
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Niobic acid nanoparticle catalysts for the aqueous phase transformation of glucose and fructose to 5-hydroxymethylfurfural†
Mariano Tapia
Reche
a,
Amin
Osatiashtiani
a,
Lee J.
Durndell
a,
Mark A.
Isaacs
a,
Ângela
Silva
b,
Adam F.
Lee
 a and
Karen
Wilson
*a
a and
Karen
Wilson
*a
aEuropean Bioenergy Research Institute, Aston University, Birmingham B4 7ET, UK. E-mail: k.wilson@aston.ac.uk
bLaboratório de Bioinorgânica e Catálise, Universidade Federal do Paraná (UFPR), Departamento de Química, CP 19081, CEP 81531-990, Curitiba, Paraná, Brasil
First published on 9th August 2016
Abstract
A family of bulk and SBA-15 supported peroxo niobic acid sols were prepared by peptisation of niobic acid precipitates with H2O2 as heterogeneous catalysts for aqueous phase glucose and fructose conversion to 5-hydroxymethylfurfural (5-HMF). Niobic acid nanoparticles possess a high density of Brønsted and Lewis acid sites, conferring good activity towards glucose and fructose conversion, albeit with modest 5-HMF yields under mild reaction conditions (100 °C). Thermally-induced niobia crystallisation suppresses solid acidity and activity. Nanoparticulate niobic acid dispersed over SBA-15 exhibits pure Brønsted acidity and an enhanced Turnover Frequency for fructose dehydration.
Introduction
Non-renewable ‘fossil’ resources have been integral to the production of energy and key chemical building blocks for humanity since the dawn of the Industrial Revolution at the end of the eighteenth century.1 Climate change, and concerns over energy security and unpredictable oil pricing, have highlighted the need to sever this societal dependency on fossil reserves, and provoked investment and academic interest in the development of sustainable technologies for the production of bio-derived fuels and commodity chemicals.2 Biomass is the most abundant biogenic source of carbon on Earth, and is widely exploited for food production through the extraction of starches, oils and sugars employing limited chemical processing. Diversion of such edible biomass components for fuels and chemicals production has sparked fierce debate over the use of biomass, and associated re-purposing of traditional agricultural land or native forests, for food versus fuels.3 Academic and commercial research has thus refocused on the transformation of non-edible lignocellulosic biomass (such as agricultural and forestry waste), for advanced biofuels and renewable chemicals production.Cellulose and hemicellulose are the key carbohydrate components of lignocellulosic biomass, whose depolymerisation, affords simple C5 and C6 sugar monomers which are amenable to subsequent valorisation to either platform chemicals or biofuels/fuel additives.4 One particular pathway, the conversion of glucose (from cellulose depolymerisation) to 5-hydroxymethylfurfural (5-HMF) has attracted much attention due to the latter's potential as a platform chemical for the synthesis of polymers, fuel additives, etc. such as levulinic acid and 2,5-furandicarboxylic acid (two of the top 10 value-added chemicals from biomass sources listed in 2004 by the U.S. Department of Energy).5 However, current routes to 5-HMF suffer from significant drawbacks which limit its potential as a platform chemical, notably high production cost due to energy inefficient separation steps and low selectivity from glucose.6 5-HMF is produced from glucose by stepwise isomerisation and dehydration steps, accompanied by the loss of three water molecules.7 Various catalysts have been tested for this reaction, including homogeneous and heterogeneous Lewis and Brønsted acids.8 While homogeneous catalysts exhibit superior activity and selectivity to their heterogeneous counterparts,9 they necessitate energy intensive separations to isolate the desired product and recover the homogeneous catalyst for recycling, with organic acid suffering comparatively poor thermal stability.8 A range of solvents have also been explored, such as ionic liquids10–14 and biphasic solutions,6,15–17 in order to suppress undesirable side reactions and thereby improve 5-HMF selectivity and glucose conversion. Yields of up to 80% 5-HMF are reported for ionic liquids13 and a 5-HMF selectivity >60% in the case of biphasic systems.15 Despite their effectiveness, such solvents are problematic for industrial applications, e.g. toxicity and energy intensive/complex separation protocols. Lignocelluosic glucose is most readily available as a dilute aqueous stream from hydrothermal processing.18 A heterogeneously catalysed process for the one-pot conversion of glucose to 5-HMF in a monophasic, aqueous media (avoiding the use of solvents such as DMSO and MIBK19) is thus highly desirable, but remains challenging due to the low activity of existing catalysts, and competing rehydration/hydrolysis, condensation to form di-D-fructose dianhydrides,20 or polymerisation to humins.21
Osatiashtiani et al. reported bifunctional sulphated zirconia catalysts for such an aqueous phase one-pot transformation,22 in which mixed Lewis–Brønsted acid character proved essential for the cascade isomerisation of glucose to fructose and the latter's dehydration to 5-HMF; these catalysts were particularly efficacious for the second step, resulting in 25% fructose conversion and 32% 5-HMF selectivity at 100 °C. Tungstated zirconia has also been explored for the aqueous phase conversion of fructose to 5-HMF, achieving 67% conversion and 40% 5-HMF selectivity, albeit at a higher reaction temperature of 130 °C.23 Stošić et al. developed ceria–niobia mixed oxides for aqueous phase fructose dehydration,24 obtaining 82% conversion and 48% selectivity at 130 °C over bulk Nb2O5. While values as high as 98% fructose conversion and 86% 5-HMF yield are reported for silver-exchanged silicotungstic acid in sub-critical water at 120 °C,25 the high solubility of heteropolyacids in polar media makes it difficult to discount possible homogeneous acid contributions to the observed catalysis.
Bulk metal oxides have also been tested in this reaction, wherein their low cost is beneficial in an industrial context. Lanziano and co-workers26 reported the use of TiO2 for glucose → 5-HMF in water, with a the maximum 5-HMF yield of 30% at 140 °C. Fifth group elements have also attracted attention, such as mesoporous tantalum pentoxide in a biphasic water/MIBK system, and (Brønsted acidic) phosphated tantalum pentoxide which promoted fructose dehydration and delivered 70% glucose conversion and 40% 5-HMF yield.27 Yang et al. investigated hydrated niobium28 and tantalum pentoxides28 in a water/2-butanol biphasic system for the dehydration of mono- and polysaccharides to 5-HMF, resulting in 99% fructose conversion and 95% selectivity to 5-HMF. Niobia and tantala are considered water-tolerant solid acids,29 with high acid strengths,30,31 and hence are well-suited to the aqueous phase dehydration of sugars; Tsang and co-workers recently demonstrated the efficacy of low area structured niobias for high temperature (180 °C) 5-HMF production from glucose, fructose and sucrose in water.32 The acidic properties of niobia were reported by Datka et al. over a range of supports including silica,33 and for mesoporous Nb-rich SBA-15 silica synthesised by a one-pot sol–gel route,34 wherein both Lewis and Brønsted character were observed. Niobia dispersed within, and impregnated upon, amorphous silica matrices has shown some promise for the continuous dehydration of fructose, reaching 20% 5-HMF selectivity at 75% conversion.35,36
The preparation of high surface area niobia materials without use of auxiliaries remains a challenge since hydrolysis of Nb5+ from NbCl5 forms hydrated niobium oxide (Nb2O5·nH2O) which in turn requires high temperatures to generate the pure oxide resulting in large crystallites. In contrast, low temperature (soft) chemical processes employing a peptised niobic acid sols prepared from ethanolic NbCl5 liberate nanoparticulate niobia at low temperature. Surprisingly, while such approaches afford highly dispersed niobia nanoparticles, their catalytic applications have yet to be explored. Here we demonstrate the first catalytic application of a high area peroxo niobic acid sol, and associated structure–function relations, in the heterogeneously catalysed aqueous phase conversion of glucose and fructose to 5-HMF, and extend the synthesis to study the impact of dispersing the niobic acid sol over SBA-15.
