
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An oxygen pool from YBaCo4O7-based oxides for soot combustion†
Guangkai
Tian
 a,
Hui
Chen
a,
Chenxi
Lu
a,
Ying
Xin
a,
Qian
Li
a,
James A.
Anderson
b and
Zhaoliang
Zhang
*a
a,
Hui
Chen
a,
Chenxi
Lu
a,
Ying
Xin
a,
Qian
Li
a,
James A.
Anderson
b and
Zhaoliang
Zhang
*a
aSchool of Chemistry and Chemical Engineering, Shandong Provincial Key Laboratory of Fluorine Chemistry and Chemical Materials, University of Jinan, No. 336, West Road of Nan Xinzhuang, Jinan 250022, PR China. E-mail: chm_zhangzl@ujn.edu.cn; Fax: +86 531 89736032; Tel: +86 531 89736032
bSurface Chemistry and Catalysis Group, Department of Chemistry, University of Aberdeen, Aberdeen AB24 3UE, UK
First published on 5th February 2016
Abstract
Soot, often referred to as black carbon emitted from diesel engines, is not only a particulate matter pollutant but also a light-absorbing agent that may affect global climate, but can be effectively controlled using a catalytic diesel particulate filter (DPF). A new YBaCo4O7+δ-type oxygen storage material is reported as an effective catalyst for soot combustion. Isotopic isothermal reactions demonstrate the activation of gaseous oxygen and subsequent oxygen storage and reaction/desorption during an oxidation process. High activity and structural stability are achieved by the substitution of Co with Al and Ga to form YBa(Co0.85Al0.075Ga0.075)4O7+δ. The specific rates at 300 °C of YBaCo4O7+δ and YBa(Co0.85Al0.075Ga0.075)4O7+δ, normalized by surface areas, are an order of magnitude higher than those of CeO2-based oxides. This kind of oxygen-storage material acts as an oxygen pool, which ensures that the accumulated soot on a DPF can be promptly combusted.
1. Introduction
Soot, often referred to as black carbon, is not only a particulate matter pollutant but also a light-absorbing agent that may affect global climate.1 Diesel engines are amongst the most abundant emission sources of soot that can be effectively controlled by a catalytic diesel particulate filter (DPF).2 Commercial catalysts are composed of noble metals (Pt and Pd) supported on ceria-based oxides,3 oxidizing NO into NO2, which is transported through a gas phase to soot aggregates where it oxidizes carbon while being reduced to NO.4 A so-called continuously regenerated trap (CRT) overcomes the problem of poor contact between soot and a catalyst. However, the limited amounts of NOx present and the need to control NOx for Euro IV standards are not ideal situations to meet the requirements of the latest generation of diesel engines, leading to a drive to develop catalysts which can produce highly reactive oxygen species from gaseous O2 molecules.5,6Among this kind of catalysts, the extensively studied soot oxidation catalysts are ceria-based oxides due to the redox properties of the Ce3+/Ce4+ couple and the capacity of ceria to exchange oxygen with a gas phase.7–12 Unfortunately, the effective utilization of such active oxygen species is limited by the extent of contact between soot and a catalyst. This limitation could be overcome by producing a sufficiently high amount of active oxygen to ensure that, at least, a proportion reaches the soot particle surface.5
In comparison with a conventional oxygen storage material, CeO2–ZrO2, YBaCo4O7 (114 structure) shows a markedly larger oxygen storage capacity (OSC), particularly at low temperatures (200–400 °C), which is just within the range of temperatures reached at the exhaust of a diesel engine.13–16 The extraordinary oxygen storage capability of the YBaCo4O7-based oxides is due to the variable valence of Co ions between Co2+ and Co3+. The limitation of YBaCo4O7+δ is that it decomposes just above 600 °C in an oxygen-containing atmosphere, which limits its applications in catalytic combustion at elevated temperatures.13 Recently, Karppinen and colleagues identified that an YBa(Co0.85Al0.075Ga0.075)4O7+δ phase, where Co is co-substituted by Al and Ga is stable up to high temperatures under oxidizing conditions,16 which creates the potential for YBa(Co0.85Al0.075Ga0.075)4O7+δ to become a more effective catalyst for catalytic combustion.
