
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Base-free oxidation of glucose to gluconic acid using supported gold catalysts†
Yueling
Cao
abc,
Xi
Liu
a,
Sarwat
Iqbal
a,
Peter J.
Miedziak
a,
Jennifer K.
Edwards
a,
Robert D.
Armstrong
a,
David J.
Morgan
 a,
Junwei
Wang
b and
Graham J.
Hutchings
*a
a,
Junwei
Wang
b and
Graham J.
Hutchings
*a
aCardiff Catalysis Institute, School of Chemistry, Cardiff University, Cardiff, CF10 3AT UK. E-mail: hutch@cardiff.ac.uk
bInstitute of Coal Chemistry, Chinese Academy of Sciences, Taiyuan 030001, PR China
cUniversity of Chinese Academy of Sciences, Beijing 100049, PR China
First published on 25th August 2015
Abstract
1% Au/TiO2 catalysts prepared by a range of preparation methods were studied for the base-free oxidation of glucose. The highest catalytic activity was observed with the catalyst prepared by the sol-immobilization method. Furthermore we have studied the effect of the post-synthesis treatments of treatment with water, or heating in air on the activity. The catalyst calcined at 250 °C showed optimal activity and selectivity. Additionally, we studied the effect of the amount of stabilising ligand in the sol-immobilisation method and observed that this is a key parameter with respect to determining the catalyst's activity.
Introduction
At present, there is a large amount of interest in the use of carbohydrates as chemical feedstocks due to their renewability and abundance.1–4D-Gluconic acid, derived from the oxidation of D-glucose, is a useful food additive and raw material for the manufacturing of medicines and biodegradable polymers.5–7 Currently, gluconic acid and its salts are produced industrially by the enzymatic oxidation of D-glucose.8 However, there are a number of drawbacks to this process including maintenance of enzyme activity, slow reaction rates and the disposal of the microbes following use.9–12 The main barrier to large-scale application of these fermentation processes is the requirement for a neutralisation step which prevents deactivation of the enzymes by the acidic products.12 A promising alternative to the enzymatic route is the direct oxidation using a heterogeneous catalyst with molecular oxygen as the oxidant. Comotti et al. have reported the oxidation of glucose both with and without hydrogen peroxide,13 there are also a number of other reports of heterogeneous catalysts, including supported Pd, Pt, Bi–Pd, Bi–Pt, Au, Au–Pd and Au–Pt nanoparticles.14–19 However strict pH control is required to obtain high rates of glucose conversion and this is achieved by the addition of a sacrificial base, typically sodium hydroxide.20–22 Therefore, from the viewpoint of green chemistry, there is a need to develop heterogeneous catalysts that can catalyse the oxidation of glucose under base-free conditions. Of the reported catalysts, Au catalysts exhibit the ability to catalyse the oxidation of glucose without pH control, thus allowing the production of gluconic acid, rather than its salt.17,19,23,24 More importantly, it has been demonstrated that, under such conditions, it is possible for the combination of acid-catalysed hydrolysis reactions and Au-catalysed glucose oxidation to occur in a single reaction vessel.25–27 Such catalytic cascade reactions are important for the conversion of disaccharides, oligosaccharides and cellulose into gluconic acid. Recently we have reported an Au/MgO catalyst for the selective synthesis of gluconic acid from glucose under mild reaction conditions.24It is well known that the activity and/or selectivity of Au catalysts is strongly dependent on several parameters such as morphology, dispersion and the interaction between the Au nanoparticles and the support (where present).28–31 According to previous reports, the particle size appears to be the most important parameter with respect to the activity and selectivity in reactions catalysed by Au.21,32,33 However, the preparation method for supported Au nanoparticles is a key factor in the synthesis of size- and structure-controlled nanoparticles. Furthermore, the synthesis of highly dispersed small Au particles is highly sensitive to any thermal pretreatment. Unfortunately, the results reported in the literature concerning these factors are often contradictory.9,34,35 Therefore, a detailed study of the influence of preparation conditions is necessary to ensure that we can obtain highly active Au catalysts reproducibly. The aim of the current study is to investigate the influence of the preparation method on Au/TiO2 catalysts for the base-free aqueous phase oxidation of glucose. TiO2 was selected as the support as it is more stable, as compared to MgO, under the base-free reaction conditions.36
Experimental methods
Catalyst preparation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, 80% hydrolyzed) and NaBH4 (0.1 M) were prepared. The requisite amount of a PVA solution was added to an aqueous Au solution. The freshly prepared solution of NaBH4 was then added to form a dark-brown sol. After 30 min of sol generation, the colloid was immobilized by adding TiO2 (Degussa P25). A small amount of H2SO4 was added under vigorous stirring to attain a pH of 1–2. After 2 h, the slurry was filtered, and the catalyst was washed thoroughly with distilled water (2 L) until the filtrate was neutral and then dried at (110 °C, 16 h, static air).
000, 80% hydrolyzed) and NaBH4 (0.1 M) were prepared. The requisite amount of a PVA solution was added to an aqueous Au solution. The freshly prepared solution of NaBH4 was then added to form a dark-brown sol. After 30 min of sol generation, the colloid was immobilized by adding TiO2 (Degussa P25). A small amount of H2SO4 was added under vigorous stirring to attain a pH of 1–2. After 2 h, the slurry was filtered, and the catalyst was washed thoroughly with distilled water (2 L) until the filtrate was neutral and then dried at (110 °C, 16 h, static air).
