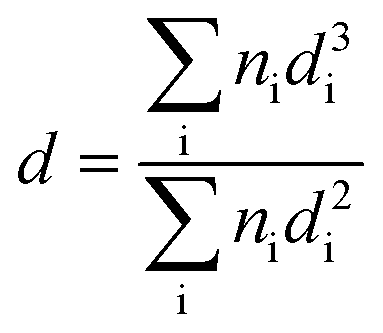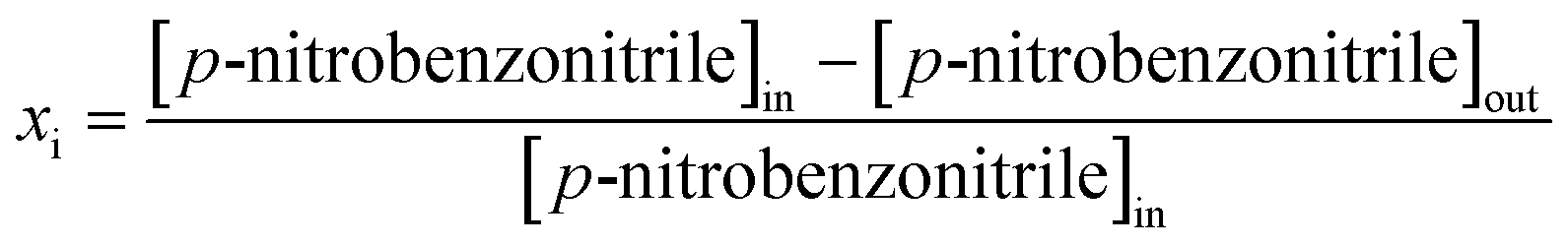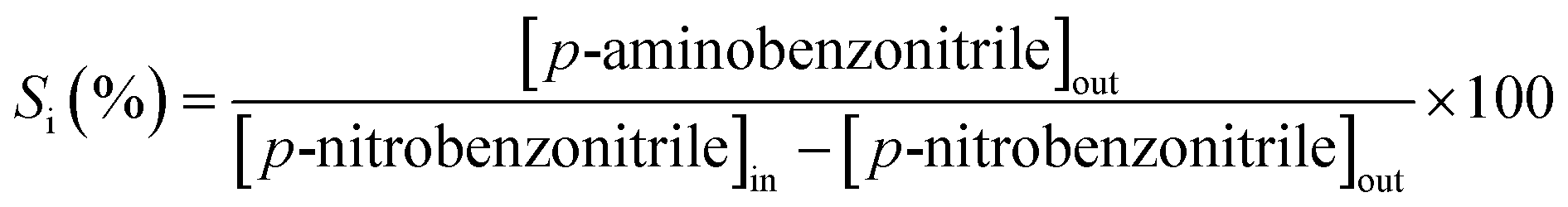Selective gas phase hydrogenation of nitroarenes over Mo2C-supported Au–Pd
Received
7th March 2016
, Accepted 12th July 2016
First published on 13th July 2016
Abstract
We report the first synthesis of Mo2C-supported Au and Au–Pd catalysts (nominal Au/Pd = 10 and 30) obtained from colloidal nanoparticles stabilised by polyvinyl alcohol (PVA). Equivalent Au/Al2O3 and Au–Pd/Al2O3 were prepared and served as benchmarks. Residual PVA was removed by thermal treatment in N2, which was monitored by thermogravimetric analysis. The catalysts were characterised in terms of temperature-programmed reduction (TPR), BET surface area, H2 chemisorption, powder X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS) and transmission electron microscopy (TEM) measurements. The reduced catalysts exhibited an equivalent metal particle size range (1–8 nm) and mean size (4–5 nm). The carbide samples showed greater H2 chemisorption capacity than the Al2O3 systems where inclusion of Pd enhanced H2 uptake. XPS measurements suggest electron transfer from Al2O3 to Au while the Au binding energy for the carbide samples is close to that of the metallic Au reference. The catalysts were tested in the gas phase hydrogenation of nitrobenzene, p-chloronitrobenzene and p-nitrobenzonitrile and delivered 100% selectivity to the target amine in each case. Inclusion of Pd served to increase selective hydrogenation rates where Au–Pd/Mo2C outperformed Au–Pd/Al2O3, a response that is attributed to increased surface hydrogen.
1. Introduction
The use of bimetallic catalysts is attracting increased research interest1 due to their enhanced activity,2,3 selectivity4 and resistance to poisoning5 in comparison with the corresponding mono-metallic systems. This has been exploited in commercial applications, notably Pd–Zn in hydrogen production,6 Pd–Au in vinyl acetate synthesis,7 Cu–Zn in methanol production8 and Fe–Cr in the water gas shift reaction.9 This study focuses on Au–Pd formulations, which to date have been used in the synthesis of hydrogen peroxide,10 NO2 decomposition,11 hydrodesulphurisation (of thiophene and dibenzothiophene),12 hydrodechlorination (of trichloroethene),13 oxidation (of glycerol,14 CO,15 benzyl alcohol, cinnamyl alcohol, 2-octen-1-ol and n-octanol)16 and hydrogenation (of 1,3-butadiene,17,18 benzaldehyde,5 naphthalene and toluene,19,20 acetylene,21 allyl alcohol2 and cinnamaldehyde22). Improved performance relative to monometallic Pd and Au catalysts has been attributed to ensemble, ligand or geometric effects.23,24 Prior studies have established chemoselectivity for supported Au in targeted –NO2 reduction of functionalised nitro-compounds.25–28 Reaction exclusivity is important in minimising waste and addressing the sustainability gap in non-selective processes using standard (Pd,29 Ru (ref. 30) or Ni (ref. 31)) transition metal catalysts. However, hydrogenation rates delivered by supported Au fall below those achieved with non-selective metal catalysts.32 It is now crucial to increase selective hydrogenation rates to address the key commercial consideration of productivity that combines activity and selectivity. Gold activation of H2 is the limiting step due to the high energy barrier for dissociative adsorption.33 We have explored the use of Mo2C as a support that can chemisorb H2 and increase surface concentration, leading to higher hydrogenation rates.34 We demonstrated higher selective hydrogenation rates over Au/Mo2C than those over Au/Al2O3 but further improvements are required in Au/Mo2C synthesis directed at decreasing the Au particle size as a critical variable that governs H2 activation.33 Preliminary data3 established that addition of Pd to Au on Al2O3 (prepared by deposition–precipitation) increased the hydrogenation activity but lower selectivities were recorded at higher Pd loading (Au/Pd < 20). We have now examined the viability of Mo2C-supported Au–Pd to elevate rates while retaining selectivity in the hydrogenation of nitroarenes.
The challenge in supported bimetallic synthesis is to exert control over surface composition and size/dispersion.35 To date, supported Au–Pd catalysts have principally been prepared by impregnation (incipient19 and wet3,5,17) and (co)-deposition.3,10,17,36,37 The synthesis procedure influences the ultimate metal loading, morphology and structure10,21 where surface segregation and formation of isolated Au and Pd particles can occur.3 The use of Au–Pd catalysts has been largely focused on Au addition to modify the catalytic properties of Pd, where Au/Pd < 1.21 This study takes the opposite approach with Pd inclusion directed at increasing selective hydrogenation rates. A range of oxides (Fe2O3,37 TiO2,38 ZrO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10 and MgO13) have been used as Au–Pd supports with the predominance of Al2O33,13,17,19,20,37 and SiO213,20,21,36,39 as carriers. In this study, we report the first preparation of Au–Pd/Mo2C (Au/Pd ≥ 10) using colloidal polyvinyl alcohol (PVA), which provides a protective layer around the metal nanoparticles preventing agglomeration in solution to generate small supported metal particles.40 The use of “protecting” agents (polyvinyl pyrrolidone (PVP)12 and PVA14,16) to minimise nanoparticle aggregation has been applied in the synthesis of supported Au,41 Pd (ref. 42) and Au–Pd.14,16 We examine the catalytic action of Au–Pd/Mo2C in the hydrogenation of nitroarenes (nitrobenzene, p-chloronitrobenzene and p-nitrobenzonitrile) where Au–Pd/Al2O3 prepared using the same method served as a benchmark.
10 and MgO13) have been used as Au–Pd supports with the predominance of Al2O33,13,17,19,20,37 and SiO213,20,21,36,39 as carriers. In this study, we report the first preparation of Au–Pd/Mo2C (Au/Pd ≥ 10) using colloidal polyvinyl alcohol (PVA), which provides a protective layer around the metal nanoparticles preventing agglomeration in solution to generate small supported metal particles.40 The use of “protecting” agents (polyvinyl pyrrolidone (PVP)12 and PVA14,16) to minimise nanoparticle aggregation has been applied in the synthesis of supported Au,41 Pd (ref. 42) and Au–Pd.14,16 We examine the catalytic action of Au–Pd/Mo2C in the hydrogenation of nitroarenes (nitrobenzene, p-chloronitrobenzene and p-nitrobenzonitrile) where Au–Pd/Al2O3 prepared using the same method served as a benchmark.
2. Experimental
2.1 Chemicals
Gold(III) chloride hydrate (HAuCl4·xH2O, 99.999%), palladium tetraamine dinitrate (Pd(NH3)4(NO3)2, 99.99%) and polyvinyl alcohol (PVA, 87–89%) were obtained from Aldrich, while molybdic acid (H2MoO4, 99%) and NaBH4 (>99%) were acquired from Merck and Fluka, respectively. All gases were of high purity (99.9%) and supplied by BOC or Air Liquide. The nitro-reactants (nitrobenzene, p-chloronitrobenzene and p-nitrobenzonitrile, Aldrich (≥98%)) and solvent (1-butanol, Riedel-de Haen, 99.8%) were used as-received without further purification.
