon precursors could also play an important role in producing the desired structure and functionality. For example, nitrogen doped hierarchically porous carbon materials (HPCs) with tunable surface area, pore volume, and pore size were synthesized from banana peel using a dual template approach where an Al-based composite served as a hard template and triblock copolymer Pluronic F127 as co-templates.190 For example, banana peel contains rich organic functional groups (–COOH, –OH, and –NH2) which strongly interacted with aluminum ions of the Al-based composite as well as with the block copolymer to form coordinated composite species. Carbonization of this composite at 800 °C followed by template removal led to a narrow range of mesopores and high nitrogen content in the HPC material.
A multifunctional catalyst consisting of tungsten carbide nanoparticles supported on a high surface area, three-dimensional mesoporous carbon (MC) can directly convert cellulose to ethylene glycol with 73% efficiency.191 The two-step process involves the hydrolysis of cellulose and hydrogenation/hydrogenolysis of cellulose-derived sugars to produce the final polyol. The MC was prepared by a nano-casting method with sucrose as the carbon precursor, and a hard template consisting of commercial silica and SBA-15. As compared to microporous activated carbon, MC has the advantage of good accessibility of the mesopores to allow the molecular diffusion of the reactant and products from the active sites. Another example of a Pt catalyst supported on a sea-urchin-like three-dimensional mesoporous carbon could provide hexitol in 80% yield from cellulose via hydrolytic hydrogenation.192 A silica template helped in achieving a very large surface area of 1570 m2 g−1 with a mesoporous area of 1340 m2 g−1. The oxygen functional groups on the carbon support effectively interact with cellulose and facilitate the transport of cellulose to the active sites within the mesopores.
Starbon is a novel family of mesoporous carbonaceous materials with chemical and surface properties intermediate between those of polysaccharides and carbons depending on the degree of carbonization. Biomass-derived Starbon was synthesized by controlled carbonization of polysaccharides without any template.193,194 Deposition of precious metals like Pd, Pt, Ru, and Rh on the Starbon offered hydrogenation of succinic acid to γ-butyrolactone, THF, and 1,4-butanediol.195 Starbon catalyst showed excellent conversion and selectivity towards target products and the catalyst could be recycled many times without any leaching of active metal species.
The hydrophilicity/hydrophobicity of the carbon materials can be easily tuned by introducing different functional groups and can be used in both aqueous and organic phase reactions, which is indeed a great advantage in the biomass conversion process. New features of carbo-catalysts in biomass valorization are yet to be revealed as realized from recent developments. For example, co-impregnation of crystalline cellulose fibers with sulfuric acid and glucose reduced the ball milling time of cellulose by 5 times to form branched α(1 → 6) glycosidic oligomers.183 This strategy to enhance the formation of soluble oligomers makes the task of a carbon-based catalyst more effective in hydrolyzing the water-soluble glucan chains to form glucose. Reacting glucose with cellulose fibers in the presence of sulfuric acid results in the prevention of re-lamination of the glucan chains, thus accelerating the formation of aqueous soluble oligomers.
8. Conclusions
Porous carbons, zeolites, metal-oxides and magnetic acidic materials containing various active sites on the surface have been explored extensively for biomass depolymerization and upgrading to chemicals and fuels. In this mini-review, we demonstrate the use of various well-defined heterogeneous catalysts and their structural-activity features to understand the importance of such features in controlling activities and defining more effective materials for upgrading biomass and biomass intermediates containing complex linkages and functionalities. To simplify the discussion, we classified the reported catalysts into subgroups such as metal oxides, supported metals, functionalized porous materials, acidic and ion-exchange resins, zeolites and carbonaceous materials. In parallel with the analysis of thermo-catalytic processes, we reviewed less energy-intensive photocatalytic pathways for upgrading carbohydrate and lignin derived oxygen functional molecules. The metal-oxide based catalysts were found to be highly effective in photocatalyzing substrates to several high value chemicals, which can be an effective strategy towards the development of energy-saving processes for biomass transformation. The effect of bifunctional metal and acid sites and their synergy in transforming multi-step cascade reactions in one pot are demonstrated with practical examples. Recently developed porous carbons and porous organic polymer materials with intrinsic features of binding sites for facilitating the subject transformation more efficiently are elucidated.As a future perspective, biomass derived carbon materials with the desired porous network and functionality is an area which is growing at a faster pace than other segments. Some of these designed carbon materials can be utilized as novel catalysts for comparatively underexplored techniques like cascading multi-step conversion of sugars to small molecules, enzyme immobilization to construct nano-enzyme catalysts, and assembling multi-functional catalyst supports. In this regard, cross-linking of organic molecules with the carbon-network derived from cellulose or lignin can also offer some unique features of such carbon-supported catalysts. Clear understanding of these issues is essential in designing and constructing catalysts with structurally diverse high surface area, regular pores and tunable composition for photochemical and chemical conversions. We believe this article will guide researchers in understanding the role of different active sites for various reactions involved in biomass transformation and the benefit of different supports such that more effective catalytic materials can be defined and designed.