In this paper, YBaCo4O7+δ and YBa(Co0.85Al0.075Ga0.075)4O7+δ were studied from the perspective of catalysis for diesel soot combustion. Their oxygen storage performance allows them to create an oxygen pool supply of active oxygen which is created from a gaseous phase.17 Although YBaCo4O7+δ has been reported as a robust catalyst for H2O2 oxidation of cyclohexene in the liquid phase,18 the present result is the first report on high-temperature oxidation reactions for this kind of non-stoichiometric transition metal oxide materials.
2. Experimental section
The YBaCo4O7-type samples were synthesized from a precursor powder prepared by an EDTA (ethylenediaminetetraacetic acid) and CA (citric acid) complex gel method,13 using Y(NO3)3·6H2O, Ba(NO3)2, Co(NO3)3·6H2O, Ga(NO3)3·6H2O and Al(NO3)3·9H2O as the starting materials. The precursor powder was calcined in a muffle at 350 °C for 2 h, then pelletized and sintered in air at 1000 °C for 24 h, followed by rapid cooling to room temperature. Further thermal treatment at 300 °C under a pure O2 flow for 2 h obtained YBaCo4O7+δ and YBa(Co0.85Al0.075Ga0.075)4O7+δ.X-ray powder diffraction (XRD) patterns were recorded on a Rigaku D/max-RC diffractometer employing Cu Kα radiation. Surface areas and pore size distributions were determined by N2 adsorption–desorption at 77 K using a Micromeritics ASAP 2020 instrument after outgassing at 300 °C for 5 h prior to analysis.
Temperature programmed desorption of O2 (O2–TPD) experiments were conducted in a fixed-bed flow reactor. A 150 mg sample was heated under a flow of high purity O2 (30 ml min−1) at 300 °C for 1 h. After cooling to room temperature, high purity He was introduced. Desorption was started at a heating rate of 2 °C min−1 in He (30 ml min−1). The desorbed O2 was monitored by a quadruple mass spectrometer (MS, OminiStar 200, Balzers).
Temperature programmed reduction with H2 (H2–TPR) experiments were performed in a quartz reactor with a thermal conductivity detector (TCD) to monitor H2 consumption. A 50 mg sample was pretreated in situ at 300 °C for 1 h under a flow of O2 and cooled to room temperature in the presence of O2. After purging with N2, TPR was conducted at 10 °C min−1 up to 700 °C under a 30 mL min−1 flow of 5 vol% H2 in N2. To quantify the total amount of H2 consumption, CuO was used as a calibration reference.
“Dynamic” OSC (DOSC) measurements with CO–O2 pulses were carried out at 200–500 °C. CO (4% CO/1% Ar/He at 300 mL min−1 for 10 s) and O2 (2% O2/1% Ar/He at 300 mL min−1 for 10 s) streams were pulsed alternately with at a frequency of 0.05 Hz. A DOSC value was obtained by integrating the CO2 formed during one CO–O2 cycle and was expressed as μmol of O per gram of catalyst (μmol [O] g−1). The concentration of CO2 was determined using a MS.
Temperature programmed oxidation (TPO) reactions were conducted in a fixed bed micro reactor consisting of a quartz tube (6 mm i. d.). Printex-U from Degussa was used as the model soot. The soot was mixed with the catalyst in a weight ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 in an agate mortar for 30 min, which resulted in a tight contact between the soot and the catalyst. A 50 mg sample of the soot/catalyst mixture was pre-treated under a flow of He (50 mL min−1) at 200 °C for 30 min to remove adsorbed species. After cooling to room temperature, a gas flow with 5 vol% oxygen in He was introduced and then TPO was initiated at a heating rate of 5 °C min−1 until 880 °C. For pure soot combustion (non-catalytic), the catalyst was substituted by silica. CO and CO2 concentrations in the effluent gas were monitored using an online gas chromatograph (GC) (SP-6890, Shandong Lunan Ruihong Chemical Instrument Corporation, China) fitted with a methanator. The ignition temperature for soot combustion was evaluated by the value of T10, which is defined as the temperature at which 10% of the soot is converted. The selectivity to CO2 is defined as the percentage CO2 outlet concentration divided by the sum of the CO2 and CO outlet concentrations.