Catalyst post-synthesis treatment
Catalyst characterization
Glucose oxidation
Reactions were carried out using a low pressure Colliver glass reactor (50 ml). Glucose (0.20 g), catalyst (0.05 g) and water (20 g) were added into the reactor, which was then purged with oxygen three times before the reactor was sealed and pressurized with oxygen (3 barg). The reaction mixture was heated to 160 °C for 1 h under constant stirring (1000 rpm), then cooled to room temperature and the products analyzed by HPLC using an Agilent 1200 fitted with a metacarb 67H column, and UV and RI detectors.Results and discussion
The effect of the catalyst preparation method on base-free glucose oxidation
1% Au/TiO2 catalysts prepared using the four methods were tested for the base free oxidation of glucose and the data is shown in Table 1. All of the catalysts were calcined in static air (250 °C, 3 h) before reaction. It is apparent that the preparation method has a marked effect on the catalyst performance with the SIM catalysts giving the best performance.| Preparation method | Conversion (%) | Yield (%) | ||
|---|---|---|---|---|
| Gluconic acid | Fructose | 5-HMF | ||
| Reaction conditions: glucose 0.20 g, water 20.0 g, catalyst 0.05 g, reaction temperature 160 °C, reaction time 1 h, O2 3 bar. | ||||
| CIM | 30.3 | 20.0 | 4.6 | Trace |
| MIM | 22.8 | 5.3 | 9.8 | 0.1 |
| DP | 61.7 | 58.0 | 2.5 | Trace |
| SIM | 71.1 | 67.3 | Trace | Trace |
The catalysts were characterized using XRD, XPS and TEM to determine the possible origin of the differences in catalyst performance we observed. The XRD patterns of the catalysts prepared by the different methods are shown in Fig. 1. The reflections associated with the Au (200), Au (220) and Au (311) (ICDD 00-004-0784) were observed in the samples prepared by the CIM. 1% Au/TiO2 prepared by MIM, presented two major reflections at 2θ = 44.4° and 64.7°, which are characteristic of the Au (200) and (220) phase respectively. No clear reflections associated with Au were present in the samples prepared by DP and SIM.
 | ||
| Fig. 1 XRD pattern of 1% Au/TiO2 catalysts prepared with different methods. | ||
Based on the XRD results it is apparent that the Au particles obtained from SIM and DP were considerably smaller than those obtained from CIM. To confirm this we examined the SIM, DP and CIM catalysts by TEM. Fig. 2a–c shows representative images and the associated particle size distributions for the CIM, SIM and DP catalysts respectively. The average particle sizes observed inversely correlate with the activity data shown in Table 1. The DP and SIM catalysts showed a similar particle size distribution with the average particle size significantly smaller than that of the CIM catalyst. This is in agreement with the XRD data as even the largest particles observed in the SIM and DP catalysts are ca. 7 nm, which would be below the XRD detection limit given the low concentration of metal in the samples.
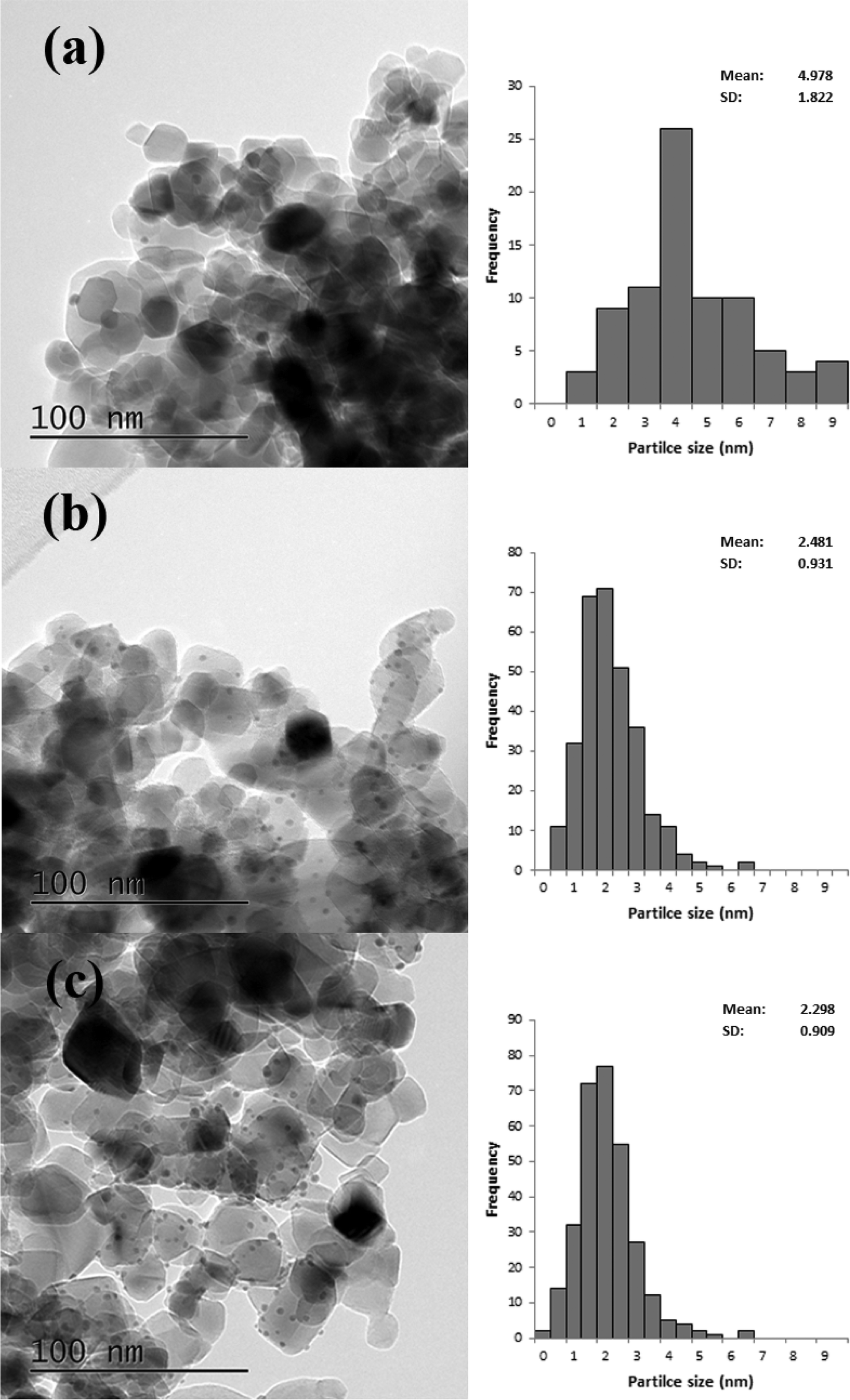 | ||
| Fig. 2 Representative TEM images and associated particle size distributions for 1% Au/TiO2 prepared by a) CIM, b) SIM and c) DP. Particle size distributions based on 300 particles. | ||
Although the largest particles observed in the CIM catalyst by TEM were ca. 9 nm, the XRD suggested that larger metal particles were also present in this catalyst. In our previous studies we found that the CIM method showed a bimodal particle size distribution with small (2–10 nm) and large (>20 nm) metal particles in the final catalyst.41
XPS spectra for Au (4f) and Cl (2p) for 1% Au/TiO2 samples prepared by the four different methods are shown in Fig. 3.