Experimental
Catalyst preparation
Pure niobic acid was synthesised using a previously reported precipitation protocol from a niobium pentachloride precursor as shown in Scheme 1.37 Briefly, 5 g of moisture sensitive NbCl5 (handled in a glove box under inert atmosphere) was dissolved in 10 cm3 of ethanol to give a yellow solution. This was subsequently added to a 0.3 M aqueous ammonia solution to form a white precipitate of niobia (Nb2O5·nH2O) which was separated by centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min. In order to minimise possible chloride contamination, the resulting precipitate was washed with distilled water until a neutral pH was obtained. Niobia was then cooled with ice and stirred for 5 min after the addition of 20 cm3 of 30 wt% H2O2 aqueous solution to initiate peptisation; the white paste-like solid niobia transformed into a clear yellow solution. The resulting solution was aged in a sealed glass vessel at 80 °C for 24 h, then the vessel opened to air and evaporated to dryness at 80 °C overnight. The yellow solid precipitated niobic acid (referred to as PNA) was ground to a powder and stored in air. The parent PNA was also calcined in static air for 2 h (ramp rate 3 °C min−1) between 300–700 °C to investigate the influence of thermal processing on its physicochemical properties.
000 rpm for 10 min. In order to minimise possible chloride contamination, the resulting precipitate was washed with distilled water until a neutral pH was obtained. Niobia was then cooled with ice and stirred for 5 min after the addition of 20 cm3 of 30 wt% H2O2 aqueous solution to initiate peptisation; the white paste-like solid niobia transformed into a clear yellow solution. The resulting solution was aged in a sealed glass vessel at 80 °C for 24 h, then the vessel opened to air and evaporated to dryness at 80 °C overnight. The yellow solid precipitated niobic acid (referred to as PNA) was ground to a powder and stored in air. The parent PNA was also calcined in static air for 2 h (ramp rate 3 °C min−1) between 300–700 °C to investigate the influence of thermal processing on its physicochemical properties.
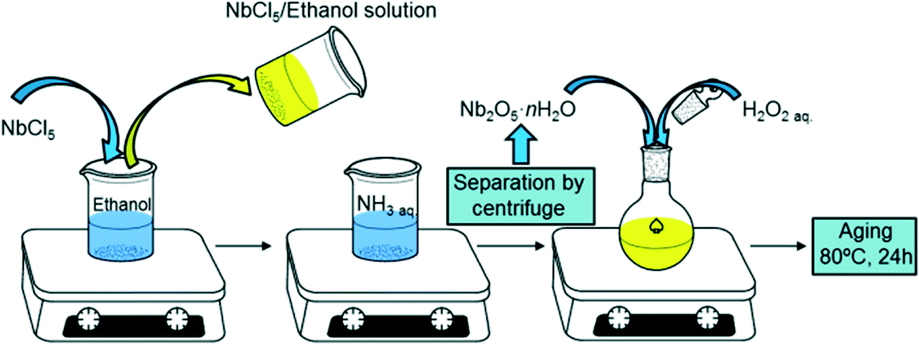 | ||
| Scheme 1 Synthesis of precipitated niobic acid. | ||
SBA-15 was prepared using the method of Zhao et al.38 In a typical synthesis, 10 g of Pluronic P123 was used as a structure directing agent dissolved in water (75.5 cm3) with HCl (2 M, 291.5 cm3) and stirred at 35 °C. 15.5 cm3 of tetraethyl orthosilicate (TEOS) was added to the solution and stirred for 20 h. The resulting gel was aged for 24 h at 100 °C without agitation. The obtained solid was filtered, washed repeatedly and dried at room temperature before calcination at 500 °C for 6 h under flowing air (ramp rate 1 °C min−1) to yield the parent SBA-15. For the preparation of supported niobic acid, 1 g of SBA-15 was dispersed in ethanol prior to the addition of an ethanolic solution containing the desired dissolved mass of NbCl5 (to achieve nominal Nb loadings of 2, 5 and 10 wt%) and subsequent precipitation and oxidation as described for the PNA; these samples were designated X% PNA/SBA-15, and they were not subject to calcination following Nb incorporation.
