DOI:
10.1039/C5CS00895F
(Review Article)
Chem. Soc. Rev., 2016,
45, 4524-4532
Silver-catalyzed carboxylation
Received
4th December 2015
First published on 18th February 2016
Abstract
Silver-catalyzed reactions are some of the important methodologies in organic chemistry. Since 2007, a new application of silver catalysts has been emerging. For the sequential carboxylation and cyclization of alkyne derivatives, such as propargyl alcohols and amines, using carbon dioxide, silver catalysts show significant reactivity under mild conditions unlike other transition metals. These developments have received much attention for the effective utilization of carbon dioxide in organic chemistry to synthesize heterocyclic compounds. Related silver-catalyzed C–C bond forming reactions with carbon dioxide have also provided the synthetic methods of the corresponding carboxylic acid derivatives. In this review, the recent studies of the silver-catalyzed carboxylation reactions using carbon dioxide are described.

K. Sekine
| Kohei Sekine was born in Yokohama, Japan, in 1988. He received PhD under the supervision of Prof. Tohru Yamada at Keio University in 2016 in the field of silver-catalyzed sequential carboxylation and cyclization using carbon dioxide. His current research interests include transition-metal-catalyzed reactions of unsaturated organic molecules and the utilization of small molecules. |

T. Yamada
| Tohru Yamada was born in Hokkaido, Japan, in 1958. He received his PhD degree in 1987 from the University of Tokyo under the guidance of Professor Teruaki Mukaiyama. In 1987, he joined Mitsui Petrochemical Ind. Ltd. In 1997, he moved to Keio University and in 2002 was promoted to a professor of Chemistry Department. In 1992 he received the Chemical Society of Japan Award for Young Chemist, in 2003 and 2011 the BCSJ Award by the Chemical Society of Japan, and in 2010 the Nissan Chemical Industries Award for Novel Reaction & Method 2010 by Synthetic Organic Chemistry, Japan. |
Introduction
Silver catalysts are widely utilized in organic reactions as Lewis acids. For example, the silver-catalyzed cyclization reactions of alkyne or allene derivatives are the promising synthetic methods of heterocyclic compounds. The silver-catalyzed or -mediated reactions have been reviewed1–5 and summarized in a book.6 It has since been revealed that silver catalysts still have an additional potential to show unique reactivity in the sequential carboxylation and cyclization reaction using carbon dioxide during the synthesis of heterocyclic compounds.
Carbon dioxide is thermodynamically stable and much less reactive than other carbon derivatives due to its high oxidation state. Therefore, strong nucleophiles or harsh reaction conditions were usually required when carbon dioxide is utilized in organic syntheses. For example, the Kolbe–Schmitt reaction, found in any organic chemistry textbook, requires high pressure carbon dioxide and high temperature, though the reaction has been employed for the commercial production of salicylic acid. Recently, carbon dioxide has been regarded as an appealing C1 resource due to its abundant supply from industries and easy handling.7 Methods to transform carbon dioxide into high-value chemicals have been developed. The transition-metal-catalyzed carboxylation reactions of various carbon nucleophiles with C–C bond formation have been actively developed for the preparation of fine chemicals.8,9
