
The graphitic carbon strengthened synergetic effect between Pt and FeNi in CO preferential oxidation in excess hydrogen at low temperature†
Limin
Chen
*ab,
Yunfeng
Bao
a,
Yuhai
Sun
a,
Ding
Ma
bc,
Daiqi
Ye
a and
Bichun
Huang
a
aGuangdong Provincial Key Laboratory of Atmospheric Environment and Pollution Control, School of Environment and Energy, South China University of Technology, Guangzhou 510006, China. E-mail: liminchen@scut.edu.cn; Tel: +86 20 39380516
bState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China
cCollege of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China
First published on 21st September 2015
Abstract
A variety of PtFeNi catalysts supported on carbon materials have been prepared and tested for CO preferential oxidation (PROX) in excess hydrogen. 100% O2 and CO conversions have been achieved over carbon black (CB) and carbon nanotube (CNT) supported PtFeNi catalysts at room temperature in a feed gas containing 1% CO, 0.5% O2 (volume ratio) and H2 balance gas. N2 adsorption, temperature-programmed desorption (TPD) and transmission electron microscopy (TEM) studies indicate that the carbon textural properties and surface chemistry determine the catalyst particle size distribution and mean size; but the mean particle size does not have a great influence on the catalytic performance within the investigated particle size range. X-ray diffraction (XRD), resistance measurements and the designed catalytic reaction results reveal the ability of graphitic carbon to capture and shuttle electrons from the noble metal to spatially different sites in the FeNi species through the π–π network, enables the indirect interactions between Pt and the FeNi species, leading to a strengthened synergistic effect, enhancing the CO oxidation activity at room temperature, increasing the Pt utilization efficiency, and apparently decreasing the Pt loading level.
1. Introduction
Proton exchange membrane fuel cells (PEMFCs) have been considered to be one of the most promising candidates to replace conventional fossil fuels, due to their high energy efficiency, environmentally-friendly characteristics and low operating temperature.1,2 However, most commercial hydrogen fuels are generally produced through various hydrocarbons or bio-alcohols, from fossil fuels or biomass, via the steam reforming and water-gas shift (WGS) reactions. The resultant H2-rich gases generally contain 0.5–1.0 vol% of CO due to the thermodynamic limitation of the WGS reaction.1–22 Unfortunately, a small amount of CO severely poisons the anode catalyst of the PEMFCs, therefore, reducing the concentration of CO to ppm level is required prior to the introduction of hydrogen to the PEMFCs.1–22 Among the various methods investigated, low temperature preferential oxidation of CO in excess H2 streams (PROX) is presently regarded as one of the most promising and cost-effective ways.1–22Among the various catalysts, Pt-based catalysts especially Fe,5–8,20,21 Co,9–14 Ni,14–19,21 Cu,11,22 and Ag23etc. promoted Pt catalysts are the most promising candidates. However, it is extremely difficult to realize the total conversion of CO at ambient temperatures with reasonable costs, which is particularly important for fuel cell applications in transportation.10,13,17,20 To the best of our knowledge, among all the supported Pt catalysts reported in the literature, there are only a few supported Pt catalysts that can accomplish the total conversion of CO at room temperature or even lower,4,5,21 mainly including our PtFe/SiO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 5 and later developed PtFeNi/CNT21 catalysts.
5 and later developed PtFeNi/CNT21 catalysts.
In order to find effective, low-cost and highly robust Pt-based catalysts, besides the promotion effects of promoters, the effects of various supports on the catalytic performance have been extensively investigated in the promoted catalytic system, including CeO2,11 TiO2,12,14 SiO2,4,5,14 Al2O3,8,11,22 zeolites10 and carbon materials6,9,13–21etc. The results indicated that some supports could help to distribute the active component uniformly,17 and some could interact with the active component24 or influence the chemical state of the active component.12 Especially, Petkov et al. reported the role of support–nanoalloy (PtCoNi) interactions on CO oxidation.14 Their investigation indicated that by controlled thermo-chemical treatment, oxygen activation sites on TiO2 supported nanoalloys could be provided both by the second/third metal sites in the nanoalloy (Type-I sites) and by the anionic oxygen deficiency sites located at the nanoalloy-support perimeter zone (Type-II sites). However, only Type-I sites can be obtained for nanoalloys on carbon supports; neither can be generated on SiO2 supported nanoalloys, resulting in the lowest activity for the silica-supported nanoalloys.
Petkov's research is based on the assumption that the promotion effects of base transition metals originates from Pt-nanoalloys. However, our extensive investigation demonstrated that the coordinatively unsaturated transition metal cations confined in nanosized Pt matrices were the main oxygen-activation sites.5–7,9,21 Therefore, Petkov's discovery could not explain the extremely high activity of SiO2 and carbon materials supported PtM (M = Fe, Ni, Co) catalysts for CO PROX and CO oxidation.5–7,9,13,21 Our previous research indicates that a 3 wt% PtFeNi/CNTs catalyst is extremely highly active, and can almost totally remove CO in the gas composition of 1 vol% CO, 0.5 vol% O2 and H2 balance even at 6 °C.21 Compared with other highly active catalysts,5,6,9,13 this catalyst consists of an evidently lower Pt loading level, operating with stoichiometric O2, a much higher concentration of H2 and nearly the same gas hourly space velocity. Our further investigation indicates that in situ formed coordinatively unsaturated FeOx and/or NiOx species confined in Pt matrices are active species and the over oxidation of Fe and Pt species seriously deactivates the catalysts.21 These discoveries are similar to those found with our previously reported PtFe/SiO2 catalysts;5 then, what leads to the extremely high activity of PtFeNi/CNTs even with apparently lower Pt loadings? Is it due to the unique properties of CNTs or the general properties of carbon?
Recently, the ability of carbon nanotubes and graphene-based systems to capture and shuttle electrons through the π–π network has been confirmed through spectroscopic studies.25,26 This feature enables the incorporation of a semiconductor and Pt nanoparticles into a single graphene or reduced graphene oxide sheet, and the resulting multifunctional photocatalyst has been demonstrated for use in photocatalytic H2 production.27,28 This photocatalyst can carry out selective catalytic processes at separate sites and tune the selectivity and efficiency of photo-catalytic reduction and oxidation processes, independently. As far as our highly active PtFeNi/CNTs catalyst is concerned, do CNTs possess the ability to capture electrons and shuttle them, and will this ability contribute to the high catalytic activity or not?