:9 in an agate mortar for 30 min, which resulted in a tight contact between the soot and the catalyst. A 50 mg sample of the soot/catalyst mixture was pre-treated under a flow of He (50 mL min−1) at 200 °C for 30 min to remove adsorbed species. After cooling to room temperature, a gas flow with 5 vol% oxygen in He was introduced and then TPO was initiated at a heating rate of 5 °C min−1 until 880 °C. For pure soot combustion (non-catalytic), the catalyst was substituted by silica. CO and CO2 concentrations in the effluent gas were monitored using an online gas chromatograph (GC) (SP-6890, Shandong Lunan Ruihong Chemical Instrument Corporation, China) fitted with a methanator. The ignition temperature for soot combustion was evaluated by the value of T10, which is defined as the temperature at which 10% of the soot is converted. The selectivity to CO2 is defined as the percentage CO2 outlet concentration divided by the sum of the CO2 and CO outlet concentrations.
Isothermal reactions at 300 °C, at which a stable and low soot conversion (<15%) was achieved, were conducted within the kinetic regime. The reaction rate for soot combustion was obtained from the slope of the conversion lines with time. Specific rates normalized by BET surface areas and turnover frequency (TOF)19 were used to characterize the activity for soot combustion.
An isotopic isothermal reaction was performed by switching the flowing gas from 1% 16O2 to 1% 18O2 diluted in Ar at 350 °C. 50 mg of a mixture of the soot and catalyst in a tight contact mode was employed. The effluent gas from the reactor was continuously monitored by a MS for all of the isotopic molecules of CO2 (at m/z = 44, 46 and 48).
3. Results and discussion
The fresh YBaCo4O7+δ and YBa(Co0.85Al0.075Ga0.075)4O7+δ were confirmed to be composed of a single phase (Fig. S1†), similar to that of hexagonal LuBaAlZn3O7 (JCPDS 40-4099).13 As the shifts in the XRD peaks for the separate substitution of Co by Al (r = 0.039 nm) and Ga (r = 0.047 nm) are opposed for each sample (Fig. S2†), the simultaneous substitution has a slight effect on the cell volume parameters. Furthermore, both ionic radii of Al and Ga are smaller than the high spin Co2+ ionic radius (r = 0.058 nm), thus Maignan et al. suggested that Al and Ga are substituted for Co3+.20 The BET surface areas are fairly low, in accordance with the high-temperature sintering preparation (Table S1†) and the highly crystalline nature of the samples.In agreement with conclusions in the literature,13,16 the O2–TPD profiles (Fig. 1) show that both YBaCo4O7+δ and YBa(Co0.85Al0.075Ga0.075)4O7+δ desorb a large amount of oxygen below 400 °C, corresponding to δ = 0.37 and δ = 0.34, respectively. The peak maximum of YBa(Co0.85Al0.075Ga0.075)4O7+δ is 50 °C lower than that of YBaCo4O7+δ, which suggests a promotion effect on O2 desorption by doping, albeit with a slight decrease of the overall oxygen storage capability.16 Above 700 °C, further desorption of O2 is observed, corresponding to the possible decomposition of the 114 structure.13 However, YBa(Co0.85Al0.075Ga0.075)4O7+δ shows less pronounced O2 desorption than YBaCo4O7+δ. After O2–TPD at about 900 °C, the Y2O3 and CoO phases are segregated from YBaCo4O7+δ, which is opposite to the stable structure of YBa(Co0.85Al0.075Ga0.075)4O7+δ (Fig. S3†).