Considering the carboxylation of simple alcohols or amines with carbon dioxide, these two reactions are in equilibrium. Therefore, the problem is that the corresponding carbonate and carbamate produced easily return back to the starting material and carbon dioxide after quenching the reactions. The transformation of the unstable intermediates into a stable product is one of the reliable solutions of the problem. With respect to atom economy, the cyclization of unsaturated organic molecules is an important methodology. Though the sequential carboxylation and cyclization of propargyl alcohols and propargyl amines using carbon dioxide have been developed, harsh conditions (high temperature or high pressure carbon dioxide) were required and substrates were limited to terminal alkynes. In 2007, the silver catalyst made a major breakthrough in the cascade carboxylation and cyclization of propargyl alcohols and carbon dioxide. Related silver-catalyzed carboxylation reactions have also been achieved. In this review, the recent studies of the silver-catalyzed carboxylation using carbon dioxide are described.
1. Silver-catalyzed synthesis of cyclic carbonates
One of the reasonable synthetic approaches of cyclic carbonates is the cyclization of readily available propargyl alcohols and carbon dioxide from the viewpoint of atom economy. The reaction of propargyl alcohols and carbon dioxide was promoted by a transition metal, carbene and phosphine as the catalyst; however, these reported methods required harsh reaction conditions, such as high carbon dioxide pressure and high reaction temperature. Moreover, terminal alkynes were only applicable for these carbonate formations, and the internal alkynes afforded sluggish reactions.
1.1 Silver-catalyzed sequential carboxylation and cyclization of propargyl alcohols
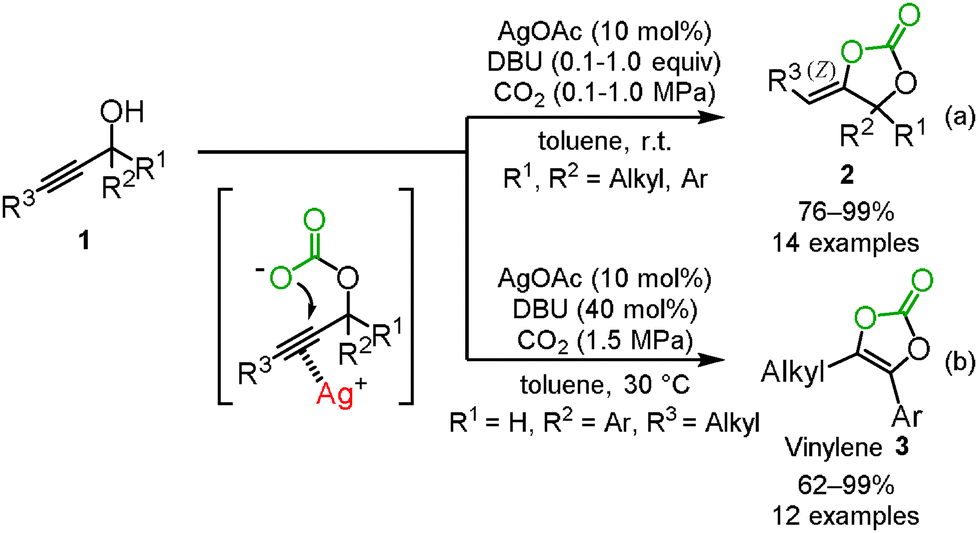
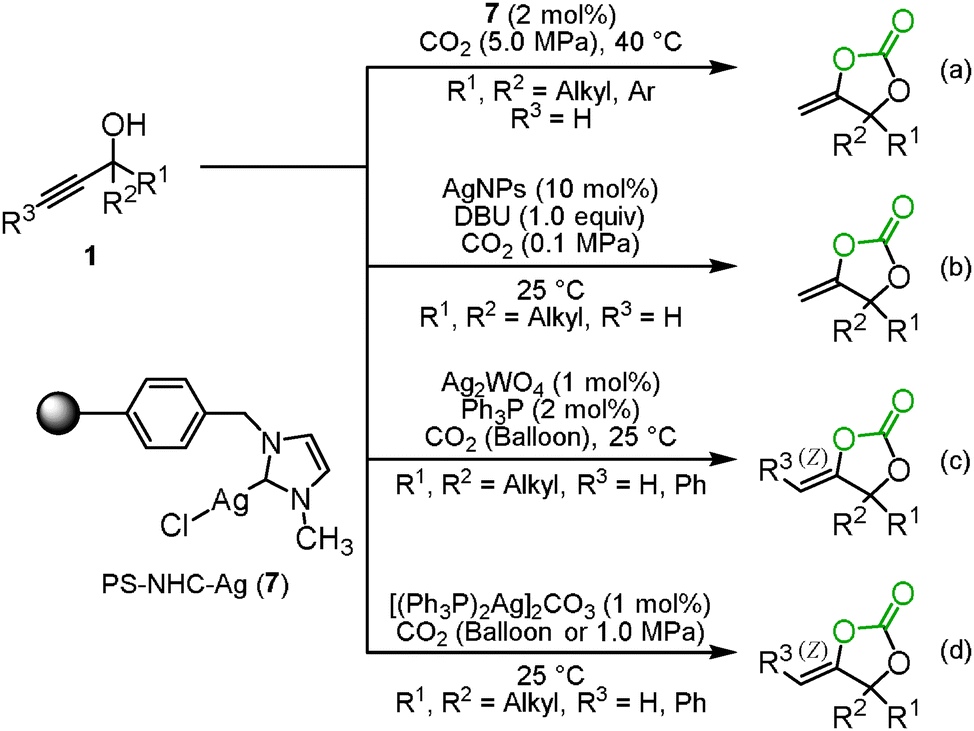
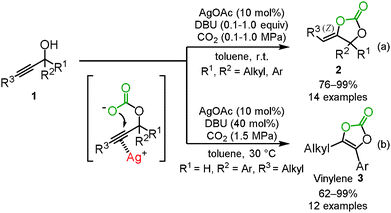
In 2007, we indeed reported the silver-catalyzed carboxylation and cyclization of propargyl alcohols 1 under mild reaction conditions (Scheme 1(a)).10 The study indicated that the combined use of silver acetate and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) was an efficient catalyst system for the incorporation of carbon dioxide into various propargyl alcohols 1 to afford the corresponding cyclic carbonates 2 in high-to-excellent yields, though other transition-metals including copper, gold, rhodium, mercury, platinum, and palladium were not effective at room temperature. It was also noted that this catalytic system could be applied to the aryl- and alkyl-substituted internal alkynes even under mild reaction conditions. The exo-alkenyl cyclic carbonates were obtained as the sole isomers and the geometry of the C–C double bond was confirmed to be the Z isomer by X-ray single crystal structure analysis or NOE. These results suggested that the silver catalyst activates the C–C triple bond from the opposite side of the carbonate to promote anti-addition through 5-exo-dig cyclization. This assumption was also supported by DFT calculations.11 The outer-sphere attack is similar to the typical reactivity of the related gold(I) catalysts,12 but only the silver catalyst is able to induce cyclization, and gold catalysis fails under these conditions.13