In order to answer the above questions and reveal the origin of the extremely high activity of the PtFeNi/CNTs catalyst compared with PtFe/SiO2 and other catalysts, four types of carbon: CNTs, carbon black (CB) with high graphitization, oxidized carbon black (CB-o), and activated carbon (AC) with less graphitization, were used to prepare PtFeNi catalysts and the effects of their catalytic properties on CO PROX were tested. CB can be utilized to exclude the unique properties of CNTs, for instance, confinement effects; CB-o can be used to investigate the negative effects of carbon surface oxidation on the ability of carbon to capture electrons and shuttle them; AC can facilitate the deliberation of the roles of the graphitization degree and the well-developed pore structure on the catalytic performance. Therefore, the effect of the properties on the catalytic performance were investigated through N2 adsorption, TPD, TEM, XRD and resistance measurements. The discovery from this research will further direct us to design and prepare more effective, economical and robust catalysts for CO PROX in H2 rich streams.
2. Experimental
2.1. Materials
Multi-walled carbon nanotubes with a 4–8 nm i.d. and a 10–20 nm o.d. were purchased from Chengdu Organic Chemicals Co., Ltd., China, which contained 0.41 wt% Fe and 0.35 wt% Ni, denoted as CNTs(FeNi). The CNTs(FeNi) purification was conducted using a procedure from the literature,21 and the obtained sample was labeled as CNTs-p. Carbon black was purchased from Acros, and marked as CB. CB was oxidized in 5% O2 balanced in He (volume ratio) at 600 °C for 3 h, and the obtained material was denoted as CB-o. Commercial granulated activated carbon (sieved through a 40–60 mesh) was obtained from Beijing Guanghua Woods Ltd., Beijing, China. The carbon was washed with boiling aqueous nitric acid (0.001 N) and dried at 120 °C overnight. Then, the sample was further treated in He at 600 °C for 3 h and the obtained sample was denoted as AC.2.2. Catalyst preparation
Pt catalysts were prepared using the conventional wetness impregnation method at room temperature using an ethanol solution of hexachloroplatinic acid (Shenyang Chemical Reagent Company, AR) according to the metal loading. The samples were further dried at 120 °C for 12 h. The Fe and Ni promoted catalysts were prepared using the sequential wetness impregnation method. For example, for the FeNi(H500)Pt catalyst, the carbon support was first impregnated with an ethanol solution of a ferric nitrate and nickel nitrate mixture followed by drying at 120 °C for 12 h and then pretreated in H2 at 500 °C for 2 h, and finally impregnated with an ethanol solution of hexachloroplatinic acid. For all PtFeNi catalysts, the loadings of Pt, Fe and Ni were 3, 0.41 and 0.35 wt% (atomic ratio, 15:7:6), respectively; for the PtFe and PtNi catalysts, the loadings of Fe and Ni were 0.75 and 0.81 wt% (the same atomic ratio, 15:13), respectively. The catalysts were pretreated in H2 at 500 °C for 2 h before the catalytic tests.
2.3. Catalytic reaction test
