
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Protection of densely populated excited triplet state ensembles against deactivation by molecular oxygen
Mikhail A.
Filatov
a,
Stanislav
Baluschev
bc and
Katharina
Landfester
c
aTrinity Biomedical Science Institute, Trinity College Dublin, 152-160 Pearse Street, Dublin 2, Ireland. E-mail: filatovm@tcd.ie
bMax Planck Institute for Polymer Research, Ackermannweg 10, D-55128 Mainz, Germany. E-mail: balouche@mpip-mainz.mpg.de
cOptics and Spectroscopy Department, Faculty of Physics, Sofia University “St. Kliment Ochridski”, 5 James Bourchier, 1164 Sofia, Bulgaria. E-mail: landfester@mpip-mainz.mpg.de
First published on 9th June 2016
Abstract
This critical review discusses different approaches towards protection of photoactive materials based on triplet excited state ensembles against deactivation by molecular oxygen though quenching and photooxidation mechanisms. Passive protection, based on the application of barrier materials for packaging, sealing, or encapsulation of the active substances, which prevent oxygen molecules from penetration and physical contact with excited states and active protection, based on the application of oxygen scavenging species are compared. Efficiencies of different approaches together with examples and prospects of their applications are outlined.
 Mikhail A. Filatov | Mikhail A. Filatov received his PhD degree at Moscow State University in 2008 in the field of organic synthesis. He further conducted postdoctoral research in the University of Burgundy, Max Planck Institute and Bulgarian Academy of Sciences, working on molecular systems for optical and biomedical applications. He is currently a Marie Curie research fellow at Trinity College Dublin where he works on new singlet oxygen sensitizers and sensors for photomedicine. His research combines multistep organic synthesis, photochemistry, optical spectroscopy, and material science to achieve new molecular devices for healthcare. He has published around 30 papers and participated in 4 patents. |
 Stanislav Baluschev | Stanislav Baluschev studied Laser Physics at the Sofia University “Saint Kliment Ochridski”, Bulgaria, where he received PhD. In 1996, he was granted a DAAD- research fellowship at Hannover University, Germany. In 1997 he was visiting scientist at the Institute of Experimental Physics, TU of Graz, Austria. In 1999 he was granted a Feinberg Research Fellowship at the Department of Complex Systems, Weizmann Institute of Science, Israel. In 2000 he started his Maria-Curie fellowship at PTB, Germany. In 2001, he joined the group of Prof. G. Wegner/Prof. K. Landfester at MPI for Polymer Research. His current research interests include nonlinear optics, atomic physics and annihilation upconversion in organic systems. |
 Katharina Landfester | Katharina Landfester joined the Max Planck Society in 2008 as one of the directors of the MPI for Polymer Research. She studied Chemistry at the Technical University of Darmstadt and in Strasbourg. In 1995, she received her doctoral degree in Physical Chemistry after working with Prof. H. W. Spiess at the MPIP. In 1996, she moved for a doctoral stay at the Lehigh University and started in 1998 at the MPI of Colloids and Interfaces in Golm. In 2003, she accepted a chair (C4) of Macromolecular Chemistry at the University of Ulm. In 2001 she was awarded the Reimund Stadler prize of the German Chemical Society (GDCh) and the prize of the Dr Hermann Schnell Foundation. |
1. Introduction
Triplet excited state chromophores are being applied in various fields, e.g. electroluminescence,1 bioimaging and molecular sensing,2 photocatalytic organic reactions,3 and triplet–triplet annihilation photon upconversion (TTA-UC).4 However, compared to fluorescence, photonic applications based on triplet excited state generation are much less developed due to a substantially higher sensitivity of triplet states towards non-emissive deactivation processes, especially ground-state triplet oxygen quenching giving singlet oxygen and ground state of the chromophore.5One of the most promising applications based on excited triplet ensembles, namely photon energy upconversion by triplet–triplet annihilation (TTA-UC), was recently extended to ultralow intensity of noncoherent excitation light (less than 10 mW cm−2), corresponding to the solar intensity at the Earth's surface.6 Such low-energy threshold allows for the development of several unique applications in material science,7 solar cell devices,8 solar fuels,9 bioimaging10 and extension of the infrared limit of oxygenic photosynthesis.11
TTA-UC process takes place in multi-chromophore systems consisting of energetically optimized pairs of sensitizer (metallated macrocycles) and emitter molecules (aromatic hydrocarbons), as shown in Fig. 1. The photon energy is absorbed by a sensitizer and stored in its triplet state, formed in the process of intersystem crossing (ISC). Further, this energy is transferred to an emitter triplet state via the process of triplet–triplet transfer (TTT). Next, the excited triplet states of two emitter molecules undergo triplet–triplet annihilation (TTA), in which one emitter molecule returns back to its singlet ground state and the other molecule gains the energy of both triplet states and is excited to the higher singlet state. As the singlet state emitter decays radiatively back to the ground state, a delayed fluorescence photon (the dark blue line, Fig. 1) bearing higher energy than that of the excitation photons is emitted.
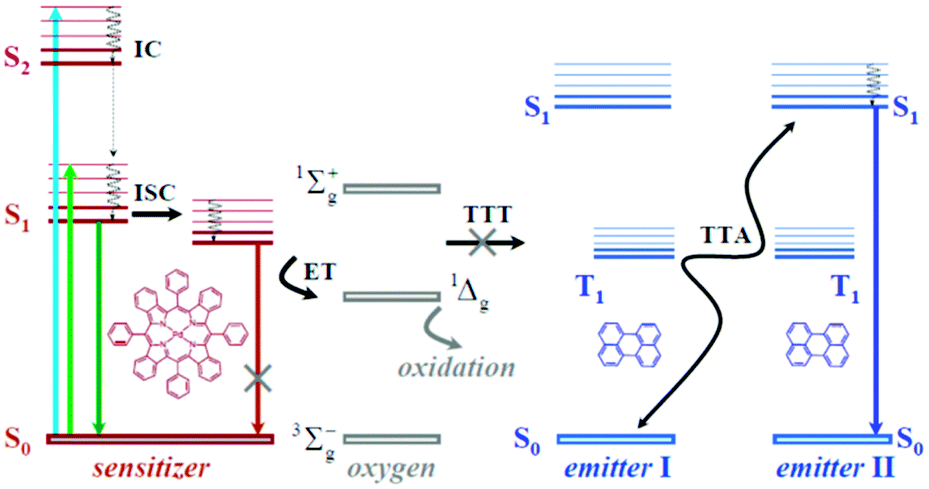 | ||
| Fig. 1 General energetic scheme of triplet–triplet annihilation upconversion. | ||
TTA in solid-state matter (organic molecular crystals)12 proceeds through direct absorption of the photons, followed by ISC in the emitter molecule. As a consequence, quadratic dependence between the excitation intensity and the intensity of the TTA-signal is observed. In the classical TTA-scheme,13 delayed fluorescence is observed at excitation intensities, comparable with those for the non-linear optical processes (MW cm−2)14 or UC in RE ion-doped systems (∼kW cm−2).15
In soft mater matrices, i.e. thin polymeric films or fluidic systems (volatile or non-volatile organic solvents) the concentration of the excited emitter triplets is orders of magnitude higher, than the concentration of excited emitter triplets states, observed in the classical TTA – experiments. Thus, in soft matter matrix the TTA-process is not only diffusion controlled and takes place in the so called “strong signal regime”, but demonstrates essential dependences on the material and environmental parameters such as degree of overlapping of the interacting energy states, matrix temperature, matrix viscosity and presence of molecular oxygen, dissolved at the solvent or adsorbed at the polymer film. It is important to notice, that all these materials and environmental parameters are strongly related, and their impact on densely populated triplet ensembles is not a linear combination of its partial impacts: for instance,16 optimization of the energy overlapping of the excited triplet states of the interacting moieties leads to drastic increase of the lifetime of delayed fluorescence (TTA-UC signal) and thus, to increased dependences on the local oxygen contamination.
Metallated macrocycles, such as porphyrins and phthalocyanines are known to possess strong phosphorescence.17 Dependence of their integral phosphorescence emission or the decay time of the phosphorescence is widely used as a thermal or oxygen sensor.18 The sensing process involves optical excitation of the molecule and registration of the decreased phosphorescence or phosphorescence decay time as a function of the increased sample temperature or oxygen content.19 The main experimental drawback of this sensing technique is that the phosphorescence emission is an integral parameter, depending simultaneously on the local temperature and local oxygen contamination.
In the presence of molecular oxygen, the energy stored in the excited states of the triplet ensembles, is being actively dissipated competing with emissive (phosphorescence) or non-emissive (triplet to triplet) energy transfer processes. The reason for this is the process energy transfer between organic molecule triplet state and ground state of molecular oxygen, leading to singlet oxygen (1Δg, Fig. 1). Singlet oxygen is a highly reactive species, leading to oxidation of the photoactive molecules, followed by further loss of efficiency. Fig. 2 illustrates both processes on an example of a palladium(II) tetraanthraporphyrin (PdTAP) – a representative of a newly explored class of near-IR light sensitizers.20 Upon interaction with molecular triplet oxygen, the excited state of PdTAP is being quenched leading to a decrease of phosphorescence intensity. Subsequently, the singlet oxygen formed takes part in a Diels–Alder type process on a parent sensitizer molecule, which leads to a loss of conjugation in the π-system and to a loss of near-IR absorption. Similar process is involved in photo-bleaching of other types of chromophores, e.g. anthracenes, perylenes, terrylenes and terrylenediimide.21
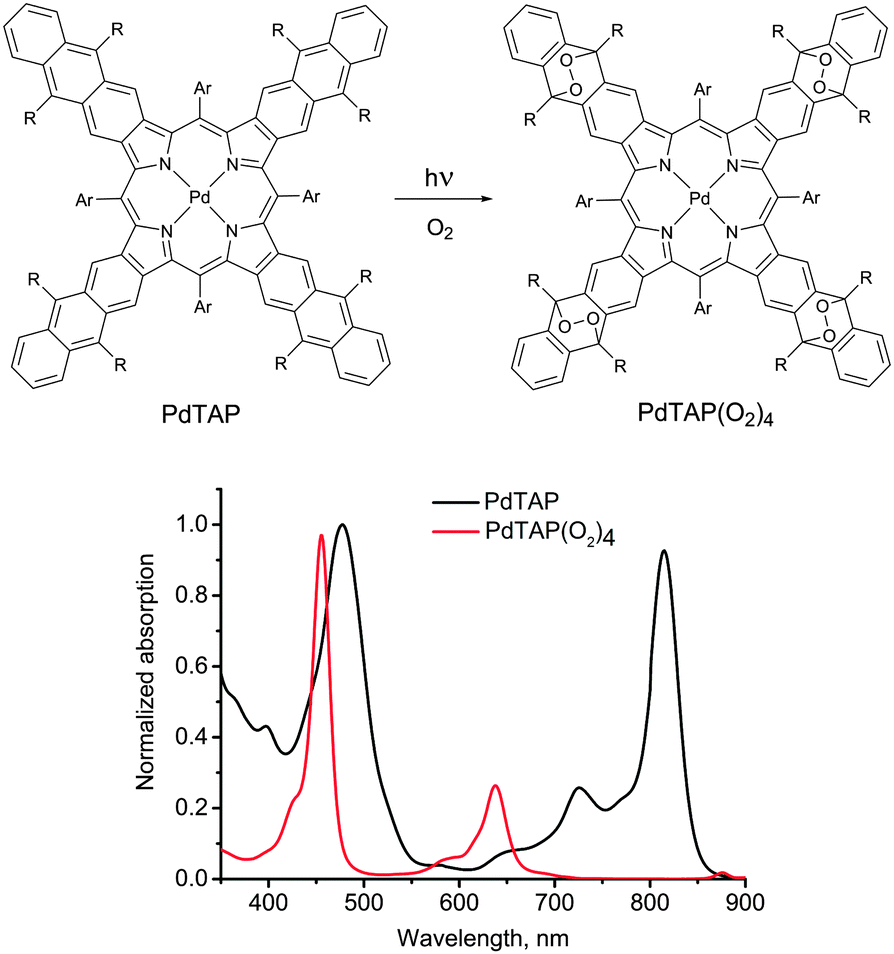 | ||
| Fig. 2 Photooxidation of tetraanthraporphyrin near-IR triplet sensitizer and change of its absorption spectrum. | ||
In the TTA based upconversion process, oxygen-induced quenching is usually a serious issue under atmospheric conditions, because the triplet energy of both the sensitizer and emitter excited states can be transferred to oxygen and generate singlet oxygen. In fact, even very small oxygen concentrations on the level of 2–10 ppm could affect the TTA-UC efficiency substantially. For example, the UC system based on platinum tetrabenzoporphyrin as a sensitizer and BODIPY as an emitter in toluene under argon exhibits a quantum yield of 15.0% (csens = 1.0 × 10−5 M, cem = 1.0 × 10−3 M, power density of 0.106 W cm−2) under deaerated condition, while in the oxygen saturated environment a 150-fold quantum yield decrease was observed.22 Thus, to fully exploit the TTA-UC process in different applications, the development of an effective protection strategy against quenching by molecular oxygen and protection against the subsequent production of highly reactive singlet oxygen is essential.
To date, several reviews on phosphorescence23 and TTA-UC24 as well as on the applications of these techniques for oxygen sensing25 have appeared, but still no accounts devoted to oxygen quenching and photooxidation protection of the excited triplet state ensembles have been published. The material presented in this review is illustrated in Fig. 3. Specifically, oxygen protection of excited triplet states can be regarded from two main perspectives: (1) passive protection, based on the application of barrier materials for packaging, sealing, or encapsulation of the active substances, which prevent oxygen molecules from penetration and physical contact with excited states, e.g. encapsulation into polymer films or nano- and microcarriers, or incorporation into supramolecular complexes and dendrimers. (2) Active protection, based on the application of oxygen scavenging species which react either with triplet or singlet oxygen to minimize the amount of oxygen available for deteriorative reactions, leading to degradation of the photoactive molecules. The material reflects works devoted both to the protection of phosphorescence and TTA-UC against oxygen quenching. We also included a discussion of the oxidative stress protection mechanisms of the natural photosystems in order to illustrate how the fundamental research in this area can contribute to the development of oxygen protection strategies in the field of organic optoelectronics.
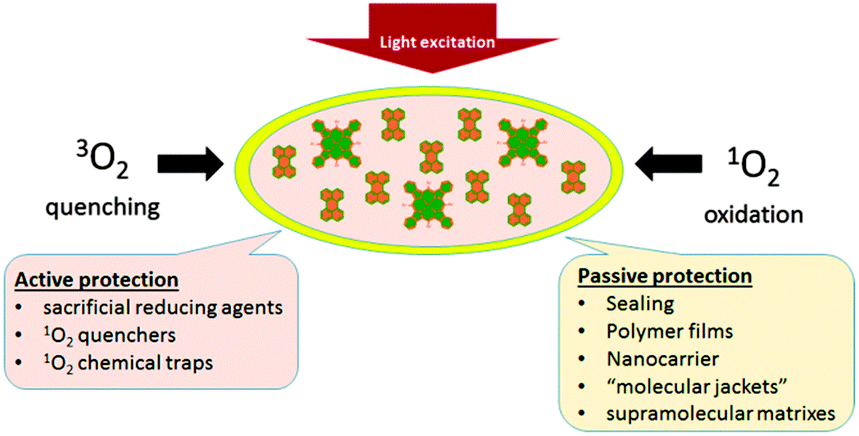 | ||
| Fig. 3 Diagram illustrating two general perspectives that can guide a research program on triplet ensembles oxygen protection. For each perspective, selected examples are provided. | ||
With the present review we intend to provide an extensive coverage of protection of both phosphorescent and TTA-UC materials against oxygen quenching with a systematic summary of the developed protection strategies and their comparison. In particular, emphasis is placed on the efficiencies of different approaches together with examples and prospects of their applications.
2. Self-protection of natural photosystems against oxidative stress and bio-inspired oxygen protection of TTA-UC
In photosynthesis, bacteriochlorophylls (BChl) play a crucial role in light harvesting and electron transport. However, some of the excited state transformations of BChl are known to be potentially dangerous for the cells due to the formation of the triplet excited state which can form the singlet oxygen. It may be formed in a variety of ways, however, a common way is by electronic energy transfer (1).| BChl + hν → 1BChl* → 3BChl* + 3O2 → BChl + 1O2 | (1) |
Since singlet oxygen is extremely reactive it may attack and oxidize proteins, lipids and nucleic acids, leading to cell destruction. All photosynthetic organisms possess special protective mechanisms to minimize the oxidative damage. Photosensitization of the triplet state of BChl leads to the formation of 1O2 unless BChl triplets are removed rapidly before 1O2 formation can take place.26 One of the conventional photoprotective mechanisms of photosynthetic organisms is based on the quenching of the triplet states by electron transfer to nearby carotenoids (Car), followed by radiationless deactivation of thus formed species. Carotenoid (Car) molecules are very effective quenchers of triplet state of BChl27 as well as of 1O2 in photosynthetic systems.28 However, despite their effectiveness in the protection, high light intensities cause a loss of photosynthetic activity in oxygenic organisms as reflected by the phenomenon of photoinhibition.29 It results in a decrease of the electron transport rate through photosystem II (PSII) and degradation of the D1 protein, an intrinsic subunit of the complex.
As shown in Fig. 4, the β-carotene bound to the PSII reaction center can act as an electron donor to BChl.30 However, if oxygen quenching of 3BChl takes place then the resulting singlet oxygen has to be removed efficiently at the point of its generation before it can diffuse away and induce uncontrolled oxidation. Two reaction sequences for an immediate scavenging of 1O2 at 3BChl are known: the turnover of the D1 protein and the oxidation of tocopherol (Fig. 4).
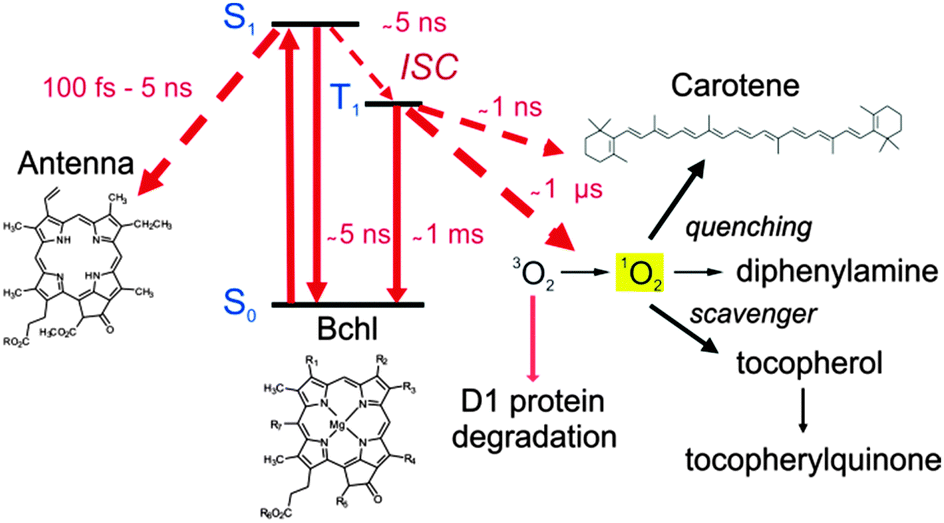 | ||
| Fig. 4 Energetic pathway of the excited singlet state of the bacteriochlorophyll molecules and singlet oxygen relaxation mechanisms. | ||
The role of α-tocopherol as an antioxidant is well known for plants and humans.31 Tocopherol is concentrated in the thylakoid membrane and plastoglobuli, playing a protective role in photosynthesis. Its concentration in plants is light dependent. Tocopherol is an effective singlet oxygen scavenger, being oxidized to tocopherylquinoned.32 The reaction proceeds via the 8-hydroperoxy-α-tocopherone, which is hydrolyzed to the quinone. Tocopherol is oxidized in PS II by singlet oxygen, but at the same time it is resynthesized by the cell. Inhibition of tocopherol synthesis lowers its concentration in the thylakoid membrane. When degraded under high light fluencies (>500 μmol photons m−2 s−1)33 and not replaced it can no longer scavenge singlet oxygen produced in the BChl triplet quenching. The D1 protein degradation is then much accelerated and the PS II activity is lowered.
Chemical quenchers of singlet oxygen, notably diphenylamines, completely protect PS II, prevent D1 protein degradation and keep tocopherol levels even at very high light intensities.34
In our research, devoted to the study of triplet–triplet annihilation upconversion and its applications, we attempted to mimic the natural function of tocopherol in the PSII, in order to decrease the sensitivity of the TTA-UC system towards oxygen quenching. Recently, we have demonstrated an efficient solubilization of TTA-UC components with low water solubility using amphiphilic block-copolymers from the family of polyoxyethanyl α-tocopheryl sebacate (PTS) as a surfactant.35 We assumed that due to hydrolysis of PTS in water some amounts of tocopherol is formed. Thus, singlet oxygen generated in the UC-system might be irreversibly scavenged by the reaction with tocopherol as illustrated in Fig. 5. It is important to notice, that binding of singlet oxygen by the sacrificial oxygen scavenger (the PTS in this case) or introducing a photo-oxidation of some of the active moieties is a competitive process. Presence of sacrificial scavenger in a concentration lower than some value, does not lead to complete protection.