 | ||
| Fig. 1 O2–TPD spectra of YBaCo4O7+δ and YBa(Co0.85Al0.075Ga0.075)4O7+δ from room temperature to 900 °C. | ||
A similar situation is observed in H2–TPR (Fig. S1†). The 114 structure of YBaCo4O7+δ is completely destroyed by H2 in TPR to 700 °C (Fig. S1a and b†). In contrast, YBa(Co0.85Al0.075Ga0.075)4O7+δ was preserved with no formation of new oxide phases (Fig. S1c and d†). The structural stability of YBa(Co0.85Al0.075Ga0.075)4O7+δ is vital to high-temperature redox reactions. As shown in Fig. 2, two peaks were observed for both YBaCo4O7+δ and YBa(Co0.85Al0.075Ga0.075)4O7+δ. The low-temperature TPR peak can be assigned to the removal of non-stoichiometric excess O accommodated within the lattice, and the values of which are slightly larger than those consumed by δ (Table 1). The 114 structure of YBaCo4O7+δ is stable at this stage (Fig. S1a and b†). The second peak corresponds to the reduction of bulk and surface Co3+ of YBaCo4O7+δ and YBa(Co0.85Al0.075Ga0.075)4O7+δ, respectively. The substitution of Co3+ by Al and Ga protects the structure from decomposition under reducing atmospheres (H2–TPR and O2–TPD).20 Furthermore, the lower TPR temperature of YBa(Co0.85Al0.075Ga0.075)4O7+δ in comparison with that of YBaCo4O7+δ coincides with the O2–TPD results.
 | ||
| Fig. 2 H2–TPR spectra of YBaCo4O7+δ and YBa(Co0.85Al0.075Ga0.075)4O7+δ from room temperature to 700 °C. | ||
Although H2–TPR and O2–TPD data may be useful in rapidly evaluating the potential OSC of the candidate materials, DOSC provides better simulation of instantaneous oscillations between lean (oxidizing) and rich (reducing) exhaust conditions during real operation and is therefore much more useful in the evaluation of the activity of OSC materials.21Fig. 3(a) shows the collected DOSC data and the corresponding transition curves at 320 °C of YBa(Co0.85Al0.075Ga0.075)4O7+δ as an example, with alternate dynamic pulses of 4% CO/1% Ar/He (10 s) and 2% O2/1% Ar/He (10 s) under 0.05 Hz given in Fig. 3(b). In comparison with CeO2–ZrO2, the normalized DOSC values by the BET surface areas of YBaCo4O7+δ and YBa(Co0.85Al0.075Ga0.075)4O7+δ are more than thirty times larger (DOSC values of YBaCo4O7+δ, YBa(Co0.85Al0.075Ga0.075)4O7+δ and Ce0.43Zr0.57O2 at 400 °C are 40.9, 41.6 and 1.3 μmol [O] m−2, respectively),22 confirming the fast responses between reduction and oxidation environments. Furthermore, the higher DOSC of YBa(Co0.85Al0.075Ga0.075)4O7+δ in comparison with that of YBaCo4O7+δ is consistent with the O2–TPD and H2–TPR results.
 | ||
| Fig. 3 DOSC of YBaCo4O7+δ and YBa(Co0.85Al0.075Ga0.075)4O7+δ (a) and enlarged transition curves of YBa(Co0.85Al0.075Ga0.075)4O7+δ (b) at 320 °C with dynamic pulses of 4% CO/1% Ar/He and 2% O2/1% Ar/He under 0.05 Hz. CO2(I) was produced by CO and surface oxygen; CO2(II) was attributed to the reaction of absorbed CO and oxygen gas.22 | ||
The catalytic activity for soot combustion was first checked by TPO (Fig. 4a). YBaCo4O7+δ and YBa(Co0.85Al0.075Ga0.075)4O7+δ decrease from a T10 value of 530 °C for non-catalytic combustion to 387 and 379 °C, respectively, confirming the catalytic effect of the YBaCo4O7+δ-type material and the higher activity of the latter than the former. In terms of the selectivity towards CO2 formation, the non-catalytic combustion is only 43.3%, while YBaCo4O7+δ and YBa(Co0.85Al0.075Ga0.075)4O7+δ yield nearly 100% CO2. After the TPO reactions, no phase decomposition occurs even for YBaCo4O7+δ (Fig. S4†), probably due to the high heating rate in 5 vol% oxygen in He.23 Furthermore, the XRD peaks of YBaCo4O7+δ and YBa(Co0.85Al0.075Ga0.075)4O7+δ after TPO shift to higher angles, suggesting a lattice shrinkage, which confirms the participation of bulk oxygen.
