Direct observation of methylcyclopentenyl cations (MCP+) and olefin generation in methanol conversion over TON zeolite
Received
27th August 2015
, Accepted 14th September 2015
First published on 16th September 2015
Abstract
The mechanism of the methanol to olefin (MTO) reaction over H-ZSM-22, a TON-type zeolite without cavities or channel intersections, has been investigated in the temperature range of 250–350 °C. For the first time, an induction period in low-temperature methanol conversion and the methylcyclopentenyl cation (MCP+) formed during this period have been observed directly and successfully. 13C magic angle spinning (MAS) NMR, 13C-labeling experiments and theoretical calculations have been employed to confirm the important active intermediates during methanol conversion at 300 °C. The reactions performed at different temperatures were comparatively studied and the differences in the reaction route for alkene formation from methanol conversion and the modes of H-ZSM-22 catalyst deactivation were revealed.
1. Introduction
Light olefins (ethene and propene) are the backbone feedstocks of the petrochemical industry and the majority of them are produced through a high energy-consuming process, the thermal cracking of naphtha. The price rise of crude oil and increasing demand of light olefins drive the non-petrochemical route development for light olefin production. The methanol-to-olefin (MTO) process has been developed as the most successful non-petrochemical route for the production of light olefins from alternative and abundant resources, natural gas or coal.1–6
Parallel to the MTO process development, intensive studies in the past three decades on the catalysis of methanol conversion over microporous solid acid catalysts have proposed more than 20 distinct reaction mechanisms to explain the generation of hydrocarbon products with C–C bonds from the C1-reactant, methanol.1,7 Among them, the direct formation of the C–C bond from C1 species derived from methanol has been proved to be unfavourable due to the high energy barrier.8,9 Alternatively, the “hydrocarbon pool” (HCP) mechanism, an indirect pathway proposed by Dahl and Kolboe, which avoids high energy barriers, has gained general acceptance based on the evidence from experimental observations and theoretical calculations.10–12 Up to date, polymethylbenzene, polymethylcyclopentadiene and their protonated analogues have been proved to be the most important reactive intermediates for olefin production.13–28 The formation of these bulky intermediates has been observed in the zeolite catalysts with wide or intersectional channels, such as Beta15 and ZSM-5 (ref. 13, 23, 25 and 27) zeolites, or in the molecular sieve with large supercage, such as DNL-6,21,29 SAPO-34 (ref. 16–20 and 26) and SSZ-13,22,26 since the accommodation and function of these intermediates require large cavity or pore structure. Over the zeolite without cage and intersectional channel, such as H-ZSM-22 zeolite with a 1-dimensional and 10-membered ring channel structure, since the HCP mechanism is too difficult to work in its narrow channel structure, methanol conversion was predicted to be suppressed over this catalyst with TON topology.30–32 But some studies indicated that methanol conversion over H-ZSM-22 presented comparable conversion to that of SAPO-34,33 and suggested the product generation over H-ZSM-22 followed the olefin methylation and cracking route,34–37 one of the routes proposed in the dual cycle mechanism put forward by Kolboe and coauthors in the study of methanol conversion over ZSM-5.38,39 The confined cyclic organics as important intermediates have not been observed in the 1-dimensional, 10-membered ring channel of H-ZSM-22 in olefin generation. Whether the methanol reaction can proceed over H-ZSM-22 via the HCP mechanism with the involvement of cyclic organic intermediates is still vague, since no any definite and direct evidence is provided for this issue.
In the present work, methanol conversion was performed over H-ZSM-22 with narrow one-dimensional 10-ring pores (0.46 nm × 0.57 nm) at the reaction temperature ranging from 250 to 350 °C to study the reaction mechanism of olefin generation. The formation of retained reactive intermediates during the low-temperature reaction was confirmed by the aid of 13C MAS NMR and GC-MS. The role of the reactive intermediates at varied reaction temperatures was determined and compared with the 13C-labeling technique. Different reaction and deactivation modes of the methanol conversion at low and high temperatures over HZSM-22 have been presented and discussed.
2. Experimental
2.1 Catalyst preparation
The K-ZSM-22 catalyst was supplied by the laboratory of DNL0802 of the Dalian Institute of Chemical Physics. After calcination at 600 °C for 10 h to remove the organic template, K-ZSM-22 was converted into NH4-ZSM-22 by subjecting to ion-exchange three times with 1 M NH4NO3 solution at 80 °C for 6 h. Subsequently, the catalyst was washed with deionized water, dried at 120 °C overnight and calcined at 550 °C for the following 4 h to achieve H-ZSM-22.
2.2. Catalyst characterization
The powder XRD patterns of H-ZSM-22 were recorded on a PANalytical X'Pert PRO X-ray diffractometer with Cu Kα radiation (λ = 1.54059 Å) at 40 kV and 40 mA. The chemical composition of the catalyst was determined using a Philips Magix-601 X-ray fluorescence (XRF) spectrometer. The morphology of the catalyst was measured by field emission scanning electron microscopy (FE-SEM, Hitachi, SU8020). N2 adsorption–desorption isotherms were obtained at 77 K on a Micromeritics ASAP 2020 system.
The acidity of the H-ZSM-22 catalyst was determined by temperature programmed desorption of ammonia (NH3-TPD) on a chemical adsorption instrument (Micromeritics AutoChem 2920). The sample was loaded in a U-shaped micro-reactor and pretreated at 650 °C for 30 min under helium atmosphere. After cooling to 100 °C, the sample was saturated with ammonia, followed by purging with helium to remove physisorbed ammonia. The ammonia desorption was carried out in helium flow (40 ml min−1) by increasing the temperature from 100 to 600 °C with a heating rate of 10 °C min−1 and monitored by a thermal conductivity detector (TCD).
The solid-state NMR spectroscopy experiments were carried out on a Bruker Avance III 600 spectrometer equipped with a 14.1 T wide-bore magnet using a 4 mm magic angle spinning (MAS) probe. The resonance frequency for 13C was 150.9 MHz. The 13C CP MAS NMR spectra were recorded at a spinning rate of 12 kHz. 4096 scans were accumulated with a contact time of 3 ms and a 2 s recycle delay.
2.3. MTO reaction
The H-ZSM-22 catalyst was pressed, sieved to 40–60 mesh and loaded in a fixed-bed quartz tubular reactor with an inner diameter of 4 mm. Prior to the reaction, the catalyst was activated at 500 °C for 40 min, and then the temperature was adjusted to the reaction temperature ranging from 250 to 350 °C. Methanol was fed by passing the carrier gas (12.4 ml min−1) through a saturator containing methanol at 14 °C, which gave a WHSV of 2.0 g g−1 h−1. Methanol conversion was performed under atmospheric pressure. The effluent products from the reactor were kept warm and analyzed by an online gas chromatograph equipped with a PoraPLOT Q-HT capillary column and an FID detector. The conversion and selectivity were calculated on CH2 basis. Dimethyl ether (DME) was considered as reactant in the calculations.
2.4.
13C MAS NMR measurement of methanol conversion
Methanol conversion over H-ZSM-22 was measured by 13C solid-state NMR employing 13C-methanol as the reactant.40 Under the identical conditions of 12C-methanol conversion, 13C-methanol was fed into the reactor for a predetermined time at a certain temperature and then the reactor was removed from the feeding line, and the catalyst was cooled very quickly by putting them into the vessel containing liquid nitrogen. Finally, the cooled catalyst was transferred to an NMR rotor in a glove box without exposure to ambient air.
2.5.
12C/13C-methanol switch experiment
In the 12C/13C-methanol switch experiments at 300 °C, after 12C-methanol was fed to the reactor by passing the carrier gas (12.4 ml min−1) through a saturator containing methanol at 14 °C for 29 and 74 min, the feeding of 12C-methanol was stopped and the feeding line was switched to 13C-methanol for further 1 min. For the 12C/13C-methanol switch experiments at 320 and 350 °C, the feeding of 12C-methanol was stopped after 29 min of reaction and the feeding line was switched to 13C-methanol for further 1 min. The isotopic distribution of the effluent products and the materials confined in the catalyst were determined by GC-MS (Agilent 7890/5975C).