2.2 Catalyst preparation
β-Mo2C was synthesised via temperature-programmed carburisation of H2MoO4 (2 g) in 20% v/v CH4 in H2 (340 cm3 min−1). The temperature was ramped at 1 K min−1 to 973 K, maintained for 1 h and decreased to ambient temperature under H2 with sample passivation in 1% v/v O2/He (30 cm3 min−1) for 1 h. The passivation step was necessary to circumvent autothermal oxidation upon contact with air.43 Gold and gold–palladium catalysts were prepared according to a colloidal method developed by Prati et al.44 for carbon supports and adopted for use with Al2O3. Gold was incorporated using an Au sol that was deposited on Mo2C (or Al2O3) with a nominal loading of 1% w/w as reported previously.18 Aqueous solutions of HAuCl4 (6 cm3, 2.5 × 10−2 M) and PVA (1.5 cm3, 2.3 × 10−6 M; Au/PVA = 50) were added to 200 cm3 distilled water with vigorous agitation for 3 min, then a freshly prepared solution of NaBH4 (4.5 cm3, 0.1 M) was added (NaBH4/Au = 3) with a resultant colour change from light yellow to red, indicating reduction of Au3+ to Au0. The sol was acidified to pH = 2 with H2SO4 (0.1 M) and 3 g of support was added to the mixture, which was maintained under vigorous stirring at ambient temperature for 2 h; water was removed using a rotary evaporator. Bimetallic synthesis followed an equivalent protocol where 1% w/w Au/Mo2C (or Au/Al2O3) was suspended in 200 cm3 distilled water to which a known amount of PVA solution (2.3 × 10−6 M; Pd/PVA = 80) was added under vigorous stirring with subsequent addition of Pd(NH3)4(NO3)2 (4.7 × 10−3 M; nominal Au/Pd = 10 or 30). Hydrogen (50 cm3 min−1) was then bubbled into the reactor for 2 h to reduce Pd on the supported Au colloids. The mixture was stirred for further 16 h and water was removed by rotary evaporation. The resultant solid was washed repeatedly and dried under vacuum at ambient temperature. The samples were heated (2 K min−1) in 600 cm3 min−1 N2 to 773 K for 1 h to remove the stabilising agents, cooled to ambient temperature and subjected to thermal reduction in 100 cm3 min−1 H2 to 773 K (at 2 K min−1). Upon cooling, the samples were passivated as above for off-line analysis. The bimetallic catalysts are labelled as Au–Pd/Mo2C (or Al2O3)-X, where X represents the nominal Au/Pd molar ratio (= 10 or 30).
2.3 Catalyst characterisation
Au, Mo, Cl and Al contents were measured by inductively coupled plasma (ICP) atomic emission spectroscopy (CNRS Centre of Chemical Analysis, Vernaison). Carbon content was determined using an Exeter CE-440 elemental analyser after sample combustion at 1873 K. Temperature-programmed reduction (TPR), BET surface area and H2 chemisorption were measured using a commercial CHEM-BET 3000 (Quantachrome) unit. The passivated samples were loaded into a U-shaped Quartz cell (10 cm × 3.76 mm i.d.) and heated in 17 cm3 min−1 (Brooks mass flow controller) 5% v/v H2/N2 at 2 K min−1 to 523 ± 1 K, which was held for 1 h. The effluent gas passed through a liquid N2 trap and H2 consumption was monitored by TCD with data acquisition/manipulation using the TPR Win™ software. The samples were swept with 65 cm3 min−1 N2 for 1.5 h, cooled to ambient temperature and subjected to H2 chemisorption using a pulse (10 μl) titration procedure. BET surface area was determined (post-TPR) in 30% v/v N2/He using pure N2 (99.9%) as an internal standard. At least three cycles of N2 adsorption–desorption in the flow mode were conducted to determine the total surface area using the standard single point method. BET area and H2 chemisorption measurements were reproducible to within ±3% and the values quoted represent the mean.
Powder X-ray diffractograms (XRD) were recorded on a Bruker/Siemens D500 incident X-ray diffractometer using Cu Kα radiation. The samples were scanned at 0.02° per step over the range 20° ≤ 2θ ≤ 85° and the diffractograms were identified using JCPDS-ICDD reference standards, i.e. Au (Card No. 04-0784), Pd (05-0681), β-Mo2C (11-0680) and δ-Al2O3 (16-394). Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) measurements were conducted using an SDT Q600 simultaneous TGA/DSC analyser (TA Instruments) that measured temporal mass and heat flow as a function of temperature. Samples were swept with air/N2 (100 cm3 min−1) and ramped (10 K min−1) from ambient temperature to 373 K with an isothermal hold (for 1 h) and subsequent heating to 773 K. Transmission electron microscopy (TEM) was performed using a JEOL 2010 electron microscope operating at 200 kV. Samples for analysis were crushed and homogeneously dispersed in ethanol by ultrasonication and a drop of the suspension was deposited on a carbon-coated copper grid and evaporated. Up to 200 individual Au particles were counted for each catalyst and the mean metal particle sizes are given as the surface area-weighted average (d) according to
| | 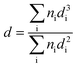 | (1) |
where
ni is the number of particles of diameter
di. XPS spectra were collected on a SPECS (Phoibos MCD 150) X-ray photoelectron spectrometer using an Al Kα (
hν = 1486.6 eV) X-ray source. The binding energies (BE) were calibrated with respect to the C–C/C–H component of the C 1s peak (BE = 284.7 eV). Spectral processing was performed using the Casa XPS software package.
2.4 Catalytic procedure
Reactions were carried out at 493 K and 1 atm, immediately after catalyst activation in H2 (60 cm3 min−1, 2 K min−1 to 523 K, held for 1 h) in a continuous flow fixed bed vertical glass reactor (i.d. = 15 mm, l = 600 mm). The catalytic reactor and operating conditions to ensure negligible heat/mass transport limitations have been fully described elsewhere.45 A layer of borosilicate glass beads served as a preheating zone, ensuring that the organic reactants were vaporised and reached the reaction temperature before coming into contact with the catalyst. Isothermal conditions (±1 K) were ensured by diluting the catalyst bed with ground glass (75 μm). The reaction temperature was continuously monitored with a thermocouple inserted in a thermowell within the catalyst bed. The reactant (nitrobenzene, p-chloronitrobenzene or p-nitrobenzonitrile) in butanol (as the carrier) was delivered via a glass/Teflon air-tight syringe and Teflon line using a microprocessor-controlled infusion pump (Model 100, kd Scientific) at a fixed flow rate. A co-current flow of the organic component and H2 (400 ≤ H2/nitroarene ≤ 2500) was maintained at GHSV = 2 × 104 h−1 with an inlet nitroarene flow (F) in the range 0.06–0.38 mmol h−1; the molar Au (nAu) to F (i.e. nAu/F) spanned the range 2 × 10−3–6 × 10−2 h. In a series of blank tests, passage of each reactant in a stream of H2 through the empty reactor did not result in any detectable conversion. The reactor effluent was frozen in a liquid nitrogen trap for subsequent analysis, which was conducted using a Perkin-Elmer Auto System XL gas chromatograph equipped with a programmed split/splitless injector and flame ionisation detector, employing a DB-1 (50 m × 0.20 mm i.d., 0.33 μm film thickness) capillary column (J&W Scientific). Repeated reactions with different samples from the same batch of catalyst delivered raw data that were reproducible to within ±6% with a carbon balance ≥95%. Taking p-nitrobenzonitrile as an example, fractional hydrogenation (xi) was obtained from| | 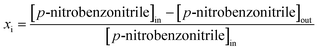 | (2) |
where selectivity (Si) to p-aminobenzonitrile (p-ABN) is given by| | 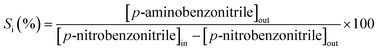 | (3) |
3. Results and discussion
3.1 Catalyst characterisation
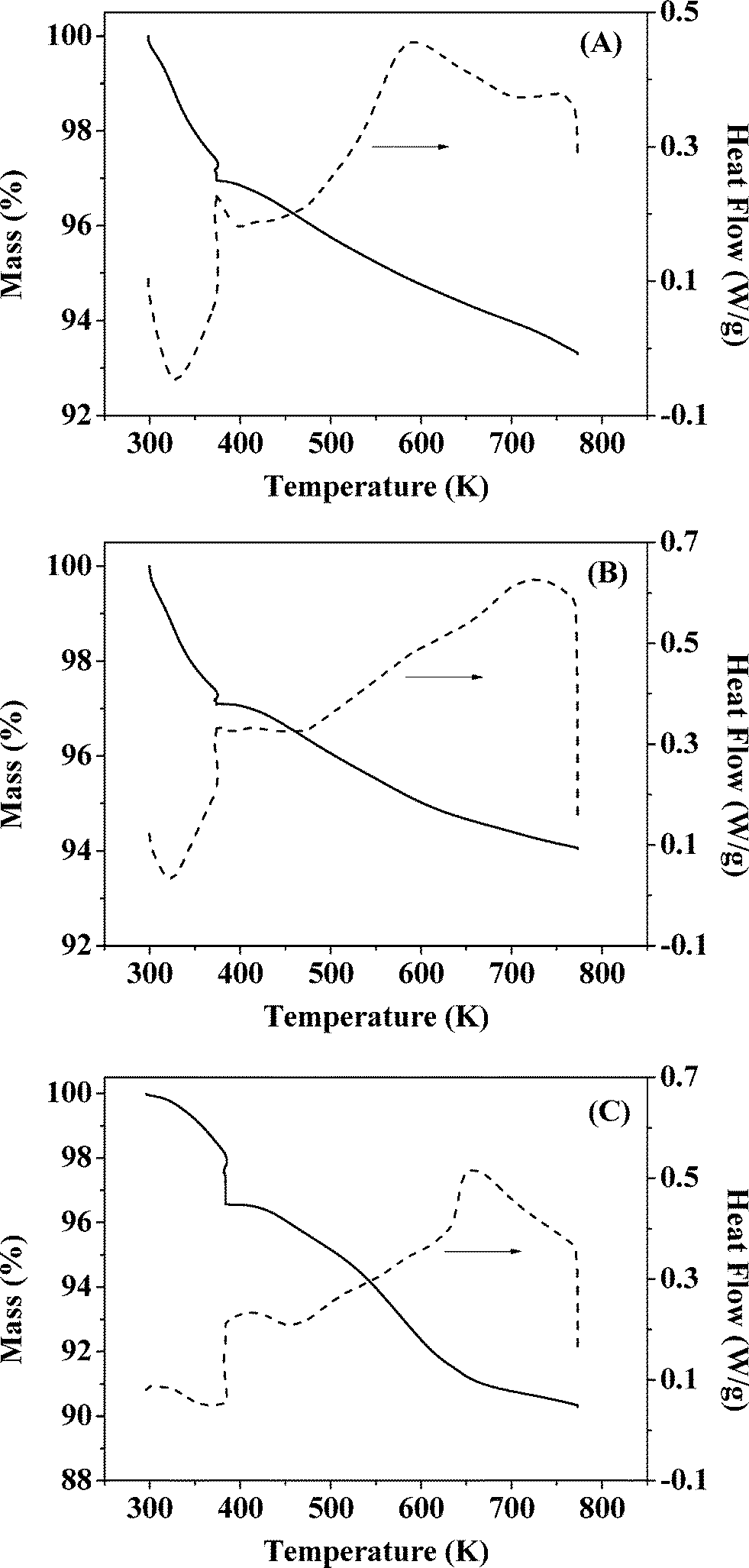
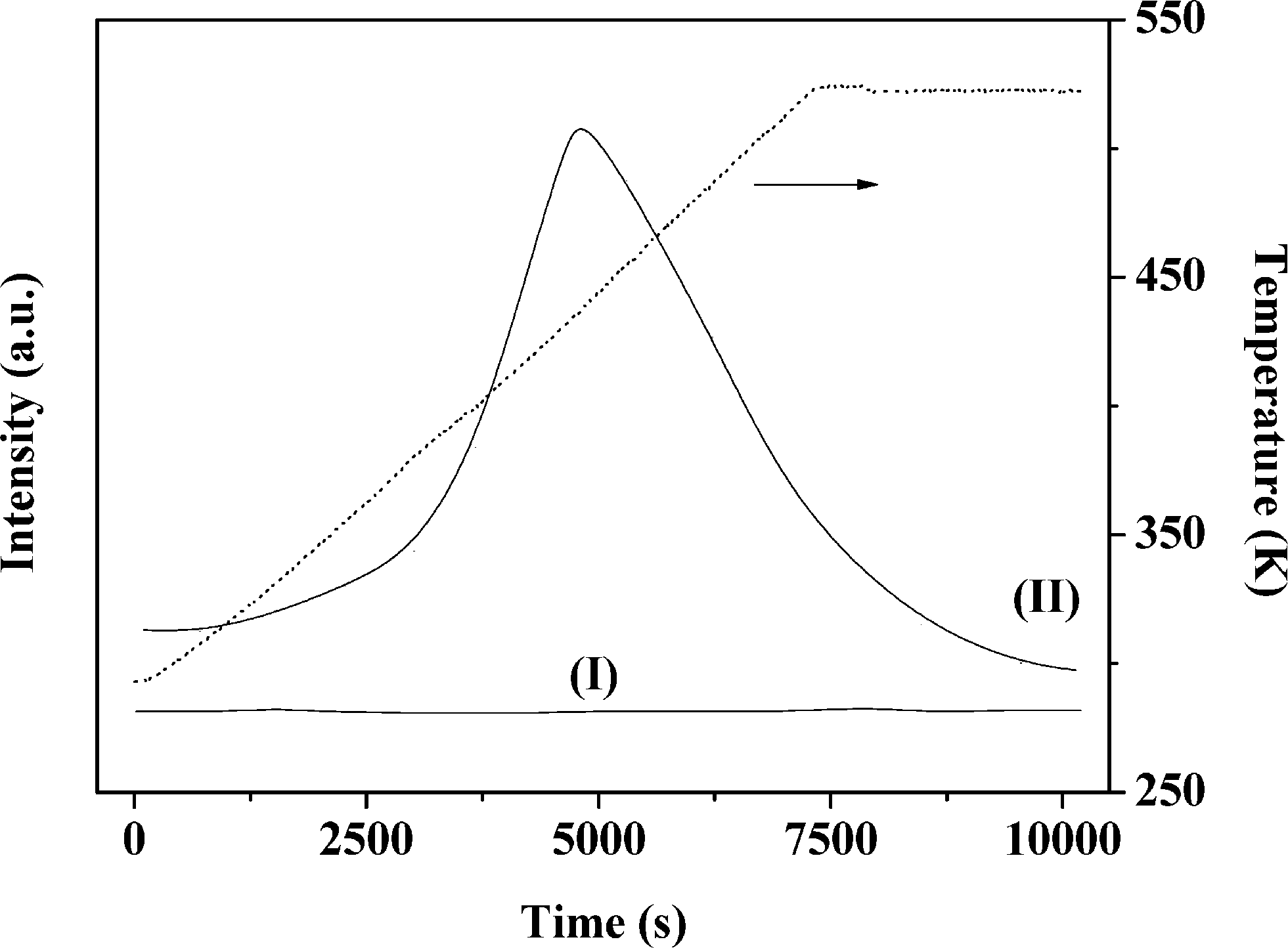
It has been shown that calcination in air (at 773 K) is effective in removing residual PVA following the synthesis of bimetallics on alumina.18 Oxidative treatment is not applicable in the case of Au–Pd/Mo2C as the support can undergo deep oxidation with irreversible structural modification at high temperatures. We examined the feasibility of thermal treatment in N2, taking Au–Pd/Al2O3-10 and Au–Pd/Mo2C-10 as representative samples. TGA-DSC analysis of Au–Pd/Al2O3-10 for treatment in air (A) and N2 (B) resulted in equivalent profiles (Fig. 1). The endothermic response at 328 K (from DSC) is due to the loss of physisorbed water while the exothermic mass loss at higher temperature (T > 400 K) can be attributed to the removal of PVA. The overall change in mass converged (at ca. 6% w/w) in both cases, suggesting equivalence for treatment in air and N2. Thermal treatment of Au–Pd/Mo2C-10 in N2 showed comparable behaviour (Fig. 1(C)) with low and high temperature endothermic and exothermic mass losses. We accordingly applied the same thermal treatment (in N2 to 773 K) for both alumina and carbide samples. Gold loadings were close to the nominal (1% w/w) value with Au/Pd atomic ratios approaching the target 30 and 10 in the bimetallic systems (Table 1). Moreover, the Mo/C ratios were close to 2, confirming the synthesis of a Mo2C phase with no significant contamination by surface free carbon, which can occlude Mo2C active sites and lower the activity.46
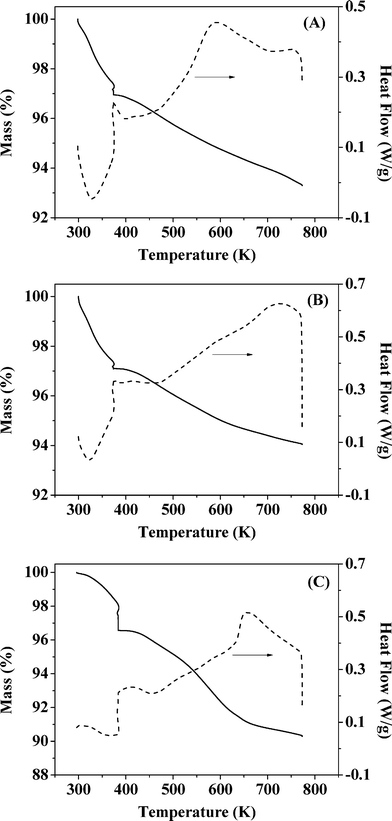 |
| | Fig. 1 TGA (solid line) and DSC (dashed line) profiles generated in the pre-treatment of (A) Au–Pd/Al2O3-10 in air, (B) Au–Pd/Al2O3-10 in N2 and (C) Au–Pd/Mo2C-10 in N2; 100 cm3 min−1 gas flow at 10 K min−1 to 373 K (held for 1 h) and then to 773 K. | |
Table 1 Gold content (% w/w), Au/Pd and Mo/C ratios, BET surface area, temperature-related H2 consumption maximum during TPR (Tmax), metal particle size range and surface area-weighted mean diameter (d), H2 chemisorption and XPS binding energies (BE) for Au 4f7/2, Pd 3d3/2 and Mo 3d5/2
|
|
Au (% w/w) |
Molar ratio |
BET area (m2 g−1) |
TPR Tmax (K) |
Particle size (nm) |
H2 chemisorption (μmol g−1) |
BE XPS (eV) |
| Au/Pd |
Mo/C |
Range |
Mean (d) |
Au 4f7/2 |
Mo 3d5/2 |
|
Pd 3d3/2.
|
| Au/Al2O3 |
0.94 |
— |
— |
104 |
— |
1–8 |
5.4 |
0.4 |
83.0 |
— |
| Au–Pd/Al2O3-30 |
0.97 |
27 |
— |
100 |
— |
1–8 |
4.6 |
0.7 |
— |
— |
| Au–Pd/Al2O3-10 |
0.95 |
9 |
— |
101 |
— |
1–7 |
4.5 |
1.2 |
83.0 |
— |
| Mo2C |
— |
— |
1.95 |
57 |
500 |
1–7 |
— |
0.4 |
— |
228.4 |
| Au/Mo2C |
0.81 |
— |
1.95 |
42 |
493 |
1–7 |
4.3 |
1.0 |
84.2 |
228.4 |
| Au–Pd/Mo2C-30 |
0.87 |
32 |
2.00 |
38 |
476 |
1–7 |
4.2 |
1.5 |
— |
— |
| Au–Pd/Mo2C-10 |
0.84 |
14 |
1.96 |
34 |
437 |
1–7 |
4.3 |
2.1 |
84.2 (340.8a) |
228.4 |
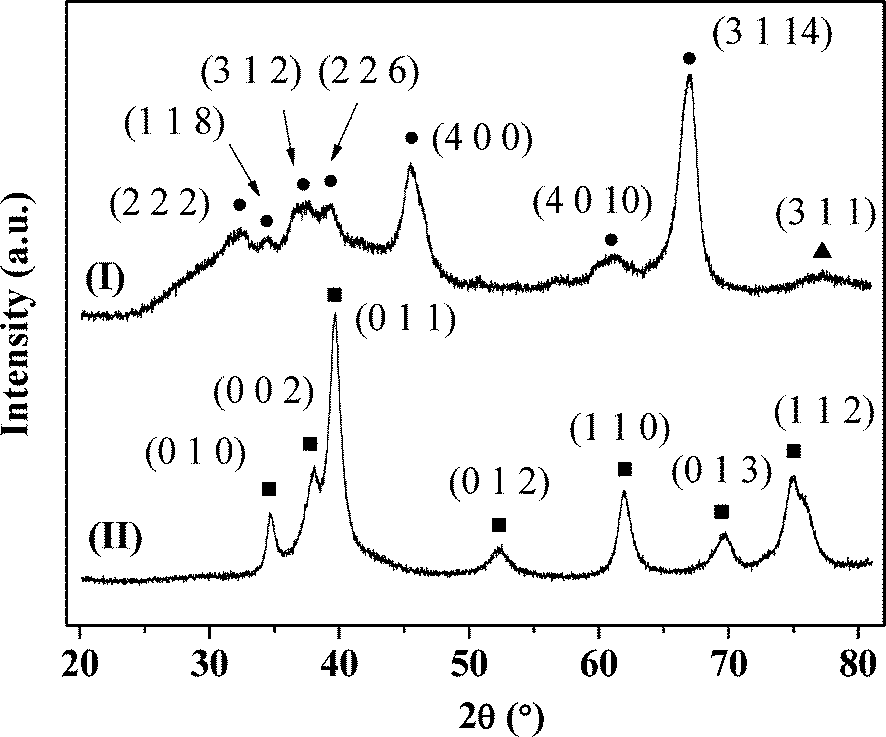
The temperature-programmed reduction (TPR) profiles of Au–Pd/Al2O3-10 and Au–Pd/Mo2C-10 are presented in Fig. 2. As the metal component was reduced by NaBH4 during preparation, there was no detectable TPR response for Au–Pd/Al2O3-10 (Fig. 2(I)). The TPR signal recorded at 437 K for Au–Pd/Mo2C-10 (II) can be ascribed to the removal of the passivation layer from the carbide support. This is facilitated by the Pd component with a lower Tmax at lower Au/Pd (Table 1). The BET surface area of the carbide systems showed a decrease from 57 to 34 m2 g−1 with increasing metal loading that was not evident for the Al2O3 samples. The XRD profile of the latter (taking Au–Pd/Al2O3-10 as a representative (Fig. 3(I)) presented peaks characteristic of δ-Al2O3.47 The signal at 2θ = 77.5° can be assigned to Au (311) but the intensity is too weak to allow any meaningful determination of Au particle size from standard line broadening. The XRD pattern of Au–Pd/Mo2C-10 (Fig. 3(II)) is consistent with that of hexagonal β-Mo2C, showing characteristic peaks due to (010), (002), (011), (012), (110), (013) and (112) planes. There was no detectable signal for bulk oxide (MoO3 or MoO2), indicating that the precursor had been fully converted to carbide and that passivation resulted in superficial (rather than bulk) oxidation. Gold detection is hampered by overlap with XRD peaks due to the carbide support but the absence of an Au (200) peak at 44.5° suggests a well-dispersed Au phase. There was no detectable diffraction peak due to Pd (notably at 40.1° (111)) in the Au–Pd samples but the Pd loading was too low for detection. The formation of bimetallic nanoparticles has been established in an earlier study using IR spectroscopy coupled with CO adsorption for Au–Pd/Al2O3 prepared by the same method.18 Villa et al. have proposed the generation of bimetallic Au–Pd particles on activated carbon when PVA is used as a protecting agent.48 The presence of isolated Pd atoms surrounded by Au atoms in bimetallic Au–Pd particles is a critical feature of various catalytic reactions using gold–palladium catalysts (e.g. hydrogenation18 and oxidation44). The IR spectra (not shown) obtained for the Au–Pd/Al2O3 samples are identical to the ones reported in a previous study.18 However, it was not possible to characterise the Au–Pd/Mo2C systems using the same technique because of strong absorption by the dark Mo2C material. At this juncture, we assume that the results obtained for the alumina-supported catalysts are applicable to the Mo2C systems.
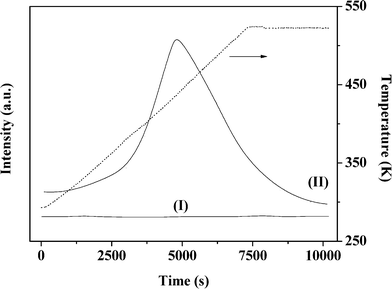 |
| | Fig. 2 Temperature-programmed reduction (TPR) profiles of Au–Pd/Al2O3-10 (I) and Au–Pd/Mo2C-10 (II). | |
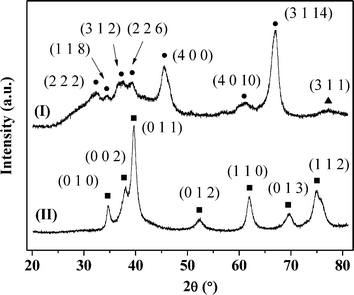 |
| | Fig. 3 XRD patterns of Au–Pd/Al2O3-10 (I) and Au–Pd/Mo2C-10 (II). Peak assignments are based on JCPDS-ICDD reference standards: δ-Al2O3 (●, 16-394); β-Mo2C (■, 11-0680); Au (▲, 04-0784). | |
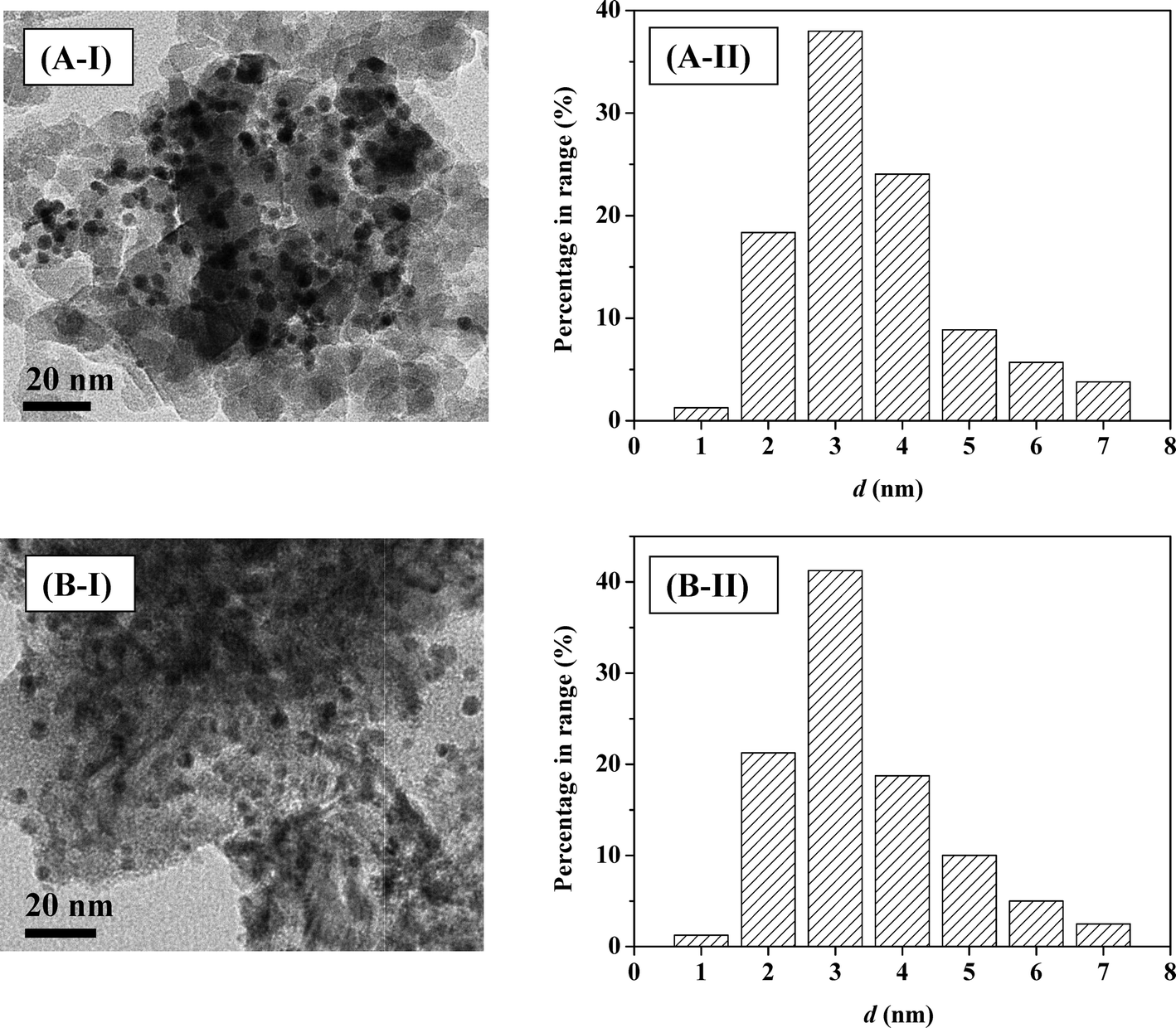
Gold particle size is important in hydrogenation applications49 where particles <5 nm are intrinsically more active in the gas phase hydrogenation of p-chloronitrobenzene and m-dinitrobenzene32 and butadiene.50 Representative TEM images (of Au–Pd/Al2O3-10 (A) and Au–Pd/Mo2C-10 (B)) are presented in Fig. 4(I), where well-dispersed pseudo-spherical particles in the 1–8 nm size range (Fig. 4(II)) can be observed. An essentially equivalent mean metal size (4–5 nm) was obtained for all the Al2O3- and Mo2C-supported systems. Previous Au/Mo2C synthesis by deposition–precipitation (with urea) generated an appreciably wider size range (4–20 nm) and larger mean size (13.4 nm).34 Application of PVA as a stabilising agent was effective in generating homogeneously dispersed nanoparticles. Hydrogen dissociation on supported Au has a higher activation energy barrier than that on conventional hydrogenation metals (Pd, Pt and Ni)49 and is dependent on Au coordination where dissociation is facilitated at edge and corner sites associated with smaller Au particles (<10 nm).33 Ambient temperature H2 chemisorption on Au/Al2O3 was low and is in agreement with previous reports.28,51 Inclusion of Pd resulted in a significant increase in H2 uptake that was enhanced at lower Au/Pd ratios as observed for catalysts prepared by deposition–precipitation.34 The higher H2 chemisorption capacity exhibited by the carbide systems cannot be attributed to metal particle size and must be due to additional chemisorption on the Mo2C support (Table 1).
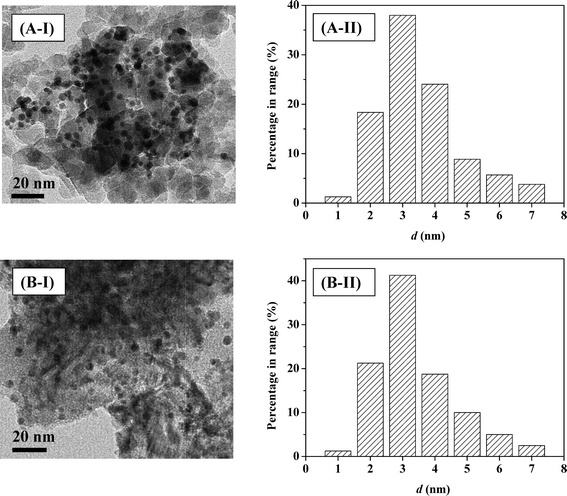 |
| | Fig. 4 Representative TEM images (I) with associated particle size distribution (II) for Au–Pd/Al2O3-10 (A) and Au–Pd/Mo2C-10 (B). | |
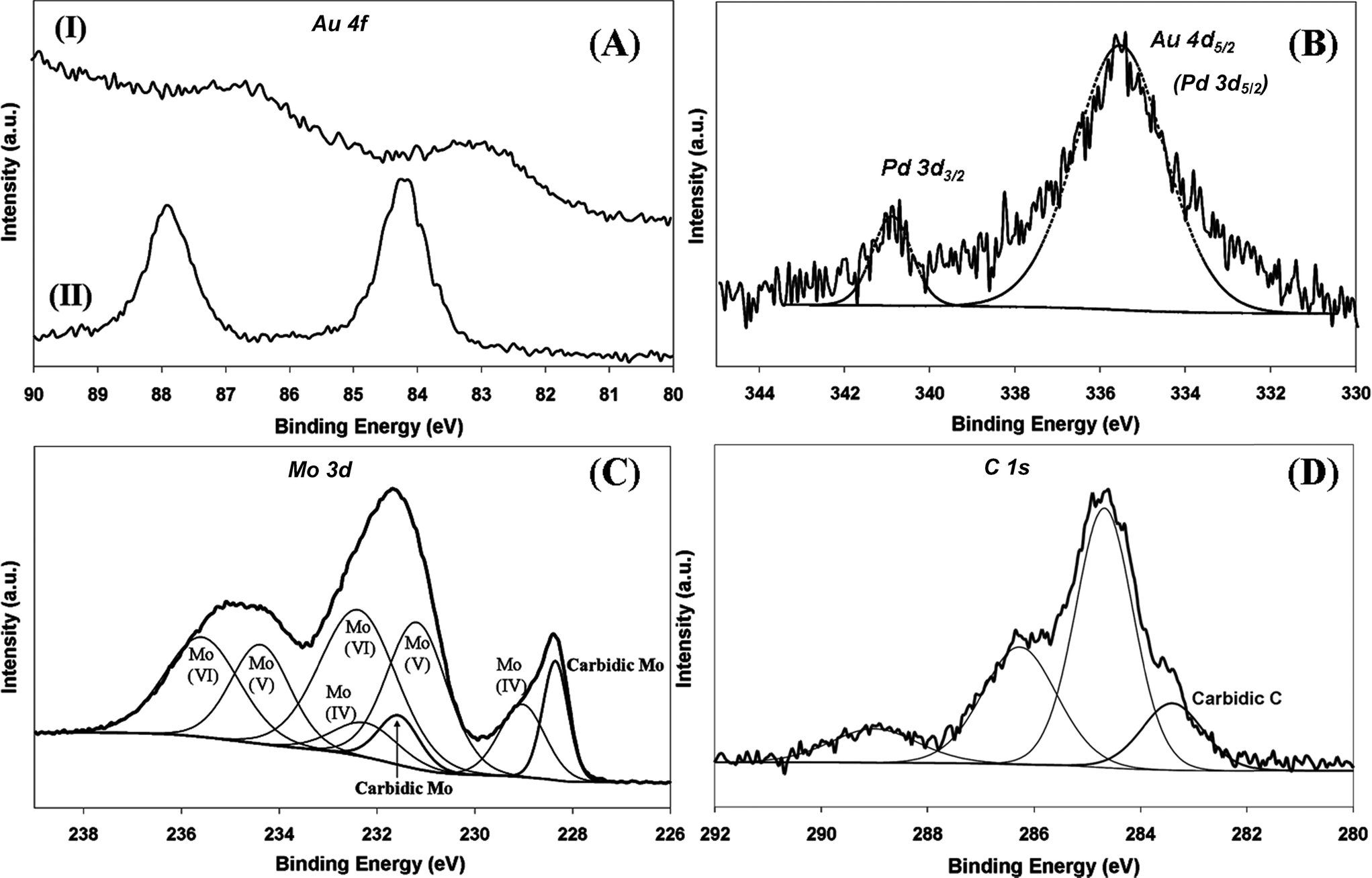
The electronic structure of the supported metal can have an impact on performance in the hydrogenation of nitroarenes28,52 as –NO2 adsorption/activation is favoured on electron-deficient sites.28,53 Electronic character was probed by XPS analysis and the resultant spectra are presented in Fig. 5. The Au 4f binding energy (BE) showed a dependence on the support but was unaffected by Pd inclusion (Table 1) where the Au 4f7/2 BE for the Al2O3-supported samples (83.0 eV) was appreciably lower than that (84.2 eV) recorded for the carbide systems (Fig. 5(A)). Previous studies reported that such a shift of the Au 4f7/2 BE to lower energy can be related to reduced screening of core holes in an assembly of low-coordinated surface Au atoms54 or to a reduced coordination number of Au atoms associated with the degree of rounding of Au nanoparticles, which is dependent on metal–support interactions.55 The equivalent particle size obtained for Au/Al2O3 and Au/Mo2C suggests that such effects alone cannot account for the variation of Au 4f7/2 BE. Taking the reference BE for metallic Au as 84.0 eV,56 the XPS results could also suggest electron transfer from support surface defects to nano-scale Au particles.57 Previous XPS studies of TiO2- and Al2O3-supported catalysts have indicated metal–support interactions that result in electron transfer from the oxide to the supported Au particles.58 In contrast, the Au 4f7/2 BE recorded for the carbide systems coincides with that of the metallic reference, suggesting negligible electron transfer. Care must be taken regarding such interpretation as charging effects in XPS measurements can differ significantly between insulating (Al2O3) and highly conductive (Mo2C) supports. We have observed a Pd 3d3/2 signal at 340.8 eV for Au–Pd/Mo2C-10 (Table 1 and Fig. 5(B)), which is close to the BE reported for metallic Pd 3d3/2 (at 340.5 ± 0.1 eV).28,53 A similar response (Pd 3d3/2 at 340.9 eV) has been recorded for Pd–Mo2C/graphitic carbon.59 The second signal at 335.7 eV (Fig. 5(B)) can be attributed to Au 4d5/2 but it should be noted that Au 4d5/2 and Pd 3d5/2 XPS signals can overlap, as reported elsewhere.60 The very low intensity of the XPS signal in the Pd 3d region in the case of Au–Pd/Al2O3-10 (not shown) precluded detection of a Pd 3d3/2 contribution. The XPS spectrum over the Mo 3d region (Fig. 5(C)) shows Mo 3d5/2 peaks at 228.4 and 231.6 eV consistent with a Mo2C phase.59,61 In addition to the carbidic Mo contribution, Mo 3d5/2 signals at higher BE can be attributed to oxidised states of Mo arising from passivation, i.e. Mo(IV) (229.0 eV),62 Mo(V) (231.2 eV)63 and Mo(VI) (232.4 eV).64 Deconvolution of the C 1s profile (Fig. 5(D)) provides evidence of carbidic (283.4 eV)65 and graphitic (284.6 eV) carbon59 with contributions (at 286.3 and 288.6 eV) due to C–O (ref. 62) and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O.66
O.66
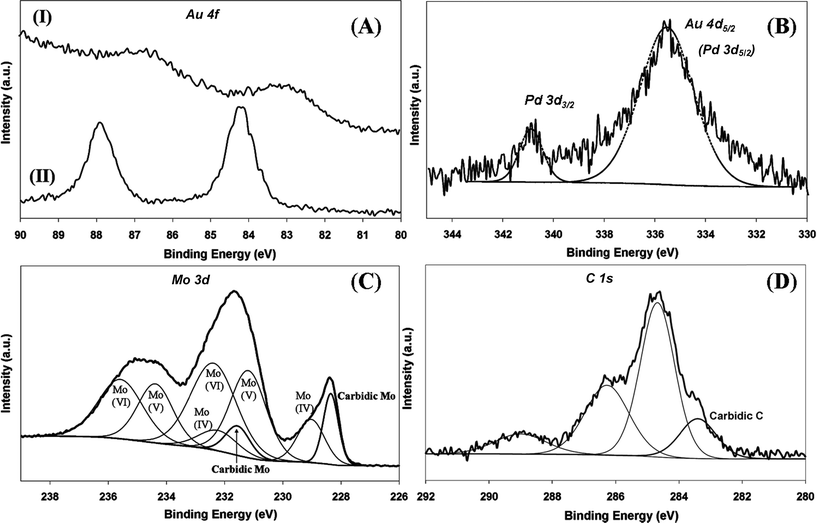 |
| | Fig. 5 XPS spectra: (A) over the Au 4f region for Au–Pd/Al2O3-10 (I) and Au–Pd/Mo2C-10 (II); (B) over the Au 4d and Pd 3d regions for Au–Pd/Mo2C-10; (C) with peak deconvolution over the Mo 3d region for Au–Pd/Mo2C-10; (D) with peak deconvolution over the C 1s region for Au–Pd/Mo2C-10. | |
3.2 Catalytic response
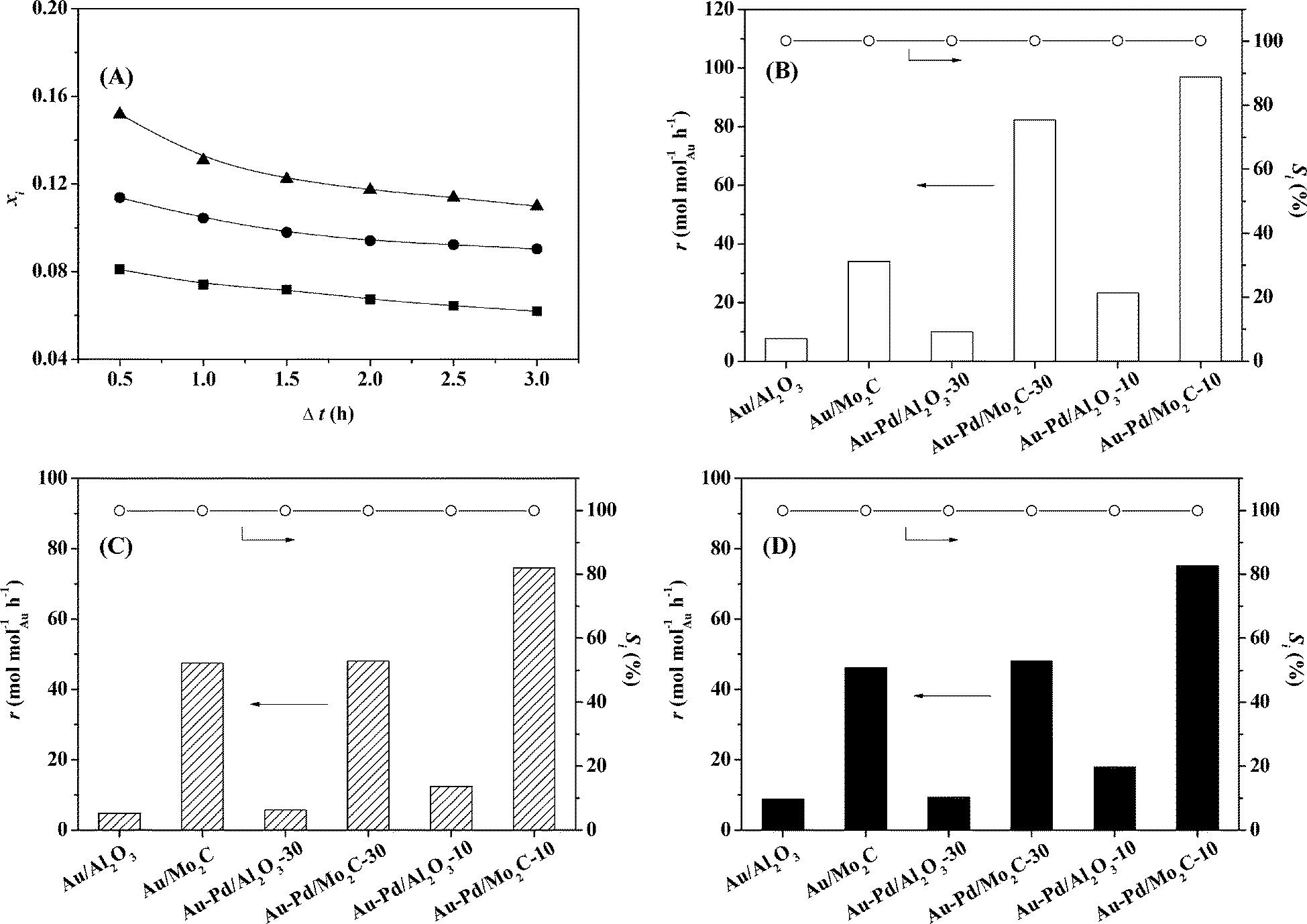
This is the first reported application of a Mo2C-supported bimetallic (Au–Pd) catalyst in (gas phase) hydrogenation where we examine nitroarene (nitrobenzene, p-chloronitrobenzene and p-nitrobenzonitrile) conversion, carrying Au/Al2O3 and Au–Pd/Al2O3 through as benchmarks. The target amine products are commercially important in the production of a diversity of agrochemicals, pharmaceuticals and fine chemicals.67,68 Initial conversion (representative time-on-stream plots shown in Fig. 6(A)) was used to obtain reaction rates (normalised with respect to molar Au loading) using the approach described previously.69 Reduction of nitrobenzene generated aniline where Au/Mo2C outperformed Au/Al2O3 and delivered a greater than three-fold higher rate (Fig. 6(B)). This can be ascribed, at least in part, to the greater amount of available surface hydrogen determined in pulse chemisorption measurements (Table 1) as H2 activation is the rate-limiting step in hydrogenation over Au. In previous work,32 we established Au particle size effects in –NO2 hydrogenation with an increase in intrinsic activity with decreasing particle size (from 10 to 3 nm). This should not be a contributory effect here given the equivalency of size distribution and mean. Alumina (without a metal additive) is inactive in hydrogenation, whereas Mo2C alone can promote H2 activation with –NO2 reduction34 and contribute directly to the higher reaction rate. Incorporation of Pd with Au resulted in increased H2 chemisorption capacity and a consequent enhanced activity with the highest values recorded for Au/Pd = 10.
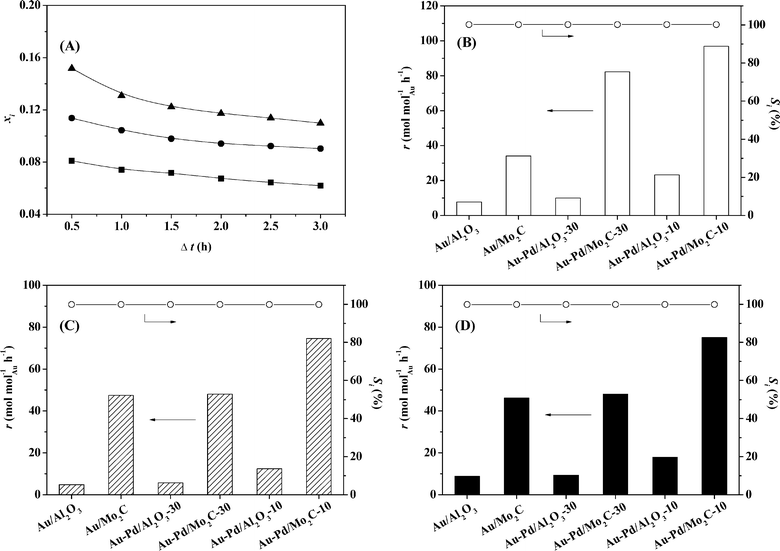 |
| | Fig. 6 (A) Variation of NB fractional conversion (xi) over Au/Mo2C (■), of p-CNB over Au–Pd/Mo2C-30 (●) and of p-NBN over Au–Pd/Mo2C-10 (▲) with time-on-stream. Reaction rate (r) with the associated selectivity (Si) to the target amines (aniline, p-chloroaniline and p-aminobenzonitrile) in the hydrogenation of (B) nitrobenzene, (C) p-chloronitrobenzene and (D) p-nitrobenzonitrile. | |
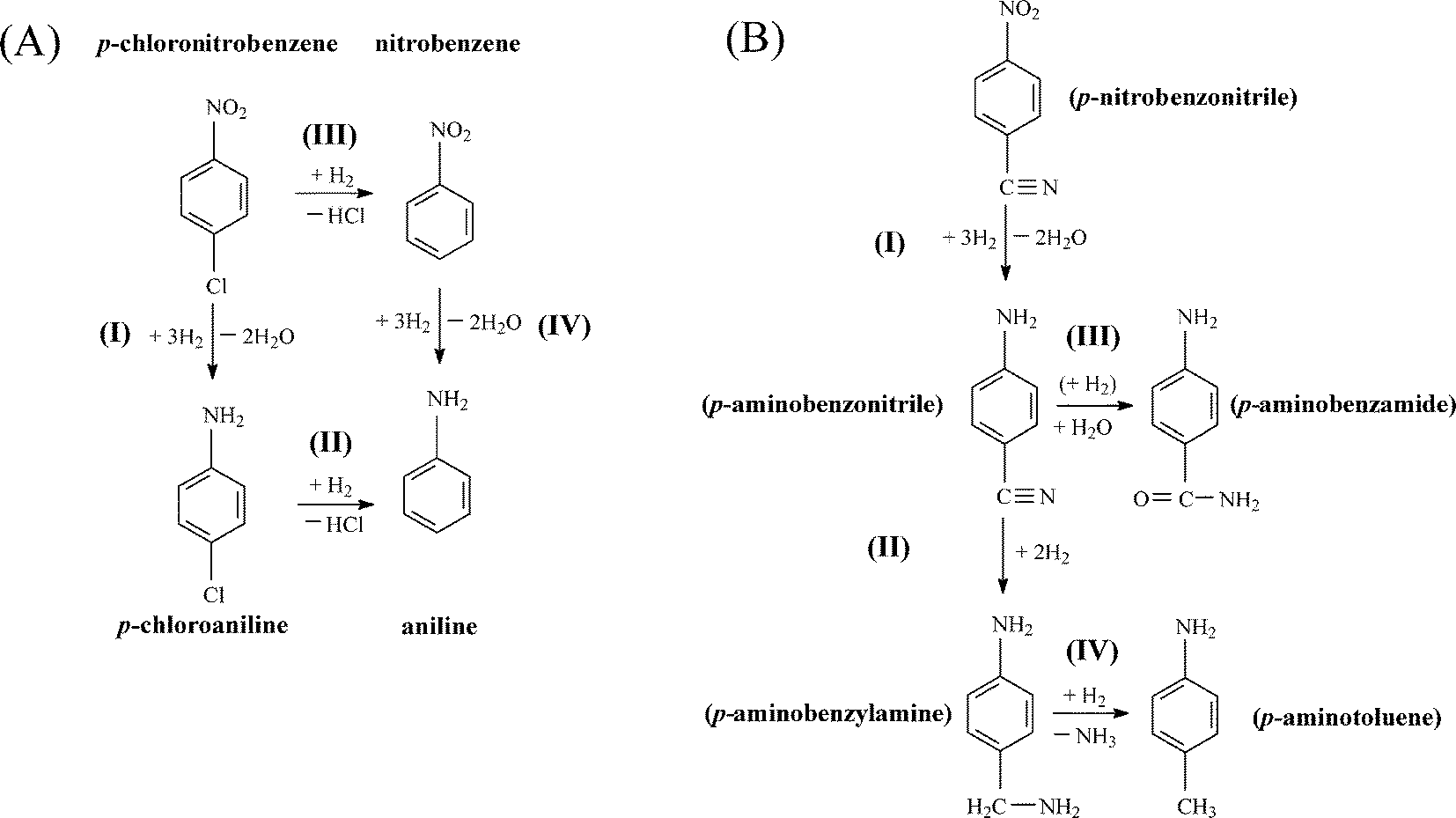
Chemoselectivity in amine production is particularly relevant in the hydrogenation of p-chloronitrobenzene70 where a range of intermediates and by-products, notably nitrobenzene and aniline, have been reported for reaction over Pd and Ni catalysts in both liquid and gas phases.28,53,71,72 A simplified reaction pathway is presented in Fig. 7(A), where the target p-chloroaniline formed via –NO2 reduction (step I) can undergo hydrodechlorination to aniline (step II) or alternatively nitrobenzene is first generated by hydrodechlorination (step III) with subsequent hydrogenation to aniline (step IV). In previous studies, reaction over Pd/Al2O3 generated nitrobenzene and aniline,28 whereas supported Au was fully selective to chloroaniline.25,27,28,34 The chemoselectivity achieved with Au catalysts can be attributed to the preferential adsorption of the reactants through the nitro group at the support (notably TiO2) and metal/support interface, where the Au (metal) sites provide reactive dissociated H2.73,74 In this study, hydrogenation of p-chloronitrobenzene resulted in exclusive p-chloroaniline formation with 100% yield over Au–Pd/Mo2C-10 and Au–Pd/Mo2C-30, i.e. full selectivity to p-chloroaniline at 100% conversion of the inlet p-chloronitrobenzene feed. This result is of importance as we previously observed that the incorporation of Pd on Au/Al2O3 (Au/Pd = 8) favoured hydrodechlorination with formation of nitrobenzene.3 Performance must be influenced by catalyst synthesis where the colloidal method with PVA used here has been shown to generate surface isolated Pd atoms (Pd monomers) surrounded by Au atoms in bimetallic particles.18 This effect appears to extend to the carbide system. In contrast, preparation by deposition–precipitation results in segregated Pd sites that promote hydrodechlorination at low Au/Pd ratios.3 This is consistent with the observation of Gao et al.7 that enhanced selectivity/activity (in H2O2/vinyl acetate synthesis) was attributed to an “ensemble effect” resulting from dilution of surface Pd by Au which was dependent on Au content. The Mo2C-supported catalysts again generated appreciably higher rates than the equivalent alumina-supported samples (Fig. 6(C)) with increased activity resulting from the inclusion of Pd.
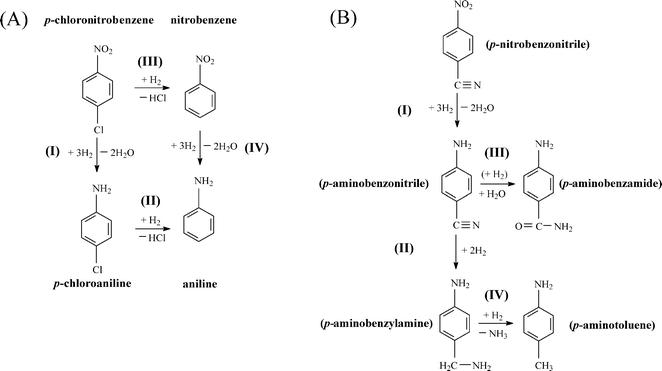 |
| | Fig. 7 Reaction pathways for (A) p-chloronitrobenzene and (B) p-nitrobenzonitrile hydrogenation. | |
Catalytic performance was further assessed in the gas phase hydrogenation of p-nitrobenzonitrile. The limited available literature67,75–77 refers to batch liquid phase reactions at elevated pressures (up to 6 MPa) where high selectivity to p-aminobenzonitrile has proved challenging. Koprivova and Cerveny76 identified 15 possible intermediates and products that involve disproportionation, reduction, condensation, hydrodenitrogenation and hydrodecarbonation reactions. A reaction pathway that includes the major reported products76,77 is presented in Fig. 7(B). Conversion of p-nitrobenzonitrile to the target p-aminobenzonitrile (step I) can be accompanied by further hydrogenation/hydration to p-aminobenzamide (step III) or hydrogenation to p-aminobenzylamine (step II)76 with subsequent hydrogenolysis to p-aminotoluene (step IV).76,77 In recent work, we have established that oxide-supported Pd and Ni are nonselective, generating p-aminotoluene as a by-product.78 Each catalyst in this study was fully selective to p-aminobenzonitrile with 100% yield over Au–Pd/Mo2C-10 and Au-Pd/Mo2C-30, exceeding the highest reported values for supported Au (96–97% over Au/TiO267 and Au/Fe2O375) operated in batch liquid phase at elevated pressure (25 bar H2). Economies of scale favour continuous processes for high throughput.25 Palladium again served as a promoter with higher rates recorded for the carbide catalysts (Fig. 6(D)). The only reported gas phase catalysis study dates back from the 1950s where Hata and Watanabe77 investigated the hydrogenation of various aromatic nitriles (including the three nitrobenzonitrile isomers) over a Ni–Cu catalyst at 523–573 K and reported aniline, toluidine and aminobenzonitrile as principal products (<32% yield). Our results represent a significant advance in the sustainable production of a target amine where reaction exclusivity at full reactant conversion removes the requirement for energy-intensive down-stream separation/purification unit operations.
4. Conclusions
We provide the first reported synthesis of Mo2C-supported (Au–Pd) bimetallic catalysts used to promote the gas phase hydrogenation of nitroarenes (nitrobenzene, p-chloronitrobenzene and p-nitrobenzonitrile). The Mo2C support was synthesised by temperature-programmed carburisation (of H2MoO4 in CH4/H2) and confirmed by XRD analysis. Bimetallic catalyst synthesis with Au and Pd colloids employed polyvinyl alcohol (PVA) as a stabiliser to avoid agglomeration and resulted in nano-scale (1–8 nm size range) supported metal particles with a mean size of 4–5 nm. The bimetallic catalysts exhibited increased H2 chemisorption capacity with higher uptake on Au–Pd/Mo2C relative to Au–Pd/Al2O3. XPS analysis suggests electron donation from Al2O3 to Au, whereas the BE recorded for Au on Mo2C (for both monometallic and Au–Pd bimetallic systems) was close to that of the metallic Au reference. All the catalysts were 100% selective in –NO2 hydrogenation to the target amine. The Mo2C-supported Au system exhibited a higher (greater than 3-fold) hydrogenation rate that was further enhanced by addition of Pd (Au/Pd from 30 to 10), which can be attributed to an increase in available surface reactive hydrogen. We achieved 100% amine yield over Au–Pd/Mo2C-10 and Au–Pd/Mo2C-30 in continuous operation. The results have established controlled preparation of well-dispersed Au–Pd on Mo2C and application in the clean continuous production of high value amines.
Acknowledgements
Financial support to Dr. X. Wang through the Overseas Research Students Award Scheme (ORSAS) is acknowledged. Dr. N. Perret also acknowledges financial support from COST Action MP0903 Nanoalloys.
References
- F. Tao, Chem. Soc. Rev., 2012, 41, 7977–7979 RSC.
- R. W. J. Scott, O. M. Wilson, S.-K. Oh, E. A. Kenik and R. M. Crooks, J. Am. Chem. Soc., 2004, 126, 15583–15591 CrossRef CAS PubMed.
- F. Cárdenas-Lizana, S. Gómez-Quero, A. Hugon, L. Delannoy, C. Louis and M. A. Keane, J. Catal., 2009, 262, 235–243 CrossRef.
- S. Chandra Shekar, J. Krishna Murthy, P. Kanta Rao, K. S. Rama Rao and E. Kemnitz, Appl. Catal., A, 2003, 244, 39–48 CrossRef CAS.
- F. Menegazzo, P. Canton, F. Pinna and N. Pernicone, Catal. Commun., 2008, 9, 2353–2356 CrossRef CAS.
- V. Dal Santo, A. Gallo, A. Naldoni, M. Guidotti and R. Psaro, Catal. Today, 2012, 197, 190–205 CrossRef CAS.
- F. Gao and D. W. Goodman, Chem. Soc. Rev., 2012, 41, 8009–8020 RSC.
- P. Gao, F. Li, F. Xiao, N. Zhao, N. Sun, W. Wei, L. Zhong and Y. Sun, Catal. Sci. Technol., 2012, 2, 1447–1454 CAS.
- D.-W. Lee, M. S. Lee, J. Y. Lee, S. Kim, H.-J. Eom, D. J. Moon and K.-Y. Lee, Catal. Today, 2013, 210, 2–9 CrossRef CAS.
- F. Menegazzo, M. Signoretto, M. Manzoli, F. Boccuzzi, G. Cruciani, F. Pinna and G. Strukul, J. Catal., 2009, 268, 122–130 CrossRef CAS.
- X. Wei, X.-F. Yang, A.-Q. Wang, L. Li, X.-Y. Liu, T. Zhang, C.-Y. Mou and J. Li, J. Phys. Chem. C, 2010, 116, 6222–6232 Search PubMed.
- M. L. Guzmán-Castillo, E. López-Salinas, J. J. Fripiat, J. Sánchez-Valente, F. Hernández-Beltrán, A. Rodríguez-Hernández and J. Navarrete-Bolaños, J. Catal., 2003, 220, 317–325 CrossRef.
- M. O. Nutt, K. N. Heck, P. Alvarez and M. S. Wong, Appl. Catal., B, 2006, 69, 115–125 CrossRef CAS.
- C. L. Bianchi, P. Canton, N. Dimitratos, F. Porta and L. Prati, Catal. Today, 2005, 102-103, 203–212 CrossRef CAS.
- F. Gao, Y. Wang and D. W. Goodman, J. Am. Chem. Soc., 2009, 131, 5734–5735 CrossRef CAS PubMed.
- A. Villa, N. Janjic, P. Spontoni, D. Wang, D. S. Su and L. Prati, Appl. Catal., A, 2009, 364, 221–228 CrossRef CAS.
- A. Hugon, L. Delannoy, J.-M. Krafft and C. Louis, J. Phys. Chem. C, 2010, 114, 10823–10835 CAS.
- N. E. Kolli, L. Delannoy and C. Louis, J. Catal., 2013, 297, 79–92 CrossRef CAS.
- B. Pawelec, A. M. Venezia, V. La Parola, E. Cano-Serrano, J. M. Campos-Martin and J. L. G. Fierro, Appl. Surf. Sci., 2005, 242, 380–391 CrossRef CAS.
- A. M. Venezia, V. L. Parola, B. Pawelec and J. L. G. Fierro, Appl. Catal., A, 2004, 264, 43–51 CrossRef CAS.
- A. Sárkány, O. Geszti and G. Sáfrán, Appl. Catal., A, 2008, 350, 157–163 CrossRef.
- X. Yang, D. Chen, S. Liao, H. Song, Y. Li, Z. Fu and Y. Su, J. Catal., 2012, 291, 36–43 CrossRef CAS.
- P. Weinberger, L. Szunyogh and B. I. Bennett, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 10154–10157 CrossRef CAS.
- A. Groß, Top. Catal., 2006, 37, 29–39 CrossRef.
- X. Wang, F. Cárdenas-Lizana and M. A. Keane, ACS Sustainable Chem. Eng., 2014, 2, 2781–2789 CrossRef CAS.
- X. Wang, N. Perret and M. A. Keane, Appl. Catal., A, 2013, 467, 575–584 CrossRef CAS.
- X. Wang, N. Perret, J. J. Delgado, G. Blanco, X. Chen, C. M. Olmos, S. Bernal and M. A. Keane, J. Phys. Chem. C, 2013, 117, 994–1005 CAS.
- X. Wang, N. Perret and M. A. Keane, Chem. Eng. J., 2012, 210, 103–113 CrossRef CAS.
- H. Liu, K. Tao, C. Xiong and S. Zhou, Catal. Sci. Technol., 2015, 5, 405–414 CAS.
- M. Pietrowski and M. Wojciechowska, Catal. Today, 2009, 142, 211–214 CrossRef CAS.
- N. Perret, F. Cárdenas-Lizana and M. A. Keane, Catal. Commun., 2011, 16, 159–164 CrossRef CAS.
-
X. Wang, M. Li and M. A. Keane, in Heterogeneous Gold Catalysts and Catalysis, ed. Z. Ma and S. Dai, The Royal Society of Chemistry, Cambridge, 2014, pp. 424–461 Search PubMed.
- E. Bus, J. T. Miller and J. A. van Bokhoven, J. Phys. Chem. B, 2005, 109, 14581–14587 CrossRef CAS PubMed.
- N. Perret, X. Wang, L. Delannoy, C. Potvin, C. Louis and M. A. Keane, J. Catal., 2012, 286, 172–183 CrossRef CAS.
- D. Wang and Y. Li, Adv. Mater., 2011, 23, 1044–1060 CrossRef CAS PubMed.
- J. K. Edwards, A. Thomas, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Green Chem., 2008, 10, 388–394 RSC.
- J. K. Edwards, B. Solsona, P. Landon, A. F. Carley, A. Herzing, M. Watanabe, C. J. Kiely and G. J. Hutchings, J. Mater. Chem., 2005, 15, 4595–4600 RSC.
- J. K. Edwards, B. E. Solsona, P. Landon, A. F. Carley, A. Herzing, C. J. Kiely and G. J. Hutchings, J. Catal., 2005, 236, 69–79 CrossRef CAS.
- J. Xu, T. White, P. Li, C. He, J. Yu, W. Yuan and Y.-F. Han, J. Am. Chem. Soc., 2010, 132, 10398–10406 CrossRef CAS PubMed.
- P. Zhao, N. Li and D. Astruc, Coord. Chem. Rev., 2013, 257, 638–665 CrossRef CAS.
- S. Yongprapat, A. Therdthianwong and S. Therdthianwong, J. Appl. Electrochem., 2012, 42, 483–490 CrossRef CAS.
- J. M. Nadgeri, M. M. Telkar and C. V. Rode, Catal. Commun., 2008, 9, 441–446 CrossRef CAS.
- Y. S. Kwon, A. A. Gromov, A. P. Ilyin, A. A. Ditts, J. S. Kim, S. H. Park and M. H. Hong, Int. J. Refract. Hard Met., 2004, 22, 235–241 CrossRef CAS.
- D. Wang, A. Villa, F. Porta, L. Prati and D. Su, J. Phys. Chem. C, 2008, 112, 8617–8622 CAS.
- F. Cárdenas-Lizana, X. Wang, D. Lamey, M. Li, M. A. Keane and L. Kiwi-Minsker, Chem. Eng. J., 2014, 255, 695–704 CrossRef.
- J.-S. Choi, G. Bugli and G. Djéga-Mariadassou, J. Catal., 2000, 193, 238–247 CrossRef CAS.
- I. Pettiti, S. Colonna, S. De Rossi, M. Faticanti, G. Minelli and P. Porta, Phys. Chem. Chem. Phys., 2004, 6, 1350–1358 RSC.
- A. Villa, D. Wang, D. Su, G. M. Veith and L. Prati, Phys. Chem. Chem. Phys., 2010, 12, 2183–2189 RSC.
-
G. C. Bond, C. Louis and D. T. Thompson, Catalysis by Gold, Imperial College Press, London, 2006 Search PubMed.
- A. Hugon, L. Delannoy and C. Louis, Gold Bull., 2008, 41, 127–138 CrossRef CAS.
- P. Claus, Appl. Catal., A, 2005, 291, 222–229 CrossRef CAS.
- S. A. Nikolaev and V. Smirnov, Gold Bull., 2009, 42, 182–189 CrossRef CAS.
- F. Cardenas-Lizana, Y. Hao, M. Crespo-Quesada, I. Yuranov, X. Wang, M. A. Keane and L. Kiwi-Minsker, ACS Catal., 2013, 3, 1386–1394 CrossRef CAS.
- A. Zwijnenburg, A. Goossens, W. G. Sloof, M. W. J. Craje, A. M. van der Kraan, L. J. de Jongh, M. Makkee and J. A. Moulijn, J. Phys. Chem. B, 2002, 106, 9853–9862 CrossRef CAS.
- J. Radnik, C. Mohr and P. Claus, Phys. Chem. Chem. Phys., 2003, 5, 172–177 RSC.
- M. Baron, O. Bondarchuk, D. Stacchiola, S. Shaikhutdinov and H. J. Freund, J. Phys. Chem. C, 2009, 113, 6042–6049 CAS.
- Z. Jiang, W. Zhang, L. Jin, X. Yang, F. Xu, J. Zhu and W. Huang, J. Phys. Chem. C, 2007, 111, 12434–12439 CAS.
- S. Arrii, F. Morfin, A. J. Renouprez and J. L. Rousset, J. Am. Chem. Soc., 2004, 126, 1199–1205 CrossRef CAS PubMed.
- R. Wang, J. Yang, K. Shi, B. Wang, L. Wang, G. Tian, B. Bateer, C. Tian, P. Shen and H. Fu, RSC Adv., 2013, 3, 4771–4777 RSC.
- C. Yang, A. K. Manocchi, B. Lee and H. Yi, J. Mater. Chem., 2011, 21, 187–194 RSC.
- R. Barthos, A. Szechenyi and F. Solymosi, Catal. Lett., 2008, 120, 161–165 CrossRef CAS.
- A. C. Lausche, J. A. Schaidle and L. T. Thompson, Appl. Catal., A, 2011, 401, 29–36 CrossRef CAS.
- M. Shimoda, T. Hirata, K. Yagisawa, M. Okochi and A. Yoshikawa, J. Mater. Sci. Lett., 1989, 8, 1089–1091 CrossRef CAS.
- Z. B. Z. Wei, P. Grange and B. Delmon, Appl. Surf. Sci., 1998, 135, 107–114 CrossRef CAS.
- T. P. St. Clair, S. T. Oyama, D. F. Cox, S. Otani, Y. Ishizawa, R.-L. Lo, K.-i. Fukui and Y. Iwasawa, Surf. Sci., 1999, 426, 187–198 CrossRef.
- S. J. Ardakani, X. Liu and K. J. Smith, Appl. Catal., A, 2007, 324, 9–19 CrossRef CAS.
- A. Corma, C. González-Arellano, M. Iglesias and F. Sánchez, Appl. Catal., A, 2009, 356, 99–102 CrossRef CAS.
- H.-U. Blaser, H. Steiner and M. Studer, ChemCatChem, 2009, 1, 210–221 CrossRef CAS.
- N. Perret, X. Wang, T. Onfroy, C. Calers and M. A. Keane, J. Catal., 2014, 309, 333–342 CrossRef CAS.
- C. Xiao, X. Wang, C. Lian, H. Liu, M. Liang and Y. Wang, Curr. Org. Chem., 2012, 16, 280–296 CrossRef CAS.
- J.-F. Su, B. Zhao and Y.-W. Chen, Ind. Eng. Chem. Res., 2011, 50, 1580–1587 CrossRef CAS.
- B. Zhao and Y.-W. Chen, J. Non-Cryst. Solids, 2010, 356, 839–847 CrossRef CAS.
- M. Boronat, P. Concepción, A. Corma, S. González, F. Illas and P. Serna, J. Am. Chem. Soc., 2007, 129, 16230–16237 CrossRef CAS PubMed.
- A. Corma, M. Boronat, S. Gonzalez and F. Illas, Chem. Commun., 2007, 3371–3373 RSC.
- A. Corma and P. Serna, Science, 2006, 313, 332–334 CrossRef CAS PubMed.
- K. Koprivova and L. Cerveny, Res. Chem. Intermed., 2008, 34, 93–101 CrossRef CAS.
- K. Hata and K.-I. Watanabe, Bull. Chem. Soc. Jpn., 1959, 32, 861–867 CrossRef CAS.
- X. Wang, Y. Hao and M. A. Keane, Appl. Catal., A, 2016, 510, 171–179 CrossRef CAS.
Footnote |
| † Present address: School of Engineering, University of Aberdeen, Aberdeen AB24 3UE, Scotland |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a