Acknowledgements
This work was supported in part by the Catalysis Center for Energy Innovation, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, and Office of Basic Energy Sciences under Award Number DE-SC0001004.References
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
- R. D. Perlack, L. L. Wright, A. Turhollow, R. L. Graham, B. Stokes and D. C. Erbach, Biomass as Feedstock for a Bioenergy and Bioproducts Industry: The Technical Feasibility of a Billion-Ton Annual Supply, Report No. DOE/Go-102995-2135, Oak Ridge National Laboratory, Oak Ridge, TN, 2005 Search PubMed.
- J. B. Binder and R. T. Raines, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4516–4521 CrossRef CAS PubMed.
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Chem. Soc. Rev., 2012, 41, 8075–8098 RSC.
- J. A. Geboers, S. Van de Vyver, R. Ooms, B. Op de Beeck, P. A. Jacobs and B. F. Sels, Catal. Sci. Technol., 2011, 1, 714–726 Search PubMed.
- Y.-C. Lin and G. W. Huber, Energy Environ. Sci., 2009, 2, 68–80 CAS.
- P. L. Dhepe and A. Fukuoka, ChemSusChem, 2008, 1, 969–975 CrossRef CAS PubMed.
- J. J. Verendel, T. L. Church and P. G. Andersson, Synthesis, 2011, 2011, 1649–1677 CrossRef.
- G. C. Bond, Heterogeneous Catalysis: Principles and Applications, Clarendon Press, Oxford, 2nd edn, 1987 Search PubMed.
- D. K. Sidiras and E. G. Koukios, Biomass, 1989, 19, 289–306 CrossRef CAS.
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974–4975 CrossRef CAS PubMed.
- J. Zhang, B. Zhang, J. Zhang, L. Lin, S. Liu and P. Ouyang, Biotechnol. Adv., 2010, 28, 613–619 CrossRef CAS PubMed.
- T. H. Kim and Y. Y. Lee, Bioresour. Technol., 2005, 96, 2007–2013 CrossRef CAS PubMed.
- M. Benoit, A. Rodrigues, Q. Zhang, E. Fourré, K. De Oliveira Vigier, J.-M. Tatibouët and F. Jérôme, Angew. Chem., Int. Ed., 2011, 50, 8964–8967 CrossRef CAS PubMed.
- V. Choudhary, S. I. Sandler and D. G. Vlachos, ACS Catal., 2012, 2, 2022–2028 CrossRef CAS.
- V. Choudhary, S. H. Mushrif, C. Ho, A. Anderko, V. Nikolakis, N. S. Marinkovic, A. I. Frenkel, S. I. Sandler and D. G. Vlachos, J. Am. Chem. Soc., 2013, 135, 3997–4006 CrossRef CAS PubMed.
- T. Dallas Swift, H. Nguyen, A. Anderko, V. Nikolakis and D. G. Vlachos, Green Chem., 2015, 17, 4725–4735 RSC.
- K.-I. Shimizu and A. Satsuma, Energy Environ. Sci., 2011, 4, 3140–3153 CAS.
- P. M. M. Blauwhoff, J. W. Gosselink, E. P. Kieffer, S. T. Sie and W. H. J. Stork, Catalysis and Zeolites: Fundamentals and Applications, Springer, Berlin, 1999 Search PubMed.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS.
- D. Zhao, J. Feng, Q. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548–552 CrossRef CAS PubMed.
- C. M. A. Parlett, K. Wilson and A. F. Lee, Chem. Soc. Rev., 2013, 42, 3876–3893 RSC.
- B. L. Su, C. Sanchez and X. Y. Yang, Hierarchically Structured Porous Materials, Wiley-VCH, Weinheim, 2012 Search PubMed.
- W.-G. Kim, X. Zhang, J. S. Lee, M. Tsapatsis and S. Nair, ACS Nano, 2012, 6, 9978–9988 CrossRef CAS PubMed.
- H. Chen, J. Wydra, X. Zhang, P.-S. Lee, Z. Wang, W. Fan and M. Tsapatsis, J. Am. Chem. Soc., 2011, 133, 12390–12393 CrossRef CAS PubMed.
- M. Watanabe, Y. Aizawa, T. Iida, R. Nishimura and H. Inomata, Appl. Catal., A, 2005, 295, 150–156 CrossRef CAS.
- X. Qi, M. Watanabe, T. M. Aida and R. L. Smith Jr, Catal. Commun., 2008, 9, 2244–2249 CrossRef CAS.
- L. Yang, Y. Liu and R. Ruan, Adv. Mater. Res., 2011, 335–336, 1448–1453 CrossRef CAS.