 |
| | Scheme 1 Silver-catalyzed incorporation of carbon dioxide into propargyl alcohols. | |
We recently found that when the secondary propargyl alcohols were employed under the present reaction conditions, the vinylene carbonates 3, which are useful as prodrugs for drug-delivery systems and are used as electrolyte additives of lithium ion batteries, were produced.14 These vinylene carbonates were produced after the isomerization of the exo-alkenyl cyclic carbonate (Scheme 1(b)).
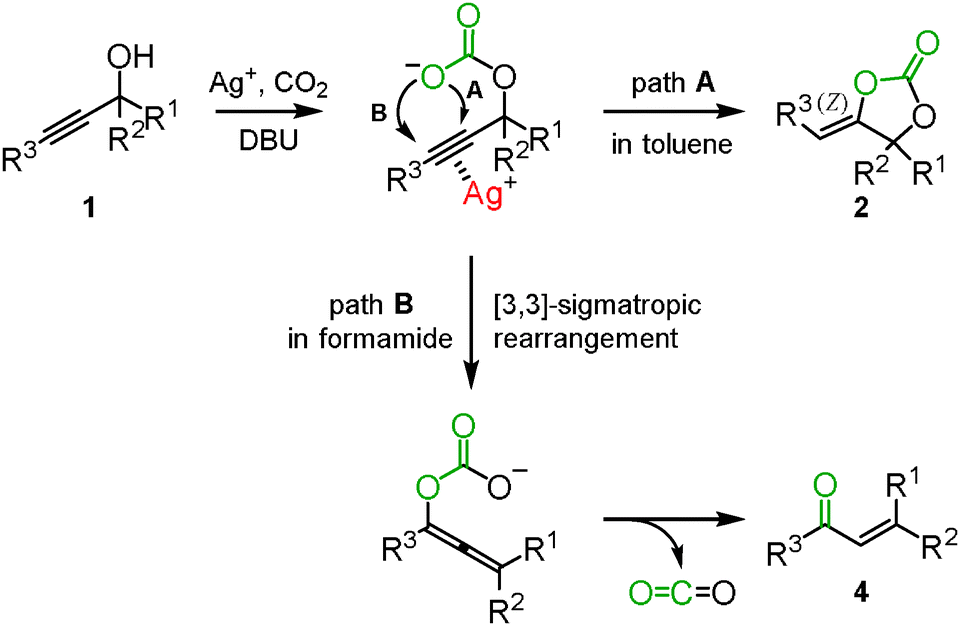
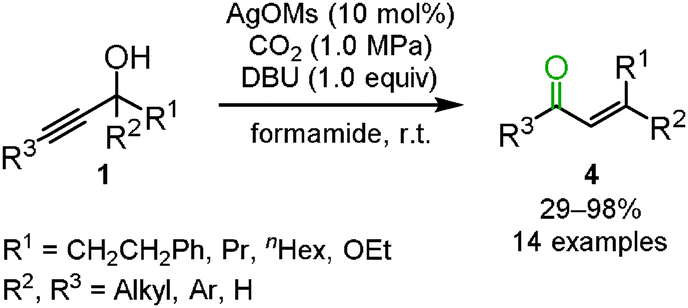
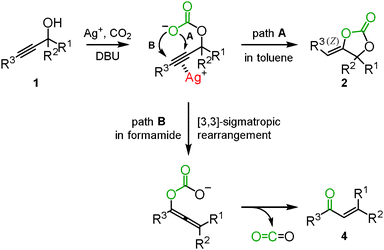
In the reactions mentioned above, the silver-catalyzed reaction of propargyl alcohols with carbon dioxide selectively afforded the corresponding cyclic carbonates in a toluene solution (path A in Scheme 2), while in dichloromethane and chlorobenzene, the corresponding α,β-unsaturated carbonyl compounds 4, generated via a Meyer–Schuster type reaction, were detected along with the cyclic carbonate. We proposed the following reaction mechanism for α,β-unsaturated carbonyl compounds: the β-carbon of propargyl alcohol would be alternatively attacked to promote the [3,3]-sigmatropic rearrangement into the allene-enolate. The α,β-unsaturated carbonyl compounds would result from the release of carbon dioxide (path B in Scheme 2). It is reasonable to assume that the polarized structure with an elongated C–O bond in the carbonate intermediate would be stabilized in a polar solvent, thus enhancing the attack on the β-carbon in the activated propargyl alcohol. In fact, DMF and DMA solvents improved the selectivity toward the enone. Finally, it was found that formamide was the best choice to selectively obtain the corresponding enone. Various tertiary and secondary propargyl alcohols were efficiently converted into the corresponding α,β-unsaturated carbonyl compounds in high yield using a catalytic amount of silver methanesulfonate with DBU and carbon dioxide (Scheme 3).15
 |
| | Scheme 2 Postulated reaction mechanism of propargyl alcohols and carbon dioxide. | |
 |
| | Scheme 3 Transformation of propargyl alcohols into enones. | |
1.2 Silver-catalyzed asymmetric synthesis of cyclic carbonate
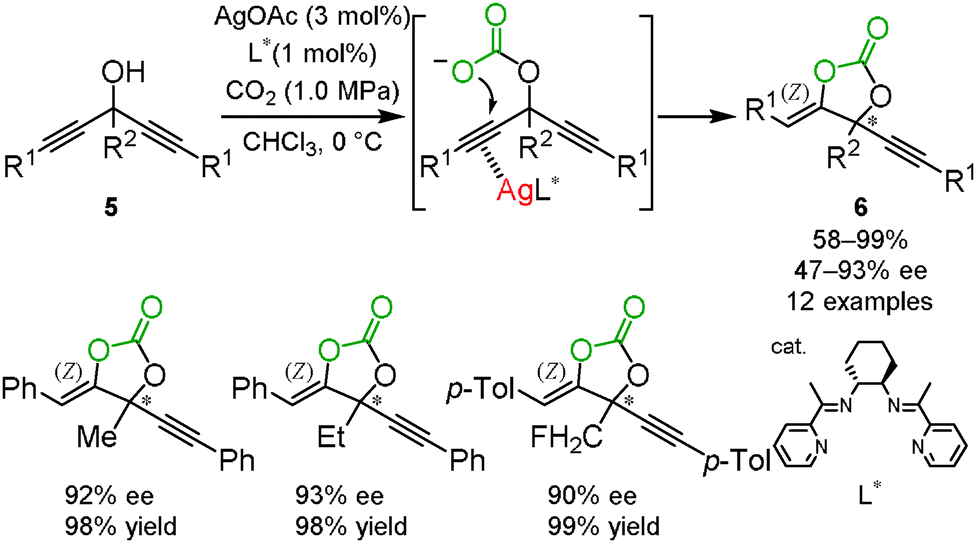
As described in Section 1.1, the silver ion activated the C–C triple bond as a π-Lewis acid. On the basis of this observation, an optically active silver catalyst, which is in rapid equilibrium between the two alkynes in bispropargyl alcohols 5, was expected to selectively promote a nucleophilic attack on one of the C–C triple bonds with asymmetric desymmetrization. We developed the asymmetric carbon dioxide incorporation into bispropargyl alcohols with desymmetrization by the combination of silver acetate and the chiral Schiff base ligand L* to afford the corresponding cyclic carbonates 6 with good-to-excellent enantiomeric excess (Scheme 4). We confirmed that (S)-cyclic carbonate was obtained in the reaction catalyzed by the complex with (R,R)-L* based on a VCD measurement in combination with the corresponding spectra predicted by DFT calculations.16
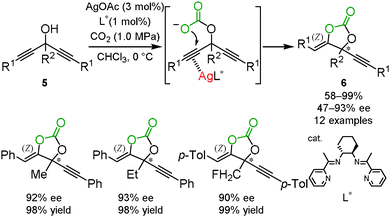 |
| | Scheme 4 Enantioselective cyclization of bispropargyl alcohols and carbon dioxide. | |
1.3 Application for highly efficient silver catalysts