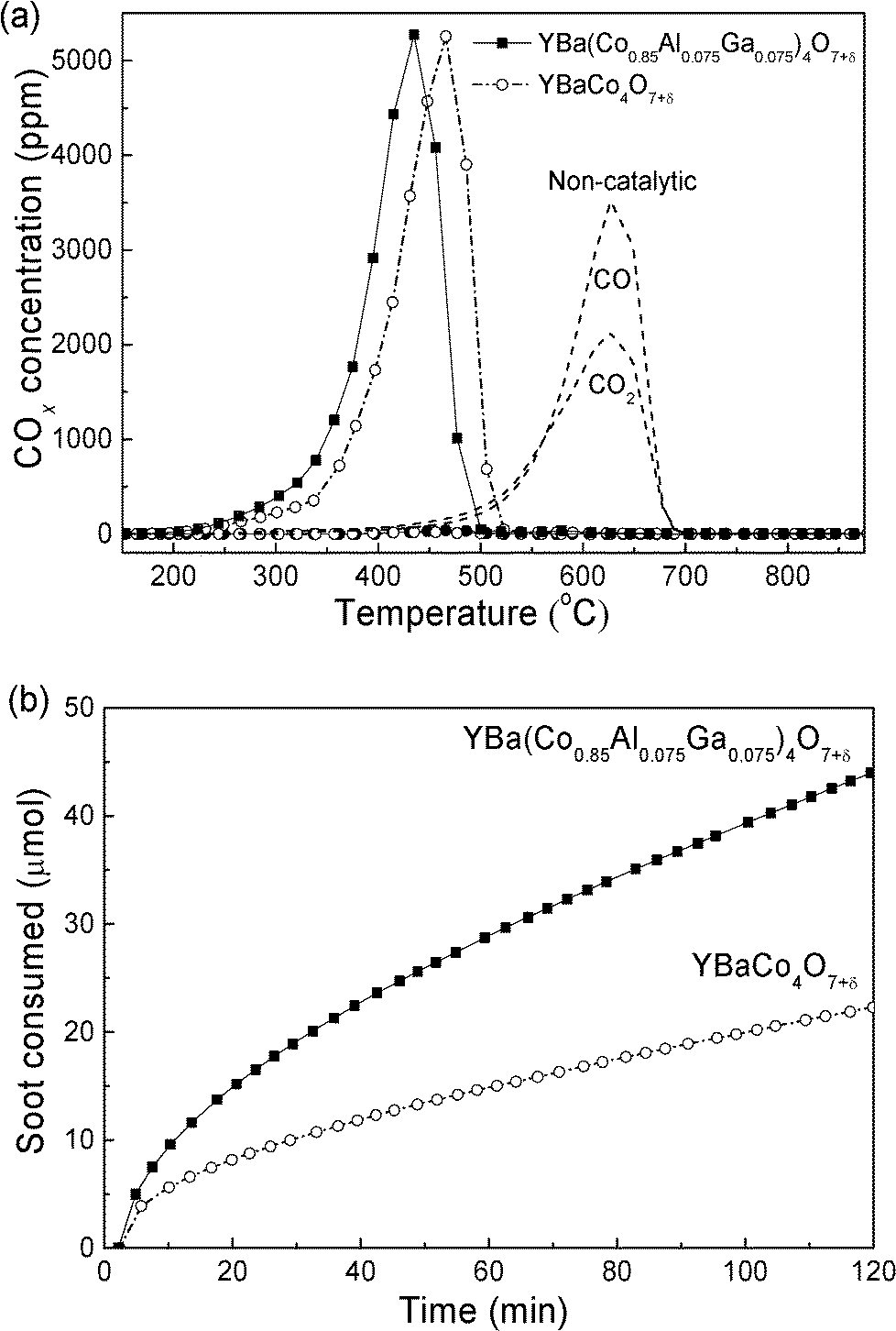 | ||
| Fig. 4 TPO patterns of COx for soot combustion with O2 over YBaCo4O7+δ and YBa(Co0.85Al0.075Ga0.075)4O7+δ (a); isothermal reactions for soot combustion at 300 °C within the kinetic regime (b) under tight contact conditions between the soot and catalysts. | ||
The intrinsic activity was further demonstrated by kinetic rates at 300 °C, which can be obtained from the slope of the lines shown in Fig. 4b. As observed in Table 1, the specific rates of YBaCo4O7+δ and YBa(Co0.85Al0.075Ga0.075)4O7+δ, normalized by BET surface areas, are an order of magnitude larger than that of Ce0.43Zr0.57O2.19–22 This is significant because 300 °C is a relevant temperature for light diesel engines. This particularly high reaction rate can ensure that the accumulated soot on the DPF can be readily combusted, leading to a lower balance point temperature (BPT) at which the rate of soot oxidation is matched with the rate of soot accumulation.24 Furthermore, both the specific rate and TOF (Table 1) of YBa(Co0.85Al0.075Ga0.075)4O7+δ are a little higher than that of YBaCo4O7+δ, confirming the higher intrinsic activity of YBa(Co0.85Al0.075Ga0.075)4O7+δ.
In order to explore the origin of the active oxygen, isotopic isothermal oxidation at 350 °C was performed (Fig. 5). Before switching from 16O2 to 18O2 (to the left of the dashed line), the main product was C16O2, confirming that the soot oxidation occurs with the bulk oxygen species. The concentration of C16O2 for YBa(Co0.85Al0.075Ga0.075)4O7+δ is much larger than that for YBaCo4O7+δ, which is again consistent with the O2–TPD, H2–TPR, DOSC, T10 and specific rates. After switching from 16O2 to 18O2 (to the right of the dashed line), the sum of the products of C16O18O, C18O2 and 16O18O still possesses higher concentrations of YBa(Co0.85Al0.075Ga0.075)4O7+δ than that of YBaCo4O7+δ. However, the concentration of C16O2 