- S. Dutta, S. De, A. K. Patra, M. Sasidharan, A. Bhaumik and B. Saha, Appl. Catal., A, 2011, 409–410, 133–139 CrossRef CAS.
- S. De, S. Dutta, A. K. Patra, A. Bhaumik and B. Saha, J. Mater. Chem., 2011, 21, 17505–17510 RSC.
- S. De, S. Dutta, A. K. Patra, B. S. Rana, A. K. Sinha, B. Saha and A. Bhaumik, Appl. Catal., A, 2012, 435–436, 197–203 CrossRef CAS.
- A. Dutta, D. Gupta, A. K. Patra, B. Saha and A. Bhaumik, ChemSusChem, 2014, 7, 925–933 CrossRef CAS PubMed.
- M. I. Alam, S. De, B. Singh, B. Saha and M. M. Abu-Omar, Appl. Catal., A, 2014, 486, 42–48 CrossRef CAS.
- C. Tagusagawa, A. Takagaki, A. Iguchi, K. Takanabe, J. N. Kondo, K. Ebitani, S. Hayashi, T. Tatsumi and K. Domen, Angew. Chem., Int. Ed., 2010, 49, 1128–1132 CrossRef CAS PubMed.
- A. Takagaki, C. Tagusagawa and K. Domen, Chem. Commun., 2008, 5363–5365 RSC.
- C. Tagusagawa, A. Takagaki, S. Hayashi and K. Domen, J. Am. Chem. Soc., 2008, 130, 7230–7231 CrossRef CAS PubMed.
- F. S. Asghari and H. Yoshida, Carbohydr. Res., 2006, 341, 2379–2387 CrossRef CAS PubMed.
- A. Dutta, A. K. Patra, S. Dutta, B. Saha and A. Bhaumik, J. Mater. Chem., 2012, 22, 14094–14100 RSC.
- R. Weingarten, Y. T. Kim, G. A. Tompsett, A. Fernández, K. S. Han, E. W. Hagaman, W. C. Conner Jr, J. A. Dumesic and G. W. Huber, J. Catal., 2013, 304, 123–134 CrossRef CAS.
- G. C. Behera and K. M. Parida, Catal. Sci. Technol., 2013, 3, 3278–3285 CAS.
- A. Jain, A. M. Shore, S. C. Jonnalagadda, K. V. Ramanujachary and A. Mugweru, Appl. Catal., A, 2015, 489, 72–76 CrossRef CAS.
- Y. Zhang, J. Wang, X. Li, X. Liu, Y. Xia, B. Hu, G. Lu and Y. Wang, Fuel, 2015, 139, 301–307 CrossRef CAS.
- Y. T. Kim, J. A. Dumesic and G. W. Huber, J. Catal., 2013, 304, 72–85 CrossRef CAS.
- P. Sun, X. Long, H. He, C. Xia and F. Li, ChemSusChem, 2013, 6, 2190–2197 CrossRef CAS PubMed.
- F. Wang, Z. Yuan, B. Liu, S. Chen and Z. Zhang, J. Ind. Eng. Chem., 2016, 38, 181–185 CrossRef CAS.
- J. C. Colmenares and R. Luque, Chem. Soc. Rev., 2014, 43, 765–778 RSC.
- R. Luque and A. M. Balu, Producing Fuels and Fine Chemicals from Biomass Using Nanomaterials, Taylor and Francis Book Inc., New Jersey, 2013 Search PubMed.
- R. Ho, J. Liebman and J. Valentine, Active Oxygen in Chemistry, Springer, Berlin, 1995 Search PubMed.
- M. Stöcker, Angew. Chem., Int. Ed., 2008, 47, 9200–9211 CrossRef PubMed.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- Z.-M. Wang, E. Sahle-Demessie and A. Aly Hassan, J. Nanotechnol., 2011, 209150 Search PubMed.
- L.-C. Chen, C.-M. Huang and F.-R. Tsai, J. Mol. Catal. A: Chem., 2007, 265, 133–140 CrossRef CAS.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS PubMed.
- A. M. Balu, B. Baruwati, E. Serrano, J. Cot, J. Garcia-Martinez, R. S. Varma and R. Luque, Green Chem., 2011, 13, 2750–2758 RSC.
- J. C. Colmenares, A. Magdziarz and A. Bielejewska, Bioresour. Technol., 2011, 102, 11254–11257 CrossRef CAS PubMed.
- J. C. Colmenares and A. Magdziarz, J. Mol. Catal. A: Chem., 2013, 366, 156–162 CrossRef CAS.
- G. Zhang, W. Choi, S. H. Kim and S. B. Hong, J. Hazard. Mater., 2011, 188, 198–205 CrossRef CAS PubMed.
- H. Kominami, H. Sugahara and K. Hashimoto, Catal. Commun., 2010, 11, 426–429 CrossRef CAS.