The catalytic reaction tests were performed in a fixed bed flow reactor. A small quartz tube containing a thermocouple was placed in the middle of the catalyst bed. Generally, 60 mg of the catalyst sample was used. A gas mixture containing 1% CO and 0.5% O2 (volume ratio) in H2 was fed at a flow rate of 25 ml min−1, with a gas hourly space velocity of 25000 ml g−1 h−1. The composition of the effluent gas was monitored on-line with a gas chromatograph (Agilent Technologies GC-6890N) equipped with PN and TDX-01 columns. The CO conversion was calculated from the change in CO concentration, and the selectivity towards CO2 was obtained from the O2 mass balance. The details were elaborated in our previous publications.29,30
2.4. Characterization
The texture properties of the carbon supports were determined using N2 adsorption–desorption isotherms at 77 K using an AS-1-MP adsorption instrument. Samples were degassed at 383 K overnight before the measurements.29 The specific surface areas were calculated using the BET equation, and the t-plot method was used to calculate the micropore volumes and areas.29 The temperature-programmed desorption (TPD) experiments were carried out in a quartz U type microreactor, connected with an on-line quadruple mass spectrometer (Balzers, OmniStar GSD300 O). The samples (60 mg) were first flushed with He (30 ml min−1) at room temperature for 2 h. Subsequently, the temperature was increased to 990 °C at a rate of 5 °C min−1. MS intensities for 2 (H2), 4 (He), 28 (CO), and 44 (CO2) were measured as a function of temperature.Transmission electron microscopy (TEM, TECNAI Spirit) was used to study the size of the metal particles. XRD measurements were carried out on a Rigaku D/Max 2500 diffractometer with a Cu Kα monochromatized radiation source at 40 kV and 250 mA. The sheet resistivities were measured by a conventional SZ-82 four-probe instrument (Suzhou Tel. Apparatus Co., China).
3. Results and discussion
3.1. Reaction results
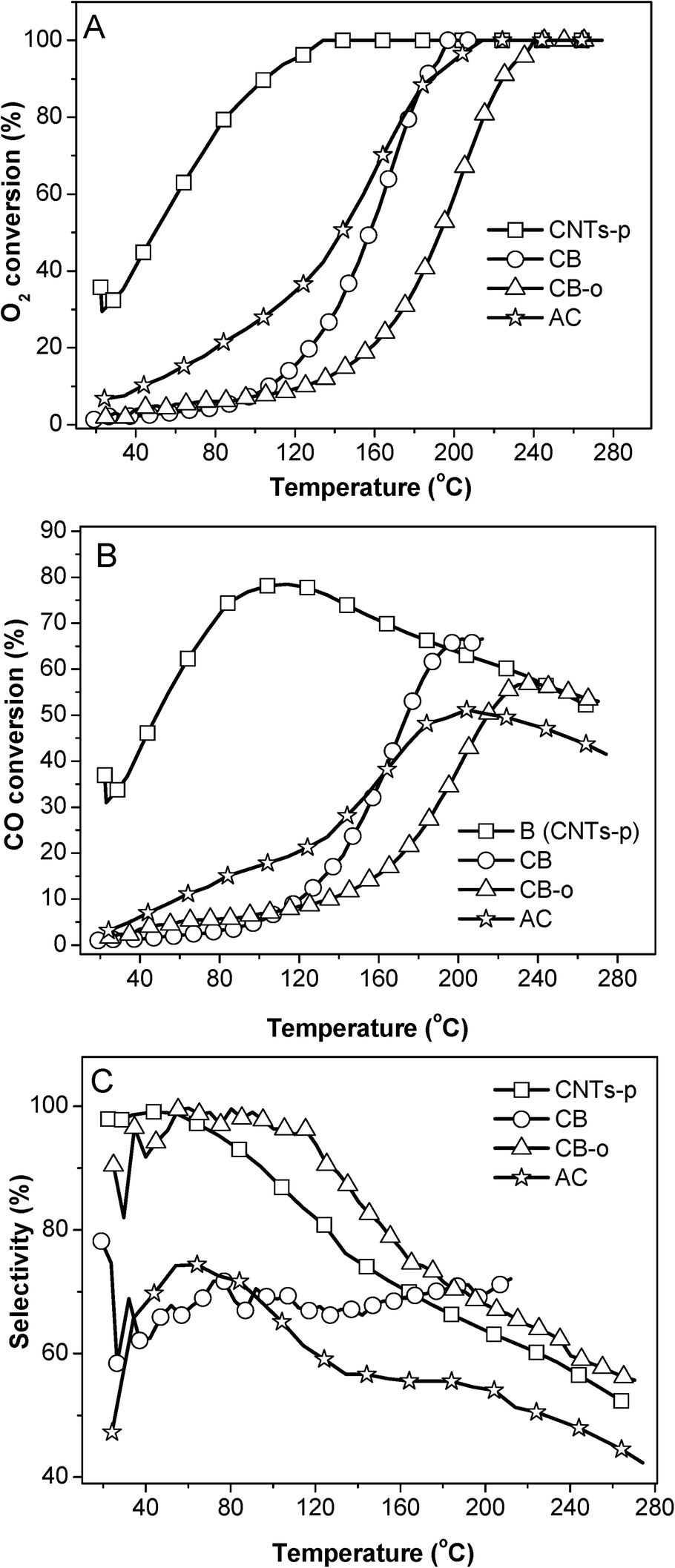 | ||
| Fig. 1 Variation in the O2 conversion (A), CO conversion (B), and selectivity (C) over different carbon supported 3 wt% Pt catalysts as a function of the reaction temperature. | ||
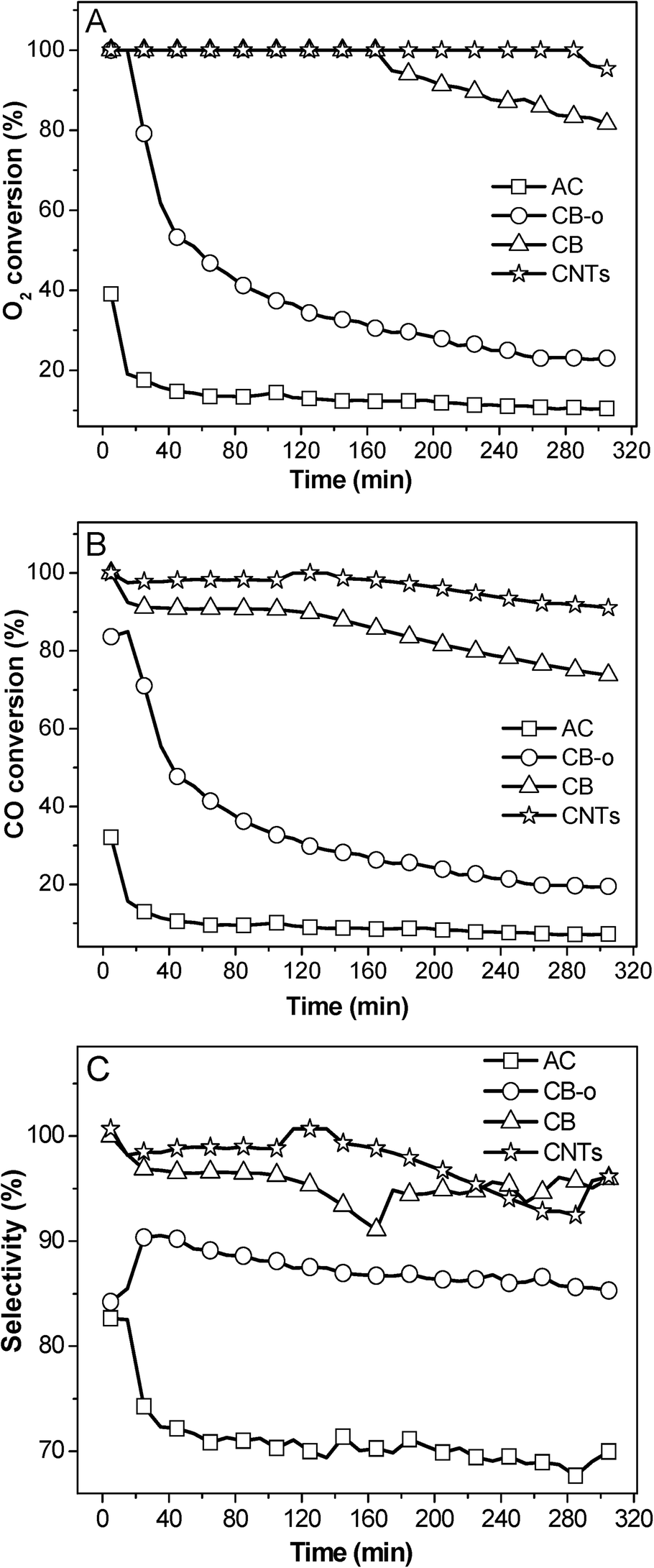 | ||
| Fig. 2 Variation in the O2 conversion (A), CO conversion (B), and selectivity (C) at 23 °C over different carbon supported Fe and Ni promoted Pt catalysts as a function of the reaction time. | ||
These results indicate that the addition of Fe and Ni can dramatically enhance the catalytic activity of Pt catalysts; additionally, the catalysts can be deactivated to some extent, which has been extensively investigated in our previous publications.5,21 In addition, carbon supports also play important roles in the catalytic performance. On one hand the same PtFeNi catalysts with different carbon supports exhibited different catalytic performances; on the other hand, Pt and PtFeNi catalysts with the same carbon supports gave different catalytic activity trends. For example, the Pt/AC catalyst presented a significantly higher activity (O2 conversion at the same temperature) than Pt/CB-o; on the contrary, the PtFeNi/AC catalyst showed a significantly lower activity (O2 conversion at 23 °C) than PtFeNi/CB-o. Furthermore, it is interesting that all PtFeNi catalysts exhibit relatively stable selectivities although there is some loss in O2 and CO reactivity. In order to elucidate the effects of the carbon supports on the catalytic performance, the catalysts have been investigated by a number of characterization methods (vide infra).