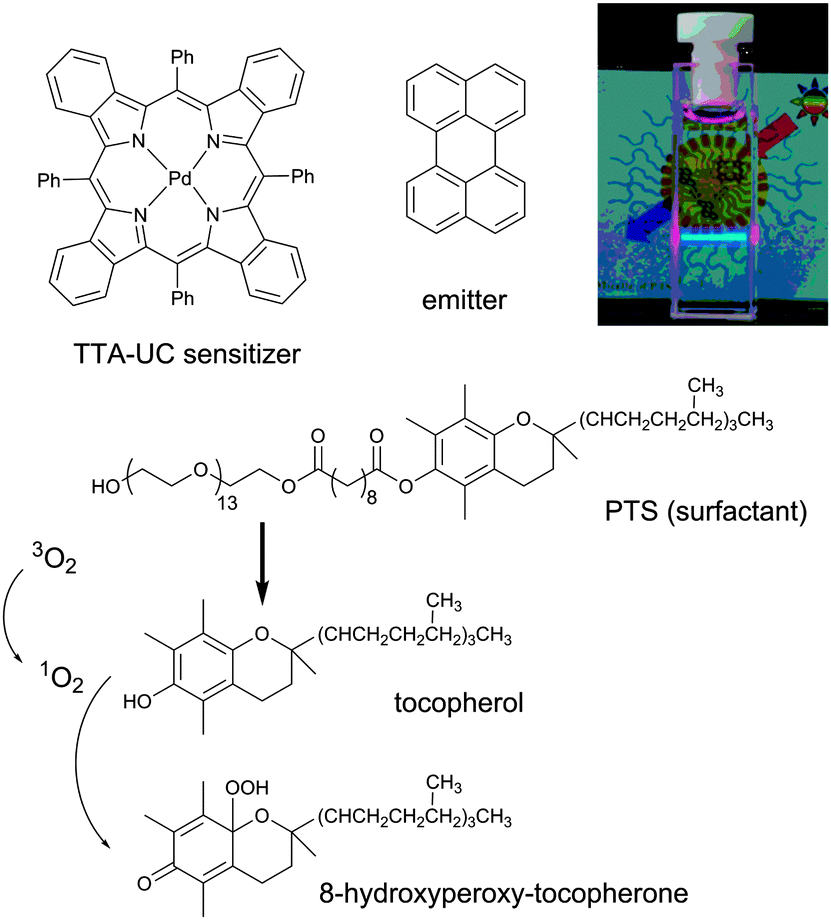 | ||
| Fig. 5 Reaction scheme of the reaction between tocopherol and photosensitized singlet oxygen and chemical structures of PTS, meso-tetraphenyl-tetrabenzoporphine palladium (PdTBP) and perylene. Inset: A photograph of the studied PTS-based micelles in water environment, excited by 635 nm laser.35 | ||
In order to prove this hypothesis we perform the following measurement: two samples of a standard UC system composed of palladium tetrabenzoporphyrin – with a low dye load (PdTBP, 1 × 10−6 M; perylene 2 × 10−5 M) and with a 20-times higher dye load (PdTBP, 2 × 10−5 M; perylene 4 × 10−4 M) in aqueous PTS solution (5%) were prepared in ambient atmosphere and in glove-box (see Fig. 6). The non-degassed sample was illuminated for a period of 5 h with very low intensity light (less than 50 μW cm−2, λexc = 633 nm, cw – laser, He–Ne). The excitation spot diameter of the low intensity light was with diameter of D ∼ 2 × 10−2 m an order of magnitude larger than the excitation spot diameter of the probing light (D ∼ 1 × 10−3 m).
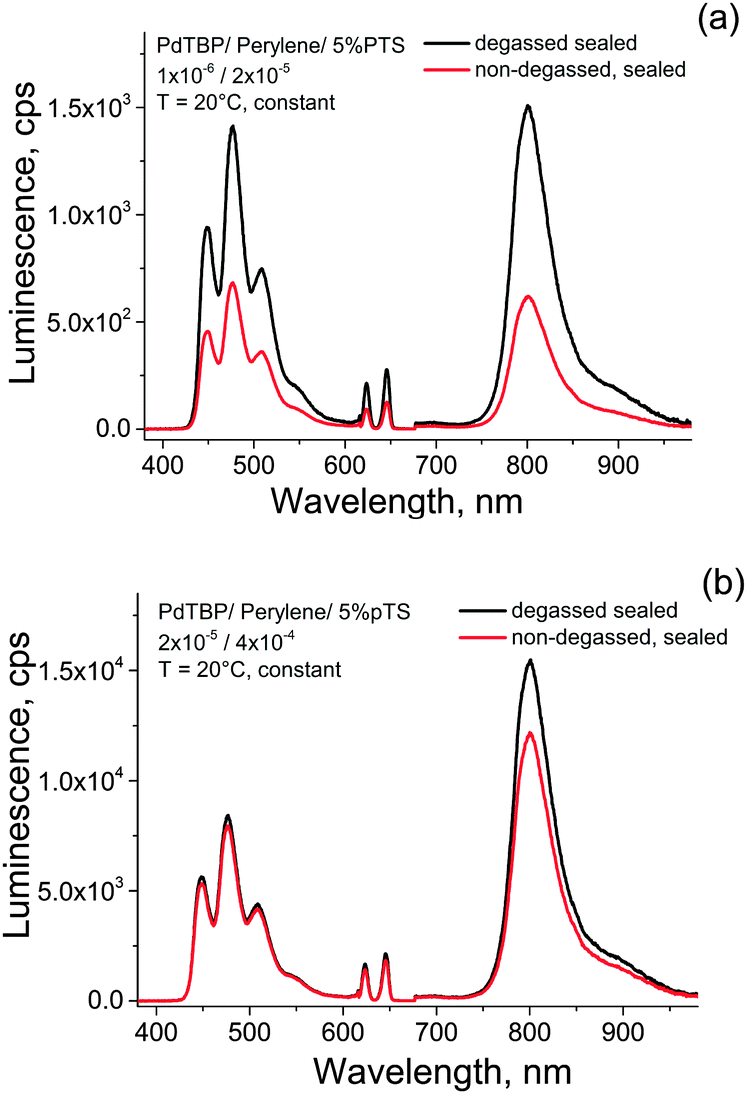 | ||
| Fig. 6 Comparison of the luminescence spectra of the UC-system PdTBP/perylene/PTS in aquatic environment for two different molecular compositions as follows: (a) 1 × 10−6/2 × 10−5/5% PTS in water and (b) 2 × 10−5/4 × 10−4/5% PTS in water. Excitation intensity of the probing light, for all measurement – 50 mW cm−2. | ||
As is shown seen in Fig. 6a, at low active dye concentrations, even after long-term illumination, the UC-fluorescence does not recover completely. This means, that a significant amount of the active dyes (most probably the sensitizer molecule is damaged by oxidation with singlet oxygen). On other hand, when the dye concentrations are relatively high the amount of the irreversible lost active dyes is negligible and the UC-fluorescence recovers. It is important to notice, that such an oxygen scavenging mechanism reported here is based on tocopherol as sacrificial agent and cannot fully replace the other oxygen protection techniques. Thus, deoxygenation of the samples before measurements is mandatory to achieve long-term protection.
3. Application of oxygen scavenging species
Usual ways to deoxygenate luminescent samples include nitrogen (or other inert gas) bubbling of the solutions, repeated freeze–thaw cycles, or preparation of samples in vacuum (glove-box). Without any doubt, degassing by inert gas purging is the most common method used in spite of the fact that it is time-consuming and inconvenient.Díaz García and Sanz-Medel were first to propose the use of sodium sulfite as an alternative to solution deoxygenation with nitrogen in the SDS micellar solution.36 The method is based on the redox reaction (2).
| 2SO32− + O2 → 2SO42− | (2) |
Heavy-atom induced phosphorescence37 of naphthalene and other aromatic hydrocarbons was studied to monitor the effectiveness and illustrate the convenience of the proposed method of deoxygenation. Time profiles of the deoxygenation reaction (2) were monitored by following the increase in phosphorescence intensity of the micellar solubilised naphthalene (Fig. 7). A gradual increase in phosphorescence intensity was observed since a diffusion rate of oxygen limits the efficiency of the quenching of phosphorescence produced by the solubilized naphthalene. This fact was ascribed to the dynamic nature of the micellar equilibria. As the sulfite ion is negatively charged, as well as SDS, its actual molar concentration is higher in the bulk solution (Gouy–Chapman layer) than in the micellar surface (Stern layer). Consequently, the oxygen consumption in the bulk phase is more efficient than in the Stern layer.
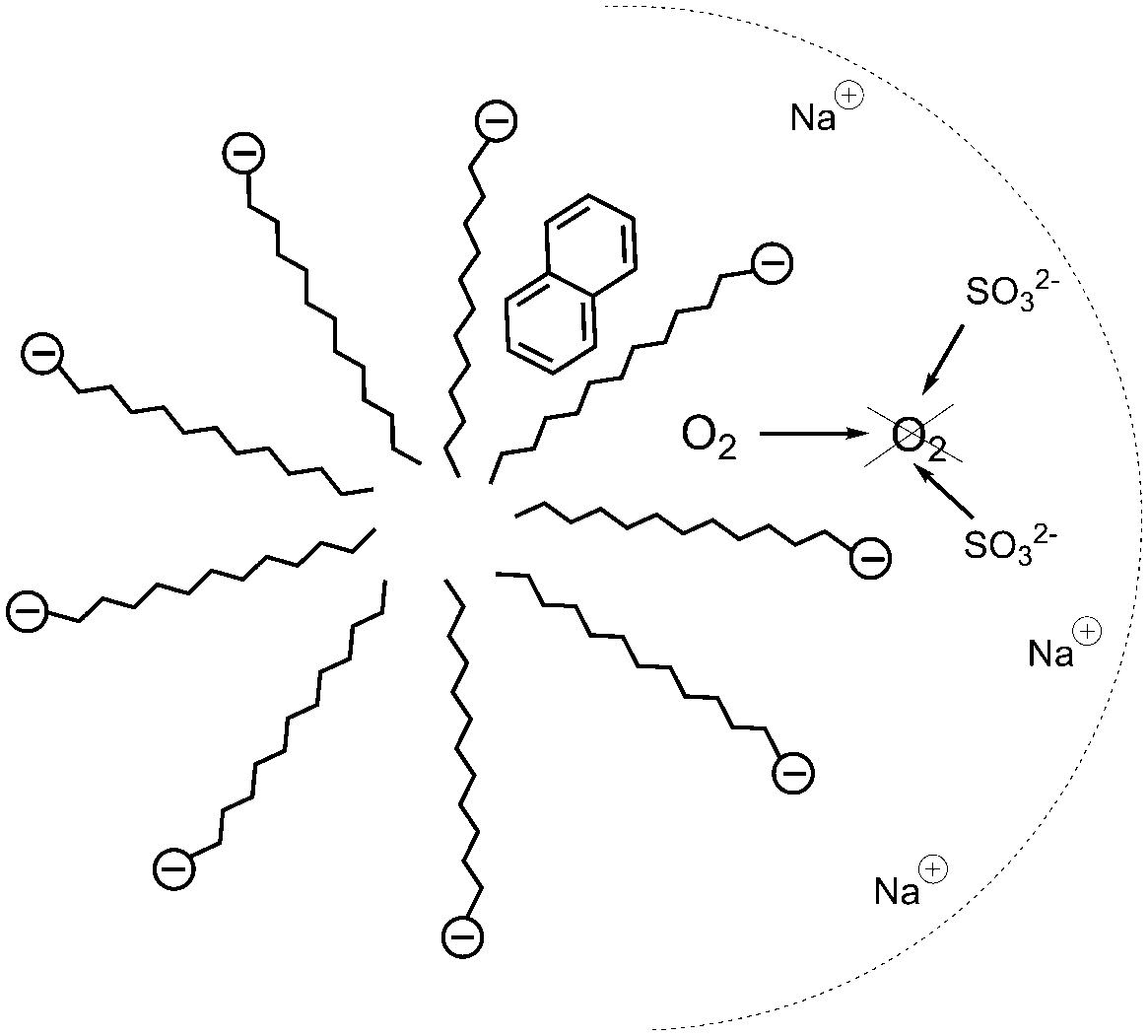 | ||
| Fig. 7 Schematic representation of the chemical deoxygenation in anionic micellar media. | ||
The concentration of sulfite is an important parameter affecting the phosphorescence intensity and must be taken into account. As a compromise between high luminescence signals and relatively short time for appearance of reproducible and convenient phosphorescence signals, a Na2S03 concentration of 10−2 M was found to be optimal for deoxygenation.
Sulfite-based O2 scavenging to observe phosphorescence signals in aqueous solutions was applied for the development of analytical methods for the detection of a variety of organic compounds (including agricultural, pharmaceutical, petroleum, and biological-related samples) and extends the technique to facile metal ion determinations in solution using room temperature phosphorescence (RTP).38
Based on these works, we explored the possibility of the application of sodium sulfite as oxygen scavenger for TTA-UC process performed in a micellar system. Sensitizer (PdTBP) and emitter (BODIPY) couple39 was found to exhibit negligible upconversion fluorescence along with residual phosphorescence in an air saturated SDS solution (Fig. 8, black line). After the addition of Na2SO3 (excess), a strong increase of the intensity of corresponding signals was immediately observed (Fig. 8), due to the deoxygenation of the solution. Such a technique allows for the protection of corresponding samples on a timescale of hours in the case the sample is open to air or for a longer time, if the sample is sealed. Nevertheless, Na2SO3 is not bio-compatible, and limits the application of sodium sulfite only to non-living objects.
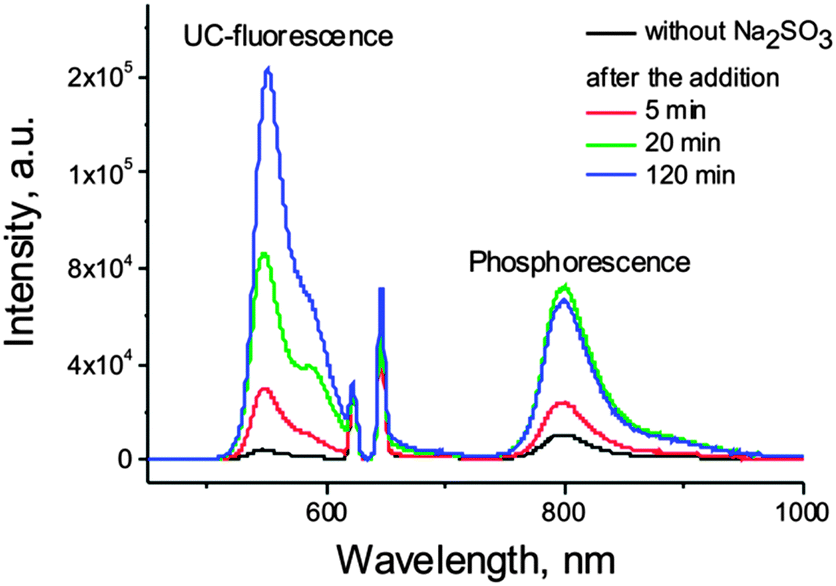 | ||
| Fig. 8 Effect of sodium sulfite on the phosphorescence and TTA-UC fluorescence in PdTBP–BODIPY micellar system. | ||
Bonnet and co-workers recently demonstrated red-to-blue photon upconversion system based on PdTBP and perylene for optical imaging of giant unilamellar vesicles lipid bilayer.40 In order to investigate the dye distributions across the membrane, the homogeneity of light emission in the lipid bilayer and the upconversion stability under imaging conditions, vesicles containing sensitizers and emitter dyes were prepared. Sodium sulfite was used as an oxygen-scavenging agent to prevent triplet states quenching. In the case addition of Na2SO3 was found to be superior with respect the common technique of deoxygenation via purging the solution with inert gas. The latter is problematic due to foam formation and mechanical damaging of the giant vesicles. A comparison of the vesicles samples deoxygenated either by bubbling Ar for 30 min or by addition of 0.3 M sodium sulfite into the buffer solution used as a media for the vesicles is shown in Fig. 9. Although for both samples the spectra are very similar, showing that Na2SO3 does not affect the photophysical processes in the system, upconversion intensities were found to be higher when using oxygen scavenger. This can be elucidated, if one to takes into account, that Ar-bubbling results in the formation of stabile small bubbles distributed into the sample, which can scatter both the excitation and emission light. Such bubbles are absent when deoxygenation is achieved using sodium sulfite oxygen scavenger.
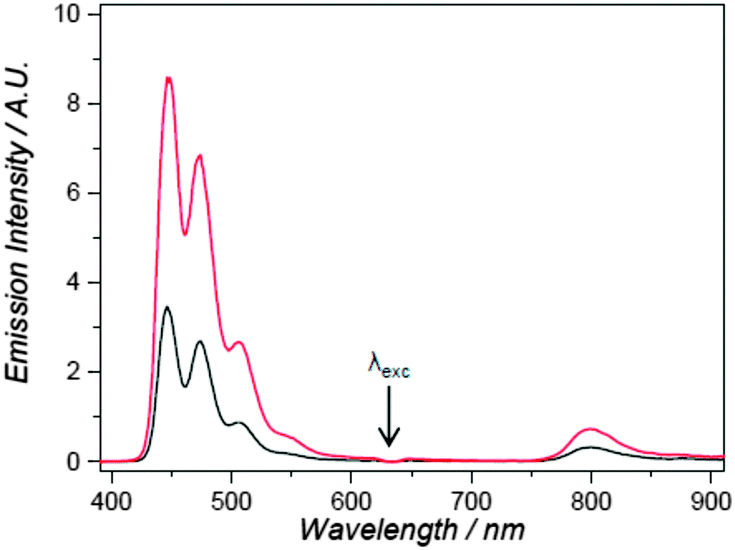 | ||
| Fig. 9 Upconversion fluorescence of perylene-PdTBP system within 1,2-dioleoyl-sn-glycero-3-phosphocholine based vesicles under 630 nm excitation. Irradiation conditions: 30 mW excitation power (4 mm beam diameter, intensity 0.24 W cm−2). Sample was deoxygenated either by bubbling Ar for 30 min (black curve) of by addition of 0.3 M sodium sulfite into the vesicles media (red curve).40 Published by The Royal Society of Chemistry. | ||
4. Incorporation of dyes into supramolecular complexes
When the molecules are incorporated into molecular aggregates, such as micelles, microemulsion droplets, and vesicles, or in cavities such as cyclodextrins (CDs), calix[n]arenas, cage compounds, and other analogues, they are often confined to a greater degree of organization compared with those in homogeneous solution. This phenomenon was used in mimicking the processes occurring in biosystems41 as well as in energy storage application.42The observation of the phosphorescence of tryptophan residues in proteins at room temperature in solutions43 initially raised an idea of the incorporation of a luminescent molecule into supramolecular complexes in order to reduce oxygen quenching processes. For example, in the case of horse liver alcohol dehydrogenase, tryptophan phosphorescence is observed even in aerated solutions at ambient temperature.44 It has been proposed, that certain protein conformations strongly inhibit quenching of tryptophan triplets by dissolved oxygen.45
The inhibition of oxygen quenching of the phosphorescence via complexation of the dye molecule with cyclodextrins (CD) was first discovered by Turro and co-workers.46 The phosphorescence of 1-bromo-4-naphthoyl group is readily quenched by oxygen in the solution. However, when this dye is complexed with γ-cyclodextrin in aqueous solution, its phosphorescence can be observed even in oxygen-saturated solutions (Fig. 10). In contrast, in the presence of α- and β-CD oxygen was observed to quench the phosphorescence, even though the dye forms complexes with β-CD.
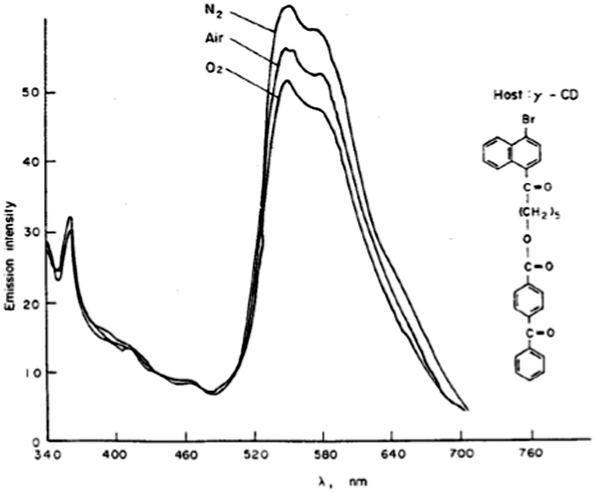 | ||
| Fig. 10 Phosphorescence spectra of 4-benzoyl-benzoic acid 6-(4-bromo-naphthalen-1-yl)-6-oxo-hexyl ester complexed with γ-cyclodextrin. The effect of added oxygen.46 Reprinted with permission from ref. 60. Copyright Wiley-VCH Verlag GmbH. | ||
Phosphorescence decay data indicated that two types of probe–cyclodextrin complexes are formed with lifetimes of 3.5 and 600 ms respectively. Oxygen completely quenches the fast decay, but only partially quenches the slow decay, indicating that the probe exists in two environments in the presence of γ-CD. This was attributed to the formation of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 2:1 complexes, giving fast and slow decays respectively. It was found that the appearance of the long-lived (>1 s) room temperature phosphorescence (RTP) in water is associated with the formation of macrostructures (aggregates) in which the dye molecules are separated from the surrounding medium by molecules of γ-CD and precipitant, which initiate the formation of the aggregate.47
:1 and 2:1 complexes, giving fast and slow decays respectively. It was found that the appearance of the long-lived (>1 s) room temperature phosphorescence (RTP) in water is associated with the formation of macrostructures (aggregates) in which the dye molecules are separated from the surrounding medium by molecules of γ-CD and precipitant, which initiate the formation of the aggregate.47
Strong phosphorescence of 1-bromo-4-(bromoacetyl)naphthalene (BBAN) was found by Jin and co-workers to be induced synergetically by β-CD and Brij30 (polyethylene glycol dodecyl ether) without removal of oxygen dissolved in the solution due to the formation of a ternary complex of β-CD, BBAN, and Brij30.48 In this system, Brij30 acts as a space-regulating molecule to enhance the stability of the complex of β-CD/BBAN and to hinder the diffusion of molecular oxygen to quench the phosphor.