Highly efficient and reusable silver catalysts for the incorporation of carbon dioxide into propargyl alcohols were developed because a relatively large amount of the silver catalyst and a stoichiometric amount of DBU were required as mentioned in Section 1.1. The polystyrene-supported N-heterocyclic carbene silver complex 7 (PS–NHC–Ag) had a good catalytic activity and could be reused up to 15 times without considerable loss of its catalytic activity, though a high pressure of carbon dioxide was required (Scheme 5(a)).17 In addition, Han and co-workers reported that silver nanoparticles (AgNPs) catalyzed the carboxylation and cyclization of propargyl alcohols and carbon dioxide and were recyclable at least 5 times (Scheme 5(b)).18
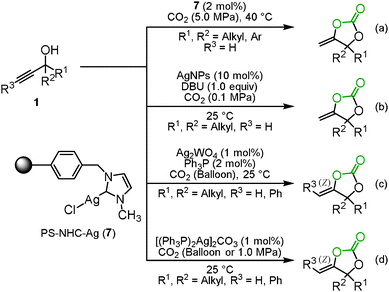 |
| | Scheme 5 Efficient silver catalysts for the carboxylation and cyclization of propargyl alcohols. | |
He's group achieved the reaction at a low catalyst loading of Ag2WO4 and Ph3P even under the balloon pressure of carbon dioxide. The activation of carbon dioxide by [WO4]2− was postulated to provide high efficiency (Scheme 5(c)).19 He and co-workers also developed the reaction which was efficiently catalyzed by the silver complex [(Ph3P)2Ag]2CO3 without any organic base (Scheme 5(d)). Based on the monitoring of the reaction by 1H and 13C NMR spectroscopy, they assumed that [(Ph3P)2Ag]2CO3 simultaneously activated the propargyl alcohol and carbon dioxide to effectively promote the reaction.20
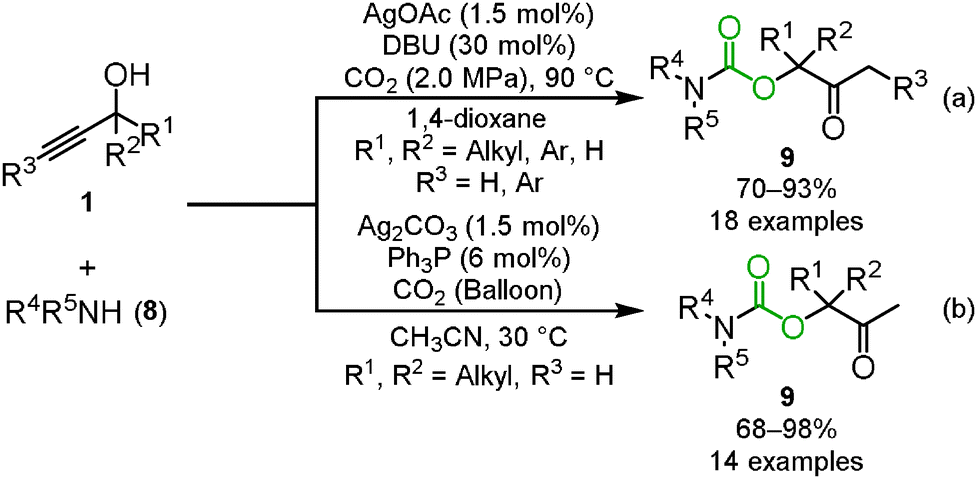
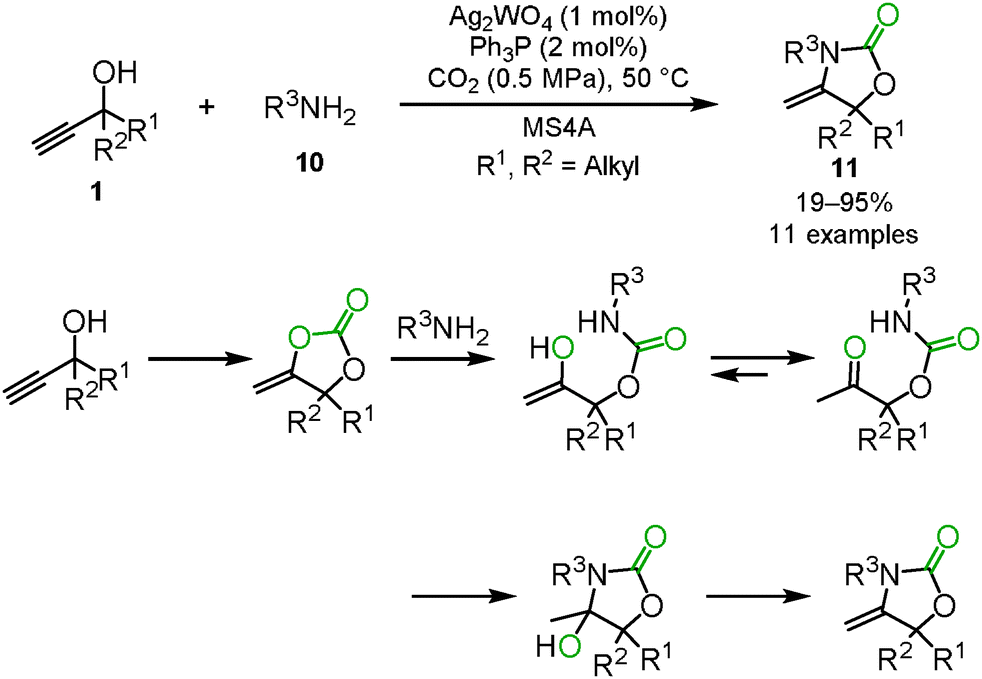
One of the applications of carbon dioxide incorporation into propargyl alcohols is the three-component reaction. The reaction between propargyl alcohol, carbon dioxide and secondary amines 8 provided the corresponding carbamates 9 through the ring-opening reaction of the cyclic carbonate by secondary amines (Scheme 6).21 In the case of primary amines 10, intramolecular nucleophilic addition and dehydration produced the corresponding oxazolidinones 11 (Scheme 7).19
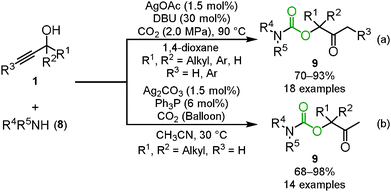 |
| | Scheme 6 Three-component reaction between propargyl alcohols, secondary amines and carbon dioxide. | |
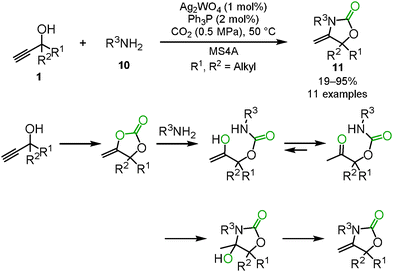 |
| | Scheme 7 Three-component reaction between propargyl alcohols, primary amines and carbon dioxide. | |
2. Transformation of amine derivatives
2.1 Propargyl amine
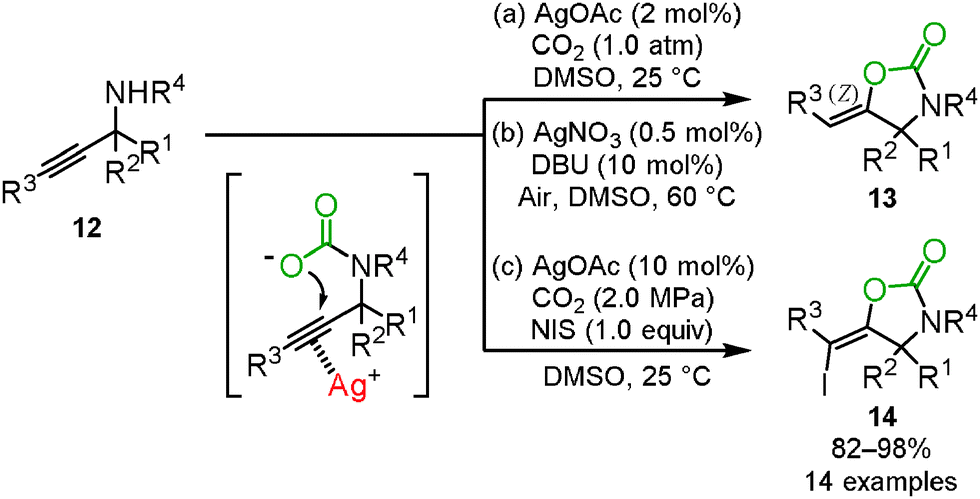
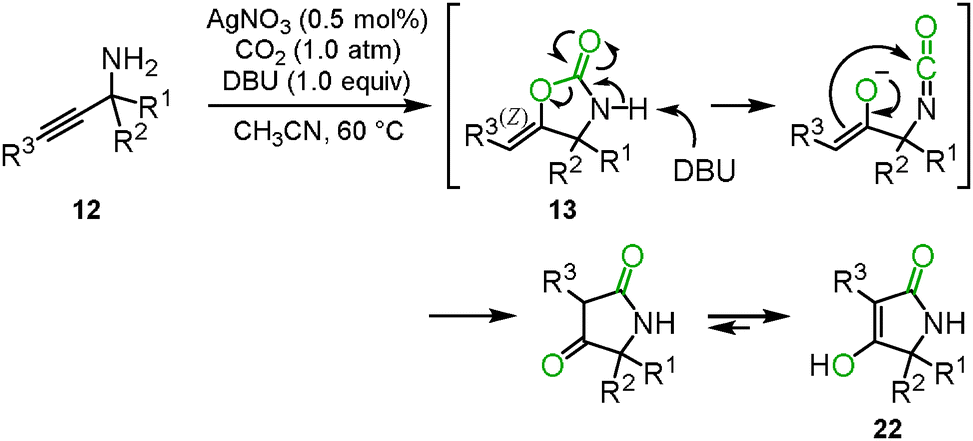
The oxazolidinone preparation from carbon dioxide is one of the most attractive synthetic methods. We reported that carbon dioxide incorporation into various propargylic amines 12 proceeded to give the corresponding oxazolidinone derivatives 13 in high yields under mild reaction conditions, i.e., 2 mol% of AgOAc and 1.0 atm of carbon dioxide (Scheme 8(a)).22
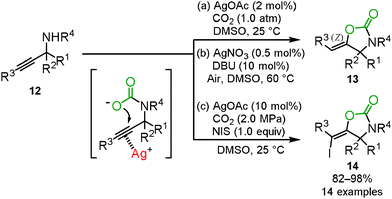 |
| | Scheme 8 Carboxylation and cyclization of propargyl amines and carbon dioxide for the synthesis of oxazolidinone derivatives. | |
Yoshida and co-workers reported that the silver/DBU catalytic system promoted the fixation of carbon dioxide into propargyl amines even under atmospheric carbon dioxide (Scheme 8(b)). DBU was found to be necessary for the reaction. They proposed that the DBU–CO2 adduct generated in situ from DBU and the atmospheric carbon dioxide promoted the reaction with the propargyl amine and carbon dioxide. This assumption was supported by the attempted reaction of propargyl amine and the DBU–CO2 adduct.23