2.6. Retained organic compound determination with GC-MS
After the reaction, the catalyst was quickly cooled down. The retained organics were analyzed following the method introduced by Guisnet.41 The discharged catalyst was dissolved in 20% HF solution, extracted with CH2Cl2 and identified by a GC-MS equipped with a HP-5 capillary column and an FID detector.
2.7. Theoretical calculations
A 126 T cluster model representing zeolite with TON topology was used to explore the accommodation of adsorbed species within ZSM-22 via theoretical calculations. The terminal Si–H was fixed with the bond length of 1.47 Å oriented along the direction of the corresponding Si–O bond. The combined theoretical method, namely ONIOM (ωB97XD/6-31G (d, p):AM1), was applied to predict the geometries of various adsorption structures. Such combined ONIOM method has been confirmed to determine the reliable adsorbed structure for the zeolite catalysis.42–44 In ONIOM simulations, the adsorbed organic species and the atoms (40 T) of the surrounding TON framework were treated as high level, and they were allowed to relax during the structure optimization, while the remaining atoms in the zeolite framework were treated as low level and were kept fixed at their crystallographic locations. In order to obtain accurate energy results, the single point energy calculations were further refined at the level of ωB97XD/6-31G (d, p).44 All adsorption energy, enthalpy and Gibbs free energy calculations were obtained from the ωB97XD/6-31G (d, p) total electronic energy and the thermal correction from ONIOM (ωB97XD/6-31G(d, p):AM1) frequency calculation at 300 °C by employing a partial Hessian vibrational analysis (PHVA).45 All the calculations were carried out by the Gaussian 09 software package.
The adsorption energy (ΔEads) of the adsorbates within the TON zeolite was defined based on eqn (1):
| | | ΔEads = Ecomplex − Ezeolite − Eadsorbate, | (1) |
where
Ecomplex represents the energy of the optimal zeolite–adsorbate complexes, and
Ezeolite and
Eadsorbate refer to the energy of optimal zeolite model and organic species in the free state. Adsorption enthalpies were calculated by adding the appropriate thermal corrections to the electronic energy differences, and Gibbs free energies were subsequently obtained with the aid of entropic contributions.
46
3. Results and discussion
3.1. Catalyst characterization
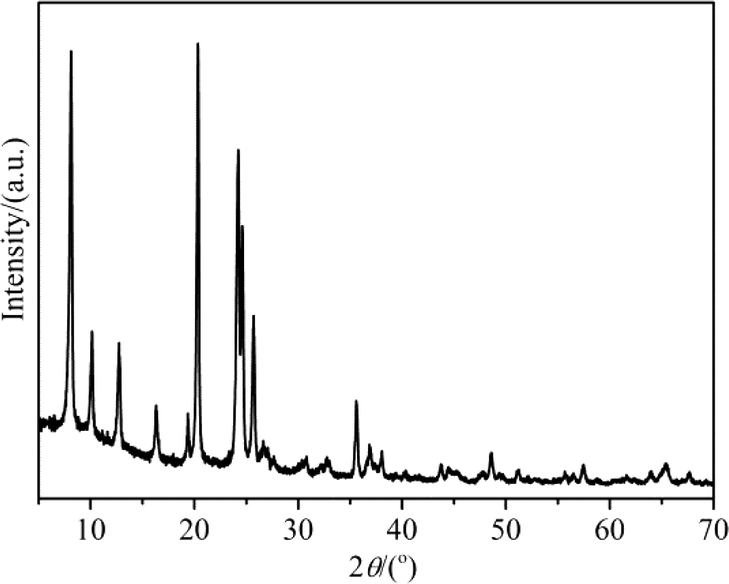
The XRD pattern of H-ZSM-22 presented in Fig. 1 showed the high crystallinity and purity of the catalyst with TON topology. The SEM image in Fig. 2 presents the rod-like crystals of H-ZSM-22 with a diameter of 40 nm and a length of 300 nm. The Si/Al ratio of H-ZSM-22 was 33.
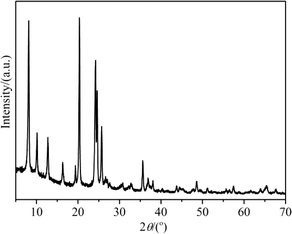 |
| | Fig. 1 XRD pattern of H-ZSM-22. | |
 |
| | Fig. 2 SEM image of H-ZSM-22. | |
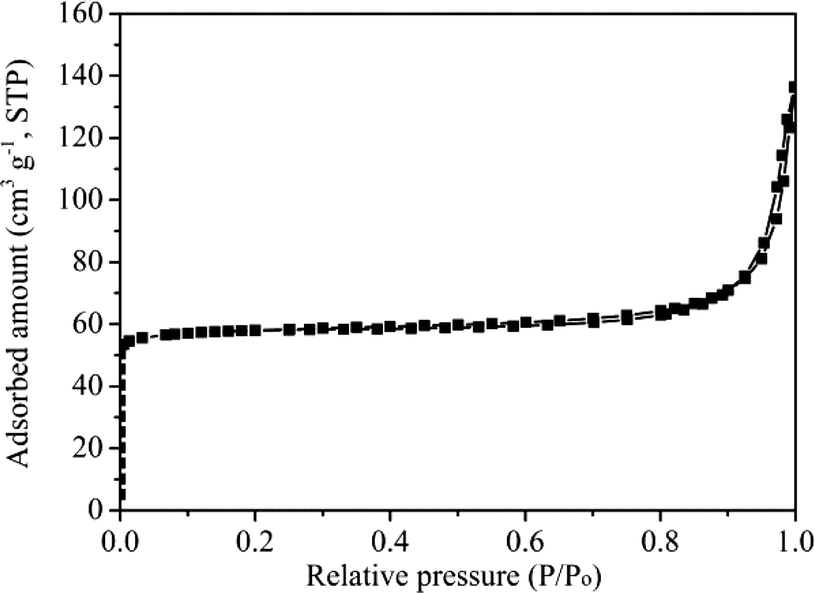
The N2 adsorption–desorption isotherm depicted in Fig. 3 indicated that the H-ZSM-22 catalyst is a typical microporous material with a Brunauer–Emmett–Teller (BET) surface area of 194 m2 g−1 and a pore volume of 0.15 cm3 g−1.
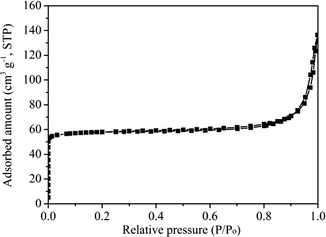 |
| | Fig. 3 N2 adsorption–desorption isotherm of H-ZSM-22. | |
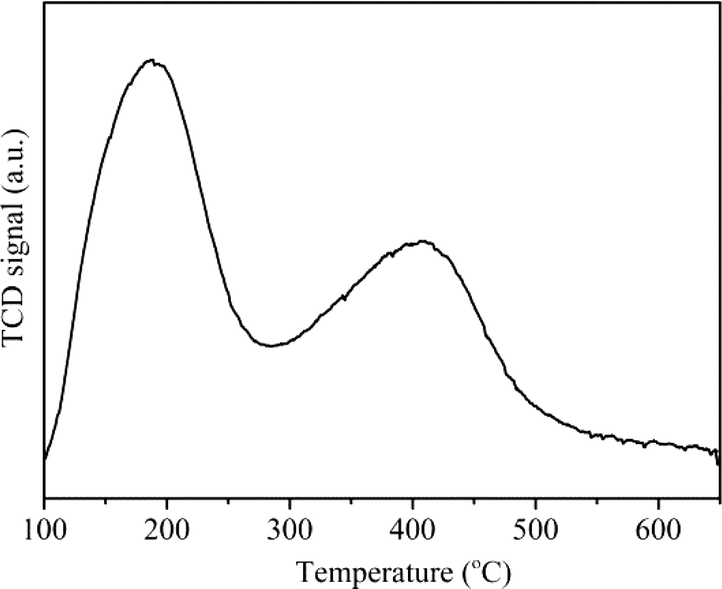
Fig. 4 shows the NH3-TPD curve of H-ZSM-22. Two desorption peaks could be observed in the temperature ranges of 100–280 °C and 280–600 °C, which represented the NH3 desorption from weak acid sites and strong acid sites, respectively.
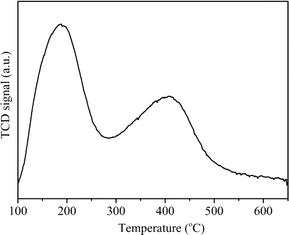 |
| | Fig. 4 NH3-TPD profile of H-ZSM-22. | |
3.2. Catalytic performance
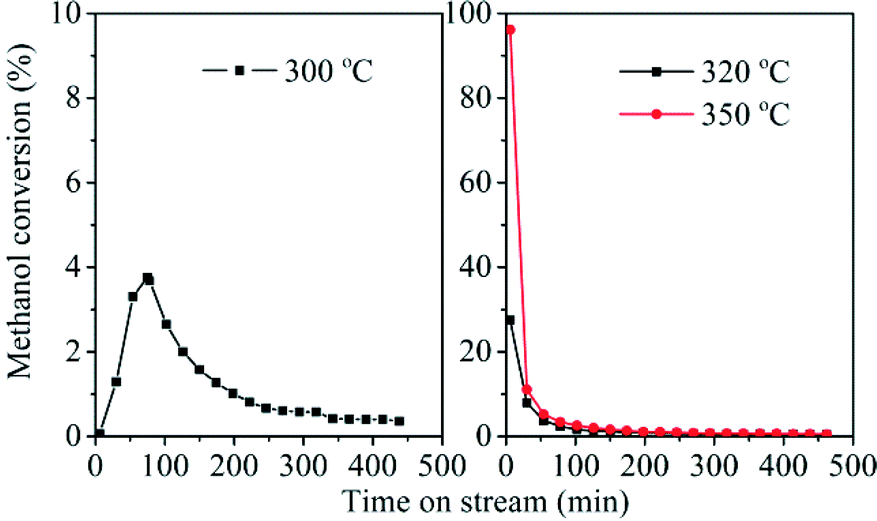
Methanol conversion over the H-ZSM-22 catalyst was performed at temperature ranging from 250 to 350 °C. When the reaction was performed at 250 °C, only methanol and dimethyl ether appeared among the effluents and no hydrocarbons were generated even if the reaction time was prolonged to 8 h. The reaction temperature of 250 °C was too low to trigger methanol conversion. For the reactions performed at temperatures higher than 250 °C, as shown in Fig. 5, the reaction temperature had a great effect on the initial methanol conversion. The initial conversion improved with temperature increase. At a time on stream (TOS) of 6 min, methanol conversion was 0.07% at 300 °C, and this value was 28% at 320 °C and 96% at 350 °C, respectively. Even at the relatively high initial conversion observed at 320 and 350 °C, the catalyst deactivated quickly with the extension of the reaction time, which was consistent with the previous report.33 To our surprise, in spite of the low initial activity at 300 °C, the conversion increased gradually from 0.07% at a TOS of 6 min to 3.78% at a TOS of 75 min and then declined slowly with reaction time, showing an apparent and typical induction period just like methanol conversion over ZSM-5 with low catalyst–reactant contact time or over SAPO-34 at relatively low temperature.47,48 For methanol conversion over zeolite or SAPO molecular sieve catalysts, the induction period is an important character of the reaction following an indirect reaction mechanism. During the induction period, formation of the important intermediates gives rise to enhanced methanol conversion and transforms a fresh and inefficient catalyst to a working catalyst over which methanol can be converted to hydrocarbons efficiently.7 The observation of induction period in H-ZSM-22-catalyzed methanol conversion implied that the low-temperature reaction over this catalyst may follow an indirect pathway, such as the HCP mechanism, especially at the beginning of the reaction. For the reaction performed at 320 and 350 °C, the high conversion occurred at the beginning of the reaction and the decrease of methanol conversion was observed with prolonged reaction time.
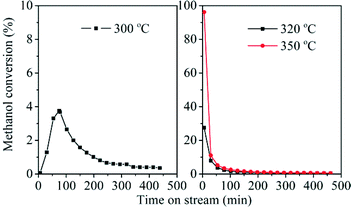 |
| | Fig. 5 Methanol conversion over H-ZSM-22 at 300, 320 and 350 °C, WHSV of methanol = 2.0 h−1, He/MeOH (in mole) = 10.1. | |
Detailed product distribution (Fig. 6) indicated that C3–C7 alkene products largely formed over H-ZSM-22 at 320 and 350 °C from the beginning of the reaction, while at 300 °C and a TOS of 6 min, methane and ethene were the dominant products and accounted for 90% and 10% of the effluent hydrocarbon products, respectively. This suggested that at the beginning of the low-temperature (300 °C) reaction, alkene methylation and cracking, the reaction route which has been proposed to be responsible for the formation of higher alkenes, might play a minor role during the induction period of methanol conversion. Meanwhile, the predominant generation of methane (H/C ratio = 4/1) in the volatile phase also implied that some hydrogen-unsaturated organic species formed at the same time and was retained in the catalyst during this period. Therefore, the observation and identification of these possible organic species formed during the induction period over H-ZSM-22 under real reaction conditions would be of great importance to reveal the underlying reaction mechanism. For the reaction performed at 350 °C, besides the alkene products, a small amount of xylene was also generated and appeared among the effluents, while the xylene products were absent in the effluents of the reaction at temperatures lower than 350 °C. Low mobility of aromatics with a relatively big size in the 1-dimensional and 10-membered ring channels at low temperature caused the retention of aromatic products as confined materials over the H-ZSM-22 catalyst.
 |
| | Fig. 6 Product distribution of methanol conversion over H-ZSM-22 catalyst at 300, 320 and 350 °C, WHSV of CH3OH = 2.0 h−1. | |
3.3. Studies of retained organic species in H-ZSM-22 by solid-state NMR and GC-MS
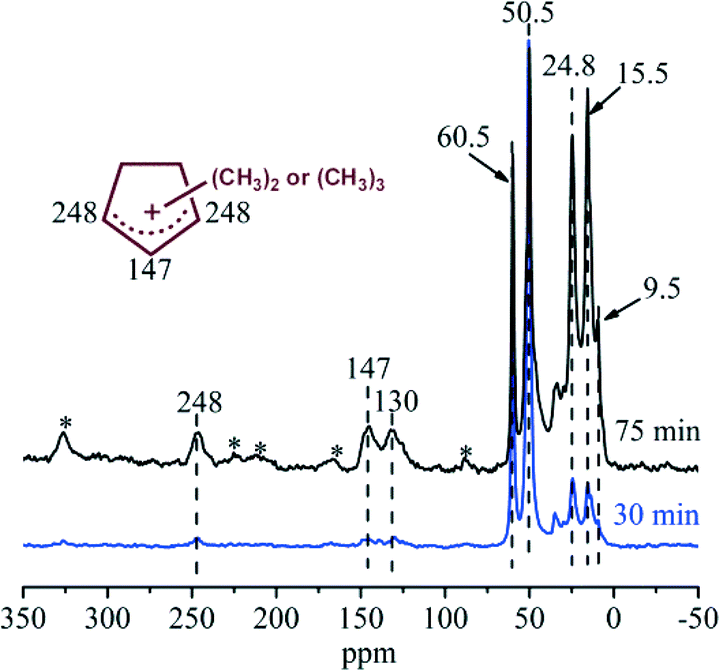
In order to capture the reaction intermediates under real working conditions, continuous-flow 13C-methanol conversion was performed over H-ZSM-22 at 300 °C. After the continuous reaction for 30 and 75 min, the catalyst was cooled by liquid nitrogen, transferred to the rotor in a glove box and then measured by 13C solid-state NMR. According to the 13C CP MAS NMR spectra shown in Fig. 7, the strong signals at 50.5 and 60.5 ppm represented the physisorbed methanol and dimethyl ether in the catalyst.13,14,19,22,23 It was very interesting to observe the resonance peaks with chemical shifts at 248 and 147 ppm in the spectrum recorded after 30 min of reaction, since they hinted the formation of the important five-membered ring cations, the polymethylcyclopentenyl cations, over H-ZSM-22.13,14,22,23 Up to now, it is the first time observation of this important reaction intermediates over H-ZSM-22, the zeolite without intersectional channel or cavity structure. These two peaks from polymethylcyclopentenyl cations were intensified at a TOS of 75 min, corresponding to the enhancement of the methanol conversion during this period. Besides the observation of polymethylcyclopentenyl cations, other cyclic organics, such as methylbenzenes with chemical shifts at 130 and 15.5 ppm also appeared.19 Even the benzenium ions, the protonated form of methylbenzenes, which are usually considered as the most active reaction centres over zeolites with large cages or channel intersection,21–23,25 cannot be captured due to their extremely high reactivity; the evidence from the NMR spectra confirmed the formation of 5-membered ring and 6-membered ring cyclic organic species during the induction period of methanol conversion over H-ZSM-22 at 300 °C. In this period, with the formation of these cyclic organics which were retained over the catalyst, methanol conversion was improved.
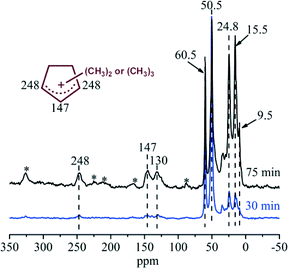 |
| | Fig. 7
13C CP MAS NMR spectra of H-ZSM-22 with retained organics after continuous-flow 13C-methanol reaction for 30 min and 75 min at 300 °C with WHSV of methanol = 2.0 h−1. The asterisk denotes spinning side bands. | |
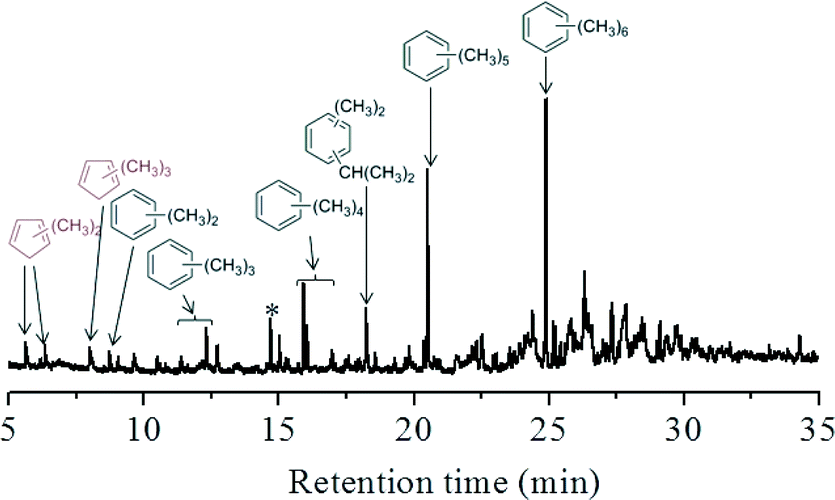
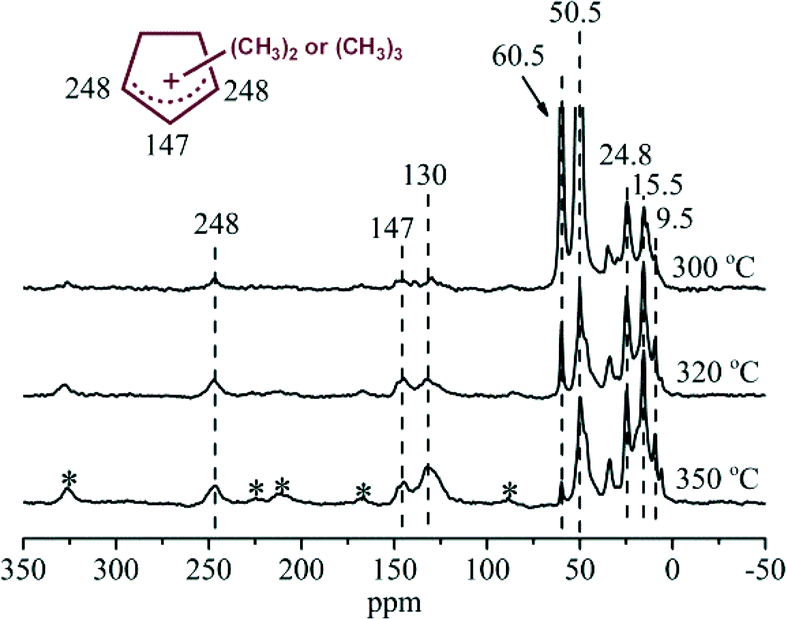
The retained organic compounds in the H-ZSM-22 catalyst were analyzed following the procedure introduced by Guisnet41 and the results are given in Fig. 8. In the GC-MS chromatogram, methylbenzenes were predominantly formed as the retained compounds after 75 min of reaction at 300 °C. Apart from them, the peaks at retention times of 5.6, 6.4 and 8.0 min were assigned to dimethylcyclopentadienes and trimethylcyclopentadienes according to the library of NIST11, and the deprotonated forms of methylcyclopentenyl cations, confirming the formation of the polymethylcyclopentenyl cations detected in the NMR measurement. To the best of our knowledge, this is the first report on the capture and confirmation of dimethylcyclopentenyl and trimethylcyclopentenyl cations and their deprotonated counterparts over H-ZSM-22 during methanol conversion. The 13C CP MAS NMR spectra in Fig. 7 showed an intensification of the signals of polymethylcyclopentenyl cations and methylbenzenes in the H-ZSM-22 catalyst from a reaction time of 30 to 75 min. Methanol conversion was also enhanced during this period. This implied a positive correlation between H-ZSM-22 catalyst reactivity and retained cyclic organic species formation as described in the HCP mechanism.21–23,25 The 13C CP MAS NMR measurements were also performed at various temperatures. The results (shown in Fig. 9) demonstrated that the five-membered ring cations could be observed in a wide temperature range, even at a high temperature of 350 °C; this has been also confirmed in our previous study.22 It was worthy to note that the resonance peak at 130 ppm representing aromatic compounds increased in intensity with temperature increase. Considering the sharp decline of methanol conversion and the retained organic species analysis after the reaction (Fig. 8), the aromatic materials, especially some bulky aromatic species, formed during the reaction at high temperature may cause the catalyst deactivation. The reactivity of the five-membered ring cations, the methylcyclopentenyl cations, would be discussed in the next section by employing 12C/13C-methanol switch experiments.
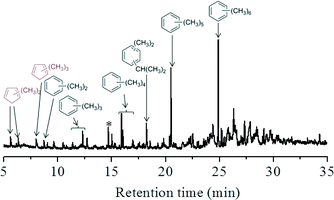 |
| | Fig. 8 GC-MS chromatogram of the extracted organics from H-ZSM-22 catalyst after continuous-flow methanol conversion at 300 °C for 75 min with WHSV of 2.0 h−1. | |
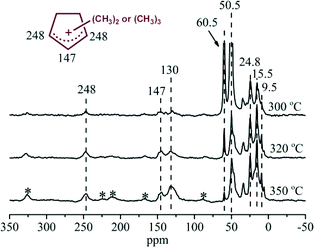 |
| | Fig. 9
13C CP MAS NMR spectra of H-ZSM-22 with retained organics after continuous-flow 13C-methanol reaction for 30 min at 300, 320 and 350 °C with WHSV of methanol = 2.0 h−1. The asterisk denotes spinning side bands. | |
3.4.