Strong and stable phosphorescence resulting from a 1:1:1 β-cyclodextrin/thiabendazole/triton X-100 supramolecular ternary inclusion complex (Scheme 1) induced by KI as a heavy atom perturber without removing dissolved oxygen from the solution was reported by Tang and co-workers.49 The formation of the inclusion complex protects the phosphorescence against varying quenching factors. Compared with the method using a chemical oxygen scavenger, this method is simpler as deoxygenation of the solution is not required. The proposed method has been successfully applied for the determination of thiabendazole (TBZ) in tap water, lake water, and pineapples.50
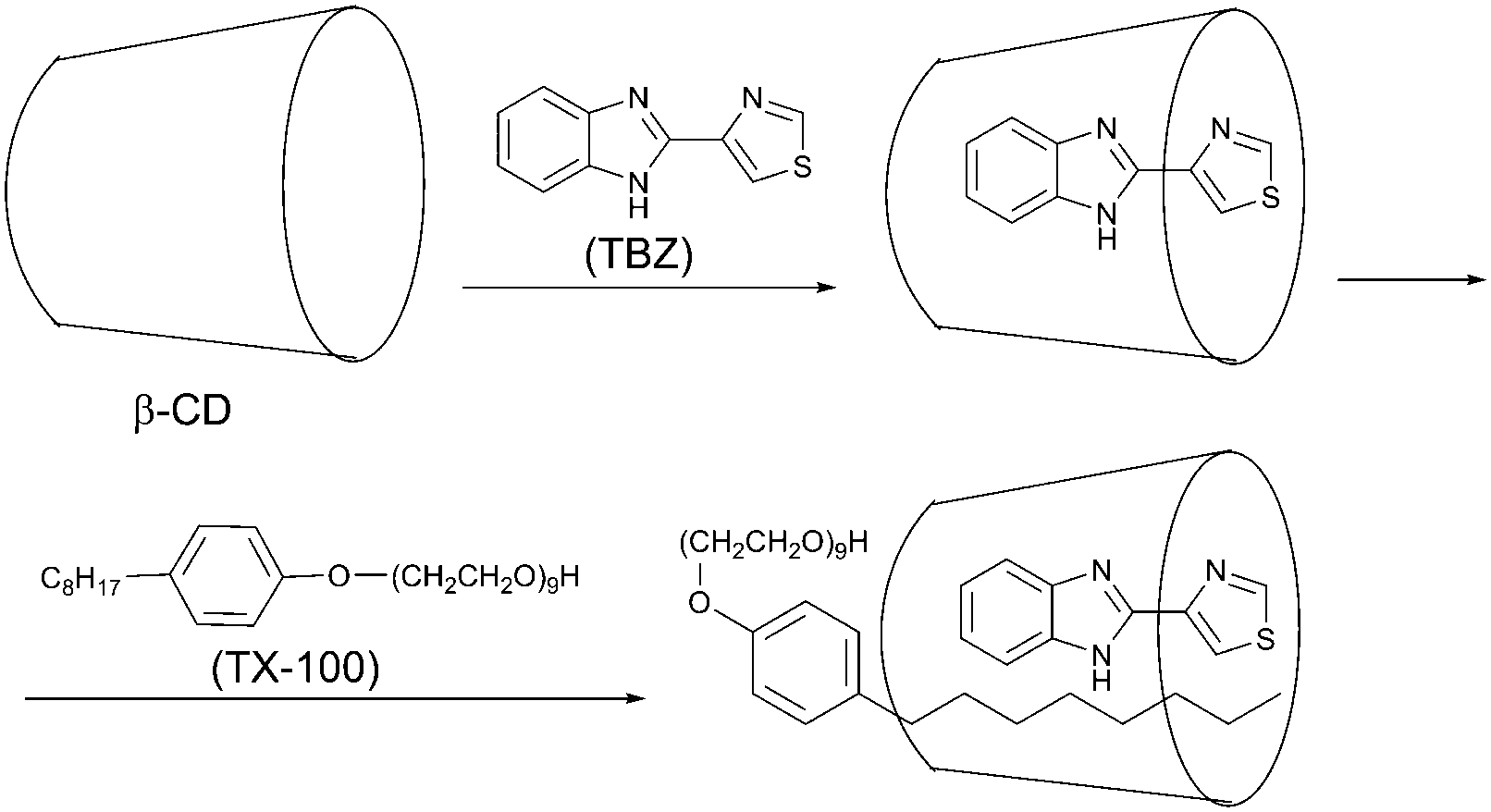 | ||
| Scheme 1 Formation of the phosphorescent inclusion complex between β-cyclodextrin, (β-CD), thiabendazole (TBZ) and triton (TX-100). | ||
Compared with other strategies of the phosphorescent samples deoxygenation, application of cyclodextrin complexes offers great advantages for the systems in which deoxygenation procedures may affect the detection of the analyte, e.g. in pH or temperature sensors in waters or biological samples.51
These examples show that the inclusion of phosphorescent dyes into cyclodextrin-based supramolecular complexes can provide a very efficient and facile way for the protection of excited triplet states. However, due to size and specific chemical environment of the host molecules, the approach is rather restricted. Particularly, no reports on its application in TTA-UC process have been made, although β-cyclodextrin has been studied by Wang and co-workers as a media to assist green-to-blue upconversion in palladium tetraphenyl porphyrin (PdTPP)–diphenylanthracene (DPA) pair. In this system a very high upconversion efficiency values (ΦUC ∼ 36%) were observed due to enhancement of triplet state lifetimes upon complexation into cyclodextrin, resulting in enhanced triplet–triplet annihilation efficiency.52 Although the behaviour of the system in oxygen-rich conditions has not been studied, the results suggest that the cyclodextrin-based TTA-UC system could provide an alternative to other oxygen protection strategies. This work may inspire others to develop new cyclodextrins or other type hosts structurally similar to cyclodextrins particularly suited to attenuate oxygen quenching in TTA-UC ensembles.
5. “Molecular jackets” around luminescent centers
5.1 Dendrimers
Phosphorescent probes based on dendrimers attracted interest in recent years because, among all types of synthetic polymeric carriers, dendrimers offer the unique advantage of molecular monodispersity.53 Optical probes for biological imaging of oxygen present an important example of the encapsulating capability of dendrimers, which plays a key role in the construction of these useful materials.Dendritic attenuation of oxygen quenching has been documented in a number of studies.54 If a chromophore is encapsulated inside a dendrimer, the latter forms a protective cage, preventing physical contacts of the core with macromolecular objects in the environment. However, protecting the chromophore from collisions with small molecules is not as straightforward because the latter can effectively diffuse through the body of the dendritic matrix. A decrease in the rate of oxygen diffusion can effectively offset an increase in its local concentration, thus lowering the apparent constant kq. Hydrophobic dendritic branches fold in polar environments (e.g., water), and as a result, their mobility becomes restricted, preventing oxygen molecules from freely reaching the phosphorescent core.55
Among dendrimers with flexible aromatic skeletons, dendritic poly(arylglycine) (AG) dendrons56 are especially well-suited for the construction of phosphorescent oxygen probes. AG dendrons offer the advantage of inexpensive starting materials, simplicity of synthesis, and chromatography-free purification. Terminal amino groups on AG dendrons can be reacted with carboxyls groups on the porphyrins, while terminal carboxyls on the dendrons provide multiple opportunities for functionalization. AG dendrons (Fig. 11) of three successive generations were developed by Vinogradov and co-workers57 other for the modification of platinum and palladium porphyrins and π-extended porphyrins. As expected, the oxygen quenching rates decrease significantly with an increase in the dendrimer generation. Overall, for such dendrimers the quenching rate drops by more than 20 times compared to parent phosphorescent porphyrins.
 | ||
| Fig. 11 Palladium(II) tetrabenzoporphyrin-based phosphorescent dendrimer. | ||
To simplify the dendrimer synthesis, dendrons can be bound to a phosphorescent molecule by means of Huisgen “click reaction”. Recently Evans and co-workers developed a new group of “clickable” phosphorescent porphyrins for sensing of tissue oxygenation. Alkynyl-substituted precursor was used to build a glutamic dendrimer by clicking eight azido-terminated glutamic dendrons (Scheme 2). Parent porphyrin precursor was found to be completely non-emissive in air saturated conditions. However, when imbedded into dendrimer structure its phosphorescence can be used visualization and quantification of tissue oxygenation of skin burns by naked eye under room lighting conditions.58
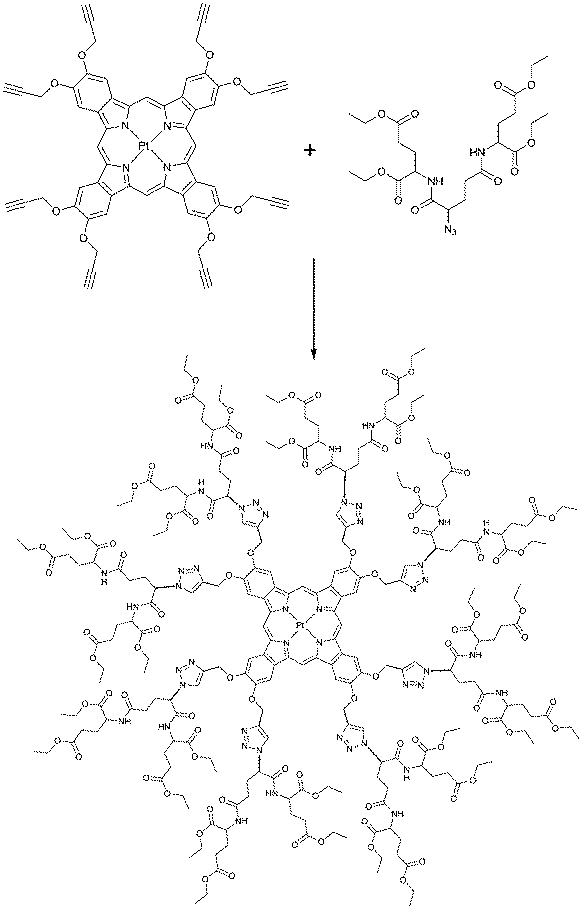 | ||
| Scheme 2 Synthesis of a phosphorescent glumatic dendrimer via click reaction. | ||
5.2 Introduction of bulky substituents into dye molecules
Decreasing triplet excited state quenching rates by means of a steric congestion caused by the introduction of bulky alkyl-groups around phosphorescent platinum porphyrin (Fig. 12) was reported by Moiseev et al.59 The comparison with the known platinum tetraphenylporphyrin PtTPP demonstrated the role of the substituents in suppressing quenching of the triplet excited states: the bimolecular rates for oxygen quenching kO2 of PtTTEPP and PtTPP triplet excited states were calculated to be 4.1 × 108 M−1 s−1 and 14.8 × 108 M−1 s−1, respectively. The difference between the two derivatives was attributed to steric effects from the ethyl groups which “screen” the molecule from interacting with the quenchers.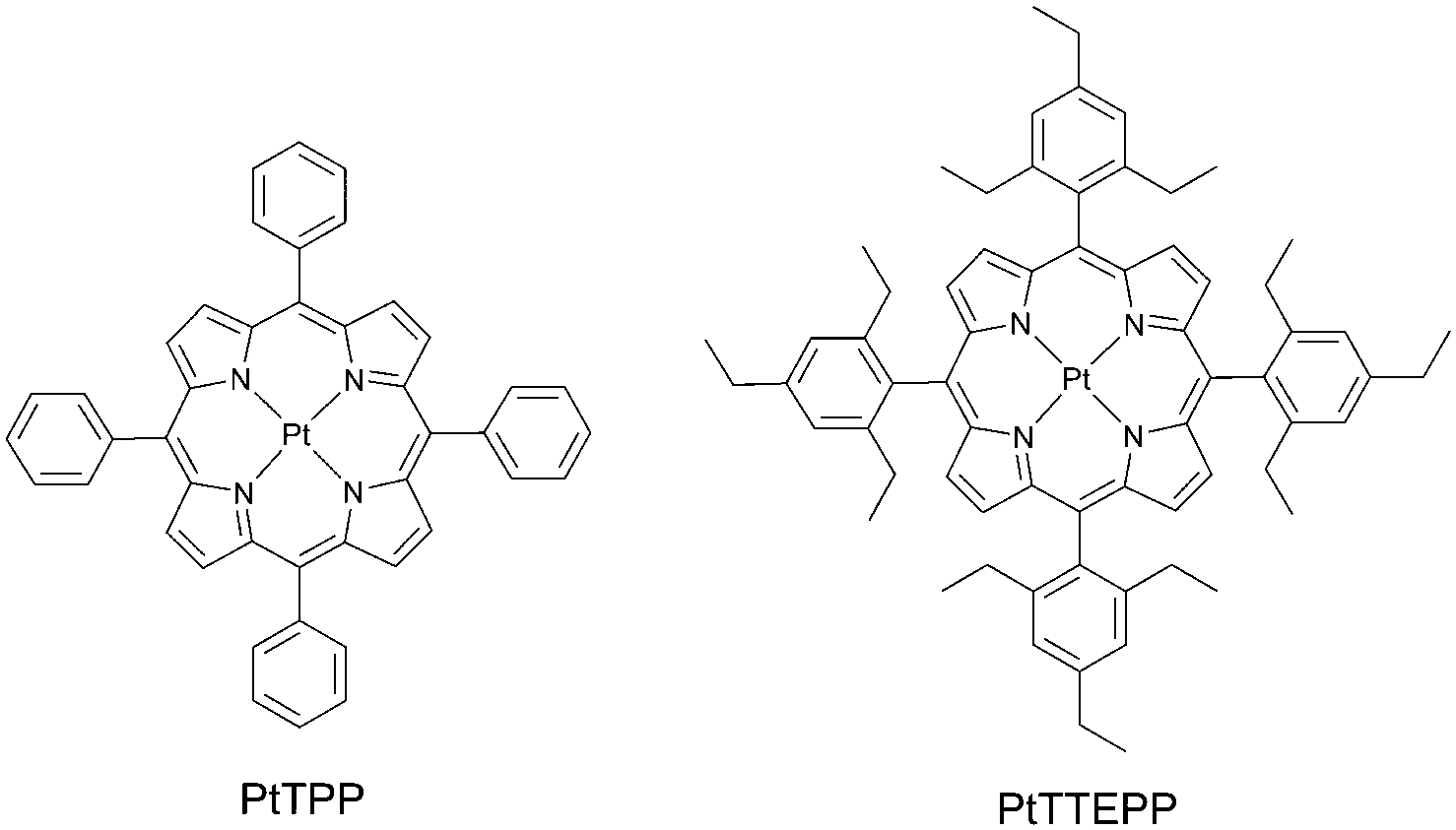 | ||
| Fig. 12 Sterically congested phosphorescent platinum porphyrin. | ||
Kimizuka and co-workers reported a TTA-UC air-operating system comprised of a solvent-free liquid sensitizer and emitter mixture (1:100 respectively) in which branched alkyl chains are appended to the PtTPBP sensitizer and DPA annihilator, respectively (Fig. 13). Due to impermeability of the hydrophobic alkyl chains, surrounding the chromophores, to oxygen, the UC efficiency of this system was found to be as high as 28% (600 mW cm−2) under aerated conditions.60
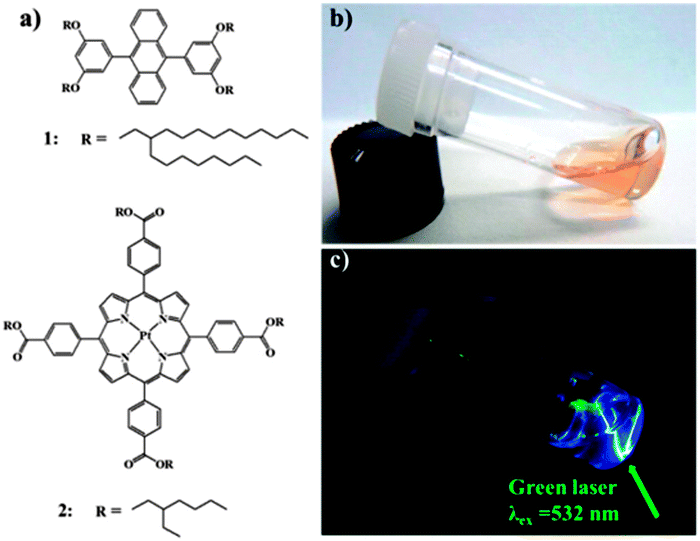 | ||
| Fig. 13 Chemical structures of liquid (a) emitter and sensitizer for TTA-UC. Photographs of the liquid mixture (b) upon irradiation with 532 nm green (c). Reprinted with permission from ref. 60. Copyright 2013 American Chemical Society. | ||
This system was found to operate almost unperturbed in the presence of air on a time scale of months. The UC intensities obtained for vacuum prepared samples were found almost identical to those obtained in air. No significant loss in the quantum efficiency within 20 days of air exposure was observed and quantum yield as high as 15% was measured even after the longer period of 80 days. These results were attributed to the suppressed diffusion of oxygen molecules into liquid chromophores, indicating an air-sealing effect of molten alkyl chains introduced around the chromophores.
5.3 Dyes bearing protective groups
In our recent work61 we reported another new strategy for protection of triplet excited states depopulation by quenching, relying on the chemical modification of the triplet sensitizer molecule. It is based on binding singlet oxygen, present in a sample, to specially designed anthracene substituents (Scheme 3) which do not affect photophysical properties. This protection strategy is of sacrificial character, and is time- and total molecular oxygen concentration limited. However, the starting porphyrin can be fully regenerated through oxygen release upon moderate heating in vacuum. Anthracene groups do not bind oxygen in its ground (triplet) state and the corresponding material is stable towards photooxidation in the course of synthetic procedures and purification. The protection is active only against singlet oxygen, thus only when sensitizer triplet states are formed.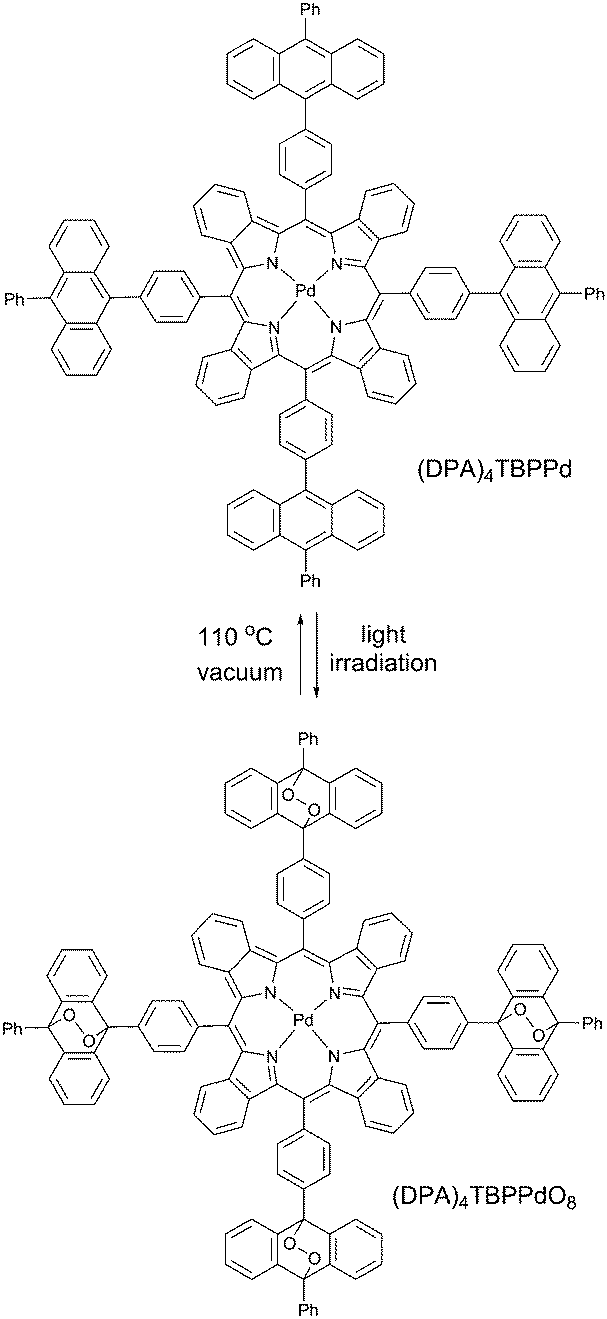 | ||
| Scheme 3 Reversible oxygen addition on a palladium tetrabenzoporphyrin molecule. | ||
Enhancement of the phosphorescence originating from the porphyrin (DPA)4TBPPd was demonstrated upon an excitation of oxygen containing sample in a local area (400 μm diameter). In the course of the measurement, the same laser beam was used for the excitation of phosphorescence as well as for local deoxygenation. The phosphorescence signal intensity of (DPA)4TBPPd rises more than 60% at continuous irradiation by laser beam during the measurement. As seen from Fig. 14 (green line) at 1 mW cm−2 excitation, the process of local oxygen scavenging needs only a few seconds. If the excitation intensity is lower, the necessary time is substantially longer (Fig. 14, black line).
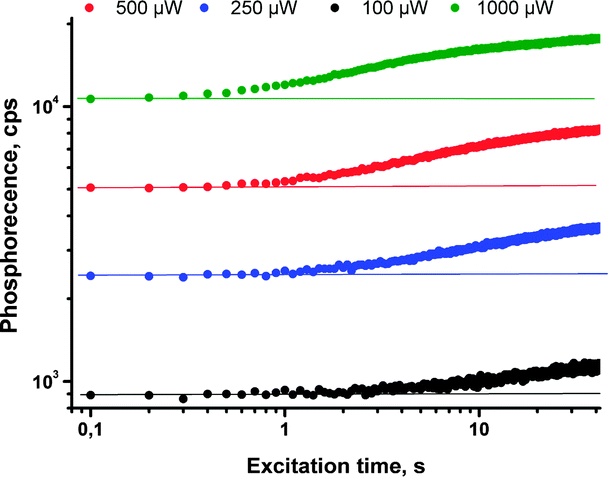 | ||
| Fig. 14 Change of phosphorescence intensity at 798 nm for the sample of (DPA)4TBPPd registered in oxygen contaminated atmosphere (100 ppm), taken at noted periods of time during continuous irradiation by red laser (toluene, 4 × 10−5 M, 633 nm).61 Published by the PCCP Owner Societies. | ||
Although such protection strategy is limited by the capacity of the sensitizer to bind four oxygen molecules, it may have a potential in applications which demand very low oxygen concentration levels. For example, most of the materials comprising the OLEDs suffer degradation effects from the presence of environmental oxygen and water, as both compounds can penetrate into the device. Much research and effort have been put into fabrication methods and proper device encapsulation to help mitigate these environmental effects. Thus, the demonstrated oxygen protection strategy on a molecular level might become complementary to those already developed.