3.2. Characterization results
3.2.1. Textural properties and surface chemistry of the carbon supports
The nitrogen adsorption/desorption isotherms of the different carbon supports are shown in the ESI† (Fig. S1) and the corresponding textural parameters are listed in Table 1. The isotherm profile of AC is a typical IV type with rich micropores and mesopores; the calculated specific surface area and mean pore diameter (MPD) are 1158.0 m2 g−1 and 2.1 nm, respectively. The isotherm profile of CB-o is similar to that of AC but has many more mesopores and fewer micropores. For CB-o, the calculated specific surface area and MPD are 210.2 m2 g−1 and 11.2 nm, respectively. In comparison with CB-o, CB has much fewer mesopores and micropores and the corresponding specific surface area and MPD are 75.8 m2 g−1 and 6.5 nm, respectively. For CNTs, the calculated specific surface area and inner pore size are 172.2 m2 g−1 and 4–8 nm, respectively.Temperature-programmed desorption (TPD) in an inert gas (He, Ar, N2) is effective in determining the different oxygen-containing groups of carbon materials.29–34 The TPD curves of CO and CO2 from different carbon supports are shown in the ESI† (Fig. S2) and the corresponding integration areas of the CO and CO2 desorption from TPD are listed in Table 1. It is clear that AC has many more acidic oxygen containing functional groups, CB-o possesses a moderate amount of oxygen containing functional groups, CNTs hold fewer oxygen containing functional groups compared with CB-o and AC, and CB hardly bears any oxygen containing functional groups.
3.2.2. Particle sizes of different carbon supported PtFeNi catalysts
Fig. 3 presents the transmission electron microscopy (TEM) images and the corresponding particle size distributions of the different carbon supported PtFeNi catalysts after activation in H2 at 500 °C for 2 h. The PtFeNi particle size distribution on AC (1.5–6 nm) is a little broader than that on the other three supports. The average particle size of the PtFeNi/AC catalyst is about 3.7 nm, which is much bigger than that of the others. The particle size distribution of the PtFeNi/CB-o catalyst is very narrow with an average particle size of 1.6 nm. The PtFeNi/CB catalyst mainly consists of particles with a diameter from 2 to 3 nm, giving an average particle size of about 2.5 nm. The Pt/CNTs(FeNi) catalyst exhibits a very narrow particle size distribution, giving an average particle size of about 1.6 nm. The average particle size of the four catalysts decreased in the following order: PtFeNi/AC > PtFeNi/CB > PtFeNi/CB-o ≈ Pt/CNTs(FeNi).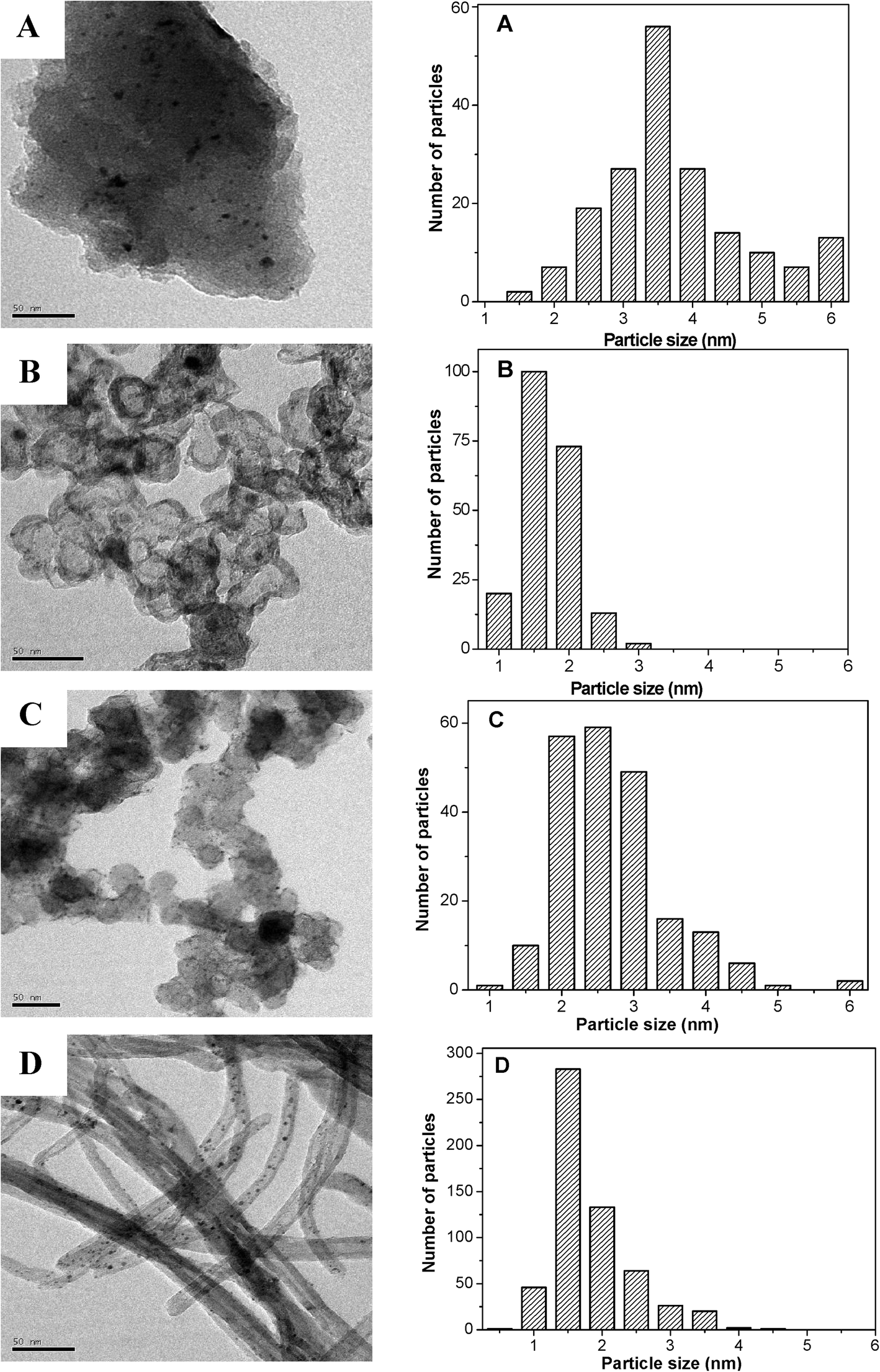 | ||
| Fig. 3 TEM images (left) and particle size distributions (right) of different carbon supported Fe and Ni promoted Pt catalysts after activation in H2 at 500 °C for 2 h. (A) AC; (B) CB-o; (C) CB, and (D) CNTs. | ||
3.3. Discussion
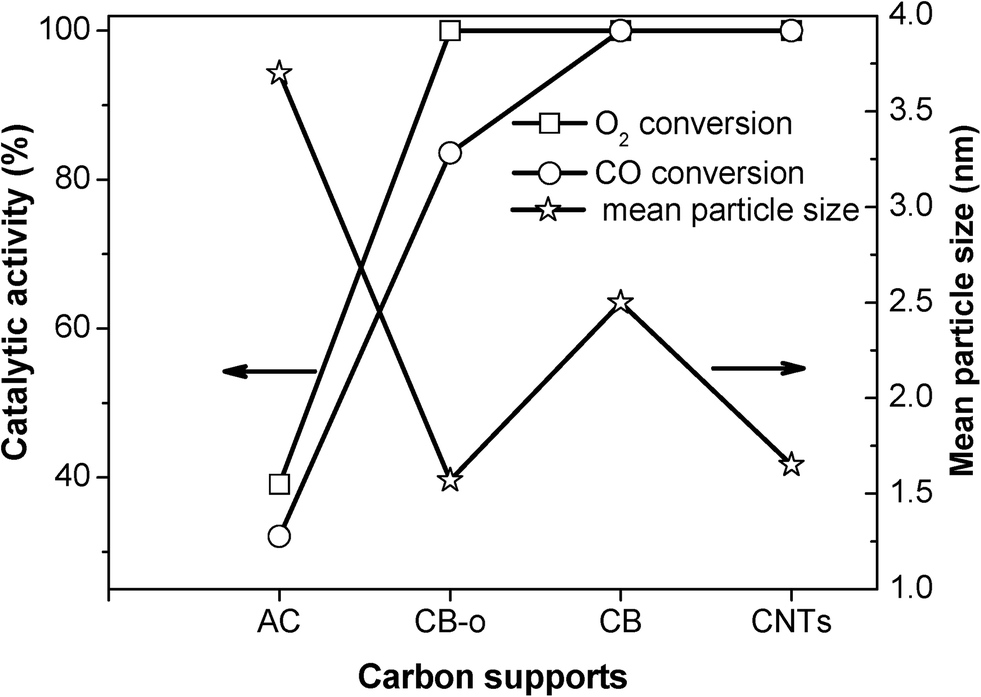 | ||
| Fig. 4 The mean particle sizes and catalytic activities of Fe and Ni promoted Pt catalysts at 23 °C as a function of the different carbon supports. | ||
The graphitization degree of carbon materials can be reflected using XRD characterization, as shown in Fig. 5. The diffraction peaks at 25.78, 42.88, 53.58 and 78.18° are characteristic of graphitic carbon (JCPDS 656212). The strong and sharp diffraction peaks for the CNTs verify the high graphitization degree of the CNT sample, which is essentially composed of coaxial graphitic cylinders. CB and CB-o show similar but slightly broadened diffraction peaks with a slightly lower intensity compared to that of the CNTs. In comparison, the diffraction peaks of AC are broadened obviously (Fig. 5d), indicating an evidently poorly crystallized graphitic structure. Generally, the high graphitization degree means that there is a complete π–π network, indicating the free movement of delocalized π-electrons on the carbon surface, i.e. the high electrical conductivity of the carbon materials.35 As far as CB-o is concerned, it displays similar diffraction features to CB; however, it bears more surface functional groups, suppressing the movement of π-electrons, resulting in a lower electrical conductivity. Therefore, the conductivity of the carbon materials was measured according to the resistivity measurement, as shown in Fig. 6. The resistivities of CNTs, CB, CB-o and AC are 0.04, 0.45, 1.35 and 141.80 Ω cm, respectively; which implies that the electron conductivity decreases in the order: CNTs > CB > CB-o > AC.
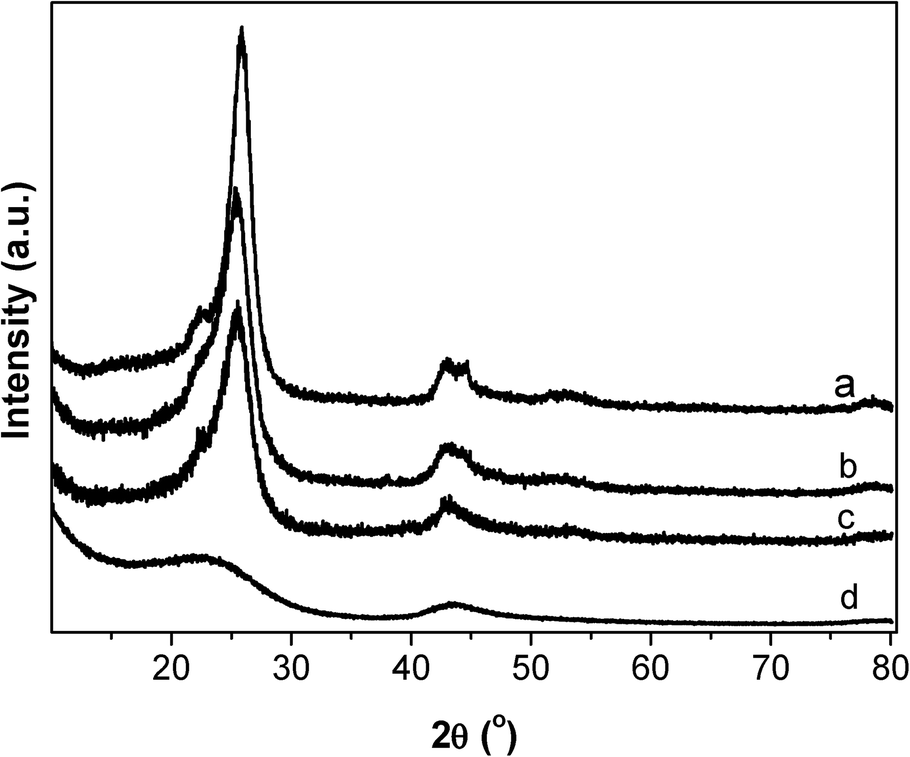 | ||
| Fig. 5 XRD patterns of carbon supports (a) CNTs, (b) CB, (c) CB-o, (d) AC. | ||
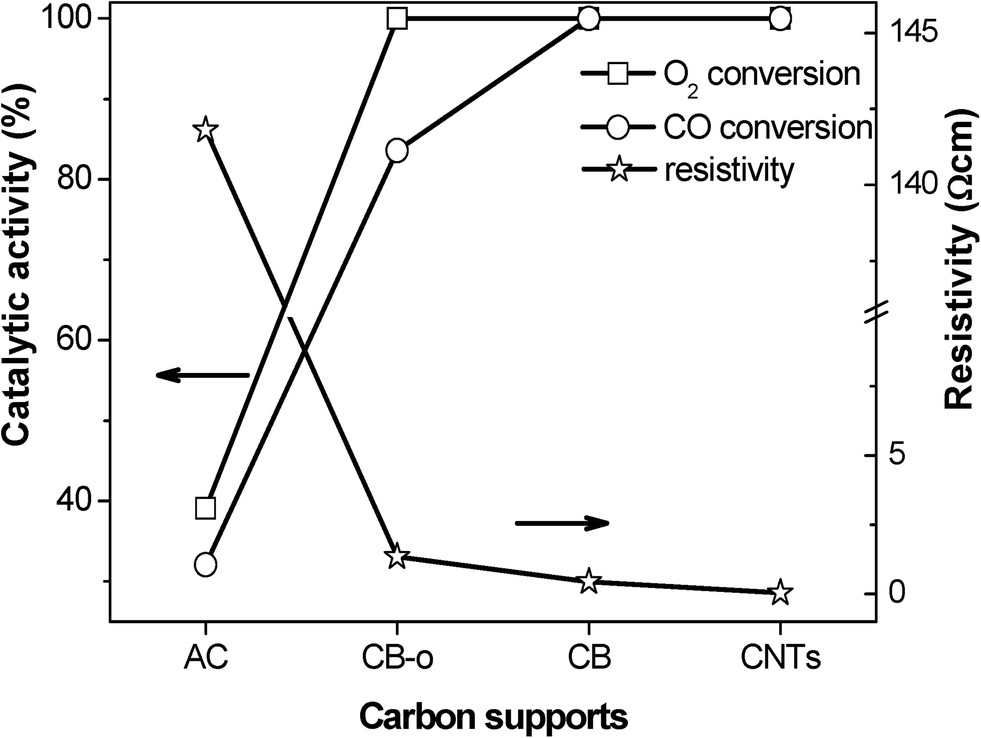 | ||
| Fig. 6 The resistivities of the supports and the catalytic activities of the Fe and Ni promoted Pt catalysts at 23 °C as a function of the different carbon supports. | ||
As mentioned in the Introduction section, carbon with a graphitic structure can capture and shuttle electrons to different sites through π–π networks. Furthermore, recent spectroscopic studies have proved the ability of graphene oxide to accept electrons from excited semiconductor nanoparticles and then reduce Ag+ at another site spatially different from that of the semiconductor nanoparticles.25 Based on this observation, a Pt-loaded graphene-Sr2Ta2O7−xNx multifunctional photocatalyst with selective catalytic processes at separate sites has been successfully demonstrated.27 Our previous research demonstrated that coordinatively unsaturated FeOx and/or NiOx confined in nanosized Pt matrices are active species for the PROX of CO and that there are interactions between Pt and the FeOx/NiOy species.5–7,21 All investigations suggest that the ability of graphitic carbon to capture electrons and shuttle them to spatially different sites through π–π networks may tune the interactions between spatially separate Pt and FeOx/NiOy species, i.e. indirect interactions between Pt and the FeOx/NiOy species. This speculation means that the higher the conductivity of the carbon materials, the stronger the indirect interaction is. Furthermore, this indirect interaction between Pt and the FeOx/NiOy species may weaken the CO adsorption on Pt and facilitate oxygen adsorption, enhancing the CO oxidation activity. On the other hand, this indirect interaction may also enable optimum CO adsorption and O2 adsorption on Pt and the FeOx/NiOy species, respectively; then the CO oxidation reaction can proceed through a chemical reaction–diffusion wave, as proposed in the previous publications.23,36 There are also other possibilities, such as metal modified carbon catalyzed CO PROX reactions, extensively studied in electro-catalysis recently.37–40 Either way will enhance the Pt utilization efficiency, leading to better or similar catalytic performances with relatively low Pt loadings. This concept is illustrated in Fig. 7 in detail.
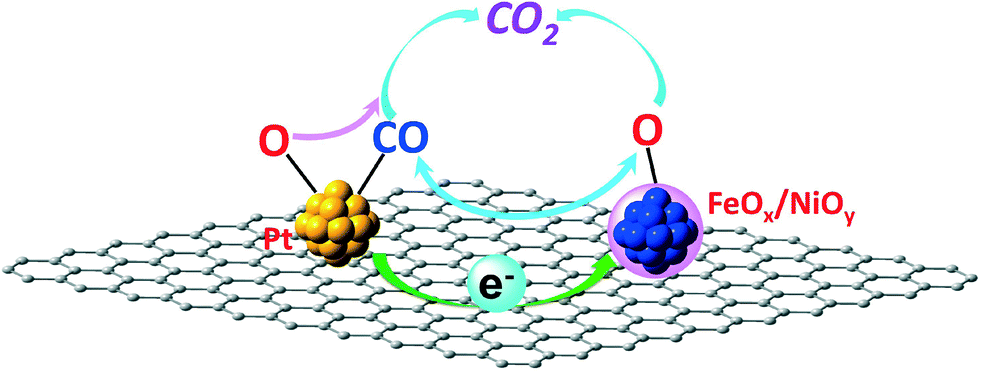 | ||
| Fig. 7 Schematic representation of the CO oxidation process on the PtFeNi catalysts tuned by the ability of graphitic carbon to capture and shuttle electrons from the noble metal to the FeOx/NiOy species through π–π networks. | ||
In order to verify the above concept, the relationship between the conductivity and the catalytic performance was investigated, as shown in Fig. 6. It is interesting that the electron conductivity of the carbon materials agrees very well with the catalytic activities of the corresponding catalysts, which can be seen from the plots of the CO and O2 conversions. The PtFeNi catalysts supported on the highly electronically conductive supports, such as CNTs, CB and CB-o, exhibit high CO conversions; however, when supported on the carbon materials with low electron conductivities, such as AC, they display low catalytic activities. Besides the high CO oxidation activity of the PtFeNi catalysts, the deactivation rate of the different carbon supported catalysts follows the order AC > CB-o > CB > CNTs. The results indicated that both the CO oxidation activity and stability agree well with the electronic conductivity of the carbon support. This implies that strong indirect interactions may lead to good catalytic performance. This observation confirms that the graphitic carbon strengthens the synergetic effect between Pt and FeNi, leading to high catalytic performance with low Pt loading levels.
It is well known that X-ray photoelectron spectroscopy (XPS) is a good way to characterize the interactions between metals and supports and/or metals and promoters. Therefore, XPS characterization was conducted. However, there is a very low signal for Fe 2p and Ni 2p due to their low loading level. Generally, different catalyst preparation methods will also influence the metal–support interactions; sometimes even changing the structure of the active phase. Thus, different catalyst preparation methods have been designed to adjust the graphitic carbon induced indirect interactions to investigate their effects on the catalytic performance.
Fig. 8 compares the catalytic performance of CB supported FeNi promoted Pt catalysts with different preparation methods. The FeNi(H500)Pt catalyst exhibits an apparently higher catalytic activity and stability compared with the FeNiPt and PtFeNi catalysts. Before being impregnated with the platinum precursor, FeNi/CB was pre-reduced with H2 which will produce elemental iron/nickel and obtain stronger metal–carbon interactions simultaneously. When impregnated with the platinum precursor, the nucleation is prone to occur around the FeNi particles, which is helpful to create active species (it shortens the distance between the Pt and FeNi nanoparticles). Therefore, this preparation method contributes to stronger indirect interactions, resulting in better catalytic performance. This example verifies that the graphitic carbon induced indirect interactions between Pt and the FeNi species play an important role in the synergistic effects. And the catalytic performance difference between CNTs and CB supported PtFeNi may be due to the unique properties of CNTs.