12C/13C-methanol switch experiments
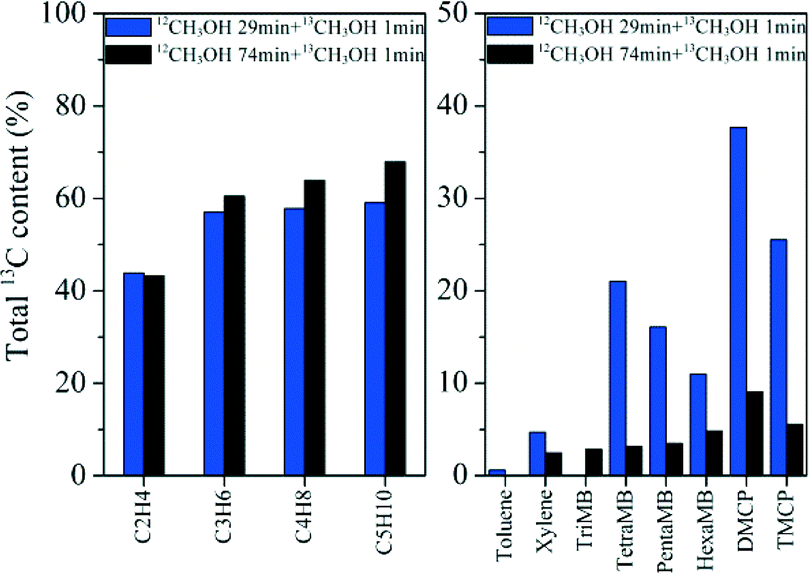
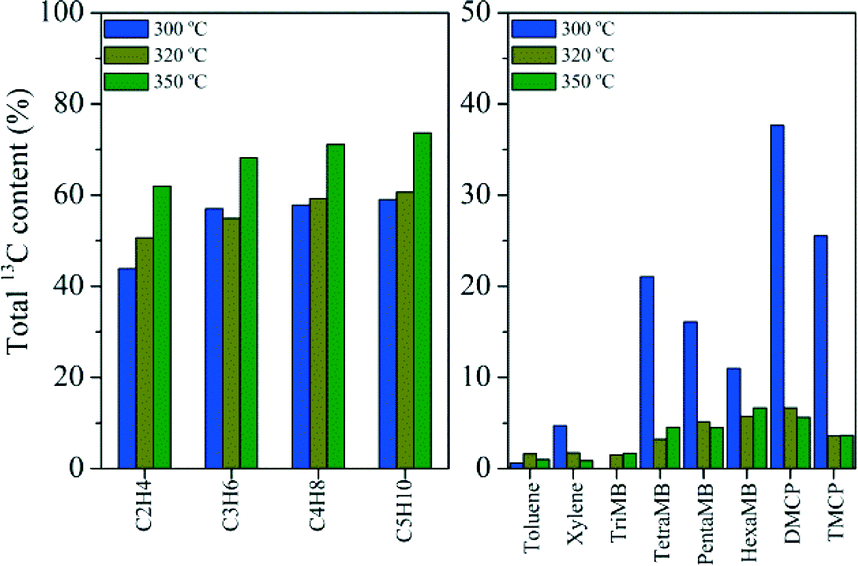
12C/13C-methanol switch experiments were performed to distinguish the active organic species retained in the catalyst and determine their role in olefin generation from methanol conversion.2 The total 13C contents of the effluent alkenes and retained organic materials on the catalyst after the switch experiments are shown in Fig. 10. Among the retained organics, tetramethylbenzene (TetraMB), dimethylcyclopentadiene (DMCP) and trimethylcyclopentadiene (TMCP) exhibited higher 13C content than other organic materials retained in H-ZSM-22 after 30 min of reaction, implying their high reactivity as important intermediates during the induction period of the reaction (Fig. 10). For the effluent products, the total 13C contents presented obvious differences in ethene and C3–C5 alkenes as reported previously.38,39 The 13C content of ethene was lower than that of the other higher alkene products, and close to that of TetraMB, DMCP and TMCP at a TOS of 30 min, implying the participation of these retained active organic species in the formation of ethene during the induction period. The appearance of more 13C atoms in higher alkene products, propene, butenes and pentenes, than ethene (Fig. 10), indicated that higher alkene generation more possibly followed the olefin methylation and cracking route,38,39 while the presence of 12C atoms in these alkene products also implied the contribution of the retained organic compounds in the H-ZSM-22 catalyst in the olefin product formation. With the prolongation of the reaction time to 75 min, the 13C content difference between the effluent hydrocarbons and the retained organic materials became dominant, thus the alkene generation was more possibly via the alkene methylation and cracking route with less involvement of confined organics. More aromatic compound formation in the 10-member ring channel of H-ZSM-22 caused the depression of space-required pathway of olefin generation with bulky cyclic organic species involvement. The retained organics in HZSM-22, such as DMCP, TMCP and TetraMB, became less involved in alkene formation with the prolongation of the reaction time. This change corresponded to the initial predominant formation of ethene and the appearance of the higher alkene products, and the formation of C3–C7 alkenes as the main products with the prolongation of the reaction time. This is also consistent with the reduced reactivity of the confined organics in H-ZSM-22 from a reaction time of 30 to 75 min which was confirmed by the 13C-labeling experiment (Fig. 10).
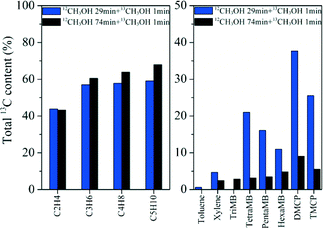 |
| | Fig. 10 The total 13C content of the effluent products (left) and retained organic materials (right) after the 12C/13C switch experiment over H-ZSM-22 at 300 °C with 12C-methanol feeding for 29 min and 74 min, respectively, followed by 1 min of 13C-methanol feeding and a WHSV of 2.0 h−1. | |
Some studies have proved that methanol conversion over H-ZSM-22 mainly follows the alkene methylation and cracking mechanism at high reaction temperatures, such as 400 or 450 °C.34–36 In the 13C CP MAS NMR experiments performed at various temperatures, five-membered ring cations also appeared over the catalyst in the reaction at 320 and 350 °C (Fig. 9). In order to elucidate the effect of reaction temperatures on the importance of alkene and aromatic-based reaction cycles for methanol conversion, 12C/13C-methanol switch experiments were performed and the isotopic distribution results are compared in Fig. 11. Different from the 13C atom scramble in the retained compounds (methylbenzenes and methylcyclopentadienes) and the effluent alkenes after reaction at 300 °C, when the reactions were performed at a relatively higher temperature, such as 320 and 350 °C, the content of the 13C atoms incorporated into the effluent was largely enhanced, meanwhile the 13C content of the retained compounds decreased a lot compared with that at 300 °C, suggesting that methanol conversion at high temperature mainly follows the methylation and cracking route. These results indicated that olefin generation from methanol conversion was very sensitive to reaction temperature. At relatively high temperature, olefin generation followed the olefin methylation and cracking route, while in the low-temperature reaction, the retained organic species over the catalyst would be more reactive and an aromatic-based cycle would be more predominant for olefin generation.