decreased gradually to a very low value due to the depletion of 16O2 in the gaseous phase. Comparatively, for both YBaCo4O7+δ and YBa(Co0.85Al0.075Ga0.075)4O7+δ, the production of C16O18O increases and then reaches a maximum, while the C18O2 production monotonically increases. This indicates that only the gaseous oxygen which has been activated by YBaCo4O7+δ and YBa(Co0.85Al0.075Ga0.075)4O7+δ can be used to oxidize the soot. In addition, the desorption of 16O18O is detected, which suggests that the desorbed 16O18O species do not interact with the soot to produce the product containing carbon, demonstrating that the intimate contact between the soot and catalysts is essential. Since the high DOSC can make up for the missing oxygen, the intrinsic activity of YBaCo4O7-based oxides is much more active than that of CeO2-based oxides.
 | ||
| Fig. 5 Isothermal reactions for soot combustion at 350 °C after 1% 16O2 was switched to 1% 18O2 in He on YBaCo4O7+δ (a) and YBa(Co0.85Al0.075Ga0.075)4O7+δ (b). | ||
Conclusions
In conclusion, a new YBaCo4O7+δ-type oxygen storage material was used to catalyze soot combustion. Isotopic isothermal reactions demonstrate the activation of gaseous oxygen and subsequent oxygen storage and reaction/desorption during the oxidation process. Higher activity and structural stability are achieved by the substitution of Co with Al and Ga to form YBa(Co0.85Al0.075Ga0.075)4O7+δ. The specific rates at 300 °C of YBaCo4O7+δ and YBa(Co0.85Al0.075Ga0.075)4O7+δ, normalized by BET surface areas, are an order of magnitude larger than that of CeO2-based oxides. This type of oxygen-storage material provides an oxygen pool, which ensures that the accumulated soot on a DPF can be readily combusted.Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (no. 21477046, 21277060 and 21547007).Notes and references
- B. Booth and N. Bellouin, Nature, 2015, 509, 167–168 CrossRef PubMed.