For the synthesis of high-valued compounds, the silver-catalyzed three-component reaction of propargylic amines, carbon dioxide, and N-iodosuccinimide was developed for the stereoselective synthesis of (E)-iodovinyloxazolidinones 14 which could be useful for the metal catalyzed coupling reactions (Scheme 8(c)).24
2.2 Benzoxazine-2-one from o-alkynylaniline and carbon dioxide
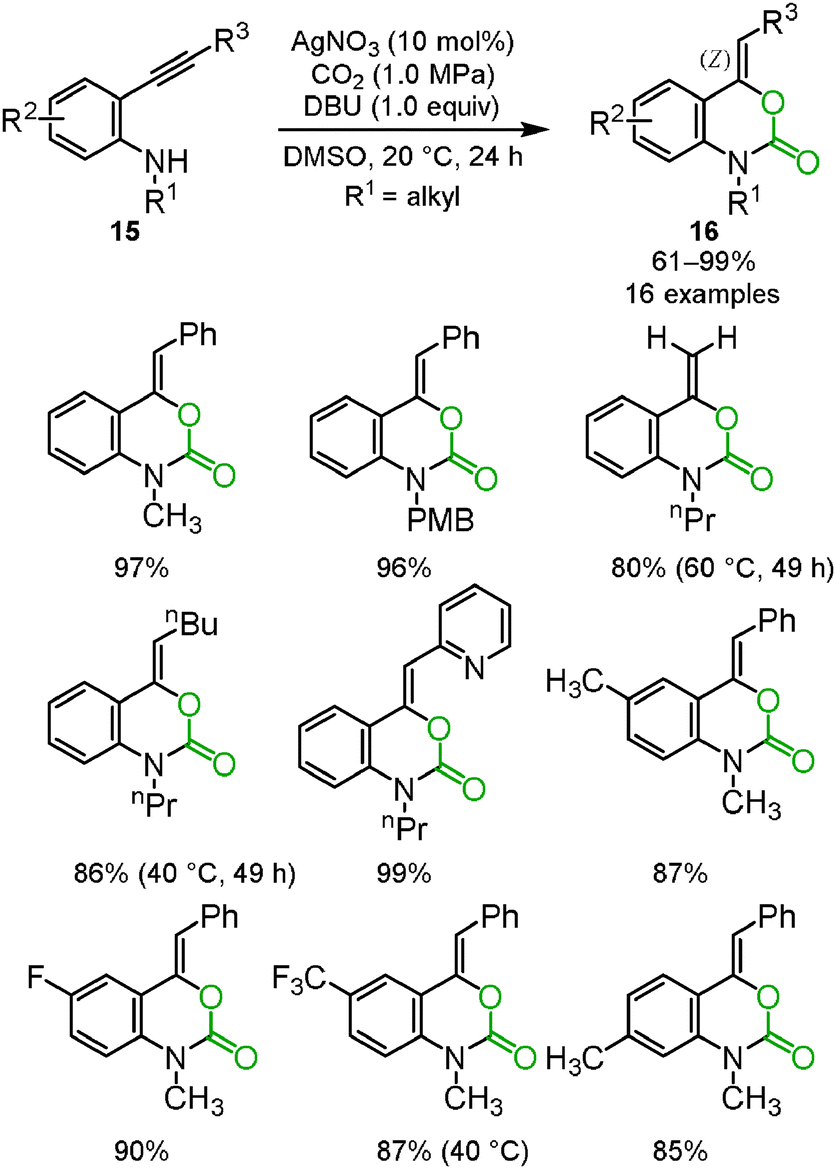
Benzoxazine-2-one derivatives 16 have attracted much attention as some of the most important heterocyclic structures in pharmaceutical science, but their synthesis methods were limited. In some cases, toxic reagents, such as phosgene or carbon monoxide, were required. We reported that the silver/DBU system promoted the incorporation of carbon dioxide into o-alkynylanilines 15 to afford the corresponding benzoxazine-2-ones 16 bearing the Z exo-olefin via 6-exo-dig cyclization. Some variations in the substituents on benzoxazine-2-ones are shown (Scheme 9).25 The detailed reaction mechanism was studied using DFT calculations by Wang, Yan and co-workers.26
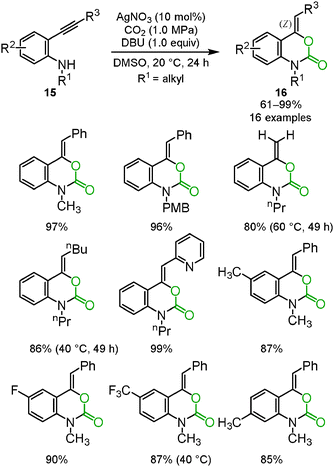 |
| | Scheme 9 Silver-catalyzed reactions of o-alkynylanilines and carbon dioxide for the synthesis of benzoxazine-2-ones. | |
2.3 Allenylamine
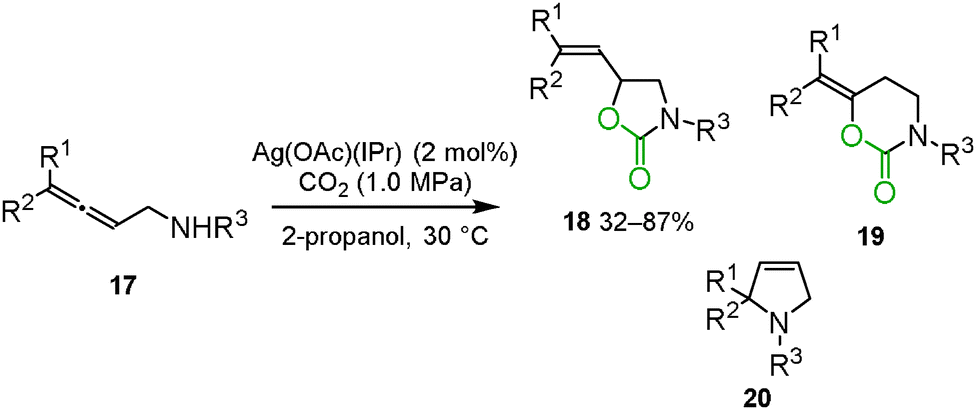
Ikariya and co-workers reported that Ag(OAc)(IPr) [IPr = 1,3-bis(2,6-diisopropylphenyl)-imidazol-2-ylidene] effectively catalyzed the carboxylation and cyclization of allenylamines 17 (Scheme 10).27 Though an intramolecular hydroamination 20 and a 6-membered ring formation 19 were competitive reactions, a 5-membered ring formation highly selectively proceeded to give the corresponding cyclic urethane 3-benzyl-5-vinyl-1,3-oxazolidin-2-one 18.
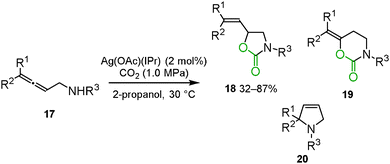 |
| | Scheme 10 Silver-catalyzed carboxylation and cyclization of allenylmethylamines. | |
In this reaction, catalytic amounts of the gold complex Au(OAc)(IPr) and the copper complex Cu(OAc)(IPr) were found to be much less effective. DFT calculations supported the difference in electronic structures between the alkenylsilver complex and the alkenylgold complex. The silver–carbon bond was suggested to be more polarized than the gold–carbon bond. This result implied that the polarization of the metal–carbon bond could be crucial to regenerate the metal cation for complete turnover of the catalytic cycle.
2.4 Rearrangement for hydroxylquinolin-2-one and tetramic acid
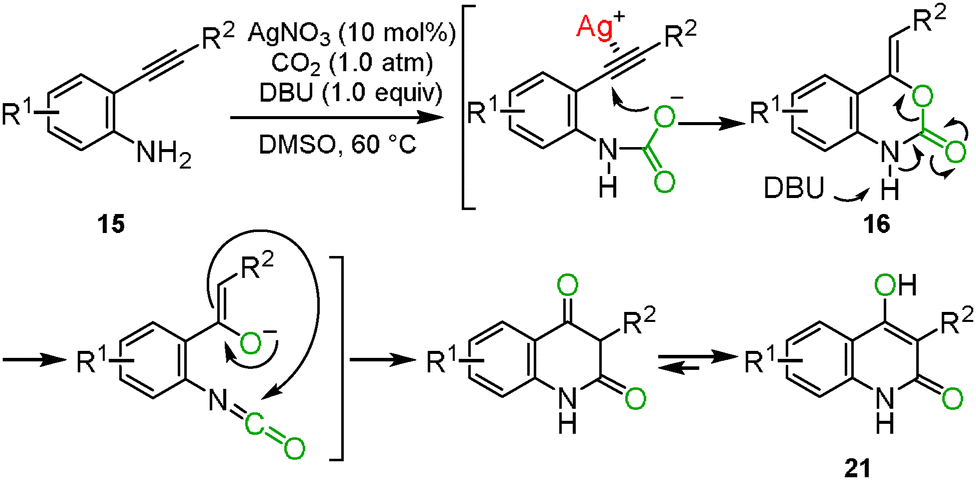
When the optimized reaction conditions as mentioned in Section 2.2 were applied to the primary o-alkynylaniline 15, a side-product was detected. Based on several analytical results, such as X-ray single crystal structure analysis, 1H NMR, 13C NMR and mass spectrometry, the side-product was identified to be 4-hydroxyquinolin-2(1H)-one 21. The possible reaction mechanism is shown in Scheme 11. First, the corresponding benzoxazin-2-one 16 would be generated from the o-alkynylaniline 15 and carbon dioxide catalyzed by the silver catalyst. In the second step, the benzoxazine 16 would immediately be deprotonated by the DBU base to generate the isocyanate and the enolate from the C–O bond cleavage of the carbamate functionality. The enolate would then attack the carbon atom of the isocyanate to afford the 1,3-diketone intermediate, which would produce the corresponding 4-hydroxyquinolin-2(1H)-one 21 after enolization. The results of isotopic labeling experiments with C18O2 and in situ IR measurements also supported this proposed reaction mechanism. The scope of the reaction system is described in Scheme 12.28
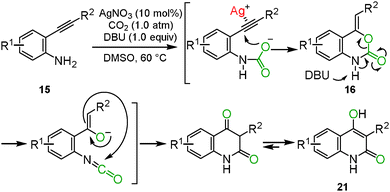 |
| | Scheme 11 Hypothetical reaction mechanism of intramolecular rearrangement. | |
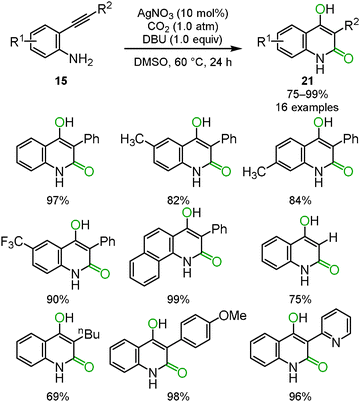 |
| | Scheme 12 Preparation of 4-hydroxyquinolin-2(1H)-one derivatives. | |
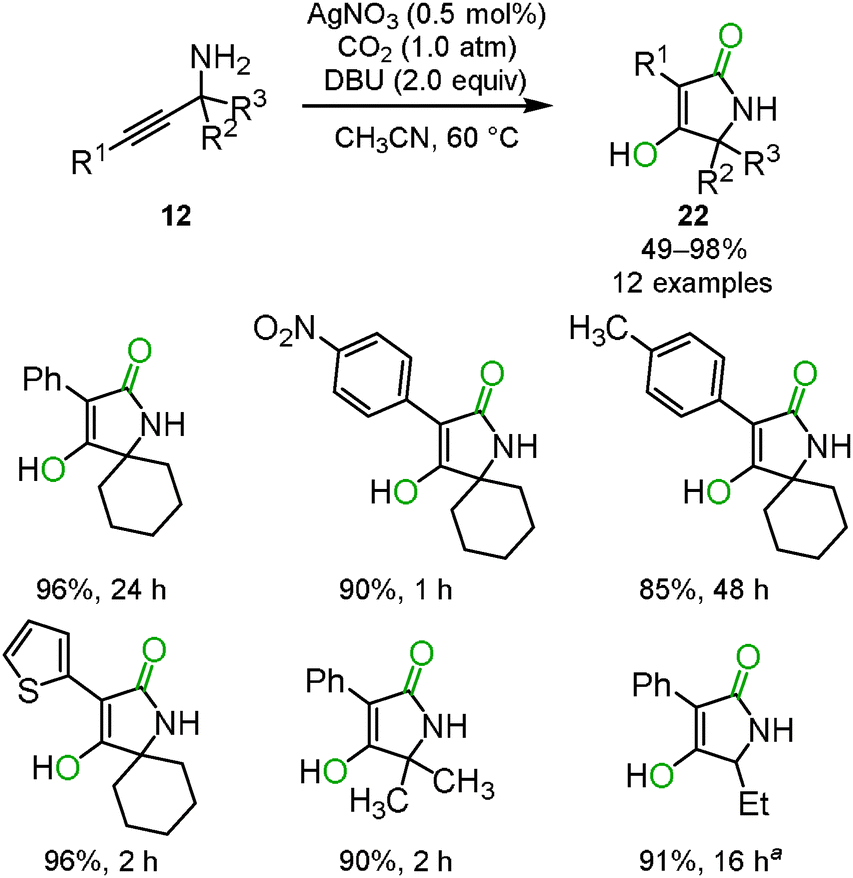
The cyclization and rearrangement system was applied to propargyl amines 12 for the synthesis of tetramic acid derivatives 22 which have been studied as biologically active heterocyclic structures for pharmaceutical and agricultural chemicals (Scheme 13). The silver/DBU system provided various multi-substituted tetramic acid derivatives 22 from easily available primary propargyl amines 12 under mild conditions, though conventional preparative approaches often required highly functionalized starting materials and harsh heating conditions in basic media (Scheme 14).29
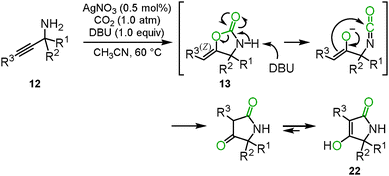 |
| | Scheme 13 New approach for tetramic acids. | |
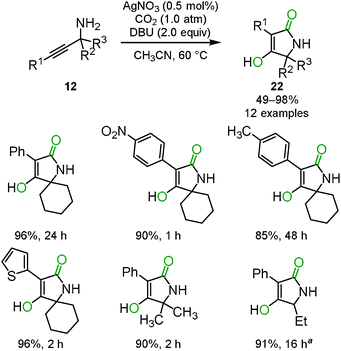 |
| | Scheme 14 Synthesis of tetramic acids by carbon dioxide incorporation and intramolecular rearrangement. | |
3. Sequential carboxylation and cyclization with C–C bond formation
Propargyl alcohols and propargyl amines were converted to the corresponding cyclic carbonates and oxazolidinones in the presence of a transition metal, organic base, phosphine, or carbene catalyst. Though cyclic carbonates and oxazolidinones are important structures in the materials and pharmaceutical sciences, their hydrolysis would readily promote decarboxylation. To incorporate carbon dioxide into organic molecules, the formation of C–C bonds between the substrate and carbon dioxide is important. Cyclization following C–C bond forming carboxylation can afford the corresponding lactones that are not easily decarboxylated. In this chapter, the silver-catalyzed C–C bond forming carboxylation and cyclization reactions are described.
3.1 Enolate
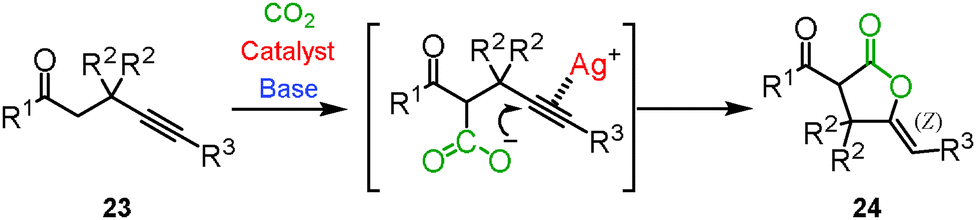
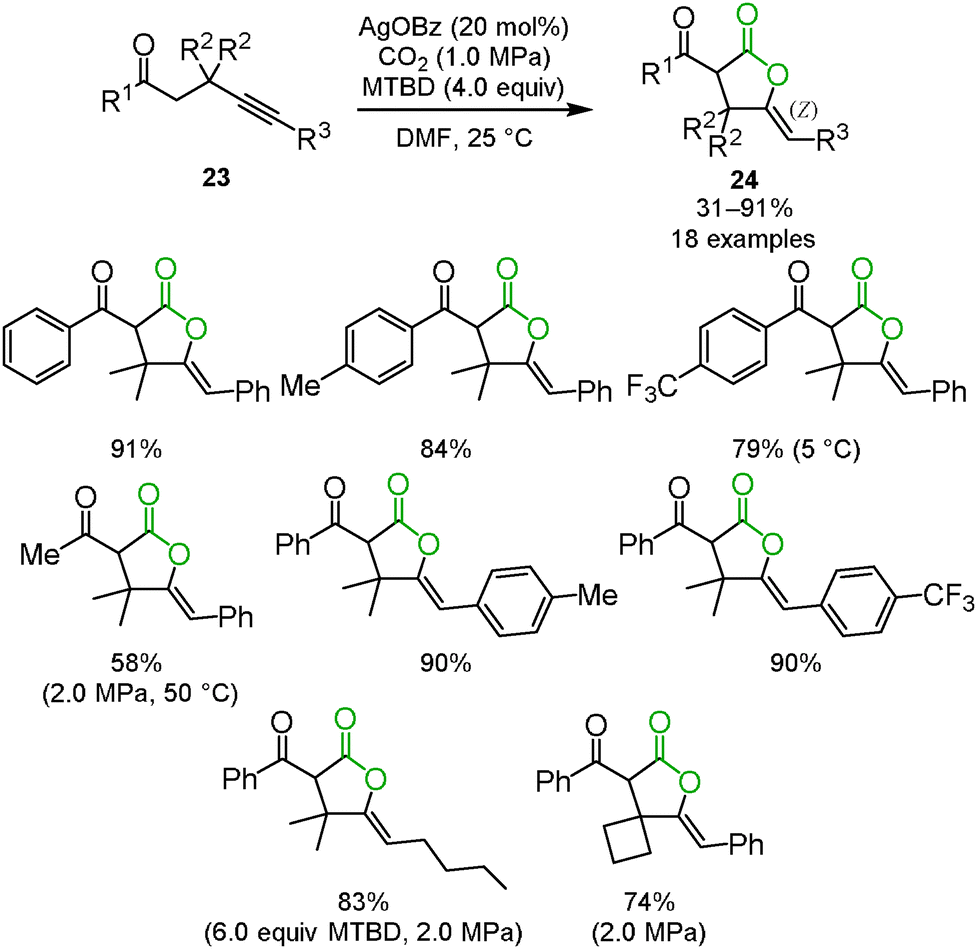
Enolate has been a promising reagent for the C–C bond forming reactions in organic chemistry. The reactions of an enolate with carbon dioxide produce the corresponding β-ketocarboxylic acids. Due to its thermodynamic instability, however, β-ketocarboxylic acid would easily revert to the starting material. Therefore, careful treatment or subsequent reduction of the product was required. A tandem reaction for the conversion of β-ketocarboxylic acid into a stable compound is one of the most reasonable methods for the reaction of an enolate and carbon dioxide. We reported that β-ketocarboxylate was trapped by the silver-activated C–C triple bond to afford the corresponding stable 5-membered lactone 24 in a one-pot synthesis (Scheme 15).30 A good scope of substrates under the optimized reaction conditions was indicated (Scheme 16). Both aromatic and aliphatic ketones were suitable for this reaction.