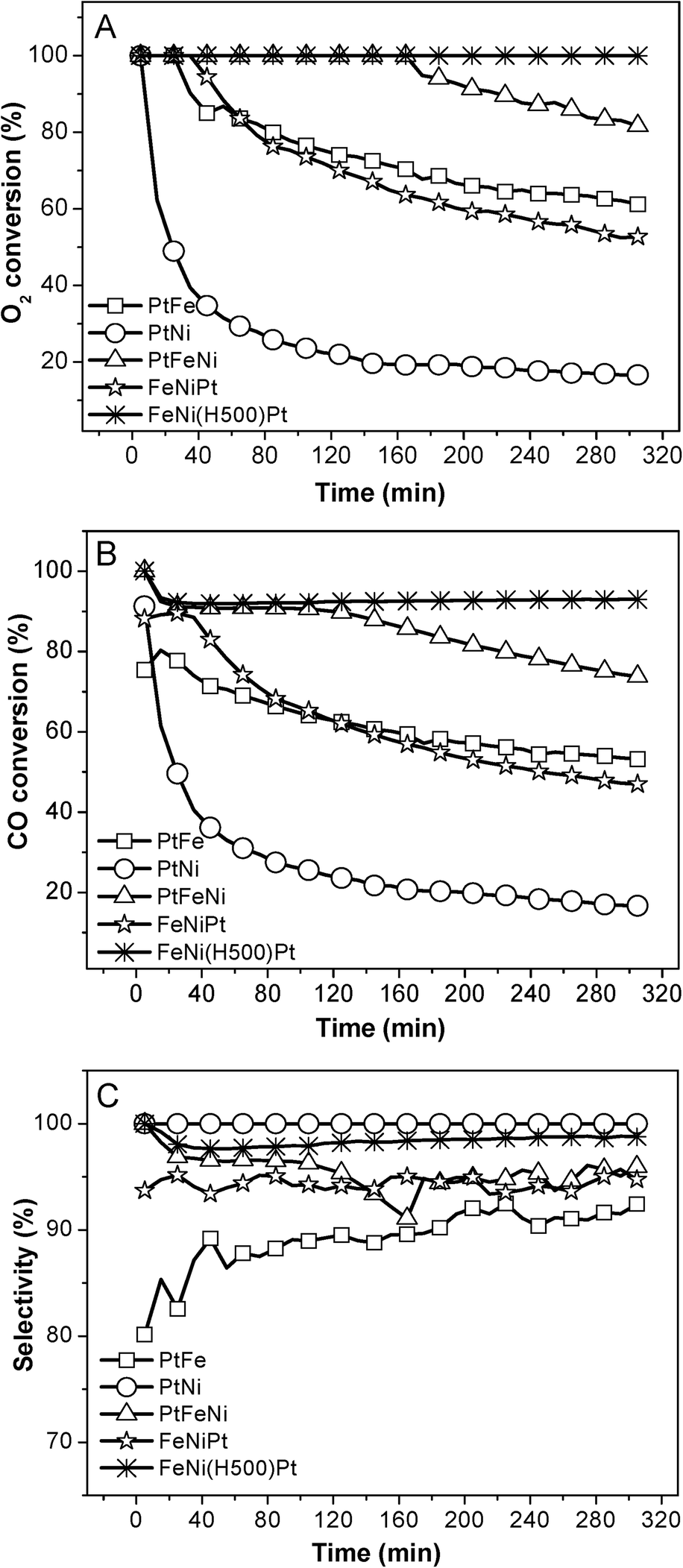 | ||
| Fig. 8 Variation in the O2 conversion (A), CO conversion (B), and selectivity (C) at 23 °C over CB supported Fe and Ni promoted Pt catalysts as a function of the reaction time. | ||
In order to further prove the important effects of the graphitic carbon induced indirect interactions on the synergistic effects, highly conductive flake graphite (FG) supported Pt and FeNi catalysts were physically mixed by grinding to prepare PtFeNi/FG catalysts and their catalytic properties for CO PROX were evaluated as shown in the ESI† (Fig. S3). This series of catalysts can effectively exclude the interfacial effects of Pt–FeNi and the alloying effects of PtFeNi on the performances; and the promotion effects should come from the graphitic carbon induced indirect interactions if they exist. It is very interesting that both the CO and O2 conversions are enhanced sharply compared with those of the Pt/FG and FeNi/FG catalysts, as shown in Fig. S3.† This is a good example to confirm the significant roles of the graphitic carbon induced indirect interactions in the synergistic effects.
Thus, carbon materials with graphitic structures can capture electrons and shuttle them to spatially different sites, which induces indirect interactions between Pt and the FeNi species, consolidating the synergistic effects, leading to high catalytic activities of PtFeNi/CNTs catalysts with much lower Pt loadings. The roles of the graphitic carbon induced indirect interactions between Pt and the transition metal species on the thermal catalytic processes have rarely been recognized, although their roles in electrochemical activity and durability of the fuel cell catalytic layers are well understood. This discovery may pave the way to further reduce the loading level of the noble metal and search for more optimal and economical catalysts for the CO PROX reaction and other thermal catalytic processes.
4. Conclusions
CNTs, CB, CB-o and AC have been selected to load PtFeNi catalysts and their catalytic properties in the PROX of CO were evaluated. Through the comparative study, it is found that the ability of graphitic carbon to capture electrons and shuttle them to different sites induces indirect interactions between Pt and the FeNi species, strengthening the synergistic effect. This graphitic carbon amplified synergistic effect leads to extremely high catalytic activity with much lower Pt loadings, when operating with stoichiometric O2, a much higher concentration of H2 and nearly the same gas hourly space velocity. This discovery will help to design more effective, economical and robust catalysts for the CO PROX reaction and other thermal catalytic processes.Acknowledgements
The authors gratefully thank Dr. Prof. Xinhe Bao and Dr. Prof. Xiulian Pan at Dalian Institute of Chemical Physics, Chinese Academy of Sciences, for their enthusiastic supervision and helpful discussions. This work is financially supported by the National Natural Science Foundation of China (21207039, 51108187), the Fundamental Research Funds for the Central Universities (Grant no. 2015zz052) and Guangdong Natural Science Foundation, China (Grant no. S2011010000737).References
- E. D. Park, D. Lee and H. C. Lee, Catal. Today, 2009, 139, 280 CrossRef CAS.
- K. Liu, A. Q. Wang and T. Zhang, ACS Catal., 2012, 2, 1165 CrossRef CAS.
- B. T. Qiao, A. Q. Wang, L. Li, Q. Q. Lin, H. S. Wei, J. Y. Liu and T. Zhang, ACS Catal., 2014, 4, 2113 CrossRef CAS.