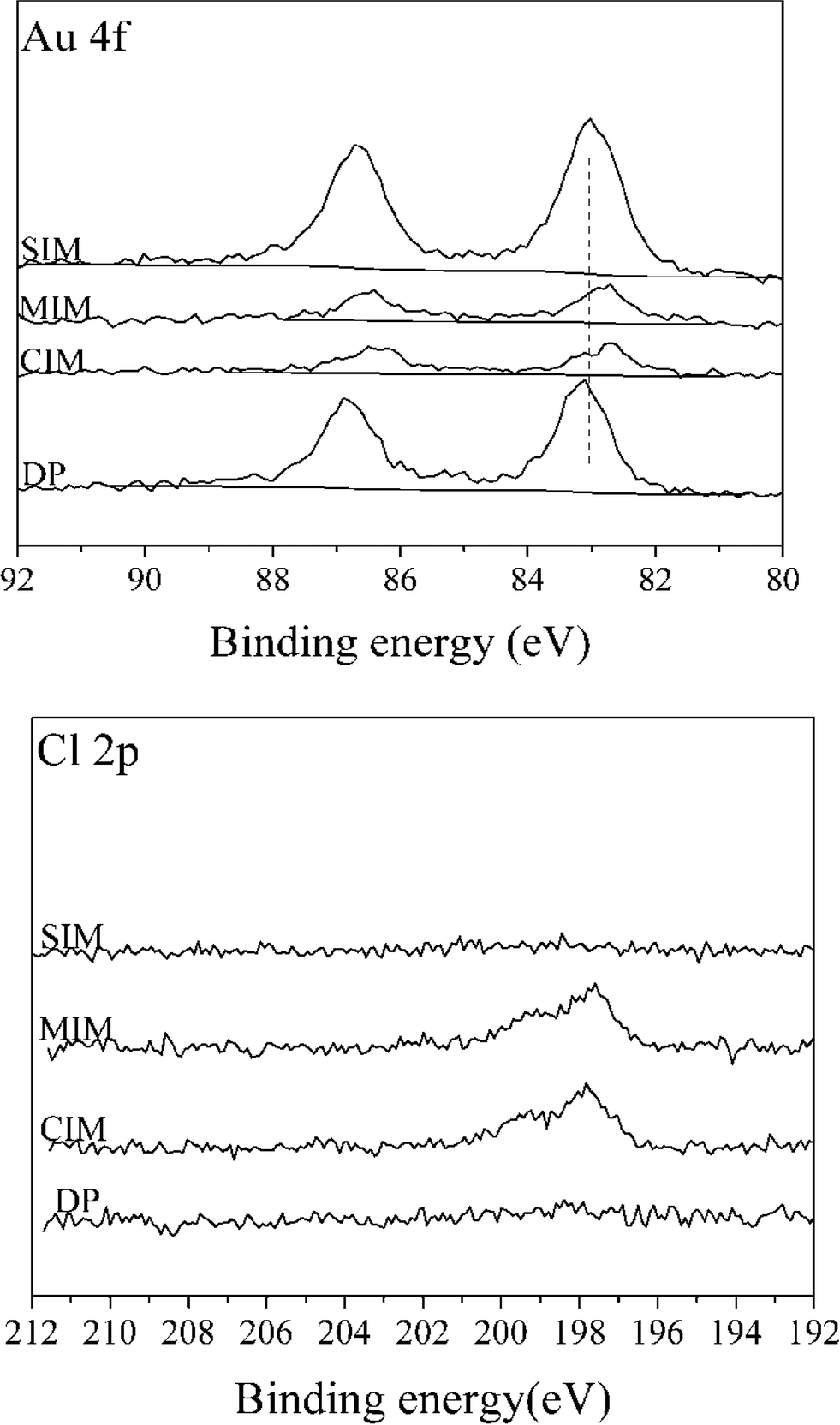 | ||
| Fig. 3 Au(4f) and Cl(2p) spectra for 1% Au/TiO2 samples prepared by different methods. | ||
Compared with the catalysts prepared by SIM and DP, the Au signal intensity of catalysts prepared by CIM and MIM was significantly lower suggesting that poorly dispersed particles are present in these catalysts, despite the smaller Au particle size suggested by the lower binding energy observed. For the catalysts prepared by the SIM and DP methods, no obvious signal associated with Cl (2p) can be detected, while Cl (2p) peaks are apparent for the samples obtained by CIM, and MIM methods. This can be further confirmed by the surface concentrations determined by XPS of a number of elements present in 1% Au/TiO2 catalysts prepared with different methods (ESI† Table S1). The variation in the amount of surface metals is not unexpected as the SIM and DP methods contain a washing step, whereas the CIM and MIM methods do not. For the catalysts prepared by CIM and MIM, the residual chloride was determined to be 0.77 wt% and 0.58 wt%, respectively. It is well known that residual chloride can affect catalytic activity in two different ways: (a) by facilitating the agglomeration of Au particles during heat treatment and (b) through poisoning of the active site.42 We therefore conclude that the large Au particle size and amount of residual chloride are the two main factors which contribute to the low catalytic activity with the CIM and MIM catalysts for this reaction.