5.4 Metal complexes with self-assembling units
Low molecular-weight gel systems have recently become a topic of growing interest because they provide a new way to access novel well-defined supramolecular architectures.62 Low molecular-weight gel materials can be produced by phase separation of gelator molecules and solvent molecules. If formed in such a way, the gelator phase does not incorporate oxygen dissolved in the solvent phase. Moreover, the gels possess crystal-like nature and deactivation of the triplet state molecules by oxygen is effectively inhibited.Shirakawa and co-workers reported gel formation and phosphorescence properties of 8-quinolinol chelates of copper, palladium and platinum bearing 3,4,5-tris(n-dodecyloxy)benzoylamide substituents (Fig. 15).63 The gelation ability of the compounds was evaluated in various organic solvents such as benzene, hexane, 1-butanol, 1,4-dioxane and others. Very low critical gelation concentrations (c.g.c.) of 0.10 mg L−1 (0.05 mM) were observed.
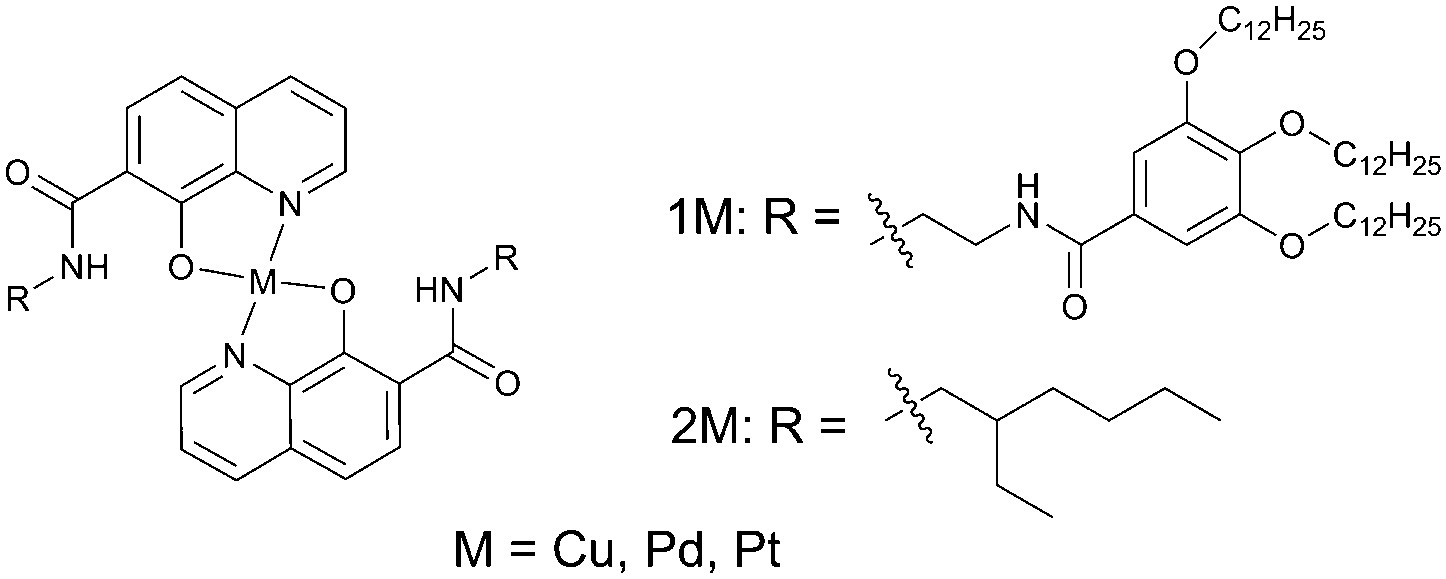 | ||
| Fig. 15 Structures of 3,4,5-tris(n-dodecyloxy)-benzoylamide substituents-appended 8-quinolinol platinum(II) chelate gelator (1Pt) and non-gelling reference compound (2Pt). | ||
A packing structure shown in Fig. 16 for platinum(II) chelate was proposed as the most likely aggregation model.
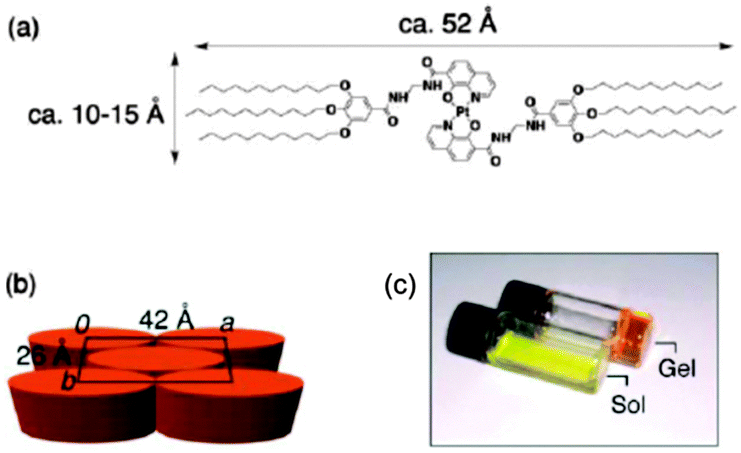 | ||
| Fig. 16 (a) Molecular structure of 8-quinolinol/platinum(II) chelate, (b) proposed packing model of the chelate molecules in the gel, (c) photograph of the gel and its heated solution (sol). Reproduced with permission from ref. 63. Copyright 2007 Wiley-VCH Verlag GmbH. | ||
The efficiency of dioxygen quenching inhibition, defined as a ratio between phosphorescence intensity under oxygen-saturated condition and intensity of the samples degassed with argon (Evs.O2), was demonstrated by the following experiment. Luminescence spectra of 1Pt and 2Pt (Fig. 15) were measured under argon atmosphere or dioxygen-saturated atmosphere in the gel or sol phase. It was found that the Evs.O2 values of 1Pt are larger than those of 2Pt in all gelling solvents. At the same time, the Evs.O2 values of 1Pt and 2Pt are almost the same in the non-gelling solvent, 1,1,2,2-tetrachloroethane (Fig. 17). These results support the idea that the enhancement of the Evs.O2 is not caused by peripheral substituents, but is due to the molecular assembling effect in the gel. Thus the gel phase insures protection of the triplet excited states from oxygen penetration and further quenching. This makes gel matrixes attractive materials for photonic applications, particularly for TTA-UC as is shown in next section.
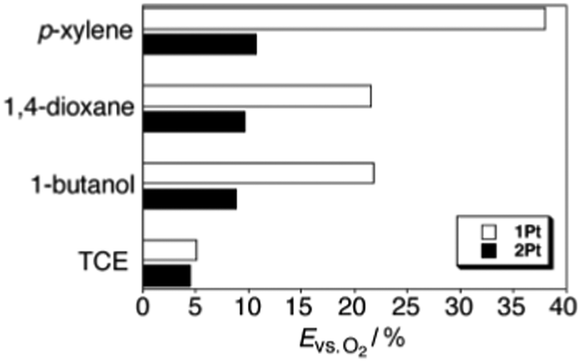 | ||
| Fig. 17
E
vs.O2 (ratio between phosphorescence intensities of oxygen-saturated and degassed samples in percent) values of 1Pt (white) and 2Pt (black) in different solvents. Reproduced with permission from ref. 63. Copyright 2007 Wiley-VCH Verlag GmbH. | ||
6. Host–guest matrixes
6.1 Molecular aggregates
In molecular aggregates, a rigid microenvironment of the excited state may result in preventing the collision with a quencher and therefore is of interest as a protection technique. Oxygen-persistent phosphorescence in molecular aggregates was first described by Jin and co-workers64 for palladium(II) meso-tetra-(4-trimethylaminophenyl)porphyrin solution in water containing sodium deoxycholate (SDC). When adding SDC into the air-saturated solution of the porphyrin, a gradual increase of the phosphorescence intensity was observed along with the increase of the lifetime, reaching 0.39 ms. Interestingly, in the absence of SDC, the porphyrin was not emissive at all even in deoxygenated aqueous solution. The effect of SDC on the phosphorescence was interpreted by assuming a formation of a ‘sandwich’ type dimers in which the porphyrin is trapped between two steroid scaffolds. The effect was found to be much less pronounced for other aggregate forming molecules, e.g. sodium dodecylsulfate and DNA.Protection of phosphorescent porphyrins from oxygen quenching in SDC aggregates was then studied in details by Zhang and co-authors.65 Three palladium(II) meso-tetraarylporphyrins tested (Fig. 18) showed no phosphorescence in air-saturated aqueous solutions. Upon titration with SDC the phosphorescence intensities of Pd-TAPP (2 × 10−6 M) displayed significant increase, while Pd-TCPP and Pd-TSPP showed a very weak increase. The phosphorescence of Pd-TAPP showed maximum value at 4 × 10−3 M SDC concentration, but at higher concentrations the phosphorescence gradually decays and completely disappears when big excess of SDC was added. This was accounted for as formation of dimer species at low concentration of deoxycholate, which then transform into larger aggregates with less rigid structure, allowing oxygen quenching of the triplet excited states. Observation of the effect of the porphyrin Pd-TAPP, possessing positively charged groups indicate electrostatic interactions between SDC and the porphyrin. Due to outstanding stability of the phosphorescence signal, Pd-TAPP was then used as a probe to detect bovine serum albumin.
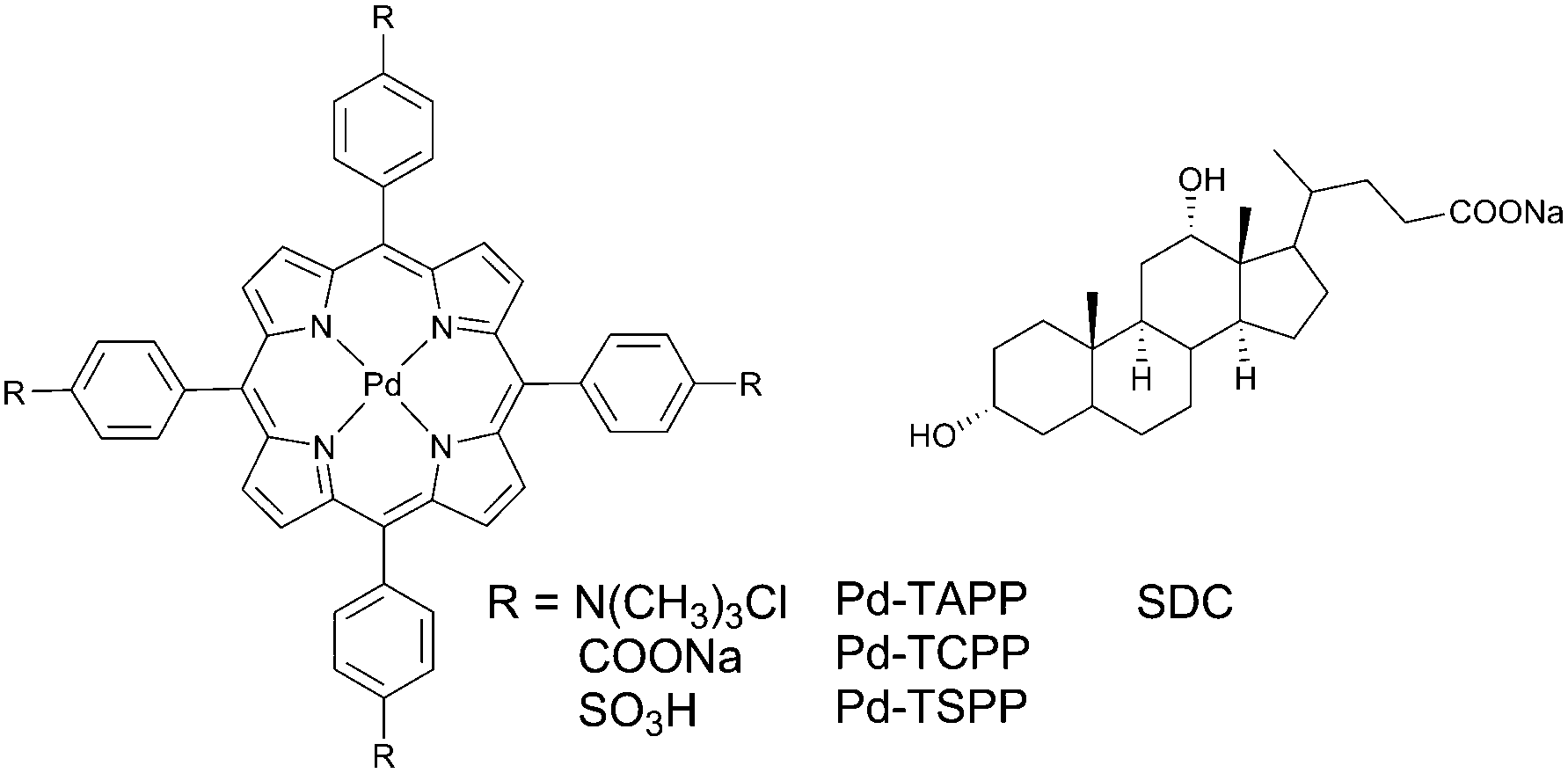 | ||
| Fig. 18 Palladium(II) porphyrin forming molecular aggregates with sodium deoxycholate. | ||
A modified approach based on using steroidal compounds as host matrixes for phosphorescent molecules was recently applied by Adachi and co-workers to develop materials with persistent emission and long lifetime (>1 s).66 Notably, in this case protective matrixes were designed not only to protect the triplet excited states from quenching, but to minimize other nonradiative deactivation pathways of triplet states. The following non-radiative processes were considered: (1) energy transfer from triplet excited states to the molecules of host matrix, (2) diffusional motion of matrix, (3) concentration quenching of guest compounds and (4) quenching by oxygen. β-Estradiol and a mixture of cholesterol with α,α,α′-tris(4-hydroxyphenyl)-1-ethyl-4-isopropylbenzene were investigated as host matrixes for a series of aromatic hydrocarbon based phosphorescent molecules (Fig. 19).
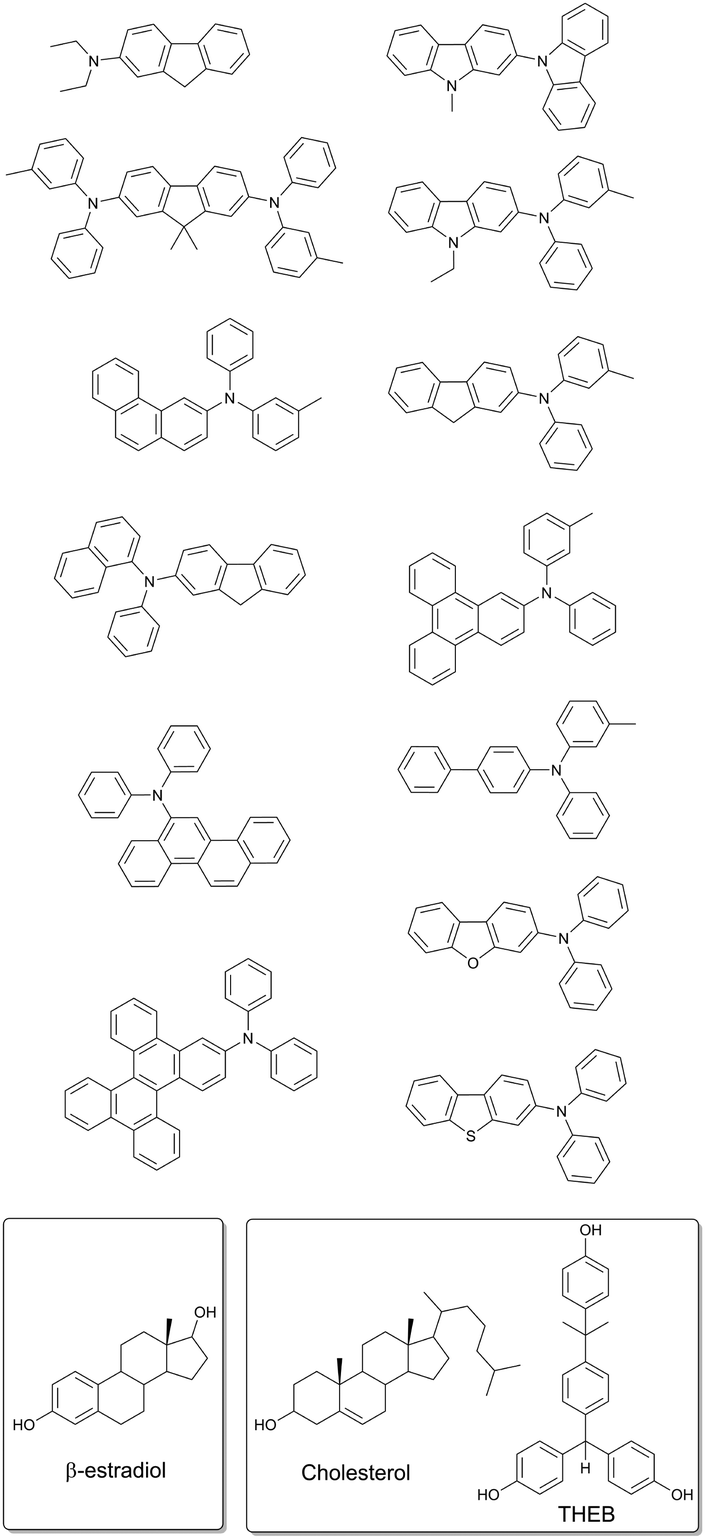 | ||
| Fig. 19 Molecular structure of the phosphorescent dyes and guest matrixes providing long time persistent emission. | ||
Oxygen was removed from the studied steroidal matrixes by heating above the melting point and further cooling. Due to the slow diffusion of oxygen, low concentration in the matrix for long periods of time are expected. It was found that phosphorescence of the studied dyes is almost not affected by oxygen quenching in β-estradiol and cholesterol matrixes. In contrast, triplet states of the same molecules were observed to be completely quenched within <1 ms time interval on air, when other amorphous materials, e.g. PMMA, were applied as the matrix.
6.2 Organogel matrixes
Supramolecular organogels are formed by the self-assembly of low-molecular-weight gelators (LMWGs), due to non-covalent interactions, such as hydrogen bonding, p–p interactions, van der Waals forces, metal–ligand coordination, hydrophobic effects and others.67Although organogels and hydrogels including phosphorescent materials have been reported, most of them turned out to be non-luminescent due to quenching of long-lived triplet states by molecular oxygen.68 So far only few reports on incorporation of phosphorescent materials into protective gel matrixes have been reported. De Cola and co-workers reported phosphorescent hydrogels based on host–guest interactions between water-soluble Pt(II) complex with attached tetraethylene glycol chains and cyclodextrins (α- and β-CD). The materials were found to be emissive and are not sensitive towards quenching, although the behavior of the system has not studied in details.69 Further Yang and co-workers reported 1,3:2,4-di-O-benzylidene-D-sorbitol (DBS) as a gelator for phosphorescent 3-bromoquinoline. The supramolecular gels was prepared by self-assembly of DBS in DMF–water mixture. It was found that deoxygenation of the samples is not required to observe phosphorescence. Moreover the emission intensities of a pre-deoxygenated samples showed similar values compared to air-saturated.70
Recently Kimizuka and co-workers developed supramolecular gel matrices inspired by the structural feature of thylakoid membranes, where supramolecular gel nanofibers was chosen as matrice-sensitizer and emitter molecules showing air-stable TTA-UC.71
As a supramolecular gelator for TTA-UC, N,N′-bis(octadecyl)-L-boc-glutamic diamide (LBG) was employed (Fig. 20). This compound is well-known for its ability to gelatinize in various solvents giving highly stable gels.72 Sensitizer (palladium(II) octaethyl porphyrin, 33 μM) and emitter (9,10-diphenylanthracene, 6.7 mM) can be incorporated into the gel by dissolving in DMF in the mixture with LGB under heating and cooling to room temperature. The gel showed good structural stability that allows a preparation of specific gel shape by injecting the hot solutions into a mould.
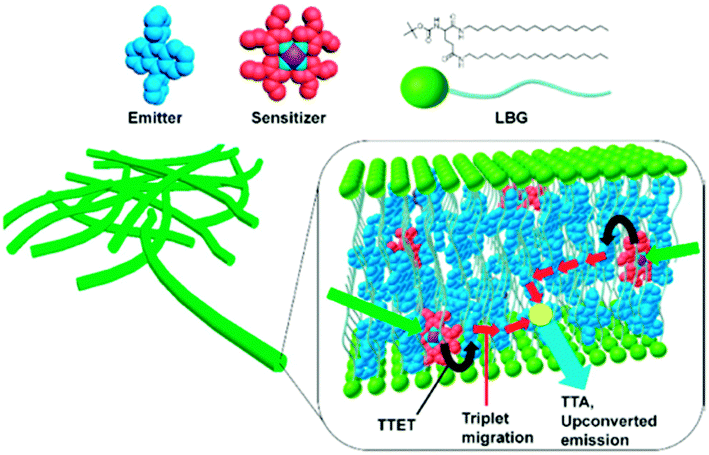 | ||
| Fig. 20 TTA-UC gel system based on N,N′-bis(octadecyl)-L-boc-glutamic diamide. Sensitizer: palladium octaethylporphyrin (red). Emitter: 9,10-diphenylanthracene (blue). Reprinted (adapted) with permission from ref. 71. Copyright 2015 American Chemical Society. | ||
Optical properties of PtOEP or DPA were found to be similar to those in DMF solutions without the gelator agent, indicating that the LBG molecules do not affect their photophysical properties. Even if prepared on air, gels show strong TTA-UC fluorescence, which can be observed by naked eyes upon excitation with 532 nm green laser. UC emission spectral shape is similar to those of the DPA excited at 375 nm. Residual phosphorescence of PtOEP at 645 nm was found to be very weak, indicating that the triplet excited state of PtOEP is efficiently quenched by triplet–triplet transfer to DPA. Quantum yield ΦUC was measured to be 2.7% for the deaerated PtOEP/DPA/LBG gel ([PtOEP] = 33 μM, [DPA] = 6.7 mM, [LBG] = 13.3 mM), which is comparable to the values of deoxygenated PtOEP–DPA system. In air saturated conditions ΦUC of 1.5% was found, confirming oxygen blocking ability of the gel system.