Catalyst characterisation
Surface areas, pore size distributions and mesopore volumes were determined by N2 porosimetry using Quantasorb Nova 4000e and 4200e porosimeters and Quantachrome Novawin 11.0 software on samples outgassed in vacuo at 120 °C for 2 h. Specific surface areas were calculated by applying the Brunauer–Emmet–Teller (BET) model over the range P/P0 = 0.05–0.30 over the linear portion of the isotherm. Pore size distributions and mesopore volumes were calculated by applying the Barrett–Joyner–Halenda (BJH) model to the desorption branch of the isotherm. X-ray diffraction (XRD) patterns were recorded on a Bruker D8 Advance diffractometer fitted with a LynxEye high-speed strip detector and a Cu Kα source (1.54 Å, 8.04 keV) with a nickel filter, calibrated against a SiO2 standard. In situ measurements were performed under air following heating to the desired temperature (ramp rate 10 °C min−1) for 60 min at each temperature. X-ray photoelectron spectroscopy (XPS) was performed using a Kratos Axis HSi photoelectron spectrometer equipped with a charge neutraliser and monochromated Al Kα (1486.6 eV) radiation. Elemental analysis was performed with a Thermo Scientific iCAP 7000 series ICP-OES and charge injection device (CID) detector, employing a CEM Discover-S & Explorer 48 Sampler Automated Microwave Synthesis System for sample digestion. Diffuse reflectance infra-red Fourier transform (DRIFT) spectra were obtained using a Thermo Scientific Nicolet iS50 FT-IR spectrometer. Samples were diluted homogeneously to 5 wt% with KBr. For ex situ pyridine adsorption, diluted samples were impregnated with neat pyridine, and excess physisorbed pyridine removed in vacuo at 70 °C for 18 h prior to room temperature measurement in vacuo. Acid site densities were quantified by propylamine chemisorption and subsequent thermal analysis on a Mettler Toledo TGA/DSC1 Star System under a N2 purge gas (60 cm3 min−1) interfaced to a ThermoStar TM GSD 301 T3 mass spectrometer. Samples were impregnated with neat propylamine and excess physisorbed propylamine removed in vacuo at 30 °C for 18 h. Propene desorption was monitored by mass spectrometry (m/z = 41) during thermogravimetric analysis of propylamine impregnated samples.Catalytic reactions
Catalytic studies of glucose and fructose were conducted at different temperatures under stirred batch conditions using a Radleys Starfish Carousel station. Reactions were performed using 0.1 g glucose or fructose (Sigma Aldrich, 99.5%), 0.1 g catalyst, and 20 cm3 deionised water. Aliquots were periodically sampled and analysed by HPLC on an Agilent 1260 Infinity Quaternary HPLC equipped with both diode array and refractive index detectors, and an Agilent Hi-Plex H column held at 65 °C, and employing a 5 mM sulphuric acid mobile phase at 0.6 cm3 min−1. Glucose conversion (XG), 5-HMF yield (Y5-HMF), selectivity to HMF (S5-HMF), fructose yield (YF) and selectivity to fructose (SF) were calculated according to eqn (1)–(5) below: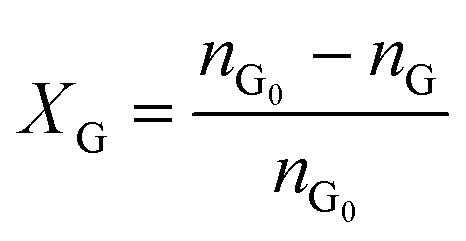 | (1) |
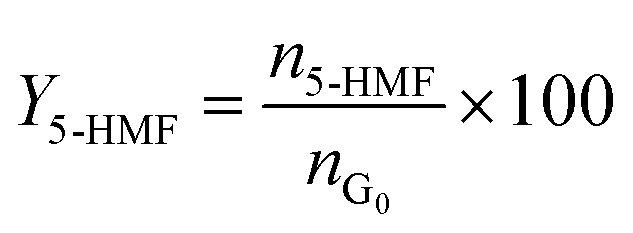 | (2) |
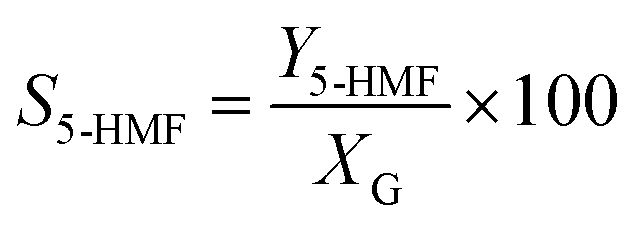 | (3) |
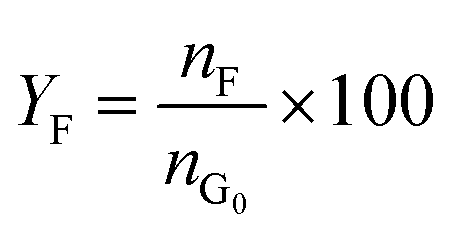 | (4) |
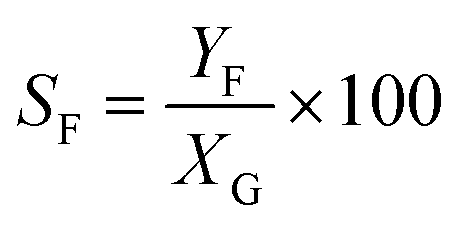 | (5) |
Results and discussion
Catalyst characterisation
Niobium oxide phase transformations have been previously reported to influence catalytic performance,39 hence the impact of calcination temperatures upon the bulk and surface structural and acidic properties of PNA were first investigated. Fig. 1 shows in situ powder XRD patterns of PNA following calcination between 100 °C to 700 °C. Reflections were absent for calcination temperatures ≤400 °C, indicative of amorphous niobium species.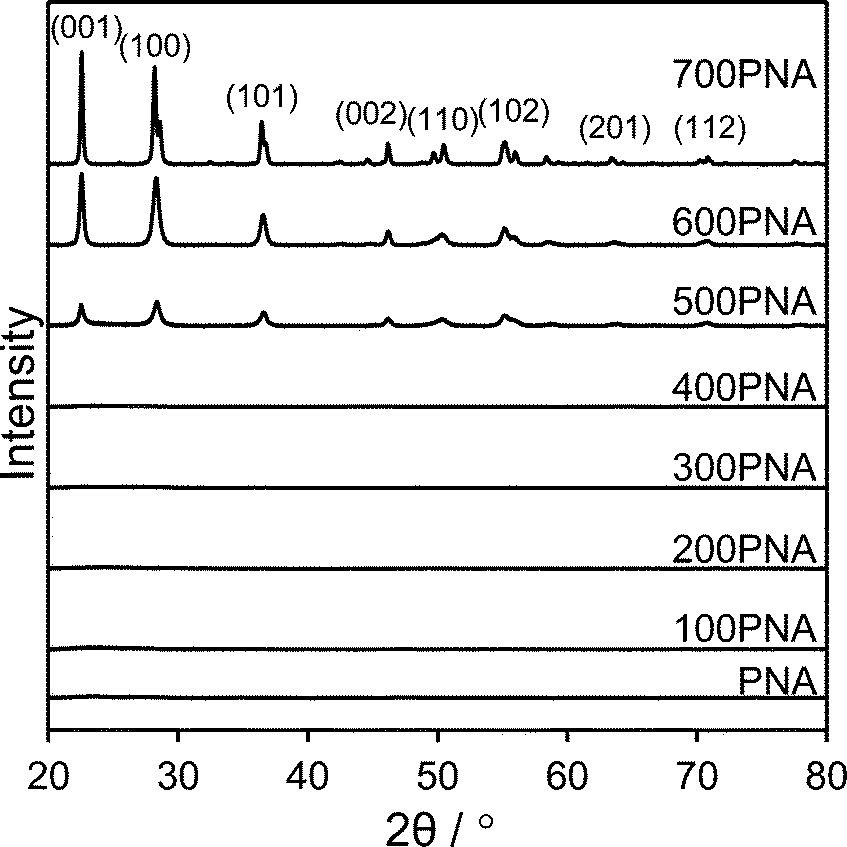 | ||
| Fig. 1 In situ XRD patterns of PNA as a function of calcination temperature. | ||
A crystalline hexagonal niobia phase emerged after calcination at 500 °C, whose intensities increased at higher temperature, consistent with crystallite sintering, concomitant with peak splitting at 700 °C due to the transition to an overwhelmingly (>90%) orthorhombic niobia phase.40 Low angle XRD patterns (Fig. S1†) of the PNA/SBA-15 samples show reflections at 2θ = 0.96, 1.55 and 1.8° characteristic of the (100), (110) and (200) planes of the p6mm SBA-15 support architecture. However, none of the PNA/SBA-15 materials exhibited wide angle reflections (Fig. S2†), suggesting that the supported niobium species were either amorphous or highly dispersed with crystallite dimensions below the instrumental limit of detection; HRTEM of the 10% PNA/SBA-15 sample (Fig. S3†) reveals the presence of ∼1 nm particles decorating the walls and mesopore channels, consistent with this hypothesis. Porosimetry revealed that both bulk PNA (Fig. S4†) and PNA/SBA-15 (Fig. S5†) samples exhibited type IV hysteresis loops associated with mesoporous materials. Since the PNA samples were prepared without a structure-directing agent, they are not expected to possess well-defined, ordered mesopores and hence we attribute their mesoporosity to the presence of interparticle voids formed during niobia precipitation. Physical properties of bulk and supported PNA materials are summarised in Table 1. Niobia crystallisation during calcination ≥500 °C coincides with a loss of surface area and interparticle pore volume.41 Niobium addition to the parent SBA-15 progressively lowered the surface area and mesopore volume of the silica support, with only a small contraction in the average mesopore diameter for the highest Nb loading, consistent with uniform dispersion throughout the pore network.