Effect of the catalyst post-synthesis treatment on base-free oxidation of glucose
When using the SIM method, part of the excess PVA used in the synthesis process can be removed either by a solvent washing step or through a heat treatment.43 However, part of the PVA still remains and requires further treatment.44 Therefore, thermal treatment (calcination at 250 °C for 3 h under static air) and a hot water washing method were used in an attempt to remove more of the PVA. The results for the post-treated catalysts for the base-free oxidation of glucose are shown in Table 2.| Treatment condition | Conversion (%) | Yield (%) | ||
|---|---|---|---|---|
| Gluconic acid | Glucaric acid | Glycolic acid | ||
| Reaction conditions: glucose 0.20 g, water 20.0 g, catalyst 0.05 g, reaction temperature 160 °C, reaction time 1 h, O2 3 bar. | ||||
| None | 31.8 | 28.7 | None | None |
| Air, 250 °C | 71.1 | 67.3 | Trace | None |
| Water | 66.4 | 63.8 | Trace | Trace |
The dried only sample exhibited 31.8% glucose conversion, whereas the activity increased to 71.1% and 66.4% after calcination and treatment with water respectively. It is possible that these treatments induce structural changes within the Au catalysts, indeed calcination has been previously reported to lead to changes in particle size.40 However, from XRD and XPS surface analysis (ESI† Fig. S1 and S2, Table S2), there was no obvious difference between dried and treated samples, TEM analysis (Fig. 4) showed that there is a small increase in the average particle size, however, this is not marked enough to be observable by XRD or XPS. As the difference in particle size observed was small we consider that additional factors may affect the change in catalytic activity. Additionally, the 1% Au/TiO2 prepared by DP showed similar activity for glucose conversion, which may further imply that the residual PVA is blocking the active site through steric effects. Villa et al., have reported that the presence of PVA can lower the activity of Au/TiO2 thorough shielding of the Au nanoparticles.44 From the increase in the activity shown in Table 2 we conclude that both calcination and reflux treatments are efficient ways to enhance the catalytic activity of SIM catalysts for base free glucose oxidation. However, as the highest activity was observed for the calcined SIM catalyst we decided to investigate this further. The effect of calcination temperature on the activity of the 1% Au/TiO2 SIM catalyst is shown in Table 3. As expected, the catalytic activity initially increased with calcination temperature reaching a maximum at 250–350 °C, then decreased gradually at 450 and 550 °C. This may be due to a compromise at the lower calcination temperatures where some PVA was removed, but not resulting in a significant increase in particle size from sintering. However, when the calcination temperature was further increased the Au particle size became too large and a decline in activity was observed.
 | ||
| Fig. 4 Representative TEM images and associated particle size distributions for 1% Au/TiO2 prepared by SIM a) no treatment, b) calcined at 250 °C and c) refluxed. Particle size distributions based on 300 particles. | ||
| Calcination temperature (°C) | Conversion (%) | Yield (%) | ||
|---|---|---|---|---|
| Gluconic acid | Glucaric acid | Glycolic acid | ||
| Reaction conditions: glucose 0.20 g, water 20.0 g, catalyst 0.05 g, reaction temperature 160 °C, reaction time 1 h, O2 3 bar. | ||||
| None | 31.8 | 28.7 | None | None |
| 250 | 71.1 | 67.3 | Trace | None |
| 350 | 69.0 | 65.4 | Trace | Trace |
| 450 | 59.2 | 53.8 | Trace | Trace |
| 550 | 46.3 | 41.6 | Trace | Trace |
The XRD patterns of 1% Au/TiO2 prepared by the SIM method, calcined at different temperatures, are shown in Fig. 5. The sample calcined at 550 °C presented one weak reflection at 44.4°, which can be attributed to the Au (200) phase. This suggests that there was a significant particle growth at this high calcination temperature. As a result, thermal treatment can be a challenging trade-off between the elimination of the stabilizer and undesirable structural changes to the final catalysts.
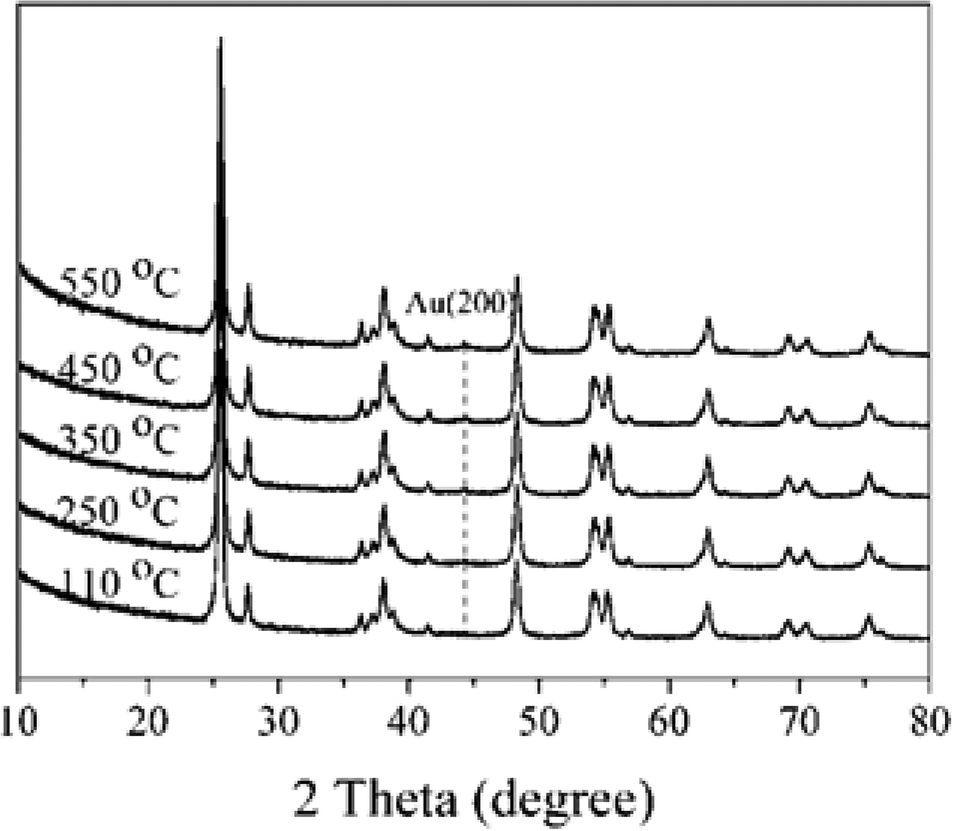 | ||
| Fig. 5 XRD patterns of 1% Au/TiO2 catalysts calcined at different temperatures. | ||
TEM analysis was also carried out on these catalysts, representative images are shown in Fig. 6 and the associated particle size distributions (PSDs) are shown in Fig. 7. As previously discussed calcination at 250 °C resulted in larger particles; however, as shown in Fig. 7 increasing the temperature to 350 °C resulted in a significant increase in particle size. The particle size was similar for the catalyst calcined at 450 °C, but increased when the calcination temperature was raised to 550 °C. This correlates well with the activity data shown in Table 3. It is clear that two factors can influence the activity namely the removal of the PVA by calcination and the increase in the average particle size which need to be balanced against each other.