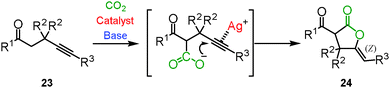 |
| | Scheme 15 Postulated reaction pathway. | |
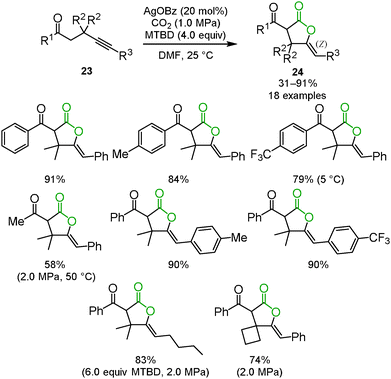 |
| | Scheme 16 C–C bond formation with carbon dioxide for lactone derivatives. | |
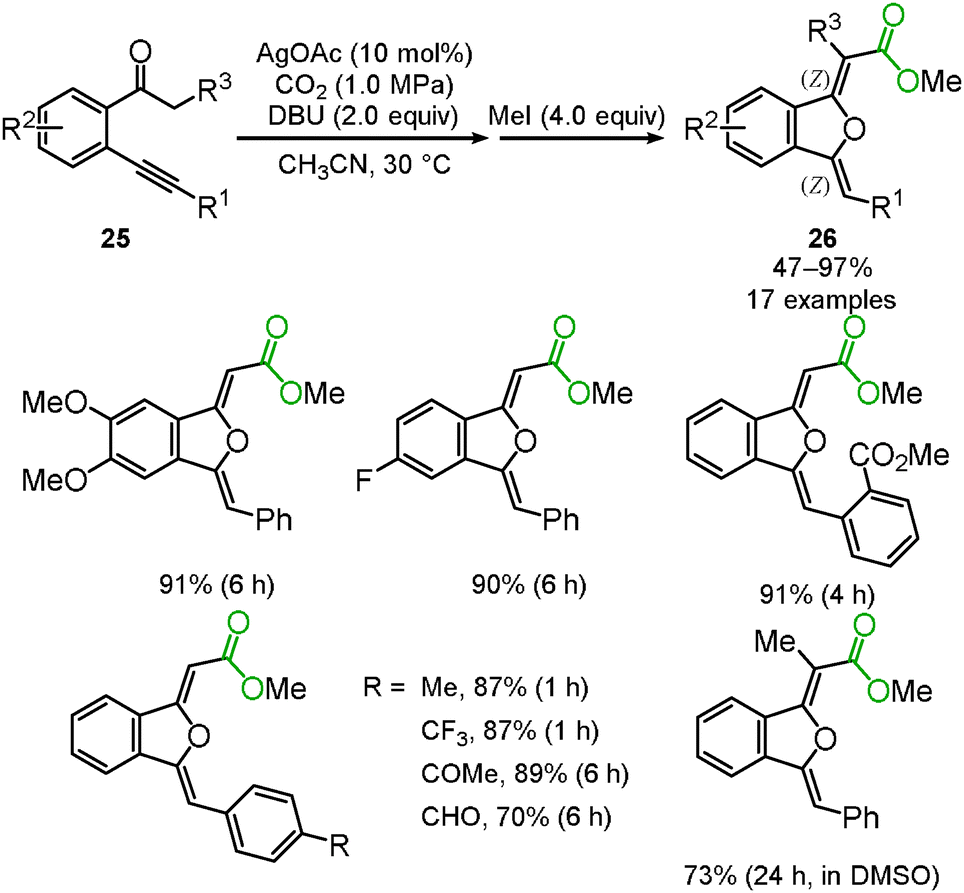
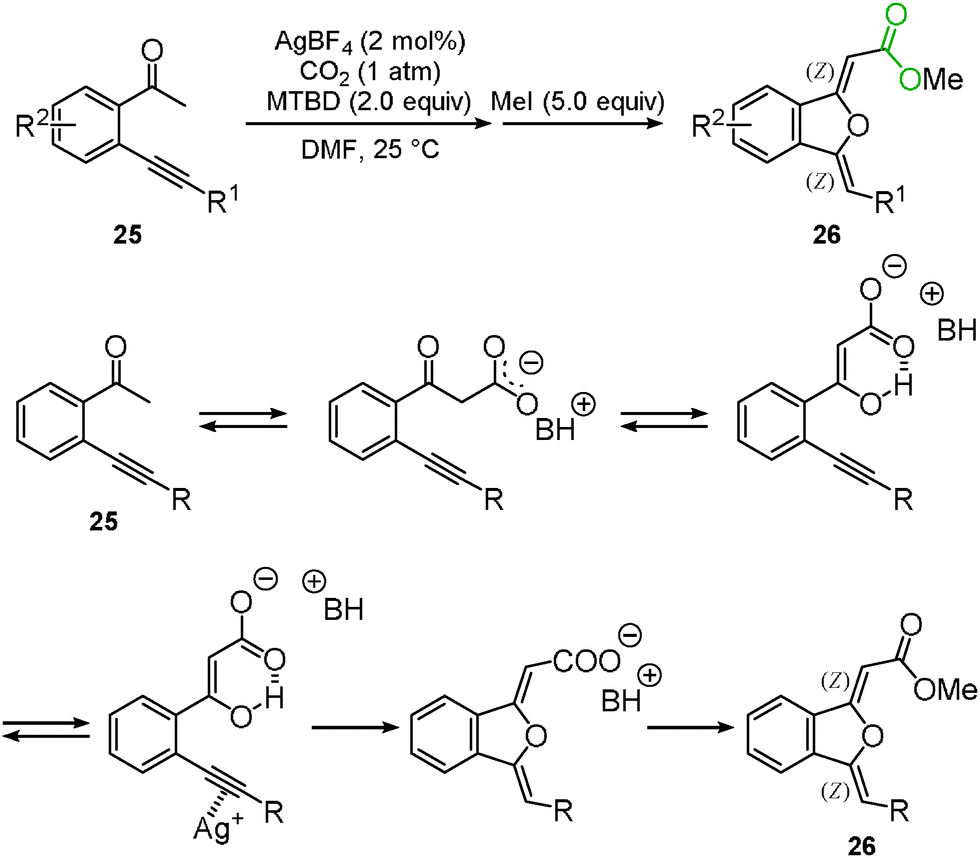
We and Lu's group achieved the transformation of β-ketocarboxylic acid to the dihydroisobenzofuran derivatives 26 at almost the same time (Schemes 17 and 18). The dihydroisobenzofurans bearing a carboxyl group were obtained by the 5-exo-dig cyclization of o-alkynylacetophenone 25 and carbon dioxide. The geometries of the two C–C double bonds were suggested to be Z isomers by NOE. The catalytic system showed the wide-range scope of the substrates; aldehydes and ketones were tolerable (Scheme 17).31 DFT calculations suggested the plausible reaction mechanism (Scheme 18).32 After the keto-enol tautomerism of the β-ketocarboxylic acid, the oxygen atom was trapped on the C–C triple bond activated by the silver catalyst. It was assumed that the geometry of the C–C double bond adjacent to the carbonyl group should be determined on the basis of the 6-membered enol form.