Other sensitizer–emitter TTA-UC pairs in LBG gel system in DMF have been prepared: PdPc(OBu)8/rubrene, PtTPBP/BPEA, and Ir(C6)2(acac)/DBP (Fig. 21). All of these UC pairs exhibited strong UC emissions in LGB gels even under air-saturated conditions.
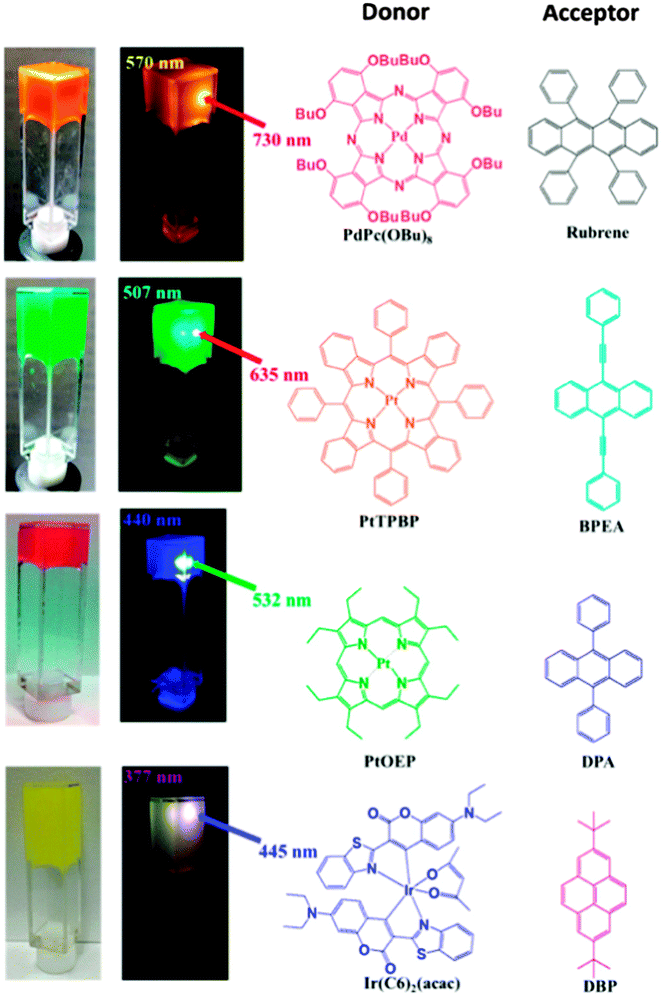 | ||
| Fig. 21 Photographs of the TTA-UC gel systems based on different sensitizer–emitter pairs obtained from air-saturated DMF solutions. Reprinted (adapted) with permission from ref. 71. Copyright 2015 American Chemical Society. | ||
Interestingly, DPA triplet excited state lifetime in the PtOEP/DPA/LBG ternary gel under deaerated and air-saturated gels was determined to be 228 μs and 178 μs respectively. At the same time, phosphorescence of PtOEP (in the absence of DPA) in LBG gel in DMF completely quenched under aerated condition. Based on this fact authors speculate that a cooperative binding of PtOEP molecules to the DPA-enriched domains in LBG nanofibers probably takes place that prevent interactions of the excited state with oxygen dissolved in the matrix. However, taking into account oxygen protection strategies discussed in previous sections, particularly in Section 5.3, one could suggest that there might be a contribution of other processes which provide overall stability of gel system under air. DPA as well as other TTA-UC emitters shown in Fig. 21 are able to react with singlet oxygen through [4+2] cycloaddition.73 Thus, since an excess of the emitter is present in the system, a part of it might be responsible for such deoxygenation process. Obviously, further works on deoxygenation mechanism are required.
A similar approach towards TTA-UC in polymer organogels was further reported by Simon and co-workers,74 based on a continuous polymer network and a liquid organic phase as a matrix for sensitizer and emitter. The matrix was found to possess good mechanical integrity, high molecular mobility and provides protection against triplet states quenching by oxygen, as was shown on an example of palladium(II) mesoporphyrin – 9,10-diphenylanthracene TTA-UC pair. Gels were prepared by cross-linking poly(vinyl alcohol) (PVA) in a 2:1 w/w DMF–DMSO mixture by reaction with hexamethylene diisocyanate (HMDI). The process was performed either on air or under air-free conditions, by stirring a DMSO solution of PVA with a DMF solution containing HMDI, sensitizer and emitter (Fig. 22). The resulting exhibited a transmittance of >95% in the visible range.
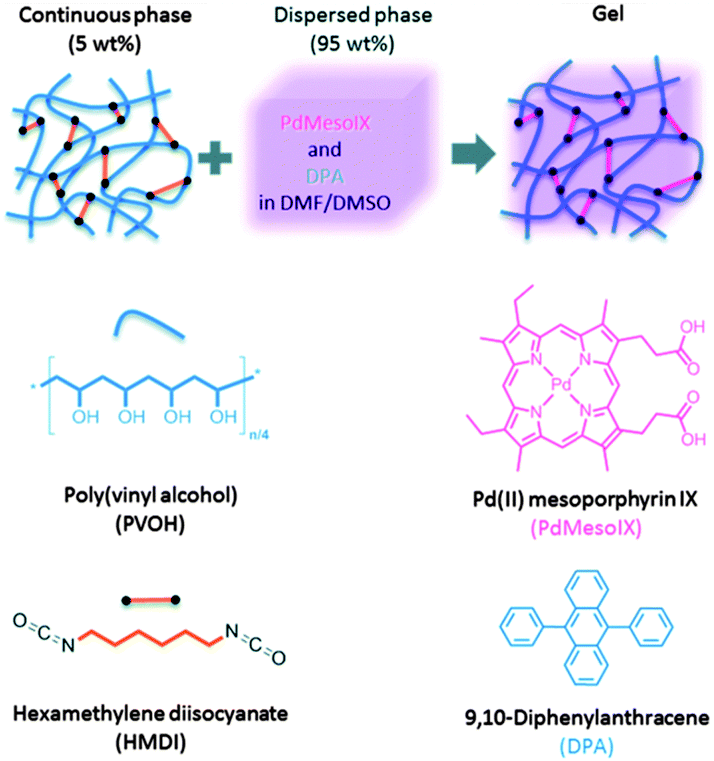 | ||
| Fig. 22 Graphic representation of the structure of the upconverting organogels and chemical structures of the components.74 Published by The Royal Society of Chemistry. | ||
When initial solutions containing TVA, HMDI, sensitizer and emitter were prepared under ambient conditions, upconversion emission of the resulting gels was found to be lower than for those prepared from air-free solutions. Thus triplet state quenching by dissolved oxygen can take place rather efficiently inside the gel matrix. However, deoxygenation of all the components prior the formation of gel, resulted in TTA-UC emission from the mould with quantum yield of 10% at 180 mW cm−2 showing good stability. The prepared materials were found to be shape-persistent (Fig. 23) when removed from the moulds.
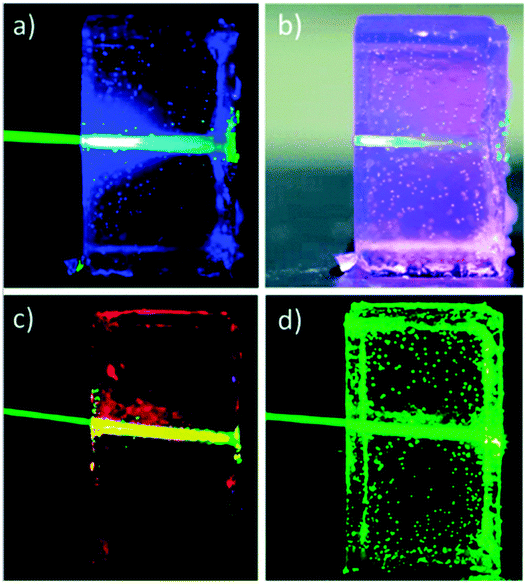 | ||
| Fig. 23 Organogels based on PVOH–HMDI/DMF/DMSO under irradiation with 543 nm laser. (a and b) Gels containing PdMesoIX (2 × 10−5 M) and DPA (10−2 M). (c and d) Gels containing only PdMesoIX (2 × 10−5 M) or DPA (10−2 M) respectively. Reproduced from ref. 74 with permission from the Royal Society of Chemistry. | ||
7. Encapsulation into polymers
A way to avoid oxygen quenching of the phosphorescence is to encapsulate phosphorescent molecules inside a solid matrix to shield them from oxygen. Selection of the encapsulation matrix is important. The matrix should have relatively low oxygen permeability and relatively high solubility of the phosphorescent molecules so that high loading of the phosphorescent molecules can be achieved and strong phosphorescence can be obtained. Different types of matrices have been studied for encapsulation of phosphorescent molecules to form phosphorescent particles, e.g. polystyrene (PS),75 polyacrylonitrile (PAN),76 and their derivatives have been commercially used.These polymeric systems, however, do not allow for maximal phosphorescence intensity at ambient conditions. The polystyrene matrix is not ideal for the encapsulation of phosphorescent molecules because of the poor solubility of many phosphorescent molecules in polystyrene, which results in low dye loading and relatively low phosphorescence. The oxygen solubility of polystyrene is also believed to be relatively high. PAN and its derivatives have been found to have low oxygen permeability but they have limited solubility for many phosphorescent molecules as well, which also limits their application for encapsulation of those phosphorescent molecules. Halogen-containing polymers and co-polymers (HCPs) reported by Song and co-workers77 as an encapsulation matrix for organic and organometallic phosphorescent molecules were shown to produce phosphorescent materials which are substantially unaffected by oxygen and water molecules at ambient conditions. On the other hand, these polymers have a particularly high loading of phosphorescent molecules to provide strong phosphorescence and high photostability. Resulting phosphorescent micro/nano-particles are suitable for a wide variety of applications such as the detection of biological molecules and species in biological assays.
TTA-UC has long been known to occur in solutions of appropriate sensitizer/emitter pairs, but it has taken considerable time to realize this process in solid materials. A comparison between the performances of the same bicomponent system in solution versus the solid state shows that, in the latter, the up-conversion efficiency drops by a factor of 100–1000.78 The origin of this difference is the large molecular mobility in solution, which strongly enhances the interaction probability and, as a consequence, enhances both the triplet–triplet energy transfer (TTET) and TTA processes. The key points for the preparation of the polymeric TTA-UC system are to maintain high translational mobility79 of the chromophores and to prevent luminescence quenching by aggregation. In the case of solid-state materials, the main focus has been made on matrix polymers with low glass transition temperatures (e.g., rubbery materials) to obtain high-efficiency solid-state systems. The larger upconversion efficiency for such materials, when compared to glassy/rigid materials, has been theorized to be due to higher segmental mobility of the elastomers and also enhanced local molecular mobility of the chromophores, which increases interaction probability and thus energy transfer and TTA processes.80 In the case of rubbery materials, low oxygen barrier properties are anticipated, due to the high free volume associated with such materials.81
Castellano and co-workers first reported rubbery poly(ethylene oxide-co-epicholorohydrin) as hosts for TTA-UC sensitizer–emitter pairs. This particular polymer was found to provide sufficient chromophores mobility that allows the bimolecular processes, i.e. triplet–triplet transfer and triplet–triplet annihilation happen efficiently.82 A fabrication technique for producing upconverting polymers which can be moulded into a variety of forms under ambient conditions with minimal processing using commercially available polyurethane precursors (Clear Flex 50, CLRFLX) was reported by the same group.83 A mixture of PdOEP and DPA was dissolved in THF along with corresponding monomer and subjected to polymerization under ambient conditions which delivered transparent rubbery solids (Fig. 24). These materials exhibited linear incident power dependence with quantum efficiencies exceeding 20%. The average lifetime of PdOEP phosphorescence in CLRFLX was 1.29 ms, similar to that determined in other rigid media including polystyrene nanoparticles (0.99 ms), PMMA (1.53 ms), and cellulose acetate (1.2 ms) but much longer than that for precast polyurethane matrices (314 μs). This indicates that CLRFLX effectively suppress deactivation of triplet excited states by dioxygen, while keeping energy transfer quenching by DPA.
 | ||
| Fig. 24 Polyurethane polymer samples containing PdOEP and DPA irradiated by 532 nm green laser. Modified a reprinted with permission from ref. 83. Copyright 2012 American Chemical Society. http://pubs.acs.org/doi/full/10.1021/cm3012414. | ||
Several other types of polymer-based upconverting solid-state materials have been reported based on photophysically inert matrices such as polyacrylates,84 poly(methyl methacrylate),85 cellulose acetate,86 and a styrene/divinylbenzene/vinylbenzyl chloride copolymer.87 Generally the protection of excited triple states in these techniques is based on the reduction of oxygen permeability. Recently we have developed a principally new approach towards polymer hosts for efficient TTA-UC based on a series of synthetic hyperbranched unsaturated poly(phosphoester)s (hbUPPEs).88 These polymers were found to scavenge singlet oxygen due to the presence of alkene double bonds in the structure (Fig. 25) but do not react with oxygen in the triplet ground state. Different branched polymers with varying branching densities were synthesized in a one-step polymerization from the respective A3-type monomers via olefin metathesis polymerization (Fig. 26).
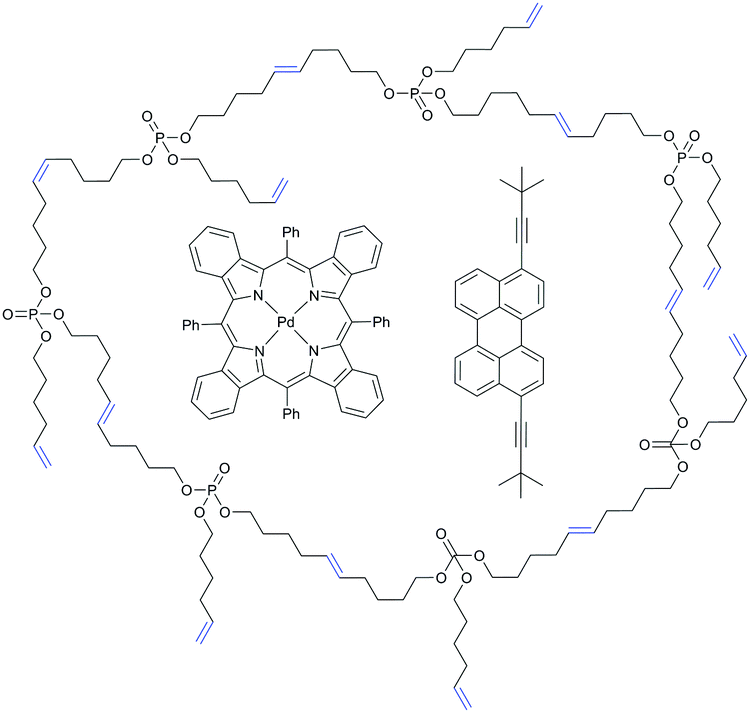 | ||
| Fig. 25 Schematic representation of palladium tetrabenzoporphyrin (sensitizer) and 3,9(10)-bis(3,3-dimethylbut-1-yn-1-yl)perylene (emitter) in a hyperbranched poly(phosphoester) matrix. | ||
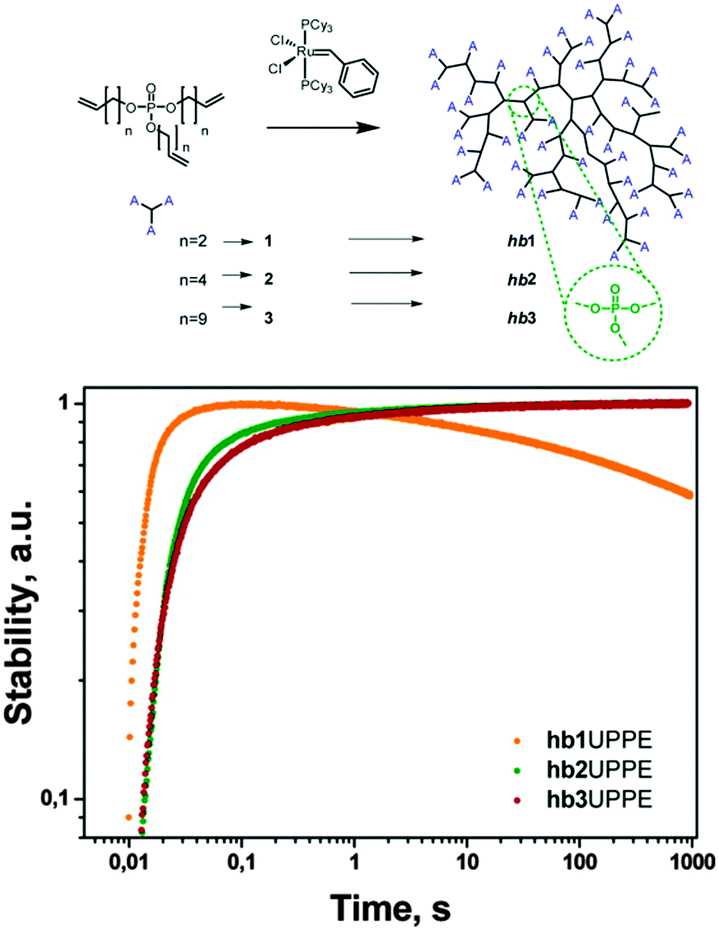 | ||
| Fig. 26 Long-term stability of the integral UC-fluorescence of the system based on PdTBP and 3,10-bis(3,3-dimethylbut-1-yn-1-yl)perylene in hb1,2,3 in an ambient atmosphere (21% O2) for continuous irradiation with an excitation intensity of 100 mW cm−2.88 Reprinted with permission from ref. 88. Copyright 2014 American Chemical Society. | ||
The hbUPPEs can be used as a matrix to dissolve sensitizers and emitters for TTA-UC without the addition of organic solvents. At the same time hbUPPEs possess optical transparency in the UV-Vis and suitable mechanical properties. The amorphous structure and low glass transition temperature impart a rubbery behaviour as well as the possibility to obtain polymer matrixes with a high dye content.
The long-term stabilities in an ambient atmosphere of the UC couples palladium tetrabenzoporphyrin (PdTBP) with 3,10-bis(3,3-dimethylbut-1-yn-1-yl)perylene (Y805) (Fig. 25) in hbUPPEs with different chain length were investigated. All polymers (hb1, hb2, and hb3) can be used as a matrix for TTA-UC with high quantum yields (QYs) in an oxygen-rich environment. The hbUPPEs demonstrate extremely efficient protection against quenching of the excited triplet states from molecular oxygen. Interestingly, hb2 and hb3 ensure almost undisturbed operation of TTA-UC in an ambient atmosphere for more than 1000 s (Fig. 17) for longer experiment times, while hb1 does not provide effective long-term protection against oxygen quenching.
The protection mechanism is based on the ability of singlet oxygen to react with olefinic double bonds.89 The particular reaction depends on the nature of the double bonds as different types of addition reactions (epoxides, dioxetanes, alcohols, etc.) can be generated as well as potential electron transfer reactions.90 The hbUPPEs inherently combine both active and passive protection strategies: namely, passive protection as a consequence of low oxygen permeability and active protection by chemical scavenging of the existing singlet oxygen while being rather inert against triplet oxygen.
8. Protective nano- and microcarriers
The idea of including interacting excited-state species into nanometer-scale shells is quite general and easily adaptable for a large number of small molecules exploited in sensitized up-conversion and other photonic applications. A fundamental benefit of this approach is that the NPs completely shield the dyes from the external environment, and especially from oxygen. In recent years, the preparation of upconverting NPs was reported in many works,91 however most of them describe the operation in the oxygen-free conditions (inert atmosphere), rather than to be suited with passive or active oxygen protection of the triplet states.Reichmanis and co-workers designed an effective approach towards low-threshold TTA-UC within uniform microcapsules fabricated through a microfluidic technique that uses a photoinduced interfacial polymerization.92 The capsules consist of a fluidic active core which provides high translational mobility of the chromophores and an elastomeric shell for mechanical integrity and passive oxygen protection. The capsules are produced via UV-initiated free-radical inverse polymerization through a microfluidic channel (Fig. 27) and their parameters, particularly the size and shell thickness, can be controlled through the UV exposure time. Trimethylolpropane triacrylate (ETPTA) monomer selected as the processable medium for the capsules showing UC emission is readily polymerized with UV light. Due to its high polarity, low viscosity, and non-volatility chromophores, molecules can be well-dispersed inside the resulting capsules at high concentrations. ETPTA retards the penetration of oxygen which quenches the excited states. The TTA-UC efficiency in bulk ETPTA remained almost constant even after 5 days in air. Moreover, within a freestanding film, the UC efficiency using a UV-cured ETPTA control matrix is higher than that observed from a commercial rubbery polyurethane film. Thus, ETPTA encapsulation can facilitate energy transfer, especially within a liquid-phase core providing highly efficient UC emission.