 | ||
| Fig. 6 Representative TEM images of 1% Au/TiO2 SIM calcined at a) 250 °C, b) 350 °C, c) 450 °C and d) 550 °C. | ||
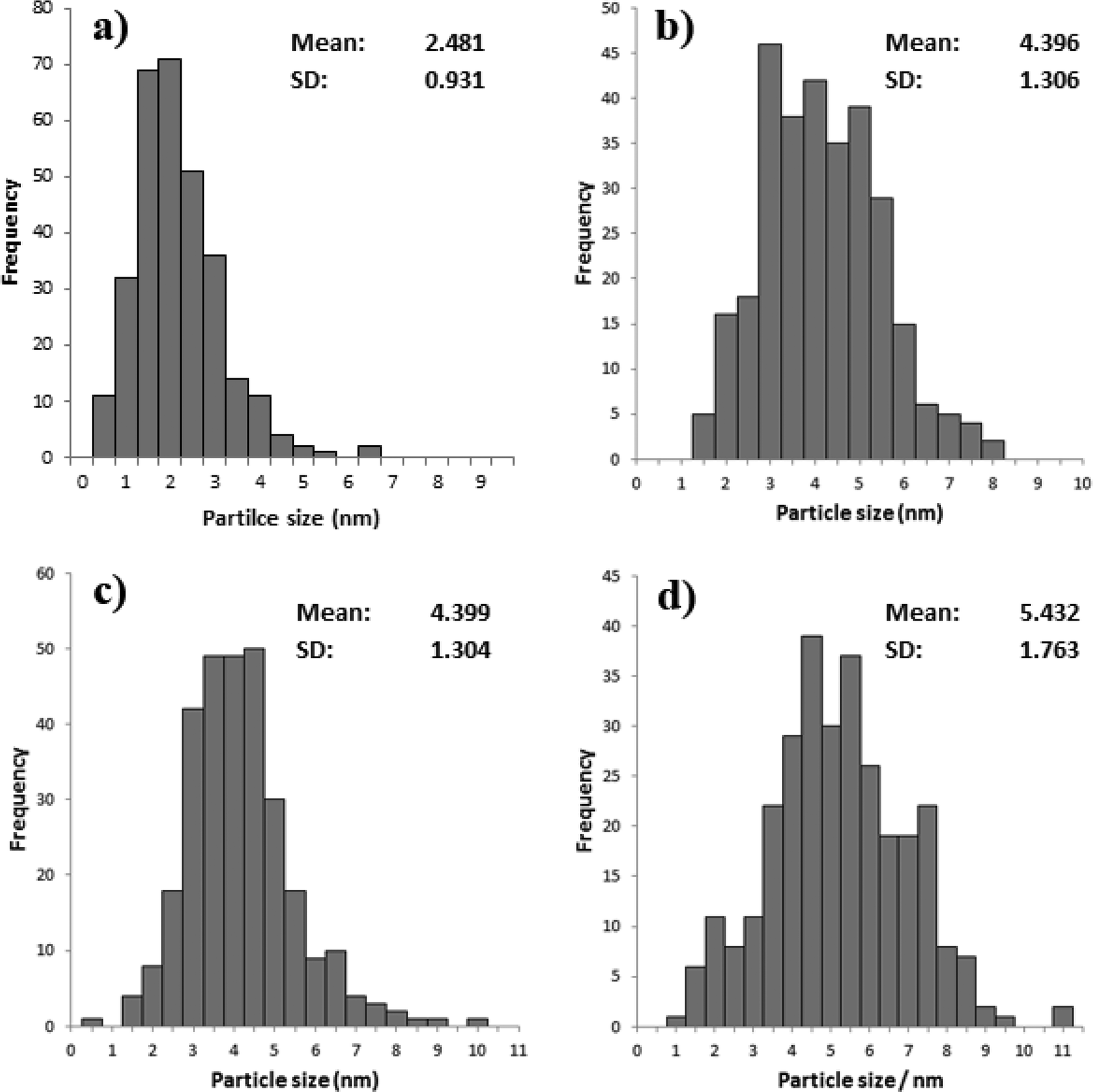 | ||
| Fig. 7 Particle size distribution for % Au/TiO2 SIM calcined at a) 250 °C, b) 350 °C, c) 450 °C and d) 550 °C. | ||
Effect of the PVA to Au ratio in the SIM preparation method
To investigate the balance between the influence of particle size and the possible ligand shielding effect we prepared 1% Au/TiO2 SIM catalysts using six PVA/Au weight ratios of 0, 0.05, 0.1, 0.6, 1.2 and 1.8. We considered the ratio of PVA to Au to be a key factor in affecting the catalytic activity as it can change both the amount of residual PVA and the metal particle size. To avoid the influence of heat treatment on the residual stabilizer and Au particle size the catalysts were prepared without further heat treatment after the drying step. The results of the effect of ratio of PVA to Au on the base-free oxidation of glucose are presented in Table 4 and reveal a volcano-type correlation for activity with the ratio of PVA to Au. The maximum activity was observed for the PVA to Au ratio of 0.1. When the ratio of PVA to Au was lower than this, the activity of catalysts decreased which may be due to a large particle size resulting from there being insufficient PVA to form colloids.45 However, when the ratio of PVA to Au was higher than 0.1, the activity of catalysts gradually decreased which was most likely due to the presence of PVA blocking active sites. This behavior is similar to what has been previously reported for PVA stabilized Au nanoparticles supported on activated carbon and TiO2.43| PVA:Au (weight ratio) |
Conversion (%) | Yield (%) | ||
|---|---|---|---|---|
| Gluconic acid | Glucaric acid | Glycolic acid | ||
| Reaction conditions: glucose 0.20 g, water 20.0 g, catalyst 0.05 g, reaction temperature 160 °C, reaction time 1 h, O2 pressure 3 bar. | ||||
| 0 | 62.9 | 60.3 | Trace | Trace |
| 0.05 | 62.5 | 60.8 | Trace | Trace |
| 0.1 | 88.4 | 84.0 | Trace | 0 |
| 0.6 | 57.6 | 54.2 | Trace | 0 |
| 1.2 | 31.8 | 28.7 | 0 | 0 |
| 1.8 | 26.7 | 10.7 | 0 | 0 |
Having found the optimum PVA/Au ratio we compared the performance of SIM catalysts with and without heat treatment (ESI† Table S3). For the dried only and calcined catalysts, the glucose conversion and gluconic acid yield were 88.4%, 89.9% and 84.0%, 86.2%, respectively. This indicates that the thermal treatment did not affect the catalytic activity when the optimum ratio of PVA to Au was used in the SIM preparation method, because there was insufficient stabilizer to create the shielding effect and therefore the removal by heat treatment did not affect the activity. Taken together, it can be concluded that the preferred weight ratio of PVA to Au is 0.1 in the preparation of Au/TiO2 catalysts by the SIM method for the base free oxidation of glucose.
To confirm this trend these catalysts were characterized in greater detail. XRD results (ESI† Fig. S3) did not show any difference between the catalysts. XPS was performed to explore the possibility of an electronic effect between Au and PVA. As shown in Fig. 8 the binding energy of Au(4f) increased with an increasing PVA/Au ratio. This may be due to the change in electronic structure as a function of cluster size, which is in agreement with previous studies, which attributed an upward shift in the Au(4f) binding energy to increasing particle size.46
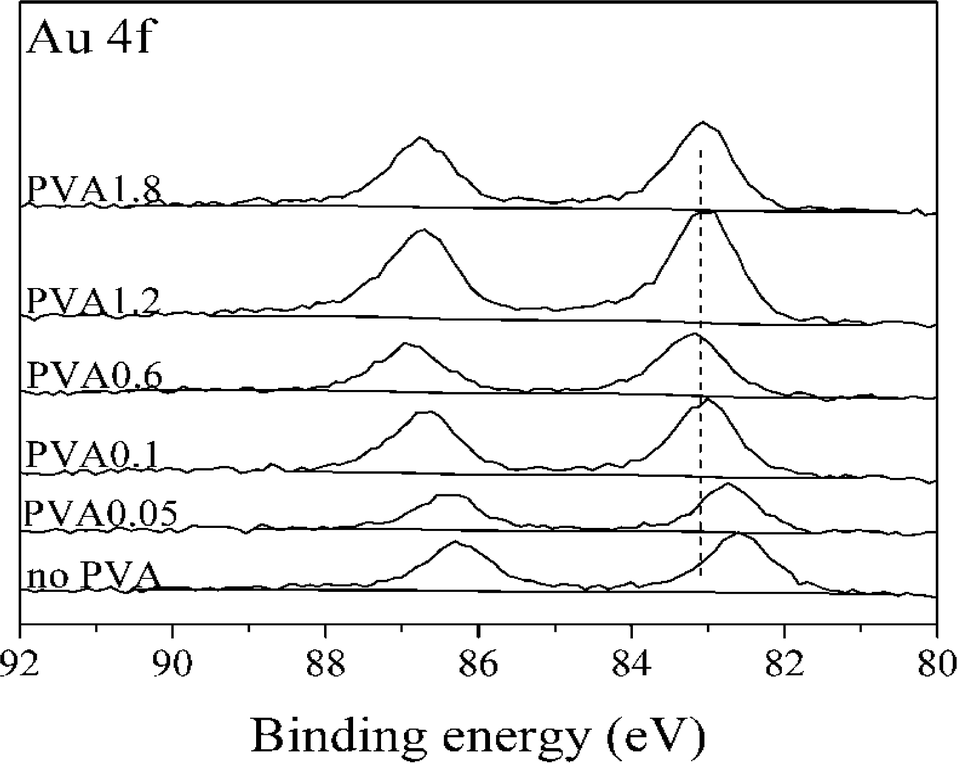 | ||
| Fig. 8 Au(4f) spectra for 1% Au/TiO2 SIM samples prepared with different ratio of PVA to Au (w/w). | ||
TEM particle size distributions (Fig. 9) of three of the catalysts confirm these conclusions. The most active catalyst (PVA/Au = 0.1) had an average particle size of 7.6 nm, whereas the catalysts with higher ratios, namely 1.2 and 1.8 both had average particle sizes of around 2.1 nm. Surprisingly, the large particles, which we would expect to be less active, were more active than the smaller particles. This further confirms that the role of PVA in shielding the active sites is significant for this reaction.
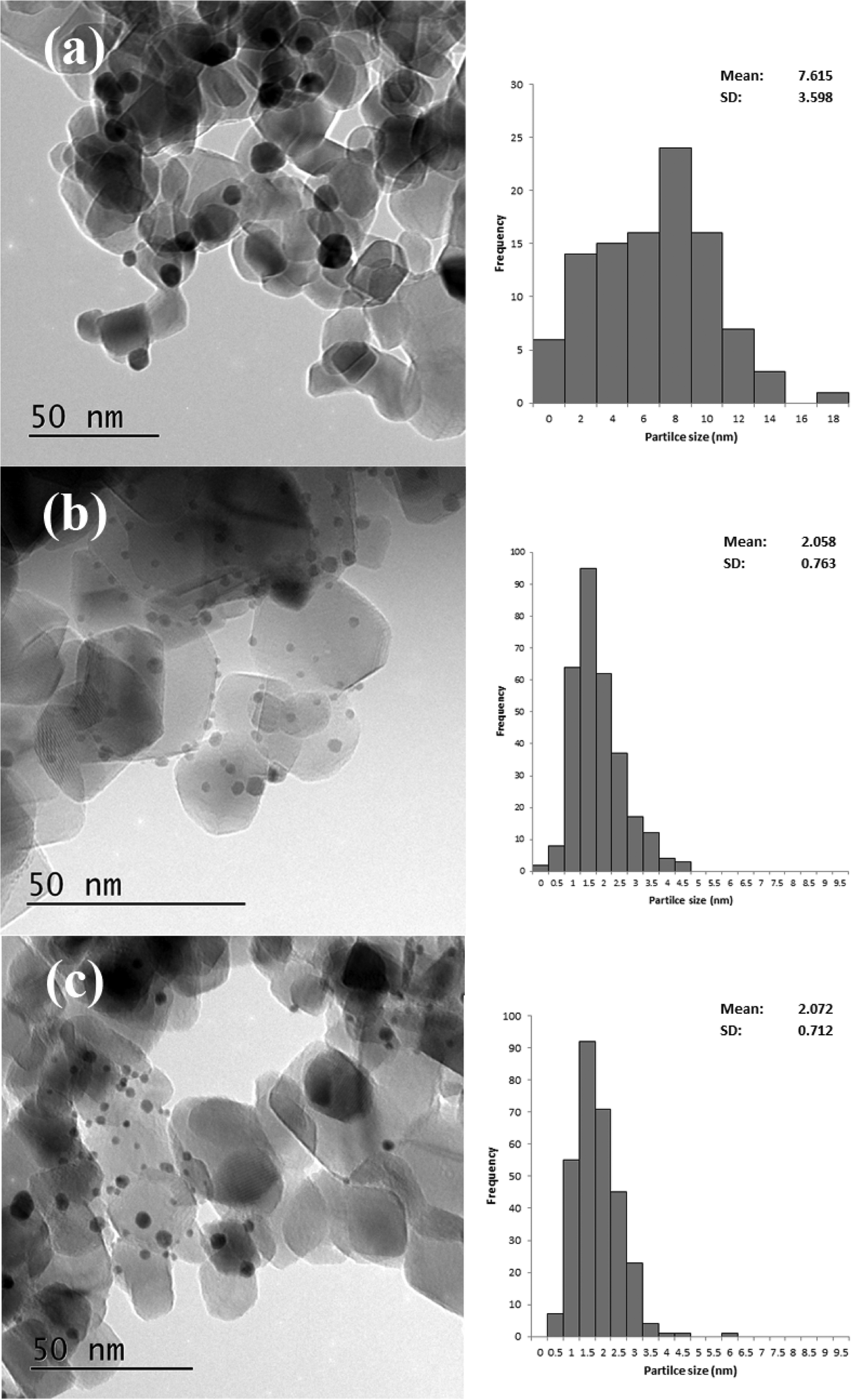 | ||
| Fig. 9 Representative TEM images and associated particle size distributions for 1% Au/TiO2 prepared by SIM with different PVA:Au ratios a) 0.1, b) 1.2 and c) 1.8. Particle size distributions based on 300 particles except for a) where 100 particles were counted. | ||
Conclusions
We have shown that Au/TiO2 catalysts are active and selective for the oxidation of glucose to gluconic acid under base-free conditions. Among the different methods for preparing the Au catalysts, sol-immobilization is considered to the favoured way to obtain Au catalysts that give high catalytic activity in this reaction. The pretreatment of catalysts has a significant influence on their catalytic activity, and the Au particle size is also shown to be an important factor.We have found that the catalytic activity is affected by a combination of factors, the most significant amongst them being the metal particle size and the amount of PVA stabilizing ligand used in the SIM method. The most active catalyst observed did not have the smallest particle size, but did use one of the lowest PVA to metal ratios in their preparation. This suggests that the amount of residual ligand is the most significant factor in determining catalytic activity. We also show that activity can be improved through the removal of the ligand using either heat treatment or refluxing, though it is preferable to minimize the amount of ligand used during the preparation. When the optimum amount of ligand was used, post preparation treatments were observed to have no significant effect upon the resulting catalytic activity or selectivity.
Acknowledgements
Yueling Cao would like to thank the China Scholarship Council (CSC, 201304910272) for financial support. Also the financial support from European Research Council: After the Goldrush, ERC-AG-291319 is gratefully acknowledged. We would also like to acknowledge the UK catalysis hub, TEM data was acquired at the Research Complex at Harwell (UK Catalysis Hub).References
- Y. Roman-Leshkov, J. N. Chheda and J. A. Dumesic, Science, 2006, 312, 1933–1937 CrossRef CAS PubMed.
- Y. Roman-Leshkov, C. J. Barrett, Z. Y. Liu and J. A. Dumesic, Nature, 2007, 447, 982–985 CrossRef CAS PubMed.
- M. S. Holm, S. Saravanamurugan and E. Taarning, Science, 2010, 328, 602–605 CrossRef CAS PubMed.
- W. R. Gunther, Y. Wang, Y. Ji, V. K. Michaelis, S. T. Hunt, R. G. Griffin and Y. Roman-Leshkov, Nat. Commun., 2012, 3, 1109 CrossRef PubMed.
- Y. Onal, S. Schimpf and P. Claus, J. Catal., 2004, 223, 122–133 CrossRef CAS.
- N. Thielecke, M. Aytemir and U. Pruesse, Catal. Today, 2007, 121, 115–120 CrossRef CAS.
- C. Baatz and U. Pruesse, J. Catal., 2007, 249, 34–40 CrossRef CAS.
- H. Hustede, H.-J. Haberstroh and E. Schinzig, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000 Search PubMed.
- H. Okatsu, N. Kinoshita, T. Akita, T. Ishida and M. Haruta, Appl. Catal., A, 2009, 369, 8–14 CrossRef CAS.
- P. Beltrame, M. Comotti, C. Della Pina and M. Rossi, J. Catal., 2004, 228, 282–287 CrossRef CAS.
- J. Bao, K. Furumoto, K. Fukunaga and K. Nakao, Biochem. Eng. J., 2001, 8, 91–102 CrossRef CAS.
- H. Kobayashi and A. Fukuoka, Green Chem., 2013, 15, 1740–1763 RSC.