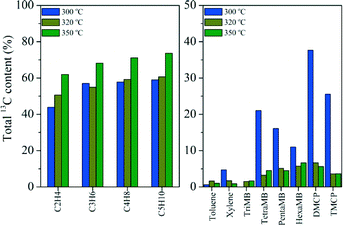 |
| | Fig. 11 The total 13C content of the effluent products (left) and retained organic materials (right) after the 12C/13C switch experiment over H-ZSM-22 at 300, 320 and 350 °C with 12C-methanol feeding for 29 min, followed by 1 min of 13C-methanol feeding and WHSV of 2.0 h−1. | |
3.5. Theoretical calculations
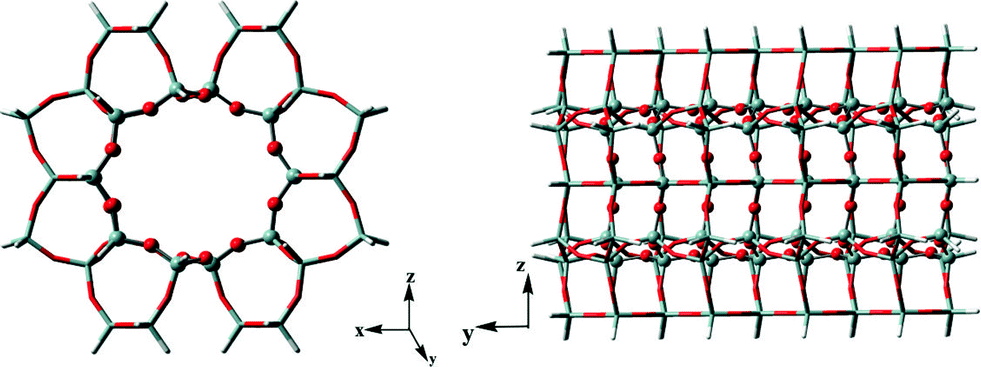
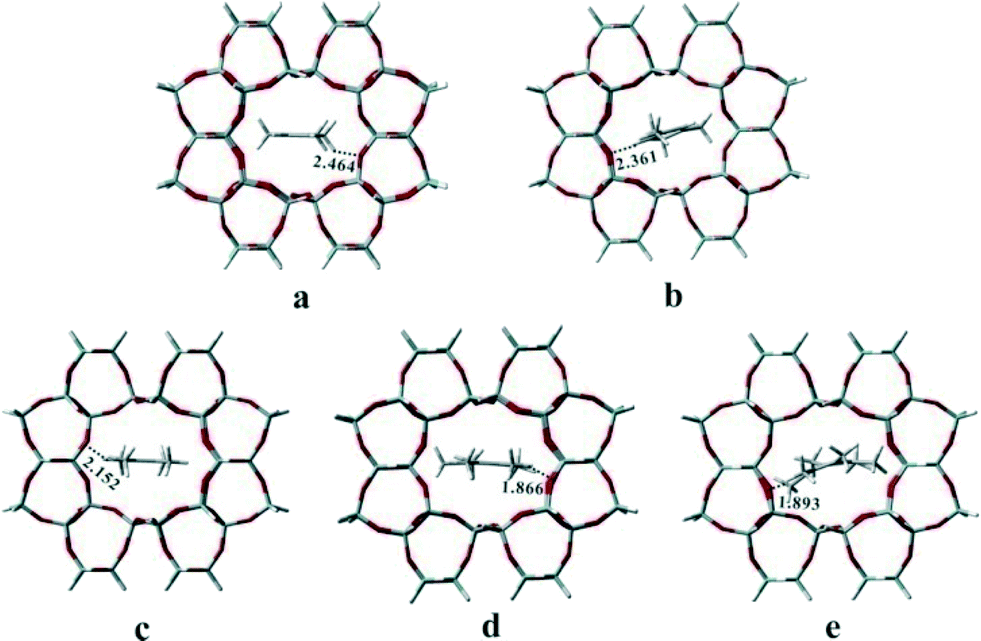
Previous studies have proved that the active HCP species and their function in olefin generation vary with the catalysts used in methanol conversion, indicative of the presence of the confinement effect from the catalyst structure.21–25 In the present work, at the beginning of the methanol reaction performed at 300 °C, the higher reactivity of DMCP, TMCP and TetraMB than that of pentamethylbenzene (PentaMB) and hexamethylbenzene (HexaMB), as presented in the 12C/13C switch experiments, also implied the presence of the spatial confinement effect imposed by the 1-dimensional and 10-membered ring channel of the H-ZSM-22 catalyst on the generation and function of these bulky HCP species. Based on the identification of the retained materials formed during methanol conversion over H-ZSM-22 as shown in Fig. 8, a 126 T cluster model (Fig. 12) representing the zeolite with TON topology was used to explore the accommodation of methylcyclopentadienes and methylbenzenes in ZSM-22 and the theoretical calculation results are detailed in Table 1. The adsorption energies of 1,2-DMCP, 1,3,5-TMCP and 1,2,4,5-TetraMB within TON zeolite were −30.59, −28.24 and −18.59 kcal mol−1, respectively, much lower than that of PentaMB (0.69 kcal mol−1) and HexaMB (47.21 kcal mol−1). Besides, the Gibbs free energies under the experimental conditions (such as 300 °C) of the adsorption of 1,2-DMCP, 1,3,5-TMCP, 1,2,4,5-TetraMB, PentaMB and HexaMB were −3.69, 1.42, 13.47, 32.59 and 84.98 kcal mol−1, respectively, which dramatically increased with the sizes of the adsorbed species in the TON zeolite. This apparently demonstrated the steric confinement repulsion effects between bulky polymethylbenzenes (i.e., PentaMB and HexaMB) and the TON framework. As given in Fig. 13, the closest distances between the hydrogen atoms of the polymethylbenzenes and the oxygen atoms of the zeolite framework decreased from 2.464 Å (1,2-DMCP), 2.361 Å (1,3,5-TMCP), 2.152 Å (1,2,4,5-TetraMB) to 1.866 Å (PentaMB) and 1.893 Å (HexaMB), indicating that the steric interaction between higher polymethylbenzenes and TON framework was much stronger than lighter polymethylbenzenes and polymethylcyclopentadienes. Therefore, the 1,2-DMCP, 1,3,5-TMCP, 1,2,4,5-TetraMB would be more energetically favourable if located inside the ZSM-22 internal channel, while the retained species with large size (i.e., PentaMB and HexaMB) were difficult to be accommodated inside the TON channel, and they were more possibly located near the pore mouth of the 10-membered ring channels towards the external surface.
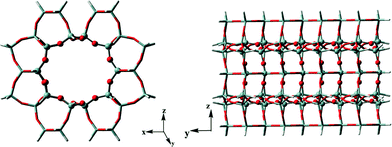 |
| | Fig. 12 A 126 T cluster model used to represent the ZSM-22 catalyst. | |
Table 1 The adsorption energies (ΔEads, with ZPE correction), adsorption enthalpies (ΔHads) and Gibbs free energies (ΔGads) of the adsorbed species within TON zeolite at 300 °C, kcal mol−1
|
|
1,2-DMCP |
1,3,5-TMCP |
1,2,4,5-TetraMB |
PentaMB |
HexaMB |
| ΔEads |
−30.59 |
−28.24 |
−18.59 |
0.69 |
47.21 |
| ΔHads |
−29.95 |
−28.22 |
−18.53 |
0.66 |
45.52 |
| ΔGads |
−3.69 |
1.42 |
13.47 |
32.59 |
84.98 |
| ΔSads |
−0.05 |
−0.05 |
−0.06 |
−0.06 |
−0.07 |
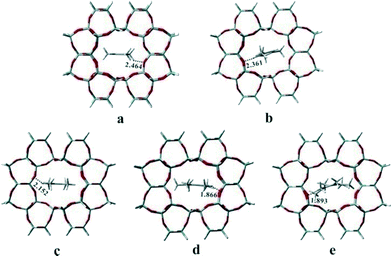 |
| | Fig. 13 Geometry structures of 1,2-dimethylcyclopentadiene (a), 1,3,5-trimethylcyclopentadiene (b),1,2,4,5-tetramethylbenzene (c), pentamethylbenzene (d) and hexamethylbenzene (e) adsorbed in the channel of TON zeolite. The closest distances (Å) between the hydrogen atoms of the polymethylbenzenes and the oxygen atoms of the TON zeolite framework are also given. | |
The results given in Fig. 13 and Table 1 theoretically demonstrated that DMCP, TMCP and TetraMB would be more preferentially accommodated from the viewpoint of thermodynamics than PentaMB and HexaMB inside the internal channel of H-ZSM-22. The function of the bulky species (PentaMB and HexaMB) detected experimentally as intermediates should require a more spacious environment that the 1-dimensional 10-membered ring channels of H-ZSM-22 cannot provide. Due to the spatial confinement effect, cyclopentenyl cations and methylbenzenes with relatively small sizes may behave as the important intermediates in the methanol reaction over H-ZSM-22 for ethene formation in low-temperature methanol conversion; this is also confirmed by their high reactivity presented in the 12C/13C-methanol switch experiment.