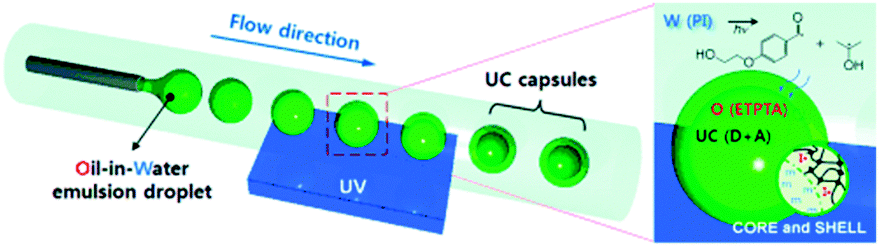 | ||
| Fig. 27 Fabrication of monodisperse capsules for photon upconversion (UC) through interfacial free radical photopolymerization; UV = ultraviolet, W = water, O = oil, PI = photoinitiator, ETPTA = ethoxylated trimethylolpropane triacrylate, m = monomer, IC = photoinitiator radical, D = donor, and A = acceptor. Reproduced with permission from ref. 92. Copyright 2012 Wiley-VCH Verlag GmbH. | ||
Liu and co-workers reported a strategy for decreasing the O2-induced upconversion quenching by introducing a reducing agents, such as oleic acid, linoleic acid, ascorbyl palmitate, or ascorbyl into the nanocapsules.93 For example, PtTPB–BDP-G system (Fig. 28) in soybean oil in the presence of O2 showed intense yellow upconversion emission with quantum yield of 5.0% (cPtTPBP = 1.0 × 10−5 M, cBDP-G = 1.0 × 10−3 M, power density of 0.106 W cm−2). The mixture of sensitizer and emitter in the same concentration in aerated toluene showed quantum yield of only 0.1%. This was attributed to the effect reducing agents in the soybean oil, which reacts with singlet oxygen initially generated within the nanocapsules.
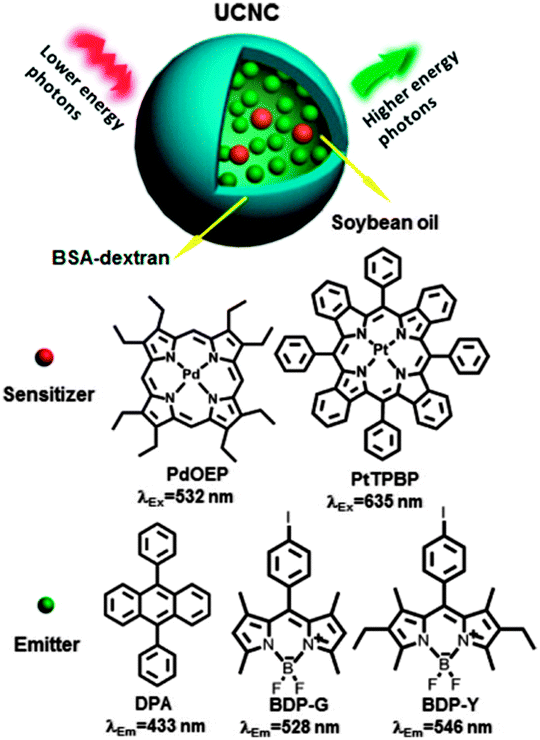 | ||
| Fig. 28 TTA-UC process in the upconversion nanocapsules and chemical structures of sensitizers (PdOEP and PtTPBP) and emitters (DPA, BDP-G, and BDP-Y). Reprinted with permission from ref. 93. Copyright 2013 American Chemical Society. | ||
The main components of soybean oil are linoleic acid and oleic acid, both of which possess oxygen scavenging capacity. Singlet oxygen scavenging in nanocapsules was then shown using other antioxidant compound, ascorbyl palmitate, in the toluene solution with BDP-G and PtTPBP. The TTA-UC fluorescence intensity was found to increase with increasing concentrations of ascorbyl palmitate. The effect of antioxidant on the phosphorescent lifetime of PtTPBP was studied. Upon continuous irradiation of PtTPBP in aerated toluene with 635 nm laser for 240 s phosphorescent lifetime increased to 32 μs in the presence of ascorbyl palmitate (2.0 × 10−3 mol L−1), which is comparable to that (40 μs) of PtTPBP in oxygen-free toluene.
Recently, we explored a method for the fabrication of ultralight upconverting mats consisting of rigid polymer nanofibers. Poly(vinyl alcohol) (PVA), known for its oxygen barrier properties was used as a matrix.94 Oxygen permeation coefficient of some PVA films was reported to be 0.09 cm3 mm m−2 day−1 atm−1 at 24 °C and 75% relative humidity.95 Due to this, PVA was found to be an excellent oxygen-barrier for solar cells.96 The PVA matrix used to immobilize and to protect the nanocapsules and TTA-UC chemical agents. The mats were prepared by simultaneously electrospinning an aqueous solution of a polymer and functional nanocapsules solutions containing a sensitizer–emitter pair (Fig. 29). The nanofibers demonstrated efficient upconversion fluorescence with λmax = 550 nm under low intensity excitation 635 nm laser (power = 5 mW). The oxygen-barrier property of the polymer matrix efficiently prevents the oxygen penetration since the emission was found to be stable in ambient atmosphere.
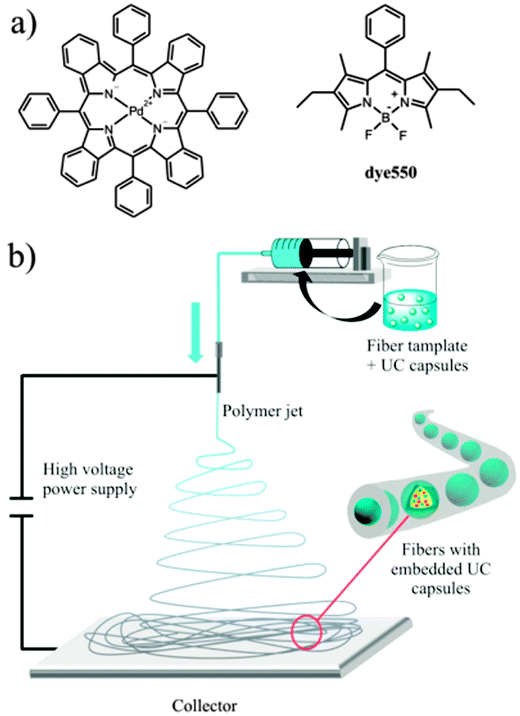 | ||
| Fig. 29 Structure of the sensitizer and emitter dyes (a) and a scheme of the colloid-electrospinning process for embedding TTA-UC nanocapsules into nanofibers (b). Reprinted (adapted) with permission from ref. 94. Copyright 2013 American Chemical Society. | ||
Wang and co-workers reported an oil-in-water (o/w) microemulsion TTA-UC system based on for 9,10-dinaphthylanthracene (emitter), palladium(II) meso-tetratolylporphyrin (sensitizer) and Tween-20 as a surfactant, efficiently operating under air without deaeration.97 Notably, these o/w microemulsions can maintain a high efficiency (Φ = 33.12%) without degassing for several days. In contrast, the UC signal of similar sensitizer–emitter pair in DMF disappears within 1.2 hour without degassing (Fig. 30). A comparison experiment of using o/w microemulsion, both with deaeration and without deaeration, confirms that the TTA-UC o/w microemulsion does not need deaeration. This indicates that triplet excited state in o/w microemulsion system are rather insensitive toward oxygen quenching.
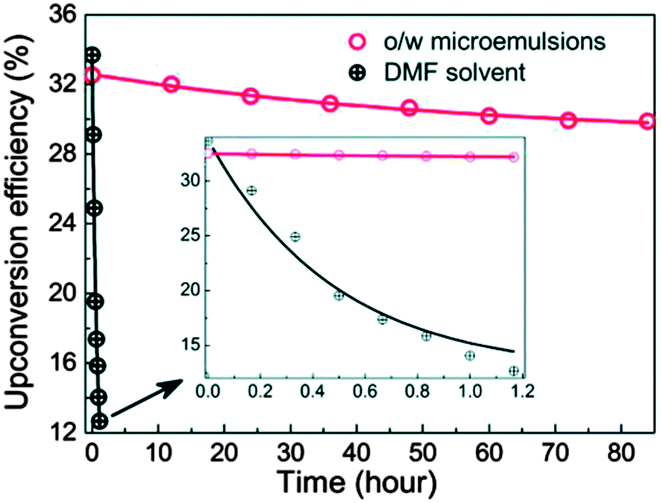 | ||
| Fig. 30 Time-dependent upconversion efficiency for 9,10-dinaphthylanthracene and palladium(II) meso-tetratolylporphyrin mixture (3.2 mM/8 mM) in o/w microemulsion (70 °C) and DMF (25 °C), respectively, under an air atmosphere (excitation – 532 nm at 60 mW cm−2 intensity). Reproduced from ref. 97 with permission from the Royal Society of Chemistry. | ||
9. Biopolymers as oxygen barriers
Nanofibrillated cellulose (NFC), cellulosic fibrils disintegrated from the plant cell walls, was first explored by Turbak and co-workers98 and Herrick with co-workers,99 and was recognized as a valuable material due to its oxygen barrier properties.100High barrier properties of NFC arises from a partly crystalline structure of microfibrils in combination with the ability of the dried films to form a dense network held together by strong inter/intra-fibrillar bonds. Due to the rather high degree of crystallinity of NFC (63 ± 8.6%),101 the permeability of NFC is expected to be limited, but, gas diffusion may nevertheless occur through the voids in the microfibril network. Fukuzumi and co-workers102 have investigated the oxygen barrier properties of cellulose nanofibers prepared by 2,2,6,6-tetramethylpiperdine-1-oxyl radical (TEMPO)-mediated oxidation and have found such films to be good barriers under dry conditions. NFC films have a potential for use in gas barrier application, although water sorption and high relative humidity can be problematic. In addition, NFC possess outstanding mechanical properties, optical transparency and non-toxicity.
NFC was recently exploited by us in the preparation of a solid state-like upconverting material operating under air.103 A direct mixing of TTA-UC components with NFC is not possible because of their hydrophobic nature. On the other hand, NFC forms aggregates in non-polar solvents, making preparation of well-dispersed systems rather complicated. Thus NFC-based capsules containing sensitizer and emitter in the liquid core were prepared by the reaction between the hydroxyl groups of NFC and cellulose nanocrystals (CNC), cross-linker (isophorone diisocyante) and water at the oil/water interface of oil droplets. A blend of NFC and CNC was used in the capsule synthesis. Resulting capsules were on average 1.2 ± 0.3 μm in diameter, with a capsule wall thickness of around 30 nm. The concentration of sensitizer and emitter in the oil core of the capsules was 7 × 10−5 M and 1 × 10−3 M, respectively.
Encapsulation the UC components into the liquid compartment of micro/submicron-sized nanocomposite shell/liquid core capsules provides the local mobility of the chromophores. NFCs are long flexible nanofibers composed of both crystalline and amorphous regions, whereas the CNCs are shorter, rod-like mainly crystalline whiskers. These capsules were then further embedded in a matrix of NFC which supplies the necessary mechanical support. In Fig. 31, typical nanocellulose-based capsules containing oil (hexadecane) cores are presented, where the fibrous capsule wall structure is shown.
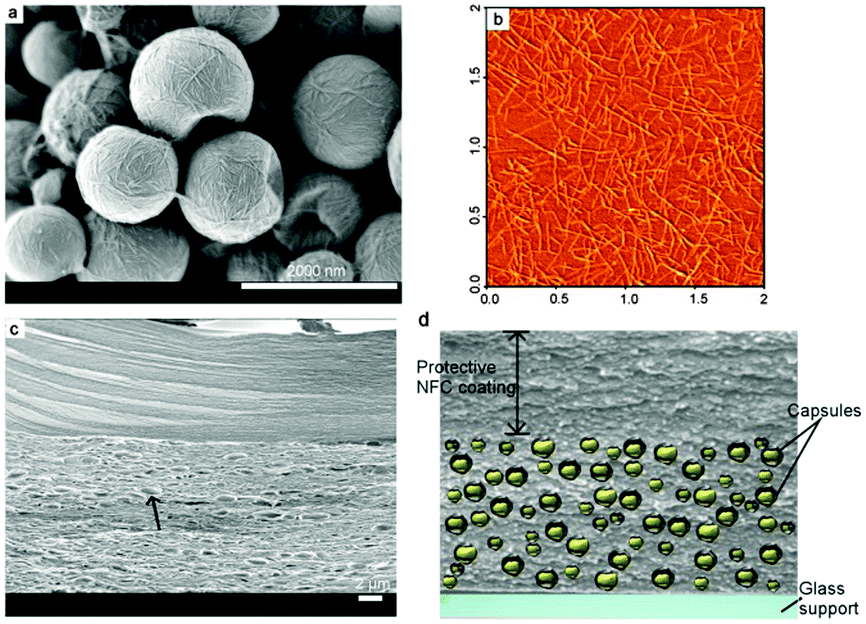 | ||
| Fig. 31 (a) SEM image of NFC/CNC-based capsules containing oil–liquid cores. (b) AFM image of individual TEMPO-NFC used as matrix material and for the protective coating. (c) Fracture surface of the synthetic leaf (SEM image) showing the two-layer structure. (d) Illustration of the cross section of the synthetic film. Reprinted (adapted) with permission from ref. 103. Copyright 2014 American Chemical Society. | ||
Three different types of films were prepared: one without and two with protective coatings of different thicknesses, namely, 4.2 and 8.8 μm. The thickness of the lower capsule layer was comparable for the three film types and is on the order of 29 ± 3 μm (films with protective coatings) to 36.5 μm (reference film). All films were supported on glass substrates. These samples were equilibrated in a nitrogen atmosphere and then exposed to dry synthetic air (20.5/79.5 vol%, O2/N2, constant and identical temperature for the synthetic air and sample).
Oxygen protection of TTA-UC in the prepared films was studied of a system comprising mixed benzo-naphthoporphyrins ensemble (five components) as sensitizer in 620–670 nm region and 3,9-bis(3,3-dimethylbutyl-1-yl)perylene as a single emitter. Such a system enable a broad-band upconversion of a whole deep-red spectral region (Δλ ∼ 70 nm) into green light with λmax = 518 nm. In oxygen free-conditions (<2 ppm) the system showed UC quantum yield of 8.2% under broadband excitation (500 mW cm−2). The change of intensity of the emission of upconverting NFC/CNC films was monitored with time. Fig. 32 shows the normalized UC fluorescence and phosphorescence of the system. In all cases, the intensity declines as more oxygen penetrates through the structure and effectively quenches the excited triplet states of the chromophores. As expected, the UC fluorescence decreased more rapidly than the phosphorescence signal. The explanation for this behavior is the fact that UC fluorescence is a consequence of the bimolecular TTA process. Both sensitizer and emitter excited triplet ensembles can be quenched by molecular oxygen; therefore, the UC fluorescence demonstrates stronger dependence on the oxygen concentration.
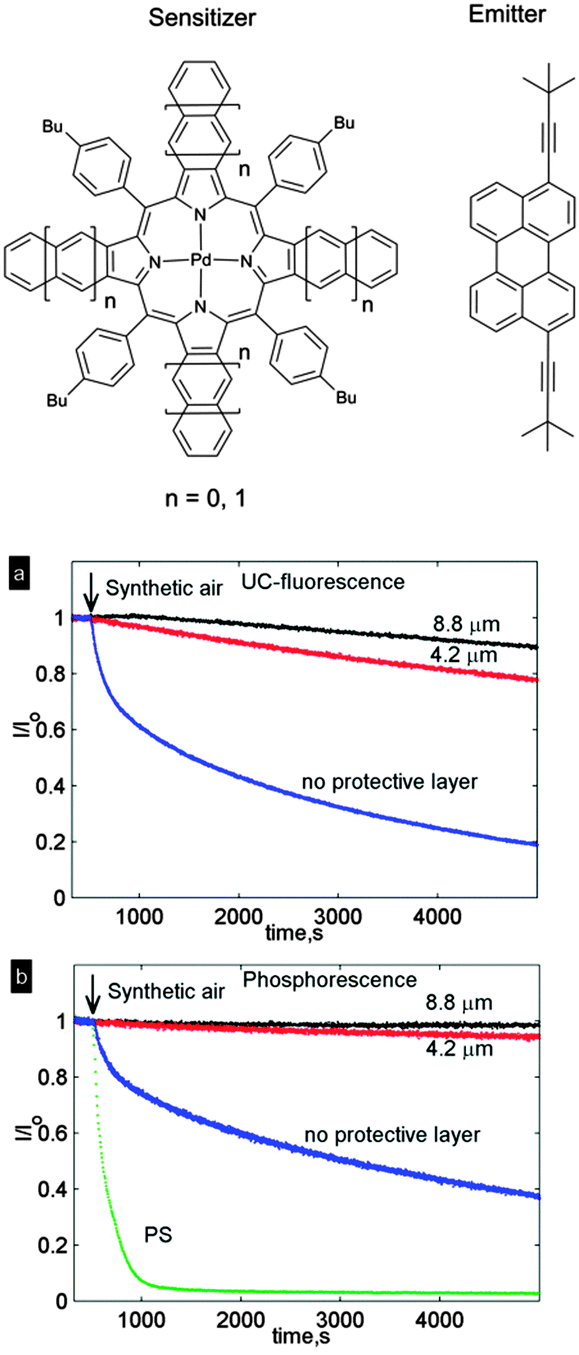 | ||
| Fig. 32 Normalized UC fluorescence at λmax = 518 nm (a) and normalized phosphorescence at λmax = 831 nm (b) as a function of time in a NFC-based film with no additional protective layer (blue) and 4.2 μm (red) or 8.8 μm (black) thickness of the oxygen protective layer. The intensities (I) were normalized by dividing with the intensity prior to air inlet (I0). Reprinted (adapted) with permission from ref. 103. Copyright 2014 American Chemical Society. | ||
To demonstrate the oxygen barrier properties of NFC films, a reference sample containing sensitizer and emitter in the same concentrations encapsulated into polystyrene was studied. In this case UC fluorescence was not observed at all, due to the low molecular mobility of the chromophores in the polymer matrix. Phosphorescence of the porphyrin sensitizers was found to decrease dramatically with time (Fig. 32b) due to oxygen quenching.
10. Protective solvents
As was shown above, various efficient approaches towards oxygen protection of triplet excited state ensembles were developed very recently to provide stable operating phosphorescent and photon upconverting systems. However, all these approaches suffer from certain limitations. Their application either require a change in the sample architecture (nanoparticle, gels, polymer films) or affect chemical and photophysical properties of the chromophores (oxygen scavengers, supramolecular complexes, dendrimers). In addition, certain types of oxygen protection strategies require extensive synthetic efforts for the preparation of corresponding chromophores or components of protective media.A straightforward, but yet unexplored in any photonic applications approach is the use of a solvent itself as an oxygen scavenger. Recently, we reported an architecturally diverse library of organophosphates (OP, Fig. 33), specially designed and optimized to enable long-term TTA-UC in ambient atmosphere.104
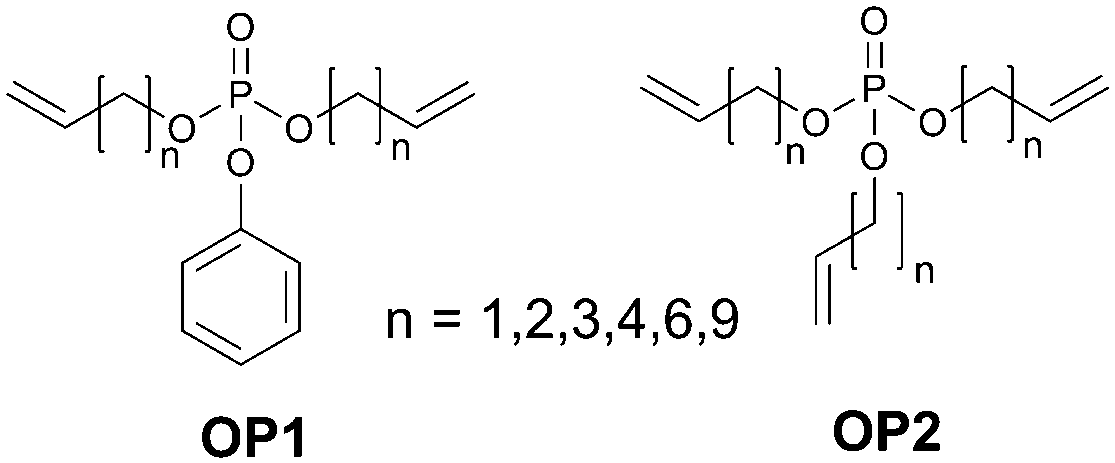 | ||
| Fig. 33 Unsaturated organophosphates protective solvents. | ||
Organic phosphates OP1 and OP2 bearing unsaturated hydrocarbon chains serves as a solvent with low viscosity, thus the efficiency of the TTA-UC process is almost identical with the efficiency in standard solvent–toluene. At the same time they provides long-term protection (over 1000 h) against the molecular oxygen quenching. Developed synthetic approach provides control of phosphate functionality, e.g. viscosity, ability to dissolve UC-dyes and oxygen scavenging properties through variation of the hydrocarbon chain length, ratio between aliphatic and aromatic parts and number of terminal double bonds.
Oxygen protection of TTA-UC was studied on two different sensitizers–emitter pairs: based on palladium tetrabenzo- and tetranaphthoporphyrins (Fig. 34). Perylene and perylenimide derivatives were used as emitters. In both cases the efficiency of the TTA-UC process in cw-excitation regime does not depend on the contamination with molecular oxygen: virtually identical UC-efficiency was observed in oxygen-free and ambient conditions. On the other hand, for both UC-couples prepared in ambient conditions using conventional solvents only vanishing UC-fluorescence intensities were observed.