- M. Comotti, C. Della Pina, E. Falletta and M. Rossi, Adv. Synth. Catal., 2006, 348, 313–316 CrossRef CAS.
- A. Abbadi and H. van Bekkum, J. Mol. Catal. A: Chem., 1995, 97, 111–118 CrossRef CAS.
- M. Besson, F. Lahmer, P. Gallezot, P. Fuertes and G. Fleche, J. Catal., 1995, 152, 116–121 CrossRef CAS.
- M. Wenkin, R. Touillaux, P. Ruiz, B. Delmon and M. Devillers, Appl. Catal., A, 1996, 148, 181–199 CrossRef CAS.
- S. Biella, L. Prati and M. Rossi, J. Catal., 2002, 206, 242–247 CrossRef CAS.
- H. Yin, C. Zhou, C. Xu, P. Liu, X. Xu and Y. Ding, J. Phys. Chem. C, 2008, 112, 9673–9678 CAS.
- M. Comotti, C. Della Pina and M. Rossi, J. Mol. Catal. A: Chem., 2006, 251, 89–92 CrossRef CAS.
- S. Hermans and M. Devillers, Appl. Catal., A, 2002, 235, 253–264 CrossRef CAS.
- A. Mirescu, H. Berndt, A. Martin and U. Pruesse, Appl. Catal., A, 2007, 317, 204–209 CrossRef CAS.
- H. Zhang and N. Toshima, Catal. Sci. Technol., 2013, 3, 268–278 CAS.
- Y. Wang, S. Van de Vyver, K. K. Sharma and Y. Roman-Leshkov, Green Chem., 2014, 16, 719–726 RSC.
- P. J. Miedziak, H. Alshammari, S. A. Kondrat, T. J. Clarke, T. E. Davies, M. Morad, D. J. Morgan, D. J. Willock, D. W. Knight, S. H. Taylor and G. J. Hutchings, Green Chem., 2014, 16, 3132–3141 RSC.
- S. Van de Vyver, J. Geboers, P. A. Jacobs and B. F. Sels, ChemCatChem, 2011, 3, 82–94 CrossRef CAS.
- X. Tan, W. Deng, M. Liu, Q. Zhang and Y. Wang, Chem. Commun., 2009, 7179–7181, 10.1039/b917224f.
- D. An, A. Ye, W. Deng, Q. Zhang and Y. Wang, Chem. – Eur. J., 2012, 18, 2938–2947 CrossRef CAS PubMed , S2938/2931-S2938/2938.
- M. Haruta, Catal. Today, 1997, 36, 153–166 CrossRef CAS.
- A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896–7936 CrossRef PubMed.
- J. Oliver-Meseguer, J. R. Cabrero-Antonino, I. Domínguez, A. Leyva-Pérez and A. Corma, Science, 2012, 338, 1452–1455 CrossRef CAS PubMed.
- A. S. K. Hashmi, Science, 2012, 338, 1434 CrossRef CAS PubMed.
- R. Zanella, S. Giorgio, C. R. Henry and C. Louis, J. Phys. Chem. B, 2002, 106, 7634–7642 CrossRef CAS.
- R. Zanella, L. Delannoy and C. Louis, Appl. Catal., A, 2005, 291, 62–72 CrossRef CAS.
- A. Wolf and F. Schuth, Appl. Catal., A, 2002, 226, 1–13 CrossRef CAS.
- C. Baatz, N. Decker and U. Pruesse, J. Catal., 2008, 258, 165–169 CrossRef CAS.
- M. Morad, M. Sankar, E. Cao, E. Nowicka, T. E. Davies, P. J. Miedziak, D. J. Morgan, D. W. Knight, D. Bethell, A. Gavriilidis and G. J. Hutchings, Catal. Sci. Technol., 2014, 4, 3120–3128 CAS.
- J. C. Pritchard, Q. He, E. N. Ntainjua, M. Piccinini, J. K. Edwards, A. A. Herzing, A. F. Carley, J. A. Moulijn, C. J. Kiely and G. J. Hutchings, Green Chem., 2010, 12, 915–921 RSC.
- M. Sankar, Q. He, M. Morad, J. Pritchard, S. J. Freakley, J. K. Edwards, S. H. Taylor, D. J. Morgan, A. F. Carley, D. W. Knight, C. J. Kiely and G. J. Hutchings, ACS Nano, 2012, 6, 6600–6613 CrossRef CAS PubMed.
- M. Sankar, E. Nowicka, R. Tiruvalam, Q. He, S. H. Taylor, C. J. Kiely, D. Bethell, D. W. Knight and G. J. Hutchings, Chem. – Eur. J., 2011, 17, 6524–6532 CrossRef CAS PubMed.
- J. A. Lopez-Sanchez, N. Dimitratos, C. Hammond, G. L. Brett, L. Kesavan, S. White, P. Miedziak, R. Tiruvalam, R. L. Jenkins, A. F. Carley, D. Knight, C. J. Kiely and G. J. Hutchings, Nat. Chem., 2011, 3, 551–556 CrossRef CAS PubMed.
- J. K. Edwards, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Faraday Discuss., 2008, 138, 225–239 RSC.
- H. S. Oh, J. H. Yang, C. K. Costello, Y. M. Wang, S. R. Bare, H. H. Kung and M. C. Kung, J. Catal., 2002, 210, 375–386 CrossRef CAS.
- J. A. Lopez-Sanchez, N. Dimitratos, C. Hammond, G. L. Brett, L. Kesavan, S. White, P. Miedziak, R. Tiruvalam, R. L. Jenkins, A. F. Carley, D. Knight, C. J. Kiely and G. J. Hutchings, Nat. Chem., 2011, 3, 551–556 CrossRef CAS PubMed.
- A. Villa, D. Wang, G. M. Veith, F. Vindigni and L. Prati, Catal. Sci. Technol., 2013, 3, 3036–3041 CAS.
- I. Pastoriza-Santos and L. M. Liz-Marzan, Langmuir, 2002, 18, 2888–2894 CrossRef CAS.
- J. Radnik, C. Mohr and P. Claus, Phys. Chem. Chem. Phys., 2003, 5, 172–177 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cy00732a |
| This journal is © The Royal Society of Chemistry 2016 |