3.6. Coke deposition and deactivation
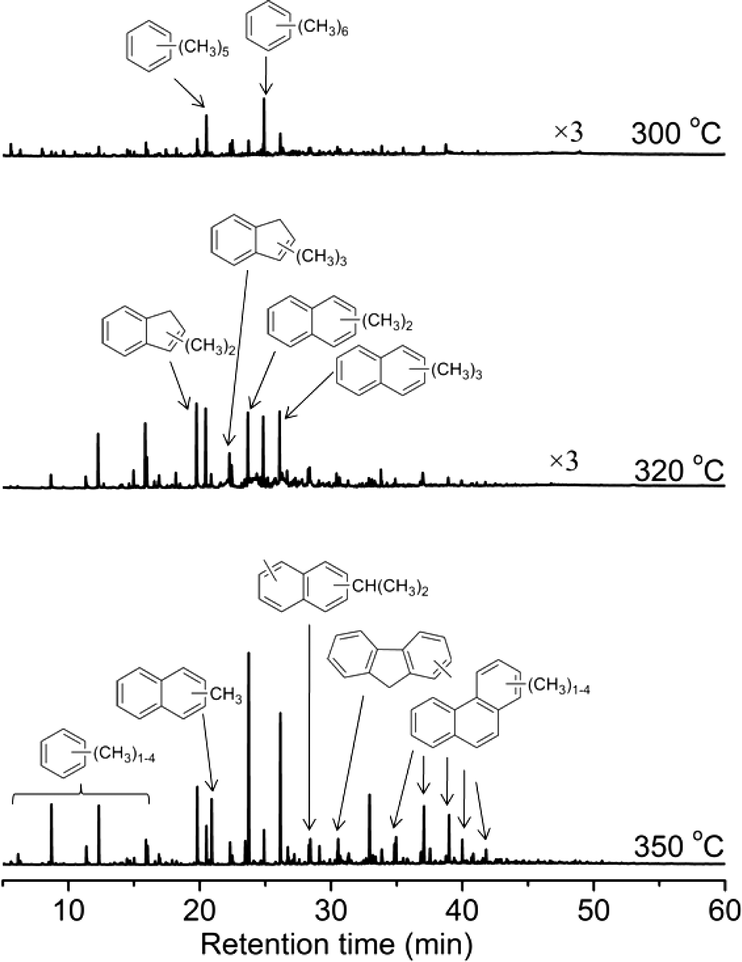
After the long-period reactions, the deactivated catalysts were discharged and destroyed in HF solution (20%), and the deposited coke species were extracted with dichloromethane and analyzed by GC-MS. Fig. 14 displays the GC-MS chromatograms after MTO reaction for 8 h at 300, 320 and 350 °C. Pentamethylbenzene and hexamethylbenzene were the dominant species retained in the deactivated catalysts at 300 °C. The isotopic switch experiment and theoretical calculations have confirmed their low reactivity during methanol conversion over the H-ZSM-22 zeolite. Under the condition of low temperature, the alkene methylation and cracking route was also not an efficient way for methanol conversion,49 therefore the low methanol conversion and catalyst deactivation occurred during the methanol reaction at 300 °C. For the reaction performed at 320 and 350 °C, methyl-substituted benzene, 1H-indene, naphthalene and phenanthrene derivatives were largely formed. The intensity of these coke species increased with the reaction temperature. The predominant formation of these bulky aromatic hydrocarbons, such as polyaromatics, resulting in the blockage of zeolite channel and coverage of acid sites, was responsible for the rapid deactivation of HZSM-22 at relatively high temperatures.37
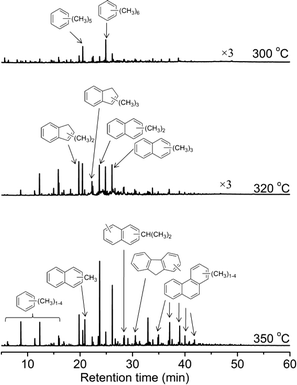 |
| | Fig. 14 GC-MS chromatograms of the organic species retained in the deactivated H-ZSM-22 after MTO reaction for 8 h at 300, 320 and 350 °C, WHSV of CH3OH = 2.0 h−1. | |
Conclusions
In summary, methanol conversion and olefin generation over HZSM-22 were studied, and for the first time the induction period and five-membered ring cations, the polymethylcyclopentenyl cations, were observed over H-ZSM-22 during methanol conversion at low temperature. The formation of the methylcyclopentenyl cations and their deprotonated products, the methylcyclopentadienes, was confirmed with the aid of 13C MAS NMR and GC-MS. The evidence from the isotopic switch experiment proved the involvement of the methylbenzenes and methylcyclopentadienes in alkene generation during methanol conversion at 300 °C. Even the alkene methylation and cracking route was important for alkene generation over the one-dimensional zeolite catalyst, H-ZSM-22; the HCP mechanism cannot be ruled out from methanol conversion, especially for ethene generation at the beginning of MTO reaction. Due to the spatial confinement effect, cyclopentenyl cations and methylbenzenes with relatively small sizes may behave as the important intermediates over H-ZSM-22 in the methanol reaction at low temperature. Olefin generation from the methanol reaction at relatively high temperatures of 320 and 350 °C mainly followed the alkene methylation and cracking route, which was also confirmed by the isotopic switch experiment. The quick deactivation of HZSM-22 resulted from the blocking of the 10-membered ring and 1-dimensional channels with the formation and deposition of the bulky aromatic compounds over the catalyst.
Acknowledgements
We thank the financial support from the National Natural Science Foundation of China (21473182, 21273005, 21273230, 21210005, 21173255 and 21103180) and the laboratory of DNL0802 for their assistance in catalyst supply.
Notes and references
- M. Stöcker, Microporous Mesoporous Mater., 1999, 29, 3–48 CrossRef.
- U. Olsbye, S. Svelle, M. Bjorgen, P. Beato, T. V. Janssens, F. Joensen, S. Bordiga and K. P. Lillerud, Angew. Chem., Int. Ed., 2012, 51, 5810–5831 CrossRef CAS PubMed.
- P. Tian, Y. Wei, M. Ye and Z. Liu, ACS Catal., 2015, 5, 1922–1938 CrossRef CAS.
- Y. Wei, D. Zhang, Z. Liu and B.-L. Su, Chin. J. Catal., 2012, 33, 11–21 CrossRef CAS.
- T. Xu, H. Song, W. P. Deng, Q. H. Zhang and Y. Wang, Chin. J. Catal., 2013, 34, 2047–2056 CrossRef CAS.
- L. Wu, Z. Y. Liu, L. Xia, M. H. Qiu, X. Liu, H. J. Zhu and Y. H. Sun, Chin. J. Catal., 2013, 34, 1348–1356 CrossRef CAS.
- J. F. Haw, W. Song, D. M. Marcus and J. B. Nicholas, Acc. Chem. Res., 2003, 36, 317–326 CrossRef CAS PubMed.
- D. Lesthaeghe, V. Van Speybroeck, G. B. Marin and M. Waroquier, Chem. Phys. Lett., 2006, 417, 309–315 CrossRef CAS.