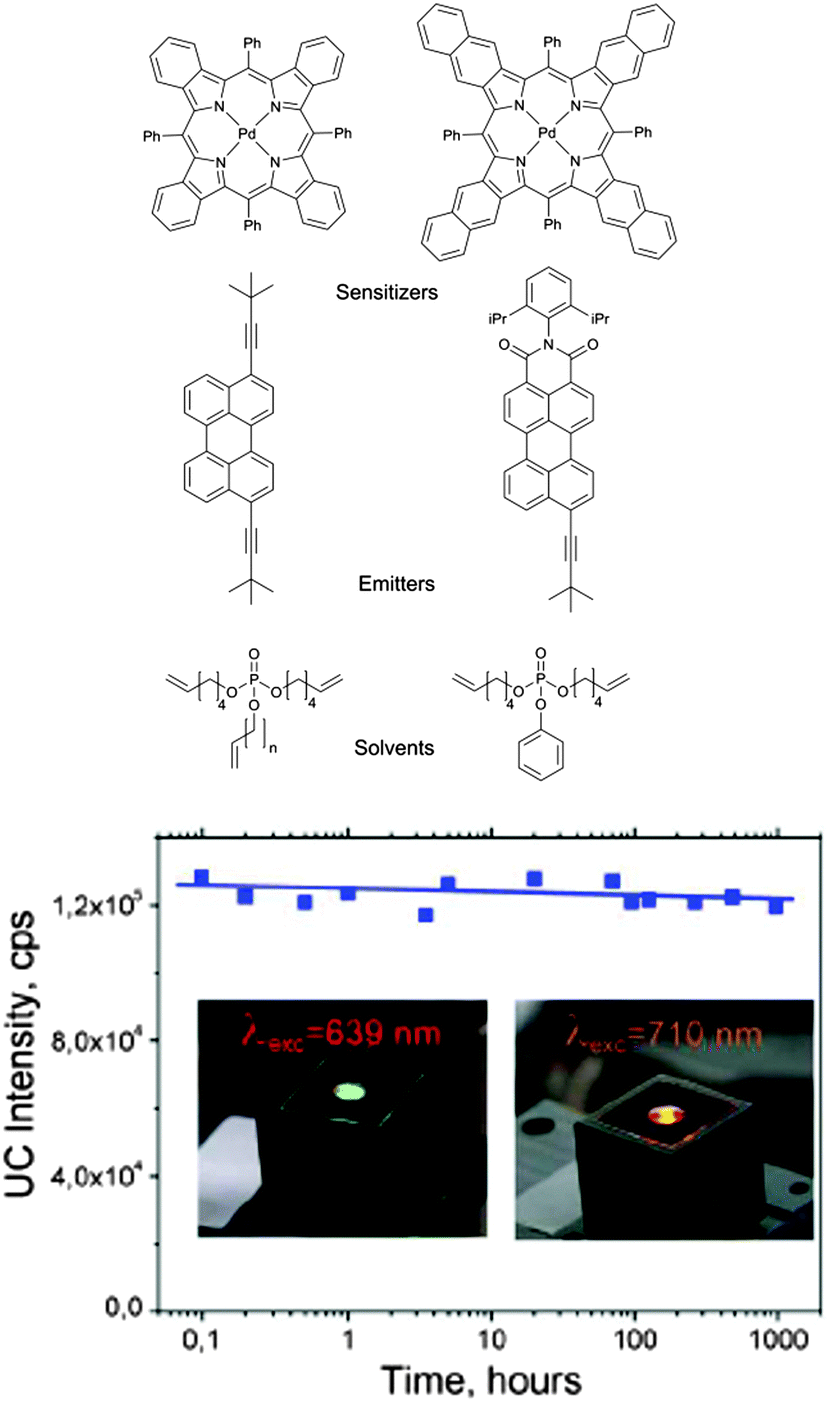 | ||
| Fig. 34 Structures of sensitizer–emitter pairs and of the protective solvents. UC-fluorescence of the samples kept under air. | ||
These experimental data suggest that during the optical excitation, dissolved oxygen is being converted into its singlet excited state due to the quenching of sensitizer triplet state. Further, generated singlet oxygen reacts with terminal C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds of the solvent forming stable peroxide compounds. In such a way, the local amount of the molecular oxygen is quickly consumed after the start of illumination through the generation of singlet oxygen and its subsequent reaction with the double bonds. Thereafter, the process of TTA-UC becomes the prevalent relaxation path of the excited sensitizer triplet state and the UC fluorescence grows-up. In oxygen-reach environment the steady-state UC-emission is reached after a significantly longer rise time in comparison with oxygen-free starting conditions. Consistently with this hypothesis, the higher the excitation intensity is, the shorter time is needed for the complete consumption of the molecular oxygen which was initially present in the optically-excited volume and therefore, the shorter rise time of the upconverted emission is observed.
C double bonds of the solvent forming stable peroxide compounds. In such a way, the local amount of the molecular oxygen is quickly consumed after the start of illumination through the generation of singlet oxygen and its subsequent reaction with the double bonds. Thereafter, the process of TTA-UC becomes the prevalent relaxation path of the excited sensitizer triplet state and the UC fluorescence grows-up. In oxygen-reach environment the steady-state UC-emission is reached after a significantly longer rise time in comparison with oxygen-free starting conditions. Consistently with this hypothesis, the higher the excitation intensity is, the shorter time is needed for the complete consumption of the molecular oxygen which was initially present in the optically-excited volume and therefore, the shorter rise time of the upconverted emission is observed.
It can be concluded, that oxygen diffusion in the organophosphates is low, so once the initial molecular oxygen is depleted in the optically-excited volume even in ambient conditions only a negligible amount of optical excitation is enough to cause reaction with the molecular oxygen diffusing into the excitation volume under steady state conditions. Therefore, the steady-state emission of the UC-fluorescence has no significant deviation from the oxygen-free UC-emission observed.
11. Summary and prospects
Optically excited densely populated triplet organic ensembles have orders of magnitude longer lifetime of their excited triplet states, than the lifetime of the optically excited singlet states. Therefore, intense interaction with the material environment of such ensembles is observed. All-optical sensors based on densely populated triplet ensembles introduce the possibility for effective control and understanding of biochemical reactions, responsible for cellular functions and/or physiological response to external stimuli. Ultimate requirement for this is unambiguous discrimination of the changes introduced by the oxygen quenching for all-other influencing parameters – local temperature, viscosity, local concentration change of the active molecules or presence of other quenching moieties. It is logically correct, that first step in this development must be the effective and sustainable protection of the organic triplet ensembles against quenching by molecular oxygen.This review has covered recent developments concerning oxygen protection of excited state triplet ensembles, including phosphorescent and TTA-UC systems. For phosphorescent materials, significant advances have been made since 1980s in tuning of their susceptibility towards oxygen quenching particularly aimed at the development of oxygen sensing techniques in biological systems.105 Currently tuning of quenching rates through the chemical modification of sensitizer molecule or incorporation into a material with defined oxygen permeability became possible. Alternatively, quenching process can be completely eliminated by means of appropriated sacrificial singlet oxygen scavengers, which open broad perspectives in material science. The current applications for these protected organic phosphors and annihilation upconversion systems include bioimaging, all-optical sensing systems, upconversion displays, organic solar cells, photocatalysis etc. For TTA-based upconversion materials, current research is focused on identifying oxygen protective materials which provide high quantum yield of the process via keeping chromophores mobility and suppressing both triplet state quenching and formation of reactive oxygen species. The mechanism and decay dynamics of the upconversion process, the accomplishment of TTA-based upconversion in aqueous media, and applications in lighting, photocatalysis, and bioimaging are also currently in the focus of research on oxygen protection of TTA-UC.
Acknowledgements
M. Filatov acknowledges the European Commission for Marie Curie Individual Fellowship (CONSORT, Grant No. 655142). S. Baluschev acknowledges the DFNI E 02/11 – SunStore-project of the Bulgarian National Science Fund for the financial support.References
- (a) Y. Sun, C. Borek, K. Hanson, P. I. Djurovich, M. E. Thompson, J. Brooks, J. J. Brown and S. R. Forrest, Appl. Phys. Lett., 2007, 90, 213503 CrossRef; (b) J. R. Sommer, R. T. Farley, K. R. Graham, Y. Yang, J. R. Reynolds, J. Xue and K. S. Schanze, ACS Appl. Mater. Interfaces, 2009, 1, 274 CrossRef CAS PubMed; (c) K. R. Graham, Y. Yang, J. R. Sommer, A. H. Shelton, K. S. Schanze, J. Xue and J. R. Reynolds, Chem. Mater., 2011, 23, 5305 CrossRef CAS; (d) F. B. Dias, K. N. Bourdakos, V. Jankus, K. C. Moss, K. T. Kamtekar, V. Bhalla, J. Santos, M. R. Bryce and A. P. Monkman, Adv. Mater., 2013, 25, 3707 CrossRef CAS PubMed; (e) F. Dumur, M. Lepeltier, B. Graff, E. Contal, G. Wantz, J. Lalevee, C. R. Mayer, D. Bertin and D. Gigmes, Synth. Met., 2013, 182, 13 CrossRef CAS; (f) C. S. Oh and J. Y. Lee, Dyes Pigm., 2013, 99, 374 CrossRef CAS; (g) A. M. Bunzli, H. J. Bolink, E. C. Constable, C. E. Housecroft, J. M. Junquera-Hernandez, M. Neuburger, E. Orti, A. Pertegas, J. J. Serrano-Perez, D. Tordera and J. A. Zampese, Dalton Trans., 2014, 43, 728 RSC.
- (a) T. V. Esipova, A. Karagodov, J. Miller, D. F. Wilson, T. M. Busch and S. A. Vinogradov, Anal. Chem., 2011, 83, 8756 CrossRef CAS PubMed; (b) A. Y. Lebedev, A. V. Cheprakov, S. Sakadzic, D. A. Boas, D. F. Wilson and S. A. Vinogradov, ACS Appl. Mater. Interfaces, 2009, 1, 1292 CrossRef CAS PubMed; (c) O. S. Finikova, A. Galkin, V. Rozhkov, M. Cordero, C. Hagerhall and S. A. Vinogradov, J. Am. Chem. Soc., 2003, 125, 4882 CrossRef CAS PubMed; (d) D. F. Wilson, W. M. F. Lee, S. Makonnen, O. S. Finikova, S. Apreleva and S. A. Vinogradov, J. Appl. Physiol., 2006, 101, 1648 CrossRef CAS PubMed; (e) Q. Zhao, M. X. Yu, L. X. Shi, S. J. Liu, C. Y. Li, M. Shi, Z. G. Zhou, C. H. Huang and F. Y. Li, Organometallics, 2010, 29, 1085 CrossRef CAS; (f) Q. Liu, B. R. Yin, T. S. Yang, Y. C. Yang, Z. Shen, P. Yao and F. Y. Li, J. Am. Chem. Soc., 2013, 135, 5029 CrossRef CAS PubMed; (g) M. Chen, Z. Lei, W. Feng, C. Y. Li, Q. M. Wang and F. Y. Li, Biomaterials, 2013, 34, 4284 CrossRef CAS PubMed; (h) K. Koren, R. Dmitriev, S. Borisov, D. Papkovsky and I. Klimant, ChemBioChem, 2012, 13, 1184 CrossRef CAS PubMed; (i) A. V. Kondrashina, R. I. Dmitriev, S. Borisov, I. Klimant, I. O'Brien, Y. M. Nolan, A. Zdanov and D. B. Papkovsky, Adv. Funct. Mater., 2012, 22, 4931 CrossRef CAS; (j) A. Fercher, S. Borisov, A. Zdanov, I. Klimant and D. B. Papkovsky, ACS Nano, 2011, 5, 5499 CrossRef CAS PubMed; (k) S. Hess, A. Becker, S. Baluschev, V. Yakutkin and G. Wegner, Macromol. Chem. Phys., 2007, 208, 2173 CrossRef CAS.
- (a) J. F. Sun, F. F. Zhong and J. Z. Zhao, Dalton Trans., 2013, 42, 9595 RSC; (b) S. Guo, L. H. Ma, J. Z. Zhao, B. Kucukoz, A. Karatay, M. Hayvali, H. G. Yaglioglu and A. Elmali, Chem. Sci., 2014, 5, 489 RSC; (c) J. F. Sun, F. F. Zhong, X. Y. Yi and J. Z. Zhao, Inorg. Chem., 2013, 52, 6299 CrossRef CAS PubMed; (d) J. Kyriakopoulos, A. T. Papastavrou, G. D. Panagiotou, M. D. Tzirakis, D. Manolis, K. S. Triantafyllidis, M. N. Alberti, K. Bourikas, C. Kordulis, M. Orfanopoulos and A. Lycourghiotis, J. Mol. Catal. A: Chem., 2014, 381, 9 CrossRef CAS; (e) K. Mori, Y. Kubota and H. Yamashita, Chem. – Asian J., 2013, 8, 3207 CrossRef CAS PubMed; (f) S. Tombe, E. Antunes and T. Nyokong, J. Mol. Catal. A: Chem., 2013, 371, 125 CrossRef CAS.
- (a) A. Turshatov, D. Busko, S. Baluschev, T. Miteva and K. Landfester, New J. Phys., 2011, 10, 083035 CrossRef; (b) C. Wohnhaas, A. Turshatov, V. Mailaender, S. Lorenz, S. Baluschev, T. Miteva and K. Landfester, Macromol. Biosci., 2011, 11, 772 CrossRef CAS PubMed; (c) S. K. Sugunan, C. Greenwald, M. F. Paige and R. P. Steer, J. Phys. Chem. A, 2013, 117, 5419 CrossRef CAS PubMed; (d) X. Cao, B. Hu and P. Zhang, J. Phys. Chem. Lett., 2013, 4, 2334 CrossRef CAS; (e) J. S. Lissau, D. Nauroozi, M. P. Santoni, S. Ott, J. M. Gardner and A. Morandeira, J. Phys. Chem. C, 2013, 117, 14493 CrossRef CAS; (f) P. C. Boutin, K. P. Ghiggino, T. L. Kelly and R. P. Steer, J. Phys. Chem. Lett., 2013, 4, 4113 CrossRef CAS; (g) S. Borisov, R. Saf, R. Fischer and I. Klimant, Inorg. Chem., 2013, 52, 1206 CrossRef CAS PubMed; (h) Y. Y. Cheng, B. Fuckel, T. Khoury, R. G. C. R. Clady, M. J. Y. Tayebjee, N. J. Ekins-Daukes, M. J. Crossley and T. W. Schmidt, J. Phys. Chem. Lett., 2010, 1, 1795 CrossRef CAS.
- C. Schweitzer and R. Schmidt, Chem. Rev., 2003, 103, 1685 CrossRef CAS PubMed.
- (a) S. Baluschev, T. Miteva, V. Yakutkin, G. Nelles, A. Yasuda and G. Wegner, Phys. Rev. Lett., 2006, 97, 143903 CrossRef CAS PubMed; (b) R. R. Islangulov, J. Lott, C. Weder and F. N. Castellano, J. Am. Chem. Soc., 2007, 129, 12652 CrossRef CAS PubMed; (c) S. Baluschev, V. Yakutkin, T. Miteva, Y. Avlasevich, S. Chernov, S. Aleshchenkov, G. Nelles, A. Cheprakov, A. Yasuda, K. Mullen and G. Wegner, Angew. Chem., Int. Ed., 2007, 46, 7693 CrossRef CAS PubMed; (d) S. Hoseinkhani, R. Tubino, F. Meinardi and A. Monguzzi, Phys. Chem. Chem. Phys., 2015, 17, 4020 RSC.
- (a) P. E. Keivanidis, S. Baluschev, T. Miteva, G. Nelles, U. Scherf, A. Yasuda and G. Wegner, Adv. Mater., 2003, 15, 2095 CrossRef CAS; (b) R. R. Islangulov, D. V. Kozlov and F. N. Castellano, Chem. Commun., 2005, 3776 RSC; (c) S. Baluschev, J. Jacob, Y. S. Avlasevich, P. E. Keivanidis, T. Miteva, A. Yasuda, G. Nelles, A. C. Grimsdale, K. Mullen and G. Wegner, ChemPhysChem, 2005, 6, 1250 CrossRef CAS PubMed; (d) T. F. Schulze, J. Czolk, Y. Y. Cheng, B. Fuckel, R. W. MacQueen, T. Khoury, M. J. Crossley, B. Stannowski, K. Lips and U. Lemmer, J. Phys. Chem. C, 2012, 116, 22794 CrossRef CAS; (e) S. Baluschev, V. Yakutkin, G. Wegner, T. Miteva, G. Nelles, A. Yasuda, S. Chernov, S. Aleshchenkov and A. Cheprakov, Appl. Phys. Lett., 2007, 90, 181103 CrossRef; (f) T. N. Singh-Rachford and F. N. Castellano, J. Phys. Chem. A, 2008, 112, 3550 CrossRef CAS PubMed; (g) T. N. Singh-Rachford, A. Haefele, R. Ziessel and F. N. Castellano, J. Am. Chem. Soc., 2008, 130, 16164 CrossRef CAS PubMed; (h) T. Miteva, V. Yakutkin, G. Nelles and S. Baluschev, New J. Phys., 2008, 10, 103002 CrossRef; (i) H. C. Chen, C. Y. Hung, K. H. Wang, H. L. Chen, W. S. Fann, F. C. Chien, P. L. Chen, T. J. Chow, C. P. Hsu and S. S. Sun, Chem. Commun., 2009, 4064 RSC; (j) T. N. Singh-Rachford and F. N. Castellano, J. Phys. Chem. Lett., 2010, 1, 195 CrossRef CAS; (k) S. M. Ji, W. H. Wu, W. T. Wu, H. M. Guo and J. Z. Zhao, Angew. Chem., Int. Ed., 2011, 50, 1626 CrossRef CAS PubMed; (l) Y. Murakami, H. Kikuchi and A. Kawai, J. Phys. Chem. B, 2013, 117, 2487 CrossRef CAS PubMed; (m) J. H. Kim, F. Deng, F. N. Castellano and J. H. Kim, Chem. Mater., 2012, 24, 2250 CrossRef CAS; (n) Q. Wang, I. W. H. Oswald, M. R. Perez, H. P. Jia, B. E. Gnade and M. A. Omary, Adv. Funct. Mater., 2013, 23, 5420–5428 CrossRef CAS.
- W. Q. Zou, C. Visser, J. A. Maduro, M. S. Pshenichnikov and J. C. Hummelen, Nat. Photonics, 2012, 6, 560 CrossRef CAS.
- K. Borjesson, D. Dzebo, B. Albinsson and K. Moth-Poulsen, J. Mater. Chem. A, 2013, 1, 8521 Search PubMed.
- (a) J. H. Kim and J. H. Kim, J. Am. Chem. Soc., 2012, 134, 17478 CrossRef CAS PubMed; (b) Q. Liu, T. S. Yang, W. Feng and F. Y. Li, J. Am. Chem. Soc., 2012, 134, 5390 CrossRef CAS PubMed; (c) S. M. Borisov, R. Saf, R. Fischer and I. Klimant, Inorg. Chem., 2013, 52, 1206 CrossRef CAS PubMed; (d) C. Wohnhaas, V. Mailander, M. Droge, M. A. Filatov, D. Busko, Y. Avlasevich, S. Baluschev, T. Miteva, K. Landfester and A. Turshatov, Macromol. Biosci., 2013, 13, 1422 CrossRef CAS PubMed; (e) J. H. Kang and E. Reichmanis, Angew. Chem., Int. Ed., 2012, 51, 11841 CrossRef CAS PubMed; (f) C. Wohnhaas, A. Turshatov, V. Mailander, S. Lorenz, S. Baluschev, T. Miteva and K. Landfester, Macromol. Biosci., 2011, 11, 772 CrossRef CAS PubMed; (g) Y. C. Simon, S. Bai, M. K. Sing, H. Dietsch, M. Achermann and C. Weder, Macromol. Rapid Commun., 2012, 33, 498 CrossRef CAS PubMed.
- M. Filatov, S. Ritz, I. Ilieva, V. Mailander, K. Landfester and S. Baluschev, SPIE Newsroom, Published Online: April 7, 2014, DOI: http://10.1117/2.1201403.005378, http://spie.org/x106642.xml.
- M. Pope and C. Swenberg, Electronic Processes in Organic Crystals, Clarendon Press Oxford, 1982 Search PubMed.
- J. B Birks, Photophysics of Aromatic Molecules, Wiley – Interscience, 1970 Search PubMed.
- Y. Shen, The Principles of Nonlinear Optics, Wiley, New York, 2002 Search PubMed.
- F. Auzel, Chem. Rev., 2004, 104, 139 CrossRef CAS PubMed.
- Next Generation of Photovoltaics: New Concepts, ed. S. Baluschev, T. Miteva and A. Luque López, Springer, Berlin, Heidelberg, 2012, pp. 157–190, ISBN-13: 978-3642233685 Search PubMed.
- A. Y. Lebedev, M. A. Filatov, A. V. Cheprakov and S. A. Vinogradov, J. Phys. Chem. A, 2008, 112, 7723 CrossRef CAS PubMed.
- M. I. J. Lorenz, S. H. Fischer and O. S. Wolfbeis, Chem. Soc. Rev., 2010, 39, 3102 RSC.
- (a) A. Fercher, S. M. Borisov, A. V. Zhdanov, I. Klimant and D. B. Papkovsky, ACS Nano, 2011, 5, 5499 CrossRef CAS PubMed; (b) S. M. Borisov, P. Lehner and I. Klimant, Anal. Chim. Acta, 2011, 690, 108 CrossRef CAS PubMed.
- M. A. Filatov, S. Baluschev, I. Z. Ilieva, V. Enkelmann, T. Miteva, K. Landfester, S. E. Aleshchnkov and A. V. Cheprakov, J. Org. Chem., 2012, 77, 11119 CrossRef CAS PubMed.