- T. Sakata, T. Kawai and K. Hashimoto, J. Phys. Chem., 1984, 88, 2344–2350 CrossRef CAS.
- J. C. Colmenares, A. Magdziarz, K. Kurzydlowski, J. Grzonka, O. Chernyayeva and D. Lisovytskiy, Appl. Catal., B, 2013, 134–135, 136–144 CrossRef CAS.
- J. C. Colmenares, A. Magdziarz, O. Chernyayeva, D. Lisovytskiy, K. Kurzydłowski and J. Grzonka, ChemCatChem, 2013, 5, 2270–2277 CrossRef CAS.
- M. Yasuda, A. Miura, R. Yuki, Y. Nakamura, T. Shiragami, Y. Ishii and H. Yokoi, J. Photochem. Photobiol., A, 2011, 220, 195–199 CrossRef CAS.
- M. Tian, J. Wen, D. MacDonald, R. M. Asmussen and A. Chen, Electrochem. Commun., 2010, 12, 527–530 CrossRef CAS.
- M. E. Himmel, S.-Y. Ding, D. K. Johnson, W. S. Adney, M. R. Nimlos, J. W. Brady and T. D. Foust, Science, 2007, 315, 804–807 CrossRef CAS PubMed.
- S. K. Hanson, R. Wu and L. A. P. Silks, Angew. Chem., Int. Ed., 2012, 51, 3410–3413 CrossRef CAS PubMed.
- B. Sedai, C. Díaz-Urrutia, R. T. Baker, R. Wu, L. A. P. Silks and S. K. Hanson, ACS Catal., 2013, 3, 3111–3122 CrossRef CAS.
- A. G. Sergeev and J. F. Hartwig, Science, 2011, 332, 439–443 CrossRef CAS PubMed.
- A. G. Sergeev, J. D. Webb and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 20226–20229 CrossRef CAS PubMed.
- J. M. Nichols, L. M. Bishop, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2010, 132, 12554–12555 CrossRef CAS PubMed.
- S. Son and F. D. Toste, Angew. Chem., Int. Ed., 2010, 49, 3791–3794 CrossRef CAS PubMed.
- J. M. W. Chan, S. Bauer, H. Sorek, S. Sreekumar, K. Wang and F. D. Toste, ACS Catal., 2013, 3, 1369–1377 CrossRef CAS.
- A. Maldotti, A. Molinari and R. Amadelli, Chem. Rev., 2002, 102, 3811–3836 CrossRef CAS PubMed.
- X. Lang, W. Ma, C. Chen, H. Ji and J. Zhao, Acc. Chem. Res., 2014, 47, 355–363 CrossRef CAS PubMed.
- S. Yurdakal, G. Palmisano, V. Loddo, V. Augugliaro and L. Palmisano, J. Am. Chem. Soc., 2008, 130, 1568–1569 CrossRef CAS PubMed.
- S. Yurdakal, G. Palmisano, V. Loddo, O. Alagoz, V. Augugliaro and L. Palmisano, Green Chem., 2009, 11, 510–516 RSC.
- C.-J. Li, G.-R. Xu, B. Zhang and J. R. Gong, Appl. Catal., B, 2012, 115–116, 201–208 CrossRef CAS.
- T. Shishido, T. Miyatake, K. Teramura, Y. Hitomi, H. Yamashita and T. Tanaka, J. Phys. Chem. C, 2009, 113, 18713–18718 CAS.
- S. R. Kadam, V. R. Mate, R. P. Panmand, L. K. Nikam, M. V. Kulkarni, R. S. Sonawane and B. B. Kale, RSC Adv., 2014, 4, 60626–60635 RSC.
- A. Tanaka, K. Hashimoto and H. Kominami, Chem. Commun., 2011, 47, 10446–10448 RSC.
- Y. Zhang, N. Zhang, Z.-R. Tang and Y.-J. Xu, ACS Sustainable Chem. Eng., 2013, 1, 1258–1266 CrossRef CAS.
- W. Zhai, S. Xue, A. Zhu, Y. Luo and Y. Tian, ChemCatChem, 2011, 3, 127–130 CrossRef CAS.
- M. Qamar, R. B. Elsayed, K. R. Alhooshani, M. I. Ahmed and D. W. Bahnemann, ACS Appl. Mater. Interfaces, 2015, 7, 1257–1269 CAS.
- Q. Wang, M. Zhang, C. Chen, W. Ma and J. Zhao, Angew. Chem., Int. Ed., 2010, 49, 7976–7979 CrossRef CAS PubMed.
- S. Higashimoto, N. Kitao, N. Yoshida, T. Sakura, M. Azuma, H. Ohue and Y. Sakata, J. Catal., 2009, 266, 279–285 CrossRef CAS.
- A. Tanaka, K. Hashimoto and H. Kominami, J. Am. Chem. Soc., 2012, 134, 14526–14533 CrossRef CAS PubMed.
- D. Tsukamoto, Y. Shiraishi, Y. Sugano, S. Ichikawa, S. Tanaka and T. Hirai, J. Am. Chem. Soc., 2012, 134, 6309–6315 CrossRef CAS PubMed.
- N. Zhang, S. Liu and Y.-J. Xu, Nanoscale, 2012, 4, 2227–2238 RSC.
- T. Ruther, A. M. Bond and W. R. Jackson, Green Chem., 2003, 5, 364–366 RSC.
- U. R. Pillai and E. Sahle-Demessie, J. Catal., 2002, 211, 434–444 CrossRef CAS.
- Z. He and X. Wang, in Catalysis for Sustainable Energy, 2012, vol. 1, p. 28 Search PubMed.
- F. Tao, S. Zhang, L. Nguyen and X. Zhang, Chem. Soc. Rev., 2012, 41, 7980–7993 RSC.
- S. C. Tsang, N. Cailuo, W. Oduro, A. T. S. Kong, L. Clifton, K. M. K. Yu, B. Thiebaut, J. Cookson and P. Bishop, ACS Nano, 2008, 2, 2547–2553 CrossRef CAS PubMed.
- G.-H. Wang, J. Hilgert, F. H. Richter, F. Wang, H.-J. Bongard, B. Spliethoff, C. Weidenthaler and F. Schüth, Nat. Mater., 2014, 13, 293–300 CrossRef CAS PubMed.
- J. Luo, H. Yun, A. V. Mironenko, K. Goulas, J. D. Lee, M. Monai, C. Wang, V. Vorotnikov, C. B. Murray, D. G. Vlachos, P. Fornasiero and R. J. Gorte, ACS Catal., 2016, 6, 4095–4104 CrossRef CAS.
- J. Luo, J. D. Lee, H. Yun, C. Wang, M. Monai, C. B. Murray, P. Fornasiero and R. J. Gorte, Appl. Catal., B, 2016, 199, 439–446 CrossRef CAS.
- S. Nishimura, N. Ikeda and K. Ebitani, Catal. Today, 2014, 232, 89–98 CrossRef CAS.
- Y. Nakagawa, K. Takada, M. Tamura and K. Tomishige, ACS Catal., 2014, 4, 2718–2726 CrossRef CAS.
- L. Yu, L. He, J. Chen, J. Zheng, L. Ye, H. Lin and Y. Yuan, ChemCatChem, 2015, 7, 1701–1707 CrossRef CAS.
- G. Bottari, A. J. Kumalaputri, K. K. Krawczyk, B. L. Feringa, H. J. Heeres and K. Barta, ChemSusChem, 2015, 8, 1323–1327 CrossRef CAS PubMed.
- B. Chen, F. Li, Z. Huang and G. Yuan, Appl. Catal., B, 2017, 200, 192–199 CrossRef CAS.
- S. Sitthisa, W. An and D. E. Resasco, J. Catal., 2011, 284, 90–101 CrossRef CAS.
- T. H. Parsell, B. C. Owen, I. Klein, T. M. Jarrell, C. L. Marcum, L. J. Haupert, L. M. Amundson, H. I. Kenttamaa, F. Ribeiro, J. T. Miller and M. M. Abu-Omar, Chem. Sci., 2013, 4, 806–813 RSC.
- B. Saha, C. M. Bohn and M. M. Abu-Omar, ChemSusChem, 2014, 7, 3095–3101 CrossRef CAS PubMed.
- P. Panagiotopoulou, N. Martin and D. G. Vlachos, J. Mol. Catal. A: Chem., 2014, 392, 223–228 CrossRef CAS.
- J. Jae, W. Zheng, A. M. Karim, W. Guo, R. F. Lobo and D. G. Vlachos, ChemCatChem, 2014, 6, 848–856 CrossRef CAS.
- M. J. Gilkey, P. Panagiotopoulou, A. V. Mironenko, G. R. Jenness, D. G. Vlachos and B. Xu, ACS Catal., 2015, 5, 3988–3994 CrossRef CAS.
- A. Fukuoka and P. L. Dhepe, Angew. Chem., Int. Ed., 2006, 45, 5161–5163 CrossRef CAS PubMed.
- H. Kobayashi, Y. Ito, T. Komanoya, Y. Hosaka, P. L. Dhepe, K. Kasai, K. Hara and A. Fukuoka, Green Chem., 2011, 13, 326–333 RSC.
- W. Deng, M. Liu, X. Tan, Q. Zhang and Y. Wang, J. Catal., 2010, 271, 22–32 CrossRef CAS.
- M. Käldström, N. Kumar, M. Tenho, M. V. Mokeev, Y. E. Moskalenko and D. Y. Murzin, ACS Catal., 2012, 2, 1381–1393 CrossRef.
- J. Pang, A. Wang, M. Zheng, Y. Zhang, Y. Huang, X. Chen and T. Zhang, Green Chem., 2012, 14, 614–617 RSC.
- G. Liang, L. He, M. Arai and F. Zhao, ChemSusChem, 2014, 7, 1415–1421 CrossRef CAS PubMed.
- S. Van de Vyver, J. Geboers, M. Dusselier, H. Schepers, T. Vosch, L. Zhang, G. Van Tendeloo, P. A. Jacobs and B. F. Sels, ChemSusChem, 2010, 3, 698–701 CrossRef CAS PubMed.
- S. Liu, M. Tamura, Y. Nakagawa and K. Tomishige, ACS Sustainable Chem. Eng., 2014, 2, 1819–1827 CrossRef CAS.
- S. Liu, Y. Okuyama, M. Tamura, Y. Nakagawa, A. Imai and K. Tomishige, ChemSusChem, 2015, 8, 628–635 CrossRef CAS PubMed.
- O. Casanova, S. Iborra and A. Corma, J. Catal., 2009, 265, 109–116 CrossRef CAS.
- B. Saha, D. Gupta, M. M. Abu-Omar, A. Modak and A. Bhaumik, J. Catal., 2013, 299, 316–320 CrossRef CAS.
- B. Saha, S. Dutta and M. M. Abu-Omar, Catal. Sci. Technol., 2012, 2, 79–81 CAS.
- N. K. Gupta, S. Nishimura, A. Takagaki and K. Ebitani, Green Chem., 2011, 13, 824–827 RSC.
- J. Artz, S. Mallmann and R. Palkovits, ChemSusChem, 2015, 8, 672–679 CrossRef CAS PubMed.
- M. Kröger, U. Prüße and K.-D. Vorlop, Top. Catal., 2000, 13, 237–242 CrossRef.
- M. L. Ribeiro and U. Schuchardt, Catal. Commun., 2003, 4, 83–86 CrossRef CAS.
- A. J. Crisci, M. H. Tucker, J. A. Dumesic and S. L. Scott, Top. Catal., 2010, 53, 1185–1192 CrossRef CAS.
- K. B. Sidhpuria, A. L. Daniel-da-Silva, T. Trindade and J. A. P. Coutinho, Green Chem., 2011, 13, 340–349 RSC.
- V. Degirmenci, E. A. Pidko, P. C. M. M. Magusin and E. J. M. Hensen, ChemCatChem, 2011, 3, 969–972 CrossRef CAS.
- H. Zhao, J. E. Holladay, H. Brown and Z. C. Zhang, Science, 2007, 316, 1597–1600 CrossRef CAS PubMed.
- E. A. Pidko, V. Degirmenci, R. A. van Santen and E. J. M. Hensen, Angew. Chem., Int. Ed., 2010, 49, 2530–2534 CrossRef CAS PubMed.
- P. A. Lay and A. Levina, in Comprehensive Coordination Chemistry II, Pergamon, Oxford, 2003, pp. 313–413 Search PubMed.
- Y.-Y. Lee and K. C. W. Wu, Phys. Chem. Chem. Phys., 2012, 14, 13914–13917 RSC.
- K. Wilson and H. Clark James, in Pure and Applied Chemistry, 2000, vol. 72, p. 1313 Search PubMed.
- A. S. Dias, M. Pillinger and A. A. Valente, J. Catal., 2005, 229, 414–423 CrossRef CAS.
- G. Akiyama, R. Matsuda, H. Sato, M. Takata and S. Kitagawa, Adv. Mater., 2011, 23, 3294–3297 CrossRef CAS PubMed.
- S. K. Kundu and A. Bhaumik, ACS Sustainable Chem. Eng., 2015, 3, 1715–1723 CrossRef CAS.
- S. Bhunia, B. Banerjee and A. Bhaumik, Chem. Commun., 2015, 51, 5020–5023 RSC.
- S. Mondal, J. Mondal and A. Bhaumik, ChemCatChem, 2015, 7, 3570–3578 CrossRef CAS.
- R. Gomes, P. Bhanja and A. Bhaumik, J. Mol. Catal. A: Chem., 2016, 411, 110–116 CrossRef CAS.
- D.-M. Lai, L. Deng, Q.-X. Guo and Y. Fu, Energy Environ. Sci., 2011, 4, 3552–3557 CAS.
- M. T. Sanz, R. Murga, S. Beltrán, J. L. Cabezas and J. Coca, Ind. Eng. Chem. Res., 2002, 41, 512–517 CrossRef CAS.
- Y. Uozumi and K. Shibatomi, J. Am. Chem. Soc., 2001, 123, 2919–2920 CrossRef CAS PubMed.
- T. Okada and M. Harada, Anal. Chem., 2004, 76, 4564–4571 CrossRef CAS PubMed.
- X. Qi, M. Watanabe, T. M. Aida and J. R. L. Smith, Green Chem., 2008, 10, 799–805 RSC.
- Z. H. Lin, C. J. Guan, X. L. Feng and C. X. Zhao, J. Mol. Catal. A: Chem., 2006, 247, 19–26 CrossRef CAS.
- N. Hartler and K. Hyllengren, J. Polym. Sci., 1962, 56, 425–434 CrossRef.
- Y. Li, H. Liu, C. Song, X. Gu, H. Li, W. Zhu, S. Yin and C. Han, Bioresour. Technol., 2013, 133, 347–353 CrossRef CAS PubMed.
- R. Rinaldi, R. Palkovits and F. Schüth, Angew. Chem., Int. Ed., 2008, 47, 8047–8050 CrossRef CAS PubMed.
- R. Rinaldi, N. Meine, J. vom Stein, R. Palkovits and F. Schüth, ChemSusChem, 2010, 3, 266–276 CrossRef CAS PubMed.
- L. Shuai and X. Pan, Energy Environ. Sci., 2012, 5, 6889–6894 CAS.
- S. Suganuma, K. Nakajima, M. Kitano, D. Yamaguchi, H. Kato, S. Hayashi and M. Hara, J. Am. Chem. Soc., 2008, 130, 12787–12793 CrossRef CAS PubMed.
- X. Qi, M. Watanabe, T. M. Aida and J. R. L. Smith, Green Chem., 2009, 11, 1327–1331 RSC.
- S.-J. Kim, A. A. Dwiatmoko, J. W. Choi, Y.-W. Suh, D. J. Suh and M. Oh, Bioresour. Technol., 2010, 101, 8273–8279 CrossRef CAS PubMed.
- J. Hegner, K. C. Pereira, B. DeBoef and B. L. Lucht, Tetrahedron Lett., 2010, 51, 2356–2358 CrossRef CAS.
- M. A. Harmer, W. E. Farneth and Q. Sun, Adv. Mater., 1998, 10, 1255–1257 CrossRef CAS.
- M. A. Harmer, W. E. Farneth and Q. Sun, J. Am. Chem. Soc., 1996, 118, 7708–7715 CrossRef CAS.
- P. Schmidt-Winkel, W. W. Lukens, D. Zhao, P. Yang, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1999, 121, 254–255 CrossRef CAS.
- D. Trong On and S. Kaliaguine, J. Am. Chem. Soc., 2003, 125, 618–619 CrossRef PubMed.
- Z. Huang, W. Pan, H. Zhou, F. Qin, H. Xu and W. Shen, ChemSusChem, 2013, 6, 1063–1069 CrossRef CAS PubMed.
- D. Liu, A. Bhan, M. Tsapatsis and S. Al Hashimi, ACS Catal., 2011, 1, 7–17 CrossRef CAS.
- T. D. Swift, H. Nguyen, Z. Erdman, J. S. Kruger, V. Nikolakis and D. G. Vlachos, J. Catal., 2016, 333, 149–161 CrossRef CAS.
- L. Ren, Q. Guo, P. Kumar, M. Orazov, D. Xu, S. M. Alhassan, K. A. Mkhoyan, M. E. Davis and M. Tsapatsis, Angew. Chem., Int. Ed., 2015, 54, 10848–10851 CrossRef CAS PubMed.
- D. Xu, G. R. Swindlehurst, H. Wu, D. H. Olson, X. Zhang and M. Tsapatsis, Adv. Funct. Mater., 2014, 24, 200 CrossRef.
- P. Bai, M. Y. Jeon, L. Ren, C. Knight, M. W. Deem, M. Tsapatsis and J. I. Siepmann, Nat. Commun., 2015, 6, 5912 CrossRef CAS PubMed.
- X. Zhang, D. Liu, D. Xu, S. Asahina, K. A. Cychosz, K. V. Agrawal, Y. Al Wahedi, A. Bhan, S. Al Hashimi, O. Terasaki, M. Thommes and M. Tsapatsis, Science, 2012, 336, 1684–1687 CrossRef CAS PubMed.
- N. Salman, C. H. Rüscher, J. C. Buhl, W. Lutz, H. Toufar and M. Stöcker, Microporous Mesoporous Mater., 2006, 90, 339–346 CrossRef CAS.
- S. Caratzoulas, M. E. Davis, R. J. Gorte, R. Gounder, R. F. Lobo, V. Nikolakis, S. I. Sandler, M. A. Snyder, M. Tsapatsis and D. G. Vlachos, J. Phys. Chem. C, 2014, 118, 22815–22833 CAS.
- M. Moliner, Y. Román-Leshkov and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6164–6168 CrossRef CAS PubMed.
- Y. Román-Leshkov, M. Moliner, J. A. Labinger and M. E. Davis, Angew. Chem., Int. Ed., 2010, 49, 8954–8957 CrossRef PubMed.
- R. Bermejo-Deval, R. S. Assary, E. Nikolla, M. Moliner, Y. Román-Leshkov, S.-J. Hwang, A. Palsdottir, D. Silverman, R. F. Lobo, L. A. Curtiss and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9727–9732 CrossRef CAS PubMed.
- V. Choudhary, A. B. Pinar, S. I. Sandler, D. G. Vlachos and R. F. Lobo, ACS Catal., 2011, 1, 1724–1728 CrossRef CAS.
- N. Rai, S. Caratzoulas and D. G. Vlachos, ACS Catal., 2013, 3, 2294–2298 CrossRef CAS.
- W. R. Gunther, Y. Wang, Y. Ji, V. K. Michaelis, S. T. Hunt, R. G. Griffin and Y. Román-Leshkov, Nat. Commun., 2012, 3, 1109 CrossRef PubMed.
- R. Bermejo-Deval, R. Gounder and M. E. Davis, ACS Catal., 2012, 2, 2705–2713 CrossRef CAS.
- R. Gounder and M. E. Davis, ACS Catal., 2013, 3, 1469–1476 CrossRef CAS.
- E. Taarning, S. Saravanamurugan, M. Spangsberg Holm, J. Xiong, R. M. West and C. H. Christensen, ChemSusChem, 2009, 2, 625–627 CrossRef CAS PubMed.
- M. S. Holm, S. Saravanamurugan and E. Taarning, Science, 2010, 328, 602–605 CrossRef CAS PubMed.
- M. Orazov and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 11777–11782 CrossRef CAS PubMed.
- A. Onda, T. Ochi and K. Yanagisawa, Green Chem., 2008, 10, 1033–1037 RSC.
- M. Dusselier, P. Van Wouwe, A. Dewaele, P. A. Jacobs and B. F. Sels, Science, 2015, 349, 78–80 CrossRef CAS PubMed.
- Y. Choi, H. Park, Y. S. Yun and J. Yi, ChemSusChem, 2015, 8, 974–979 CrossRef CAS PubMed.
- J. C. Yori, S. A. D'Ippolito, C. L. Pieck and C. R. Vera, Energy Fuels, 2007, 21, 347–353 CrossRef CAS.
- V. Kocherbitov and V. Alfredsson, J. Phys. Chem. C, 2007, 111, 12906–12913 CAS.
- J. Pang, A. Wang, M. Zheng and T. Zhang, Chem. Commun., 2010, 46, 6935–6937 RSC.
- M. G. Mazzotta, D. Gupta, B. Saha, A. K. Patra, A. Bhaumik and M. M. Abu-Omar, ChemSusChem, 2014, 7, 2342–2350 CrossRef CAS PubMed.
- P. Dornath, H. J. Cho, A. Paulsen, P. Dauenhauer and W. Fan, Green Chem., 2015, 17, 769–775 RSC.
- M. Kitano, D. Yamaguchi, S. Suganuma, K. Nakajima, H. Kato, S. Hayashi and M. Hara, Langmuir, 2009, 25, 5068–5075 CrossRef CAS PubMed.
- D. Yamaguchi and M. Hara, Solid State Sci., 2010, 12, 1018–1023 CrossRef CAS.
- A. V. Filikov and N. F. Myasoedov, J. Phys. Chem., 1986, 90, 4915–4916 CrossRef CAS.
- E. Keren and A. Soffer, J. Catal., 1977, 50, 43–55 CrossRef CAS.
- D. Wang, W. Niu, M. Tan, M. Wu, X. Zheng, Y. Li and N. Tsubaki, ChemSusChem, 2014, 7, 1398–1406 CrossRef CAS PubMed.
- S. De, A. M. Balu, J. C. van der Waal and R. Luque, ChemCatChem, 2015, 7, 1608–1629 CrossRef CAS.
- R.-L. Liu, W.-J. Ji, T. He, Z.-Q. Zhang, J. Zhang and F.-Q. Dang, Carbon, 2014, 76, 84–95 CrossRef CAS.
- Y. Zhang, A. Wang and T. Zhang, Chem. Commun., 2010, 46, 862–864 RSC.
- D. S. Park, D. Yun, T. Y. Kim, J. Baek, Y. S. Yun and J. Yi, ChemSusChem, 2013, 6, 2281–2289 CrossRef CAS PubMed.
- V. Budarin, J. H. Clark, J. J. E. Hardy, R. Luque, K. Milkowski, S. J. Tavener and A. J. Wilson, Angew. Chem., Int. Ed., 2006, 45, 3782–3786 CrossRef CAS PubMed.
- R. J. White, V. Budarin, R. Luque, J. H. Clark and D. J. Macquarrie, Chem. Soc. Rev., 2009, 38, 3401–3418 RSC.
- R. Luque, J. H. Clark, K. Yoshida and P. L. Gai, Chem. Commun., 2009, 5305–5307 RSC.
| This journal is © The Royal Society of Chemistry 2016 |