- Barry A. A. L. van Setten, M. Makkee and J. A. Moulijn, Catal. Rev.: Sci. Eng., 2001, 43, 489–564 CAS.
- S. Sumiya, L. Wang and G. Zhang, SAE [Tech. Pap.], 2012, 2012-28-0009 Search PubMed.
- K. Leistner, A. Nicolle and P. Da Costa, J. Phys. Chem. C, 2012, 116, 4642–4654 CAS.
- I. Atribak, F. E. López-Suárez, A. Bueno-López and A. García-García, Catal. Today, 2011, 176, 404–408 CrossRef CAS.
- E. Obeid, L. Lizarraga, M. N. Tsampas, A. Cordier, A. Boréave, M. C. Steil, G. Blanchard, K. Pajot and P. Vernoux, J. Catal., 2014, 309, 87–96 CrossRef CAS.
- Q. Liang, X. Wu, D. Weng and H. Xu, Catal. Today, 2008, 139, 113–118 CrossRef CAS.
- J. Liu, Z. Zhao, J. Wang, C. Xu, A. Duan, G. Jiang and Q. Yang, Appl. Catal., B, 2008, 84, 185–195 CrossRef CAS.
- E. Aneggi, C. de Leitenburg and A. Trovarelli, Catal. Today, 2012, 181, 108–115 CrossRef CAS.
- A. Buenolopez, K. Krishna, M. Makkee and J. Moulijn, J. Catal., 2005, 230, 237–248 CrossRef CAS.
- K. Harada, T. Oishi, S. Hamamoto and T. Ishihara, J. Phys. Chem. C, 2014, 118, 559–568 CAS.
- A. Bueno-López, Appl. Catal., B, 2014, 146, 1–11 CrossRef.
- M. Karppinen, H. Yamauchi, S. Otani, T. Fujita, T. Motohashi, Y. H. Huang, M. Valkeapää and H. Fjellvåg, Chem. Mater., 2006, 18, 490–494 CrossRef CAS.
- S. Kadota, M. Karppinen, T. Motohashi and H. Yamauchi, Chem. Mater., 2008, 20, 6378–6381 CrossRef CAS.
- Y. Jia, H. Jiang, M. Valkeapää, H. Yamauchiä, M. Karppinen and E. I. Kauppinen, J. Am. Chem. Soc., 2009, 131, 4880–4883 CrossRef CAS PubMed.
- O. Parkkima, H. Yamauchi and M. Karppinen, Chem. Mater., 2013, 25, 599–604 CrossRef CAS.
- K. Sato, M. Yamaguchi, S. Fujita, K. Suzuki and T. Mori, Catal. Commun., 2006, 7, 132–135 CrossRef CAS.
- O. Parkkima, A. Silvestre-Albero, J. Silvestre-Albero and M. Karppinen, Catal. Lett., 2014, 145, 576–582 CrossRef.
- Z. Zhang, D. Han, S. Wei and Y. Zhang, J. Catal., 2010, 276, 16–23 CrossRef CAS.
- A. Maignan, V. Caignaert, V. Pralong, D. Pelloquin and S. Hébert, J. Solid State Chem., 2008, 181, 1220–1226 CrossRef CAS.
- M. Boaro, F. Giordano, S. Recchia, V. D. Santo, M. Giona and A. Trovarelli, Appl. Catal., B, 2004, 52, 225–237 CrossRef CAS.
- Z. Zhang, Y. Fan, Y. Xin, Q. Li, R. Li, J. A. Anderson and Z. Zhang, Environ. Sci. Technol., 2015, 49, 7989–7995 CrossRef CAS PubMed.
- T. Komiyama, T. Motohashi, Y. Masubuchi and S. Kikkawa, Mater. Res. Bull., 2010, 45, 1527–1532 CrossRef CAS.
- J. Oi-Uchisawa, A. Obuchi, S. Wang, T. Nanba and A. Ohi, Appl. Catal., B, 2003, 43, 117–129 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Textural properties and XRD patterns of the samples. See DOI: 10.1039/c5cy02181b |
| This journal is © The Royal Society of Chemistry 2016 |