- S. Huang, K. Hara, Y. Okubo, M. Yanagi, H. Nambu and A. Fukuoka, Appl. Catal., A, 2009, 365, 268 CrossRef CAS.
- Q. Fu, W. X. Li, Y. X. Yao, H. Y. Liu, H. Y. Su, D. Ma, X. K. Gu, L. M. Chen, Z. Wang, H. Zhang, B. Wang and X. H. Bao, Science, 2010, 328, 1141 CrossRef CAS PubMed.
- H. Xu, Q. Fu, Y. X. Yao and X. H. Bao, Energy Environ. Sci., 2012, 5, 6313 CAS.
- X. G. Guo, Q. Fu, Y. X. Ning, M. M. Wei, M. R. Li, S. Zhang, Z. Jiang and X. H. Bao, J. Am. Chem. Soc., 2012, 134, 12350 CrossRef CAS PubMed.
- A. Tomita, K. Shimizu, K. Kato and Y. Tai, Catal. Commun., 2012, 17, 194 CrossRef CAS.
- H. Xu, Q. Fu, X. G. Guo and X. H. Bao, ChemCatChem, 2012, 4, 1645 CrossRef CAS.
- C. X. Wang, L. H. Zhang and Y. T. Liu, Appl. Catal., B, 2013, 136, 48 CrossRef.
- J. Kugai, T. Moriya, S. Seino, T. Nakagawa, Y. Ohkubo, H. Nitani and T. A. Yamamoto, Int. J. Hydrogen Energy, 2013, 38, 4456 CrossRef CAS.
- H. R. Zheng, H. Y. Yang, R. R. Si, W. X. Dai, X. Chen, X. X. Wang, P. Liu and X. Z. Fu, Appl. Catal., B, 2011, 105, 243 CrossRef CAS.
- C. Wang, B. D. Li, H. Q. Lin and Y. Z. Yuan, J. Power Sources, 2012, 202, 200 CrossRef CAS.
- L. Yang, S. Y. Shan, R. Loukrakpam, V. Petkov, Y. R. Bridgid, N. Wanjala, M. H. Engelhard, J. Luo, J. Yin, Y. S. Chen and C. J. Zhong, J. Am. Chem. Soc., 2012, 134, 15048 CrossRef CAS PubMed.
- K. Tanaka, M. Shou and Y. Z. Yuan, J. Phys. Chem. C, 2010, 114, 16917 CAS.
- H. W. Yang, C. Wang, B. D. Li, H. Q. Lin, K. Tanaka and Y. Z. Yuan, Appl. Catal., A, 2011, 402, 168 CrossRef CAS.
- S. H. Lu, C. Zhang and Y. Liu, Int. J. Hydrogen Energy, 2011, 36, 1939 CrossRef CAS.
- S. H. Lu and Y. Liu, Appl. Catal., B, 2012, 111–112, 492 CrossRef CAS.
- C. Zhang, W. Lv, Q. H. Yang and Y. Liu, Appl. Surf. Sci., 2012, 258, 7795 CrossRef CAS.
- H. Zhang, D. R. Lin, G. T. Xu, J. B. Zheng, N. W. Zhang, Y. H. Li and B. H. Chen, Int. J. Hydrogen Energy, 2015, 40, 1742 CrossRef CAS.
- L. M. Chen, D. Ma, Z. Zhang, Y. Y. Guo, D. Q. Ye and B. C. Huang, Catal. Lett., 2012, 142, 975 CrossRef CAS.
- T. Komatsu, M. Takasaki, K. Ozawa, S. Furukawa and A. Muramatsu, J. Phys. Chem. C, 2013, 117, 10483 CAS.
- L. M. Chen, D. Ma, Z. Zhang, Y. Y. Guo, D. Q. Ye and B. C. Huang, ChemCatChem, 2012, 4, 1960 CrossRef CAS.
- Y. H. Kim, E. D. Park, H. C. Lee, D. Lee and K. H. Lee, Catal. Today, 2009, 146, 253–259 CrossRef CAS (Pt, Rh, Ru/Al2O3).
- I. V. Lightcap, T. H. Kosel and P. V. Kamat, Nano Lett., 2010, 10, 577 CrossRef CAS PubMed.
- P. V. Kamat, J. Phys. Chem. Lett., 2012, 3, 663 CrossRef CAS PubMed.
- A. Mukherji, B. Seger, G. Q. Lu and L. Wang, ACS Nano, 2011, 5, 3483 CrossRef CAS PubMed.
- Q. J. Xiang, J. G. Yu and M. Jaroniec, J. Am. Chem. Soc., 2012, 134, 6575 CrossRef CAS PubMed.
- L. M. Chen, D. Ma and X. H. Bao, J. Phys. Chem. C, 2007, 111, 2229 CAS.
- L. M. Chen, D. Ma, X. Y. Li and X. H. Bao, Catal. Lett., 2006, 111, 133 CrossRef CAS.
- B. W. Zhong, H. Y. Liu and X. M. Gu, ChemCatChem, 2014, 6, 1553 CrossRef CAS.
- S. Kundu, Y. M. Wang, W. Xia and M. Muhler, J. Phys. Chem. C, 2008, 112, 16869 CAS.
- Z. L. Fan, W. Chen, X. L. Pan and X. H. Bao, Catal. Today, 2009, 147, 86 CrossRef CAS.
- L. Perini, C. Durante and M. Favaro, Appl. Catal., B, 2014, 144, 300 CrossRef CAS.
- D. Sebastian, I. Suelves, R. Moliner and M. J. Lazaro, Carbon, 2010, 48, 4421 CrossRef CAS.
- W. X. Huang, X. H. Bao, H. H. Rotermund and G. Ertl, J. Phys. Chem. B, 2002, 106, 5645 CrossRef CAS.
- G. K. Ramesha, J. F. Brennecke and P. V. Kamat, ACS Catal., 2014, 4, 3249 CrossRef CAS.
- J. Deng, P. J. Ren, D. H. Deng, L. Yu, F. Yang and X. H. Bao, Energy Environ. Sci., 2014, 7, 1919 CAS.
- D. H. Deng, L. Yu. X. Chen, G. X. Wang, L. Jin, X. L. Pan, J. Deng, G. Q. Sun and X. H. Bao, Angew. Chem., Int. Ed., 2013, 52, 371 CrossRef CAS PubMed.
- W. J. Zhou, J. Zhou, Y. C. Zhou, J. Lu, K. Zhou, L. J. Yang, Z. H. Tang, L. G. Li and S. W. Chen, Chem. Mater., 2015, 27, 2026 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cy01091h |
| This journal is © The Royal Society of Chemistry 2016 |