| Catalyst | Crystallite sizea/nm | Surface areab/m2 g−1 | Pore diameterc/nm | Pore volumec/cm3 g−1 | Nb contentd/wt% |
|---|---|---|---|---|---|
| a XRD. b BET. c BJH. d ICP. | |||||
| PNA | — | 284 | 3.4 | 0.35 | 61 |
| 300PNA | — | 273 | 3.4 | 0.29 | 60 |
| 500PNA | 23 | 112 | 3.8 | 0.20 | 61 |
| 700PNA | 52 | 89 | 2.0 | 0.11 | 61 |
| SBA-15 | — | 1040 | 3.9 | 1.18 | 0 |
| 2% PNA/SBA-15 | — | 916 | 3.9 | 0.96 | 3 |
| 5% PNA/SBA-15 | — | 830 | 3.9 | 0.93 | 5 |
| 10% PNA/SBA-15 | — | 709 | 3.4 | 0.76 | 12 |
Surface analysis of the PNA/SBA-15 materials confirmed the presence of niobium with loadings from 3.3 to 16.5 wt% Nb, in good agreement with bulk elemental analysis and hence consistent with uniform dispersion throughout the SBA-15 mesopore network. High resolution spectra revealed a single spin–orbit split doublet with a Nb 3d5/2 binding energy ≥207.3 eV, characteristic of a Nb5+ chemical environment, for all loadings (Fig. S6†). While the 12% PNA/SBA-15 spectra was identical to that of the parent PNA sample, Nb 3d peaks shifted by ∼0.7 eV to higher binding energy for lower loadings, evidencing a strong interaction with the silica support and appearance of (electron deficient) interfacial Nb–O–Si environments, as previously reported for conformal alumina42 and zirconia43 films grown over SBA-15 architectures.
Surface acidity was probed by both pyridine and propylamine adsorption. The nature of acid sites in the PNA materials was first examined through DRIFTS following pyridine treatment (Fig. S7 and S8†). Vibrational bands attributable to Lewis acid sites were observed at 1444 and 1607 cm−1, together with a 1540 cm−1 band characteristic of a pyridinium ion coordinated to Brønsted sites and a band at 1487 cm−1 which is common to both Lewis and Brønsted acid sites.44 The relative Brønsted:Lewis acid character (determined from the 1444 cm−1 and 1540 cm−1 peak areas) in Fig. 2 shows a monotonic decrease in Brønsted acidity with calcination temperature. The total acid site density shows a similar strong temperature dependence, falling from ∼1.6 mmol g−1 in the as-precipitated pure material, to essentially zero by 700 °C. This thermally induced loss in acidity can be understood largely in terms of the coincident sintering of PNA crystallites and the attendant loss of external surface area; the preferential disappearance of Brønsted acidity follows that previously that reported by Foo and co-workers,45 wherein 700 °C calcination of a commercial HY-340 niobic acid (of unknown preparative method) eliminated all Brønsted character, due to the loss of water of hydration, but left a small number of Lewis acid sites. In contrast, niobic acid supported over SBA-15 exhibited pure Brønsted acidity for all loadings (Fig. S7†). This may arise from the coordination of Lewis base sites in the precipitated niobic acid nanoparticles with surface hydroxyls on the SBA-15. Total acid densities for the PNA/SBA-15 materials increased almost linearly with bulk Nb loading (Fig. S9†), consistent with the evolution of a conformal niobic acid adlayer. While solvents are expected to impact on the absolute or ‘effective’ acid site loading, these intrinsic acid site measurements can be considered a reliable guide to assess trends in acidic properties of PNA and PNA/SBA-15 materials. Indeed, previous acid site measurements comparing adsorption of basic probe molecules from vapour or solvent on bulk niobium phosphate46 demonstrate minimal impact on the Brønsted:Lewis ratio, and only a moderate decrease in total acidity for bulk niobia.
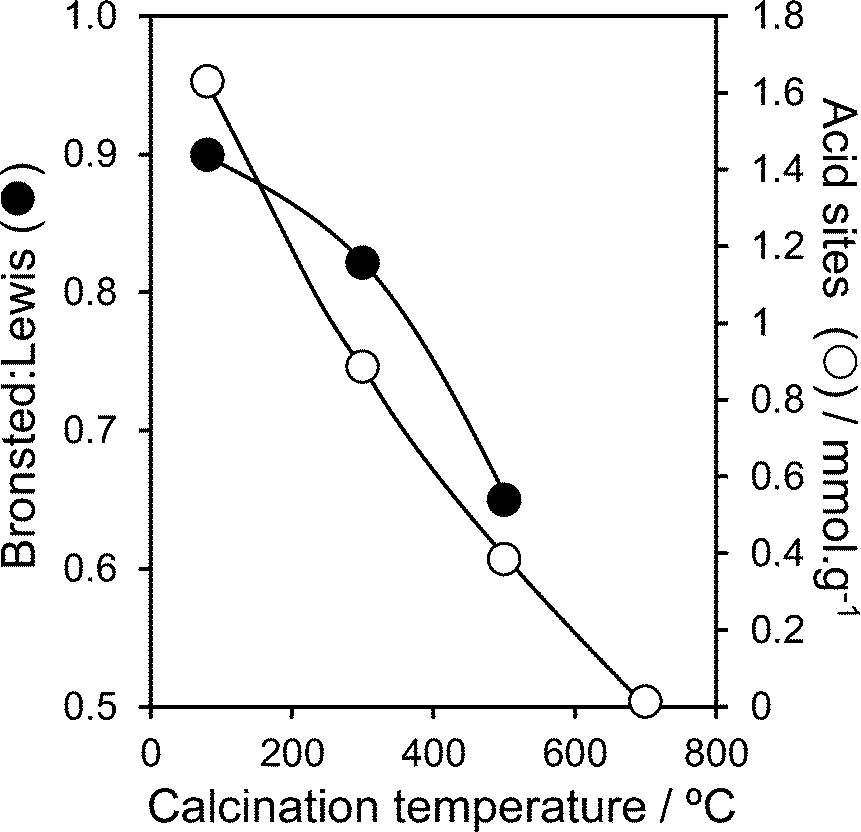 | ||
| Fig. 2 Brønsted:Lewis character and total acid site density of PNA as a function of calcination temperature. | ||
Reactivity of bulk niobic acid (PNA)
The temperature dependent, aqueous phase reaction of glucose and fructose over the parent PNA was first explored, since it is known that undesirable side-reactions that lead to humins are favoured at higher temperature. Fig. 3 shows that conversion of both glucose and fructose was very temperature sensitive, increasing exponentially from 16 → 50% and 20 → 90% respectively upon increasing the reaction temperature from 80 °C to 110 °C.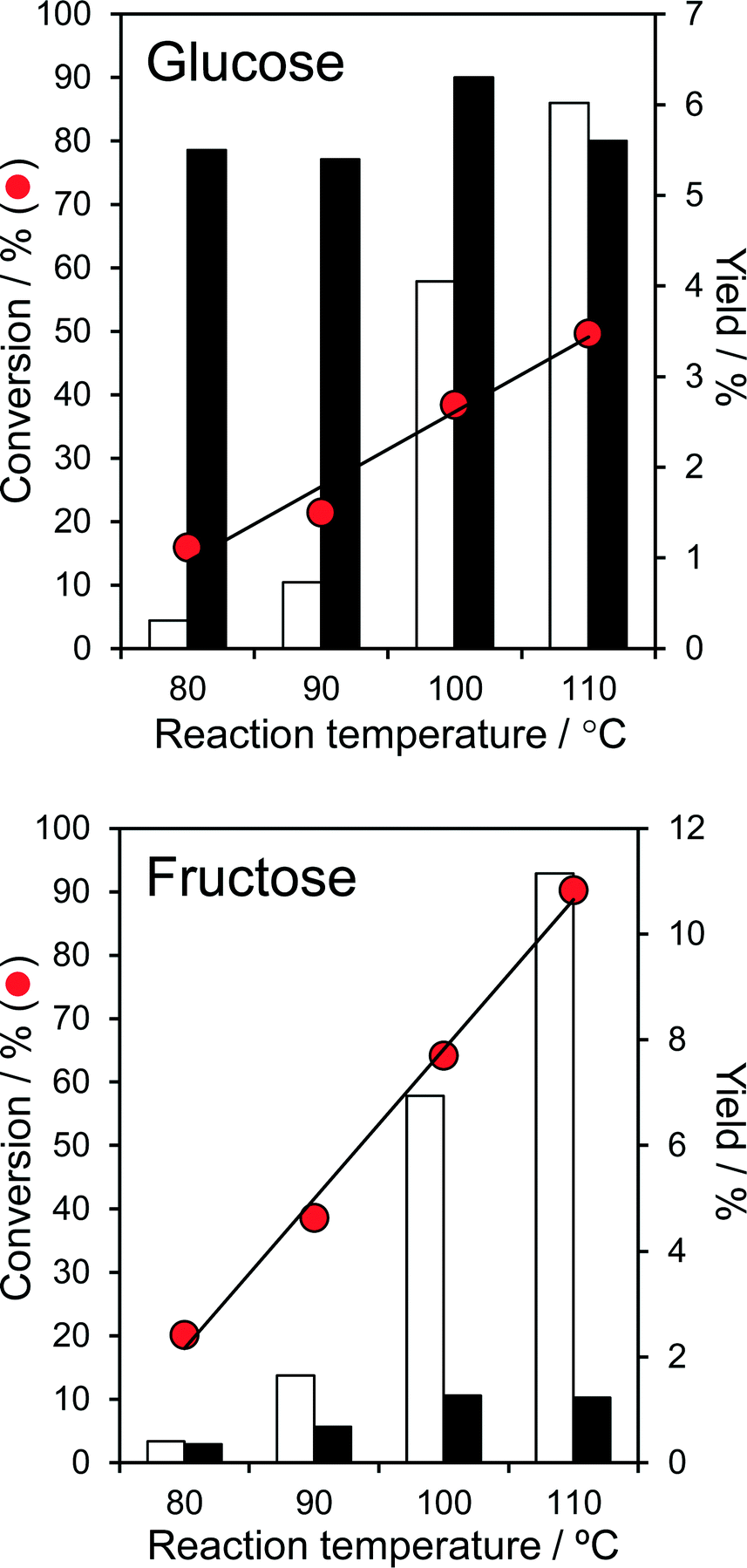 | ||
| Fig. 3 Aqueous phase conversion of glucose and fructose over parent PNA and corresponding yields of 5-HMF (white) and fructose/glucose (black) as a function of reaction temperature: 0.1 g glucose or fructose, 0.1 g PNA, 20 cm3 water, 6 h. | ||
These conversions are comparable or superior to literature values for fructose over tungstated zirconia,22 25% CeO2/75% Nb2O524 and Ag3PW12O40,47 and glucose over sulphated zirconia,22 sulfated mesoporous niobia,48 and TiO2 or ZrO249 at similar fructose/glucose concentrations, but at lower temperature and in the absence of any co-solvents. The higher reactivity of fructose (to predominantly 5-HMF) is in accordance with previous reports that its solid acid catalysed dehydration is more facile than glucose isomerisation.47,49 While the absolute 5-HMF yield from glucose was unsurprisingly small, in light of the modest glucose conversions, this increased almost exponentially with reaction temperature, reaching 6% at 110 °C (comparable to the performance of TiO2 and ZrO2 at 200 °C49). 5-HMF yields from direct fructose dehydration were approximately double those obtained by the stepwise isomerisation and subsequent dehydration of glucose, but again exhibited an exponential reaction temperature dependence; glucose isomerisation to fructose is clearly the rate-determining step in 5-HMF production over PNA. The strong temperature sensitivity of the turnover frequencies (TOFs) for both glucose and fructose over PNA (Fig. S10†) are reflected in their significant apparent activation energies of 79 kJ mol−1 and 87 kJ mol−1 respectively. While the 5-HMF yields observed from both glucose and fructose are far lower than reported in DMSO, it must be noted that DMSO catalyses significant fructose dehydration >100 °C. Indeed, many recent solid acid studies of 5-HMF synthesis are conducted in DMSO, despite the work of Musau and Munavu which showed this solvent alone delivers a 92% 5-HMF yield from fructose in 2 h at 150 °C!50 In the present low temperature, aqueous phase study, the modest 5-HMF yields (despite 90% fructose conversion) cannot be attributed to self-poisoning, since the spiking of a fructose solution with 1 equivalent of 5-HMF had no impact on fructose conversion. Furthermore, only trace levulinic acid was observed as a minor by-product of reactively-formed 5-HMF rehydration.
The impact of PNA pre-calcination on reactivity was subsequently explored at 100 °C, conditions under which good glucose and fructose conversions and modest 5-HMF yields were obtained over the parent amorphous PNA. Glucose conversion fell significantly for PNA calcined above 300 °C, coincident with the crystallisation of hexagonal niobia and concomitant drop in surface area. The retention of significant Lewis acidity for calcination temperatures between 300–500 °C enabled glucose isomerisation to fructose to proceed essentially unperturbed, whereas the preferential loss of Brønsted acidity over this temperature range systematically suppressed the dehydration of reactively-formed fructose to 5-HMF (Fig. 4). Calcined PNA displayed a similar reactivity toward fructose, with the loss in Brønsted acidity ≥300 °C progressively decreasing fructose dehydration to 5-HMF yield, with little impact on its (undesired) Lewis catalysed isomerisation to glucose. The transition to orthorhombic niobia after 700 °C calcination, and further loss in surface area and acid density, rendered the PNA material inert towards both glucose and fructose. As-prepared, amorphous PNA proved optimal for both glucose isomerisation to fructose, and the latter's subsequent dehydration to 5-HMF; thermal processing and associated crystallisation destroys niobic acid. Despite the fall in glucose and fructose conversion upon calcination, the corresponding TOFs systematically increase, approximately doubling for PNA pre-calcined at 500 °C (Fig. S11†). This trend reflects the steep fall in acid loading following calcination, i.e. presence of fewer, but more active catalytic sites, for both isomerisation and dehydration steps, albeit propylamine thermal desorption experiments showed no shift in the desorption temperature of reactively-formed propene (a measure of acid strength51) between the parent and calcined PNAs.
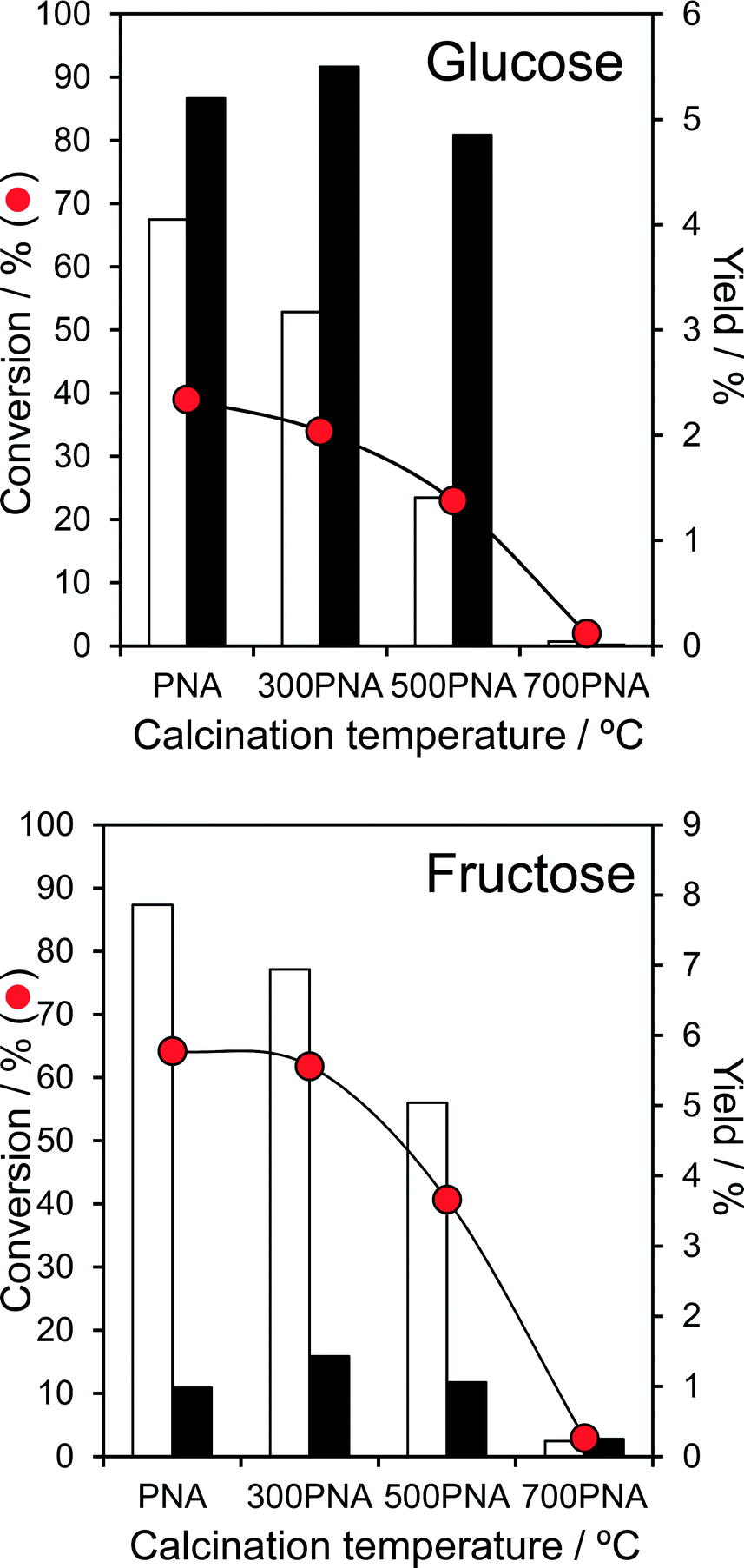 | ||
| Fig. 4 Aqueous phase conversion of glucose and fructose over parent PNA and corresponding yields of 5-HMF (white) and fructose/glucose (black) as a function of pre-calcination temperature: 0.1 g glucose or fructose, 100 °C, 0.1 g PNA, 20 cm3 water, 6 h. | ||
In all cases, the low selectivity towards 5-HMF from either glucose or fructose appears attributable to humin formation, di-D-fructose dianhydrides,20 or the rehydration of 5-HMF favoured by the aqueous media, although under our reaction conditions PNA forms <5% levulinic acid (Fig. S12†) through the latter pathway (in contrast to DMSO wherein >40% of 5-HMF is lost under our reaction conditions).
Reactivity of PNA/SBA-15
In light of the comparatively low surfaces areas and large crystallite sizes of the bulk PNA materials, we attempted to improve the performance of the most active as-precipitated amorphous niobic acid through its dispersion over a high area mesoporous SBA-15 support. The resulting PNA/SBA-15 catalysts, with Nb loadings from 2 to 10 wt%, were tested for the aqueous phase conversion of glucose and fructose to 5-HMF at 100 °C. Negligible conversion of glucose was observed for all silica supported materials, which is unsurprising since these possess no Lewis acidity. In contrast, the fructose conversion increased almost linearly with Nb loading from 6% (2% PNA/SBA-15) to 18% (10% PNA/SBA-15) (Fig. S13†), consistent with the formation of a two-dimensional niobic acid overlayer and linear increase in the acid loading seen in Fig. S9.† 5-HMF yields mirrored the fructose conversion, but selectivity to this desired furanic platform chemical remained low at around 12%. Associated initial rates and TOFs for fructose conversion over parent PNA and PNA/SBA-15 catalysts are compared in Fig. 5, which reveals an inverse correlation between Nb loading and reactivity, evidencing the superior performance of highly dispersed niobic acid (per acid site/mass of niobium), presumably due to the latter's superior Brønsted acidity.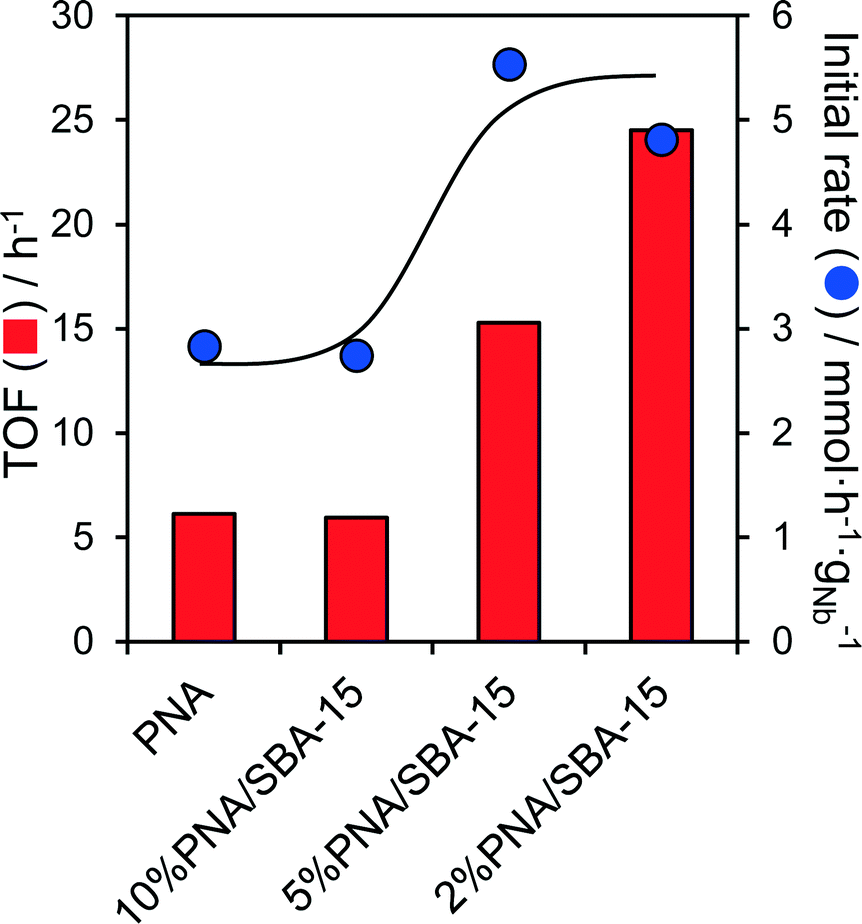 | ||
| Fig. 5 Comparison of turnover frequencies and activities for the aqueous phase conversion of fructose normalised to Brønsted acid loadings over parent PNA and PNA supported on SBA-15 as a function of Nb loading: 0.1 g fructose, 0.1 g PNA or X% PNA/SBA-15, 100 °C, 20 cm3 water, 6 h. | ||
Indeed acid site measurements reveal that PNA/SBA-15 materials contain a small proportion of weaker Brønsted acid sites (Fig. S14†), which are believed to be more selective towards 5-HMF formation from fructose due to a reduced tendency for side reactions.32 This evolution of acid sites may reflect a transition from highly dispersed niobia species comprising corner shared NbO6 octahedra with weakly acidic terminal hydroxyls, to more extended structures with a higher proportion of more strongly acidic bridged hydroxyls spanning edge or face shared NbO6 octahedra.32
Conclusions
Successful synthesis of niobic acid nanoparticles and niobic acid functionalised SBA-15, possessing higher surface areas and acid loadings than by conventional sol–gel or precipitation routes, is reported through the peptisation of a niobic acid sol with H2O2. Calcination induces nanoparticle sintering, surface dehydration and the crystallisation of orthorhombic niobia, resulting in the loss of acid sites and Brønsted character. In contrast, Nb2O5/SBA-15 exhibits pure Brønsted acidity independent of Nb loading. Nanoparticulate niobic acid is active for the aqueous phase isomerisation of glucose to fructose, and subsequent fructose dehydration to 5-HMF under mild reaction conditions. Niobia crystallisation and the concomitant loss of (particularly Brønsted) acid sites lowers activity, notably for fructose dehydration. Niobic acid nanoparticles dispersed over SBA-15 possess pure Brønsted acidity and enhanced turnover frequencies for fructose dehydration to 5-HMF, but are unable to isomerise glucose due to absence of Lewis acid sites. Although the absolute 5-HMF yields of nanoparticulate niobic acid from glucose and fructose are modest compared to those obtained employing pure or bi-phasic water/DMSO or MIBK solvent systems, the latter processes are not readily compatible with reforming dilute aqueous C5–C6 sugar streams, while few conventional solid acids exhibit the excellent water tolerance of the niobia nanomaterials in the present work. Niobic acid nanoparticles are promising solid acids for the aqueous phase conversion of waste-derived sugars to platform chemicals.Acknowledgements
We thank the Engineering and Physical Sciences Council for (EP/K014706/1, EP/K000616/2, EP/K014676/1 and EP/K014749/1) for financial support, the Royal Society and British Council Global Innovation Initiative for GB3-Net.Notes and references
- W. Steffen, J. Grinevald, P. Crutzen and J. McNeill, Philos. Trans. R. Soc., A, 2011, 369, 842–867 CrossRef PubMed.
- R. A. Sheldon, Green Chem., 2014, 16, 950–963 RSC.
- S. Srinivasan, Renewable Energy, 2009, 34, 950–954 CrossRef.
- S. G. Wettstein, D. M. Alonso, E. I. Gürbüz and J. A. Dumesic, Curr. Opin. Chem. Eng., 2012, 1, 218–224 CrossRef CAS.
- T. Werpy, G. Petersen, A. Aden, J. Bozell, J. Holladay, J. White, A. Manheim, D. Eliot, L. Lasure and S. Jones, Top value added chemicals from biomass. Volume 1-Results of screening for potential candidates from sugars and synthesis gas, DTIC Document, 2004 Search PubMed.
- B. Saha and M. M. Abu-Omar, Green Chem., 2014, 16, 24–38 RSC.
- W. Haworth and W. Jones, J. Chem. Soc., 1944, 667–670 RSC.
- A. A. Rosatella, S. P. Simeonov, R. F. Frade and C. A. Afonso, Green Chem., 2011, 13, 754–793 RSC.
- R. D. M. Erica Farnetti and J. Kaspar, in Inorganic and Bio-inorganic Chemistry, ed. I. Bertiniaytexasphl, 2009, vol. 2, pp. 50–87 Search PubMed.
- S. Hu, Z. Zhang, J. Song, Y. Zhou and B. Han, Green, 2009, 11, 1746–1749 RSC.
- H. Abou-Yousef, E. B. Hassan and P. Steele, J. Fuel Chem. Technol., 2013, 41, 214–222 CrossRef CAS.
- I.-J. Kuo, N. Suzuki, Y. Yamauchi and K. C.-W. Wu, RSC Adv., 2013, 3, 2028–2034 RSC.
- X. Guo, Q. Cao, Y. Jiang, J. Guan, X. Wang and X. Mu, Carbohydr. Res., 2012, 351, 35–41 CrossRef CAS PubMed.
- H. Zhao, J. E. Holladay, H. Brown and Z. C. Zhang, Science, 2007, 316, 1597–1600 CrossRef CAS PubMed.
- Y. Román-Leshkov, J. N. Chheda and J. A. Dumesic, Science, 2006, 312, 1933–1937 CrossRef PubMed.
- I. Jiménez-Morales, J. Santamaría-González, A. Jiménez-López and P. Maireles-Torres, Fuel, 2014, 118, 265–271 CrossRef.
- A. J. Crisci, M. H. Tucker, J. A. Dumesic and S. L. Scott, Top. Catal., 2010, 53, 1185–1192 CrossRef CAS.
- K. Tekin, S. Karagöz and S. Bektaş, Renewable Sustainable Energy Rev., 2014, 40, 673–687 CrossRef CAS.
- J. N. Chheda, Y. Roman-Leshkov and J. A. Dumesic, Green Chem., 2007, 9, 342–350 RSC.
- P. Carniti, A. Gervasini and M. Marzo, Catal. Commun., 2011, 12, 1122–1126 CrossRef CAS.
- S. J. Dee and A. T. Bell, ChemSusChem, 2011, 4, 1166–1173 CrossRef CAS PubMed.
- A. Osatiashtiani, A. F. Lee, D. R. Brown, J. A. Melero, G. Morales and K. Wilson, Catal. Sci. Technol., 2014, 4, 333–342 CAS.
- R. Kourieh, V. Rakic, S. Bennici and A. Auroux, Catal. Commun., 2013, 30, 5–13 CrossRef CAS.
- D. Stošić, S. Bennici, V. Rakić and A. Auroux, Catal. Today, 2012, 192, 160 CrossRef.
- A. H. Jadhav, H. Kim and I. T. Hwang, Bioresour. Technol., 2013, 132, 342–350 CrossRef CAS PubMed.
- C. S. Lanziano, F. Rodrigues, S. Rabelo, R. Guirardello, V. T. Da Silva and C. B. Rodella, Chem. Eng. Trans., 2014, 37, 589–594 Search PubMed.
- I. Jiménez-Morales, M. Moreno-Recio, J. Santamaría-González, P. Maireles-Torres and A. Jiménez-López, Appl. Catal., B, 2014, 154, 190–196 CrossRef.
- F. Yang, Q. Liu, M. Yue, X. Bai and Y. Du, Chem. Commun., 2011, 47, 4469–4471 RSC.
- T. Okuhara, Chem. Rev., 2002, 102, 3641–3666 CrossRef CAS PubMed.
- T. Ushikubo and K. Wada, Appl. Catal., 1990, 67, 25–38 CrossRef CAS.
- I. Nowak and M. Ziolek, Chem. Rev., 1999, 99, 3603–3624 CrossRef CAS PubMed.
- H. T. Kreissl, K. Nakagawa, Y.-K. Peng, Y. Koito, J. Zheng and S. C. E. Tsang, J. Catal., 2016, 338, 329–339 CrossRef CAS.
- J. Datka, A. Turek, J. Jehng and I. Wachs, J. Catal., 1992, 135, 186–199 CrossRef CAS.
- M. Trejda, A. Tuel, J. Kujawa, B. Kilos and M. Ziolek, Microporous Mesoporous Mater., 2008, 110, 271–278 CrossRef CAS.
- P. Carniti, A. Gervasini and M. Marzo, J. Phys. Chem. C, 2008, 112, 14064–14074 CAS.
- P. Carniti, A. Gervasini and M. Marzo, Catal. Today, 2010, 152, 42–47 CrossRef CAS.
- N. Uekawa, T. Kudo, F. Mori, Y. J. Wu and K. Kakegawa, J. Colloid Interface Sci., 2003, 264, 378–384 CrossRef CAS PubMed.
- D. Zhao, J. Feng, Q. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548–552 CrossRef CAS PubMed.
- E. Nikishina, E. Lebedeva and D. Drobot, Inorg. Mater., 2012, 48, 1243–1260 CrossRef CAS.
- M. Paulis, M. Martın, D. Soria, A. Dıaz, J. Odriozola and M. Montes, Appl. Catal., A, 1999, 180, 411–420 CrossRef CAS.
- J. M. Jehng and I. E. Wachs, Chem. Mater., 1991, 3, 100–107 CrossRef CAS.
- J. J. Creasey, C. M. A. Parlett, J. C. Manayil, M. A. Isaacs, K. Wilson and A. F. Lee, Green Chem., 2015, 17, 2398–2405 RSC.
- A. Osatiashtiani, A. F. Lee, M. Granollers, D. R. Brown, L. Olivi, G. Morales, J. A. Melero and K. Wilson, ACS Catal., 2015, 5, 4345–4352 CrossRef CAS.
- G. Busca, Catal. Today, 1998, 41, 191–206 CrossRef CAS.
- G. S. Foo, D. Wei, D. S. Sholl and C. Sievers, ACS Catal., 2014, 4, 3180–3192 CrossRef CAS.
- P. Carniti, A. Gervasini, S. Biella and A. Auroux, Catal. Today, 2006, 118, 373–378 CrossRef CAS.
- C. Fan, H. Guan, H. Zhang, J. Wang, S. Wang and X. Wang, Biomass Bioenergy, 2011, 35, 2659–2665 CrossRef CAS.
- E. L. S. Ngee, Y. Gao, X. Chen, T. M. Lee, Z. Hu, D. Zhao and N. Yan, Ind. Eng. Chem. Res., 2014, 53, 14225–14233 CrossRef CAS.
- X. Qi, M. Watanabe, T. M. Aida and R. L. Smith Jr, Catal. Commun., 2008, 9, 2244–2249 CrossRef CAS.
- R. M. Musau and R. M. Munavu, Biomass, 1987, 13, 67–74 CrossRef CAS.
- M. V. Juskelis, J. P. Slanga, T. G. Roberie and A. W. Peters, J. Catal., 1992, 138, 391–394 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional structural and reactivity data. See DOI: 10.1039/c6cy01129b |
| This journal is © The Royal Society of Chemistry 2016 |