- D. Lesthaeghe, V. Van Speybroeck, G. B. Marin and M. Waroquier, Angew. Chem., Int. Ed., 2006, 45, 1714–1719 CrossRef CAS PubMed.
- I. M. Dahl and S. Kolboe, Catal. Lett., 1993, 20, 329–336 CrossRef CAS.
- I. M. Dahl and S. Kolboe, J. Catal., 1994, 149, 458–464 CrossRef CAS.
- I. M. Dahl and S. Kolboe, J. Catal., 1996, 161, 304–309 CrossRef CAS.
- P. W. Goguen, T. Xu, D. H. Barich, T. W. Skloss, W. Song, Z. Wang, J. B. Nicholas and J. F. Haw, J. Am. Chem. Soc., 1998, 120, 2650–2651 CrossRef CAS.
- J. F. Haw, J. B. Nicholas, W. Song, F. Deng, Z. Wang, T. Xu and C. S. Heneghan, J. Am. Chem. Soc., 2000, 122, 4763–4775 CrossRef CAS.
- M. Bjørgen, U. Olsbye, D. Petersen and S. Kolboe, J. Catal., 2004, 221, 1–10 CrossRef.
- W. Song, J. F. Haw, J. B. Nicholas and C. S. Heneghan, J. Am. Chem. Soc., 2000, 122, 10726–10727 CrossRef CAS.
- B. Arstad and S. Kolboe, J. Am. Chem. Soc., 2001, 123, 8137–8138 CrossRef CAS PubMed.
- J. F. Haw and D. M. Marcus, Top. Catal., 2005, 34, 41–48 CrossRef CAS.
- W. Wang, Y. Jiang and M. Hunger, Catal. Today, 2006, 113, 102–114 CrossRef CAS.
- B. P. C. Hereijgers, F. Bleken, M. H. Nilsen, S. Svelle, K.-P. Lillerud, M. Bjørgen, B. M. Weckhuysen and U. Olsbye, J. Catal., 2009, 264, 77–87 CrossRef CAS.
- J. Li, Y. Wei, J. Chen, P. Tian, X. Su, S. Xu, Y. Qi, Q. Wang, Y. Zhou, Y. He and Z. Liu, J. Am. Chem. Soc., 2012, 134, 836–839 CrossRef CAS PubMed.
- S. Xu, A. Zheng, Y. Wei, J. Chen, J. Li, Y. Chu, M. Zhang, Q. Wang, Y. Zhou, J. Wang, F. Deng and Z. Liu, Angew. Chem., Int. Ed., 2013, 52, 11564–11568 CrossRef CAS PubMed.
- C. Wang, Y. Chu, A. Zheng, J. Xu, Q. Wang, P. Gao, G. Qi, Y. Gong and F. Deng, Chem. – Eur. J., 2014, 20, 12432–12443 CrossRef CAS PubMed.
- J. Chen, J. Li, C. Yuan, S. Xu, Y. Wei, Q. Wang, Y. Zhou, J. Wang, M. Zhang, Y. He, S. Xu and Z. Liu, Catal. Sci. Technol., 2014, 4, 3268–3277 CAS.
- M. J. Wulfers and F. C. Jentoft, ACS Catal., 2014, 4, 3521–3532 CrossRef CAS.
- S. J. Kim, H. G. Jang, J. K. Lee, H. K. Min, S. B. Hong and G. Seo, Chem. Commun., 2011, 47, 9498–9500 RSC.
- H.-G. Jang, H.-K. Min, S. B. Hong and G. Seo, J. Catal., 2013, 299, 240–248 CrossRef CAS.
- G. Seo, J.-H. Kim and H.-G. Jang, Catal. Surv. Asia, 2013, 17, 103–118 CrossRef CAS.
- J. Li, Y. Wei, J. Chen, S. Xu, P. Tian, X. Yang, B. Li, J. Wang and Z. Liu, ACS Catal., 2015, 5, 661–665 CrossRef CAS.
- Z.-M. Cui, Q. Liu, W.-G. Song and L.-J. Wan, Angew. Chem., Int. Ed., 2006, 45, 6512–6515 CrossRef CAS PubMed.
- Q. Wang, Z.-M. Cui, C.-Y. Cao and W.-G. Song, J. Phys. Chem. C, 2011, 115, 24987–24992 CAS.
- Z. M. Cui, Q. Liu, Z. Ma, S. W. Bian and W. G. Song, J. Catal., 2008, 258, 83–86 CrossRef CAS.
- S. Teketel, S. Svelle, K.-P. Lillerud and U. Olsbye, ChemCatChem, 2009, 1, 78–81 CrossRef CAS.
- S. Teketel, U. Olsbye, K.-P. Lillerud, P. Beato and S. Svelle, Microporous Mesoporous Mater., 2010, 136, 33–41 CrossRef CAS.
- J. Li, Y. Wei, Y. Qi, P. Tian, B. Li, Y. He, F. Chang, X. Sun and Z. Liu, Catal. Today, 2011, 164, 288–292 CrossRef CAS.
- J. Li, Y. Wei, G. Liu, Y. Qi, P. Tian, B. Li, Y. He and Z. Liu, Catal. Today, 2011, 171, 221–228 CrossRef CAS.
- J. Wang, J. Li, S. Xu, Y. Zhi, Y. Wei, Y. He, J. Chen, M. Zhang, Q. Wang, W. Zhang, X. Wu, X. Guo and Z. Liu, Chin. J. Catal., 2015, 36, 1392–1402 CrossRef CAS.
- S. Svelle, F. Joensen, J. Nerlov, U. Olsbye, K.-P. Lillerud, S. Kolboe and M. Bjørgen, J. Am. Chem. Soc., 2006, 128, 14770–14771 CrossRef CAS PubMed.
- M. Bjorgen, S. Svelle, F. Joensen, J. Nerlov, S. Kolboe, F. Bonino, L. Palumbo, S. Bordiga and U. Olsbye, J. Catal., 2007, 249, 195–207 CrossRef CAS.
-
A. George, G. K. Olah, S. Prakash, J. Sommer and A. Molnar, Superacid Chemistry, Wiley, 2nd edn, 2009 Search PubMed.
- M. Guisnet, L. Costa and F. R. Ribeiro, J. Mol. Catal. A: Chem., 2009, 305, 69–83 CrossRef CAS.
- D. Lesthaeghe, B. De Sterck, V. Van Speybroeck, G. B. Marin and M. Waroquier, Angew. Chem., Int. Ed., 2007, 46, 1311–1314 CrossRef CAS PubMed.
- H. Fang, A. Zheng, J. Xu, S. Li, Y. Chu, L. Chen and F. Deng, J. Phys. Chem. C, 2011, 115, 7429–7439 CAS.
- X. Yi, Y. Byun, Y. Chu, A. Zheng, S. B. Hong and F. Deng, J. Phys. Chem. C, 2013, 117, 23626–23637 CAS.
- A. Ghysels, T. Verstraelen, K. Hemelsoet, M. Waroquier and V. Van Speybroeck, J. Chem. Inf. Model., 2010, 50, 1736–1750 CrossRef CAS PubMed.
- J. Van der Mynsbrugge, J. De Ridder, K. Hemelsoet, M. Waroquier and V. Van Speybroeck, Chem. – Eur. J., 2013, 19, 11568–11576 CrossRef CAS PubMed.
- Y. Wei, D. Zhang, F. Chang and Z. Liu, Catal. Commun., 2007, 8, 2248–2252 CrossRef CAS.
- Y. Wei, C. Yuan, J. Li, S. Xu, Y. Zhou, J. Chen, Q. Wang, L. Xu, Y. Qi, Q. Zhang and Z. Liu, ChemSusChem, 2012, 5, 906–912 CrossRef CAS PubMed.
- S. Ilias, R. Khare, A. Malek and A. Bhan, J. Catal., 2013, 303, 135–140 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.