- (a) B. D. Rihter, M. E. Kenney, W. E. Ford and M. A. J. Rodgers, J. Am. Chem. Soc., 1993, 115, 8146 CrossRef CAS; (b) R. Schmidt, W. Drews and H. D. Brauer, J. Phys. Chem., 1982, 86, 4909 CrossRef CAS; (c) W. Fudickar and T. Linker, J. Am. Chem. Soc., 2012, 134, 15071 CrossRef CAS PubMed; (d) T. Christ, F. Kulzer, P. Bordat and T. Basche, Angew. Chem., Int. Ed., 2001, 40, 4192 CrossRef CAS; (e) K. Naito, T. Tachikawa, S.-C. Cui, A. Sugimoto, M. Fujitsuka and T. Majima, J. Am. Chem. Soc., 2006, 128, 16430 CrossRef CAS PubMed.
- R. Ziessel, T. N. Singh-Rachford, A. Haefele and F. N. Castellano, J. Am. Chem. Soc., 2008, 130, 16164 CrossRef PubMed.
- (a) J. Zhao, S. Ji, W. Wu, W. Wu, H. Guo, J. Sun, H. Sun, Y. Liu, Q. Li and L. Huang, RSC Adv., 2012, 2, 1712 RSC; (b) H. Xiang, J. Cheng, X. Ma, X. Zhoua and J. J. Chruma, Chem. Soc. Rev., 2013, 42, 6128 RSC.
- (a) T. N. Singh-Rachford and F. N. Castellano, Coord. Chem. Rev., 2010, 254, 2560–2573 CrossRef CAS; (b) P. Ceroni, Chem. – Eur. J., 2011, 17, 9560 CrossRef CAS PubMed; (c) J. Zhao, S. Ji and H. Guo, RSC Adv., 2011, 1, 937 RSC; (d) J. Zhao, W. Wu, J. Suna and S. Guo, Chem. Soc. Rev., 2013, 42, 5323 RSC; (e) G. Chen, H. Qiu, P. N. Prasad and X. Chen, Chem. Rev., 2014, 114, 5161 CrossRef CAS PubMed; (f) H. Xu, R. Chen, Q. Sun, W. Lai, Q. Su, W. Huang and X. Liu, Chem. Soc. Rev., 2014, 43, 3259 RSC; (g) J. Zhou, Q. Liu, W. Feng, Y. Sun and F. Li, Chem. Rev., 2015, 115, 395 CrossRef CAS PubMed.
- For recent examples, see: (a) D. B. Papkovsky and R. I. Dmitriev, Chem. Soc. Rev., 2013, 42, 8700 RSC; (b) A. Y. Lebedev, A. V. Cheprakov, S. Sakadzic, D. A. Boas, D. F. Wilson and S. A. Vinogradov, ACS Appl. Mater. Interfaces, 2009, 1, 1292 CrossRef CAS PubMed; (c) S. M. Borisov, C. Larndorfer and I. Klimant, Adv. Funct. Mater., 2012, 22, 4360 CrossRef CAS.
- P. R. Ogilby, Chem. Soc. Rev., 2010, 39, 3181 RSC.
- H. A. Frank and R. J. Cogdell, Photochem. Photobiol., 1996, 63, 257 CrossRef CAS PubMed.
- O. Hirayama, K. Nakamura, S. Hamada and Y. Kobayasi, Lipids, 1994, 29, 149 CrossRef CAS PubMed.
- N. Adir, H. Zer, S. Shochat and I. Ohad, Photosynth. Res., 2003, 76, 343 CrossRef CAS PubMed.
- J. De Las Rivas, A. Telfer and J. Barber, Biochim. Biophys. Acta, 1993, 1142, 155 CrossRef CAS.
- S. Munne-Bosch and L. Alegre, Crit. Rev. Plant Sci., 2002, 21, 31–57 CrossRef CAS.
- W. C. Neely, J. M. Martin and S. A. Barker, Photochem. Photobiol., 1988, 48, 423D428 Search PubMed.
- E.-M. Aro, S. McCaffery and J. M. Anderson, Plant Physiol., 1993, 103, 835 CAS.
- A. Trebst, B. Depka and H. Hollander-Czytko, FEBS Lett., 2002, 516, 156–160 CrossRef CAS PubMed.
- A. Turshatov, D. Busko, S. Baluschev, T. Miteva and K. Landfester, New J. Phys., 2011, 13, 083035 CrossRef.
- M. E. Díaz García and A. Sanz-Medel, Anal. Chem., 1986, 58, 1436 CrossRef.
- (a) V. Ramamurthy, J. V. Caspar, D. F. Eaton, E. W. Kuo and D. R. Corbin, J. Am. Chem. Soc., 1992, 114, 3882 CrossRef CAS; (b) A. S. Carreteroa, A. S. Castilloa and A. F. Gutiérreza, Crit. Rev. Anal. Chem., 2005, 35, 3 CrossRef.
- M. R. Fernández de la Campa, Y. Ming Liu, M. E. Díaz García and A. Sanz Medel, Anal. Chim. Acta, 1990, 238, 297 CrossRef.
- A. Turshatov, D. Busko, Y. Avlasevich, T. Miteva, K. Landfester and S. Baluschev, ChemPhysChem, 2012, 13, 3112 CrossRef CAS PubMed.
- S. H. C. Askes, N. López Mora, R. Harkes, R. I. Koning, B. Koster, T. Schmidt, A. Kros and S. Bonnet, Chem. Commun., 2015, 51, 9137 RSC.
- (a) H. Yamamura, K. Suzuki, K. Uchibori, A. Miyagawa, M. Kawai, C. Ohmizo and T. Katsu, Chem. Commun., 2012, 48, 892 RSC; (b) Z. Fan, C. H. Diao, H. B. Song, Z. L. Jing, M. Yu, X. Chen and M. J. Guo, J. Org. Chem., 2006, 71, 1244 CrossRef CAS PubMed; (c) J. Deng, X. Liu, L. Ma, C. Cheng, W. Shi, C. Nie and C. Zhao, ACS Appl. Mater. Interfaces, 2014, 6, 21603 CrossRef CAS PubMed.
- F. Zeng, W. Wang, A. Wang, K. Yuan, Z. Jin and Y. Yang, ACS Appl. Mater. Interfaces, 2015, 7, 26257 CAS.
- Y. Kai and K. Imakubo, Photochem. Photobiol., 1979, 29, 261 CrossRef CAS.
- M. L. Saviotti and W. C. Galley, Proc. Natl. Acad. Sci. U. S. A., 1974, 71, 4154 CrossRef CAS.
- M. W. Geiger and N. J. Turro, Photochem. Photobiol., 1975, 22, 273 CrossRef CAS PubMed.
- N. J. Turro, T. Okubo and G. C. Weed, Photochem. Photobiol., 1983, 37, 149 CrossRef CAS.
- (a) V. B. Nazarov, V. I. Gerko and M. V. Alfimov, Izv. Akad. Nauk, Ser. Khim., 1996, 1014 ( Russ. Chem. Bull. , 1996 , 45 , 969 ) CAS (Engl. Transl.); (b) V. B. Nazarov, V. I. Gerko and T. G. Vershinnikova, Izv. Akad. Nauk, Ser. Khim., 1995, 1966 ( Russ. Chem. Bull. , 1995 , 44 , 1886 ) CAS (Engl. Transl.).
- J.-J. Wu, Y. Wang, J.-B. Chao, L.-N. Wang and W.-J. Jin, J. Phys. Chem. B, 2004, 108, 8915 CrossRef CAS.
- B. Tang, X. Wang, G. Wang, Y. Wang and Z. Chen, Analyst, 2005, 130, 1038 RSC.
- A. S. Carretero, C. C. Blanco, R. E. Fernandez and A. F. Gutierrez, J. Anal. Chem., 1998, 360, 605 Search PubMed.
- R. E. Brewster, M. J. Kidd and M. D. Schuh, Chem. Commun., 2001, 1134 RSC.
- C. Ye, J. Wang, X. Wang, P. Ding, Z. Lianga and X. Tao, Phys. Chem. Chem. Phys., 2016, 18, 3430 RSC.
- Dendrimers and Other Dendritic Polymers, ed. J. M. J. Frechet and D. A. Tomalia, Wiley, New York, 2001 Search PubMed.
- (a) R. H. Jin, T. Aida and S. Inoue, J. Chem. Soc., Chem. Commun., 1993, 1260 RSC; (b) R. Sadamoto, N. Tomioka and T. Aida, J. Am. Chem. Soc., 1996, 118, 3978 CrossRef CAS; (c) M. Kimura, K. Nakada, Y. Yamaguchi, K. Hanabusa, H. Shirai and N. Kobayashi, Chem. Commun., 1997, 1215 RSC; (d) J. Issberner, F. Vogtle, L. DeCola and V. Balzani, Chem. – Eur. J., 1997, 3, 706 CrossRef CAS; (e) F. Vogtle, M. Plevoets, M. Nieger, G. C. Azzellini, A. Credi, L. De Cola, V. De Marchis, M. Venturi and V. Balzani, J. Am. Chem. Soc., 1999, 121, 6290 CrossRef; (f) J. M. Riley, S. Alkan, A. D. Chen, M. Shapiro, W. A. Khan, W. R. Murphy and J. E. Hanson, Macromolecules, 2001, 34, 1797 CrossRef CAS.
- V. Rozhkov, D. Wilson and S. Vinogradov, Macromolecules, 2002, 35, 1991 CrossRef CAS.
- S. A. Vinogradov, Org. Lett., 2005, 7, 1761 CrossRef CAS PubMed.
- A. Y. Lebedev, A. V. Cheprakov, S. Sakadzic, D. A. Boas, D. F. Wilson and S. A. Vinogradov, ACS Appl. Mater. Interfaces, 2009, 1, 1292 CAS.
- E. Roussakis, Z. Li, N. H. Nowell, A. J. Nichols and C. L. Evans, Angew. Chem., Int. Ed., 2015, 54, 14728 CrossRef CAS PubMed.
- A. G. Moiseev, E. A. Margulies, J. A. Schneider, F. Beanger-Garieyb and D. F. Perepichka, Dalton Trans., 2014, 43, 2676 RSC.
- P. Duan, N. Yanai and N. Kimizuka, J. Am. Chem. Soc., 2013, 135, 19056 CrossRef CAS PubMed.
- M. A. Filatov, E. Heinrich, D. Busko, I. Z. Ilieva, K. Landfestera and S. Baluschev, Phys. Chem. Chem. Phys., 2015, 17, 6501 RSC.
- (a) P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133 CrossRef CAS PubMed; (b) J. H. van Esch and B. L. Feringa, Angew. Chem., Int. Ed., 2000, 39, 2263 CrossRef CAS; (c) S. Shinkai and K. Murata, J. Mater. Chem., 1998, 8, 485 RSC.
- (a) M. Shirakawa, N. Fujita, T. Tani, K. Kanekob and S. Shinkai, Chem. Commun., 2005, 4149 RSC; (b) M. Shirakawa, N. Fujita, T. Tani, K. Kaneko, M. Ojima, A. Fujii, M. Ozaki and S. Shinkai, Chem. – Eur. J., 2007, 13, 4155 CrossRef CAS PubMed.
- G.-R. Li, J.-J. Wu, W.-J. Jin and J.-W. Xie, Talanta, 2003, 60, 555 CrossRef CAS PubMed.
- Y.-T. Wang, X.-W. Wang and Y. Zhang, Photochem. Photobiol., 2011, 87, 772 CrossRef CAS PubMed.
- S. Hirata, K. Totani, J. Zhang, T. Yamashita, H. Kaji, S. R. Marder, T. Watanabe and C. Adachi, Adv. Funct. Mater., 2013, 23, 3386 CrossRef CAS.
- (a) P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133 CrossRef CAS PubMed; (b) N. Kimizuka, M. Shimizu, S. Fujikawa, K. Fujimura, M. Sano and T. Kunitake, Chem. Lett., 1998, 967 CrossRef CAS; (c) L. Gao, D. Xu and B. Zheng, Chem. Commun., 2014, 50, 12142 RSC.
- C. A. Strassert, M. Mauro and L. De Cola, Photophysics of soft and hard molecular assemblies based on luminescent complexes, Academic Press, 2001 Search PubMed.
- N. Kumar Allampally, M. Bredol, C. A. Strassert and L. De Cola, Chem. – Eur. J., 2014, 20, 16863 CrossRef PubMed.
- H. Wang, H. Wang, X. Yang, Q. Wang and Y. Yang, Langmuir, 2015, 31, 486 CrossRef CAS PubMed.
- P. Duan, N. Yanai, H. Nagatomi and N. Kimizuka, J. Am. Chem. Soc., 2015, 137, 1887 CrossRef CAS PubMed.
- Y. G. Li, T. Y. Wang and M. H. Liu, Soft Matter, 2007, 3, 1312 RSC.
- J.-M. Aubry, C. Pierlot, J. Rigaudy and R. Schmidt, Acc. Chem. Res., 2003, 36, 668 CrossRef CAS PubMed.
- R. Vadrucci, C. Weder and Y. C. Simon, Mater. Horiz., 2015, 2, 120 RSC.
- T. Miteva, G. Nelles, S. Balouchev and V. Yakutkin, EP 2 067 837 B1, 2007; T. Miteva, G. Nelles, S. Balouchev and V. Yakutkin, US 7,969,646 B2, 2011.
- (a) T. C. O'Riordan, A. E. Soini and D. B. Papkovsky, Anal. Biochem., 2001, 290, 366 CrossRef PubMed; (b) T. C. O'Riordan, A. E. Soini, J. T. Soini and D. B. Papkovsky, Anal. Chem., 2002, 74, 5845 CrossRef; (c) J. M. Kuerner, I. Klimant, C. Krause, H. Preu, W. Kunz and O. S. Wolfbeis, Bioconjugate Chem., 2001, 12, 883 CrossRef CAS.
- X. Song, L. Huang and B. Wu, Anal. Chem., 2008, 80, 5501 CrossRef CAS PubMed.
- A. Monguzzi, R. Tubino and F. Meinardi, J. Phys. Chem. A, 2009, 113, 1171 CrossRef CAS PubMed.
- T. N. Singh-Rachford, J. Lott, C. Weder and F. N. Castellano, J. Am. Chem. Soc., 2009, 131, 12007 CrossRef CAS PubMed.
- (a) S. Balouchev, T. Miteva, P. Keivanidis, J. Lupton, G. Nelles and A. Yasuda, US 7,683,363 B2, 2010; (b) A. Monguzzi, F. Bianchi, A. Bianchi, M. Mauri, R. Simonutti, R. Ruffo, R. Tubino and F. Meinardi, Adv. Energy Mater., 2013, 3, 680 CrossRef CAS; (c) Y. C. Simon and C. Weder, J. Mater. Chem., 2012, 22, 20817 RSC.
- (a) X. Lu and M. A. Winnik, Chem. Mater., 2001, 13, 3449 CrossRef CAS; (b) V. M. Litvinov, O. Persyn, V. Miri and J. M. Lefebvre, Macromolecules, 2010, 43, 7668 CrossRef CAS.
- J.-H. Kim, F. Deng, F. N. Castellano and J.-H. Kim, Chem. Mater., 2012, 24, 2250 CrossRef CAS.
- R. R. Islangulov, J. Lott, C. Weder and F. N. Castellano, J. Am. Chem. Soc., 2007, 129, 12652 CrossRef CAS PubMed.
- A. Monguzzi, F. Bianchi, A. Bianchi, M. Mauri, R. Simonutti, R. Ruffo, R. Tubino and F. Meinardi, Adv. Energy Mater., 2013, 3, 680 CrossRef CAS.
- (a) S. H. Lee, J. R. Lott, Y. C. Simon and C. Weder, J. Mater. Chem. C, 2013, 1, 5142 RSC; (b) P. B. Merkel and J. P. Dinnocenzo, J. Lumin., 2009, 129, 303 CrossRef CAS; (c) P. B. Merkel and J. P. Dinnocenzo, J. Phys. Chem. A, 2008, 112, 10790 CrossRef CAS PubMed.
- A. Monguzzi, R. Tubino and F. Meinardi, J. Phys. Chem. A, 2009, 113, 1171 CrossRef CAS PubMed.
- A. Monguzzi, M. Frigoli, C. Larpent, R. Tubino and F. Meinardi, Adv. Funct. Mater., 2012, 22, 139 CrossRef CAS.
- F. Marsico, A. Turshatov, R. Pekoz, Y. Avlasevich, M. Wagner, K. Weber, D. Donadio, K. Landfester, S. Baluschev and F. R. Wurm, J. Am. Chem. Soc., 2014, 136, 11057 CrossRef CAS PubMed.
- D. P. Jason, G. O. Brian, P. B. Justin, K. H. Mohammad, R. Jo Ann, S. W. Jeffrey, W. R. James and N. Sergei, High-Oxygen Barrier Materials Based on Hyperbranched Aliphatic Polyesters. Polymer Degradation and Performance, American Chemical Society, Washington, DC, 2009, vol. 1004, pp. 17–30 Search PubMed.
- (a) P. Di Mascio, S. Kaiser and H. Sies, Arch. Biochem. Biophys., 1989, 274, 532 CrossRef CAS PubMed; (b) C. S. Foote, Acc. Chem. Res., 1968, 1, 104 CrossRef CAS.
- (a) A. Monguzzi, M. Frigoli, C. Larpent, R. Tubino and F. Meinardi, Adv. Funct. Mater., 2012, 22, 139 CrossRef CAS; (b) K. K. Ng and G. Zheng, Chem. Rev., 2015, 115, 11012 CrossRef CAS PubMed.
- J.-H. Kang and E. Reichmanis, Angew. Chem., Int. Ed., 2012, 51, 11841 CrossRef CAS PubMed.
- Q. Liu, B. R. Yin, T. S. Yang, Y. Yang, Z. Shen, P. Yao and F. Y. Li, J. Am. Chem. Soc., 2013, 135, 5029 CrossRef CAS PubMed.
- C. Wohnhaas, K. Friedemann, D. Busko, K. Landfester, S. Baluschev, D. Crespy and A. Turshatov, ACS Macro Lett., 2013, 2, 446 CrossRef CAS.
- L. K. Massey, Permeability Properties of Plastics and Elastomers: A Guide to Packaging and Barrier Materials, William Andrew, New York, 2003 Search PubMed.
- J. Gaume, P. Wong-Wah-Chung, A. Rivaton and J.-L. Gardette, RSC Adv., 2011, 1, 1471 RSC.
- C. Ye, B. Wang, R. Hao, X. Wang, P. Ding, X. Tao, Z. Chen, Z. Liang and Y. Zhou, J. Mater. Chem. C, 2014, 2, 8507 RSC.
- A. F. Turbak, F. W. Snyder and K. R. Sandberg, J. Appl. Polym. Sci.: Appl. Polym. Symp., 1983, 37, 815 CAS.
- F. W. Herrick, R. L. Casebier, J. K. Hamilton and K. R. Sandberg, J. Appl. Polym. Sci.: Appl. Polym. Symp., 1983, 37, 797 CAS.
- (a) J. Huang, H. Zhu, Y. Chen, C. Preston, K. Rohrbach, J. Cumings and L. Hu, ACS Nano, 2013, 7, 2106 CrossRef CAS PubMed; (b) J. T. Korhonen, P. Hiekkataipale, J. Malm, M. Karppinen, O. Ikkala and R. H. A. Ras, ACS Nano, 2011, 5, 1967 CrossRef CAS PubMed; (c) J. Wang, Q. Cheng, L. Lin and L. Jiang, ACS Nano, 2014, 8, 2739 CrossRef CAS PubMed; (d) H. Zhu, Y. Li, Z. Fang, J. Xu, F. Cao, J. Wan, C. Preston, B. Yang and L. Hu, ACS Nano, 2014, 8, 3606 CrossRef CAS PubMed; (e) M. M. Hamedi, A. Hajian, A. B. Fall, K. Hakansson, M. Salajkova, F. Lundell, L. Wagberg and L. A. Berglund, ACS Nano, 2014, 8, 2467 CrossRef CAS PubMed.
- C. Aulin, S. Ahola, P. Josefsson, T. Nishino, Y. Hirose, M. Osterberg and L. Wagberg, Langmuir, 2009, 25, 7675 CrossRef CAS PubMed.
- H. Fukuzumi, T. Saito, T. Iwata, Y. Kumamoto and A. Isogai, Biomacromolecules, 2009, 10, 162 CrossRef CAS PubMed.
- (a) A. J. Svagan, D. Busko, Y. Avlasevich, G. Glasser, S. Baluschev and K. Landfester, ACS Nano, 2014, 8, 8198 CrossRef CAS PubMed; (b) A. A. Svagan, K. Landfester, J. Risbo and S. Balouchev, EP 2865443 A1, WO 2015/059179 A1, 2015.
- A. Turshatov, F. Marsico, D. Busko, Y. Avlasevich, M. A. Filatov, F. R. Wurm, S. Balouchev and K. Landfester, EP2851407A1, WO 2015/044129 A1, 2015.
- (a) J. N. Demas, B. A. DeGraff and P. B. Coleman, Anal. Chem., 1999, 71, 793A CrossRef CAS PubMed; (b) Y. Amao, Microchim. Acta, 2003, 143, 1 CrossRef CAS; (c) D. B. Papkovsky and T. C. O'Riordan, J. Fluoresc., 2005, 15, 569 CrossRef CAS PubMed; (d) L. Huynh, Z. U. Wang, J. Yang, V. Stoeva, A. Lough, I. Manners and M. A. Winnik, Chem. Mater., 2005, 17, 4765 CrossRef CAS; (e) J. F. Fernandez-Sanchez, T. Roth, R. Cannas, M. K. Nazeeruddin, S. Spichiger, M. Graetzel and U. E. Spichiger-Keller, Talanta, 2007, 71, 242 CrossRef CAS PubMed; (f) S. M. Borisov and I. Klimant, Anal. Chem., 2007, 79, 7501 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |