
Kinetics and thermodynamics of polymethylbenzene formation over zeolites with different pore sizes for understanding the mechanisms of methanol to olefin conversion – a computational study†
Abstract
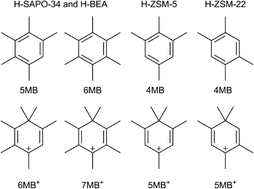
On the basis of density functional theory including dispersion correction (ωB97XD), the thermodynamics and kinetics of the formation of polymethylbenzene intermediates in methanol to olefin conversion over zeolites with different pore sizes have been systematically computed. The agreement between the experimental and theoretical adsorption enthalpies of the several polymethylbenzenes over H-FAU reasonably validates the applied models and methods, and reveals the importance of dispersion correction in the space confinement and electrostatic stabilization of the zeolite framework. The free energies of the stepwise formation of the polymethylbenzenes show that the most favorable active hydrocarbon pool intermediates are pentamethylbenzene and hexamethylbenzene over H-BEA and H-SAPO-34, as well as tetramethylbenzene over H-ZSM-5 and H-ZSM-22. These stable polymethylbenzenes are also precursors for the formation of geminal methylated cationic intermediates on the basis of kinetic and thermodynamic analyses. The agreement of the thermodynamic and kinetic results on the favorable intermediates validates the use of Gibbs free reaction energies to estimate the primary component of the intermediates in the various zeolites. All these pore-size-dependent differences among the zeolites show their enhanced confinement effect, which is mainly influenced by the short-range electrostatic potential including stabilization and repulsion.
 Please wait while we load your content...
Please wait while we load your content...