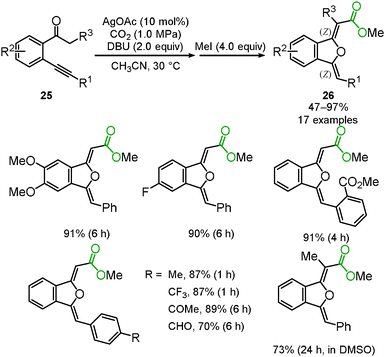 |
| | Scheme 17 Silver-catalyzed carboxylation and cyclization of o-alkynylacetophenones. | |
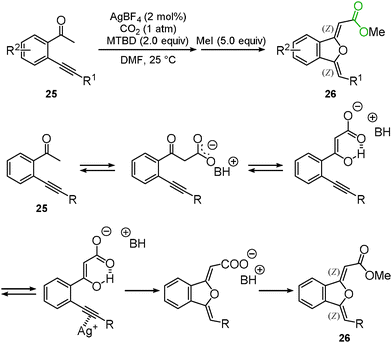 |
| | Scheme 18 Possible reaction mechanism. | |
3.2 Allylsilane
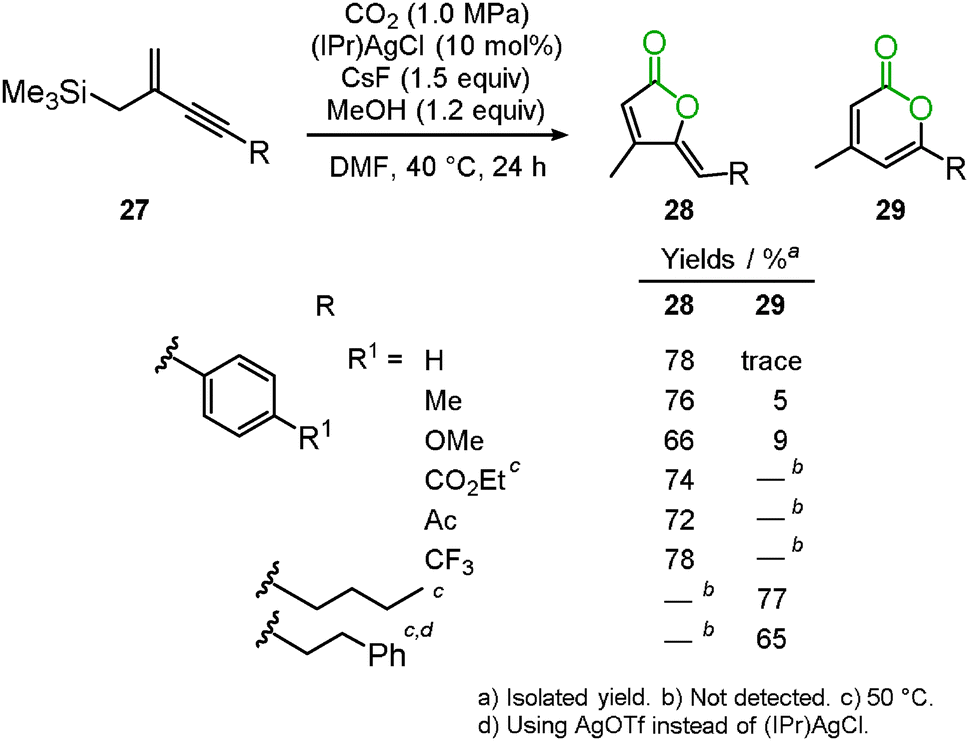
Recently, the silver-catalyzed cascade carboxylation and cyclization of the trimethyl(2-methylenebut-3-yn-1-yl)silane derivatives 27 were developed.33 The allylsilane compound is one of the useful reagents for new C–C bond formation. For example, Hosomi–Sakurai allylation has been used to provide homoallyl alcohols, which are an important framework for the total synthesis of natural products and medicinal compounds. Though allylsilane compounds have the additional potential for carbon dioxide incorporation, few systems involving the Lewis acid mediated carboxylation have been reported. The present reaction was promoted by a silver salt and CsF to afford the corresponding 2-furanones 28 and 2-pyrones 29 in good-to-high yields (Scheme 19). When aromatic ring-substituted alkynes were used, 2-furanone derivatives were selectively obtained via 5-exo-dig cyclization, whereas the reaction of the alkyl-substituted alkynes produced 2-pyrone derivatives with high selectivity.
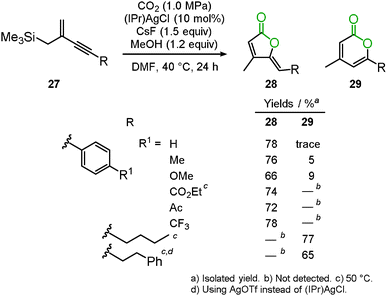 |
| | Scheme 19 Silver-catalyzed reaction for the synthesis of 2-furanone and 2-pyrone. | |
4. Other reactions
4.1 Carboxylation of terminal alkynes
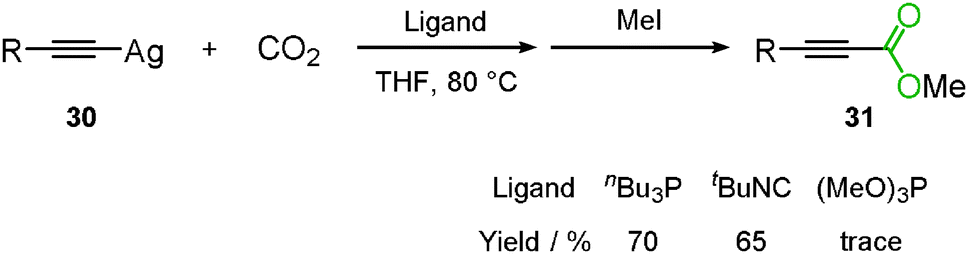
The fundamental study of the carboxylation of terminal alkyne was reported by Saegusa's group. At this stage, it was found that silver acetylide 30 reacted with carbon dioxide using the appropriate ligand, nBu3P, to produce the corresponding propiolic acid methyl ester 31 after methylation (Scheme 20).34
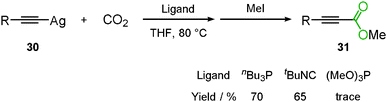 |
| | Scheme 20 Reaction of silver acetylide and carbon dioxide. | |
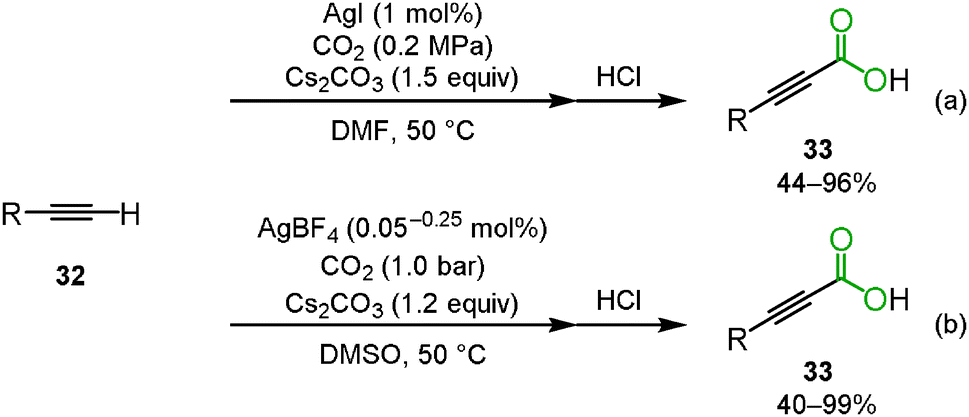
Lu and co-workers reported the silver-catalyzed carboxylation of terminal alkynes 32 using Cs2CO3 under 0.2 MPa of carbon dioxide in DMF to afford the corresponding propiolic acids 33 (Scheme 21(a)).35 In addition, Gooßen and co-workers found that the same reaction proceeded at the lower catalyst loading of AgBF4 (0.05–0.25 mol%) in DMSO (Scheme 21(b)).36,37 Zhang's group developed a highly efficient and reusable N-heterocyclic carbene polymer-supported silver nanoparticle catalyst (poly-NHC–Ag). They found that the carboxylation of terminal alkynes was effectively promoted using 0.3 mol% of poly-NHC–Ag and 1.2 equiv. of Cs2CO3 in DMSO even under 1 atm of carbon dioxide.38
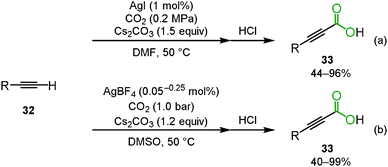 |
| | Scheme 21 Silver-catalyzed carboxylation of terminal alkynes. | |
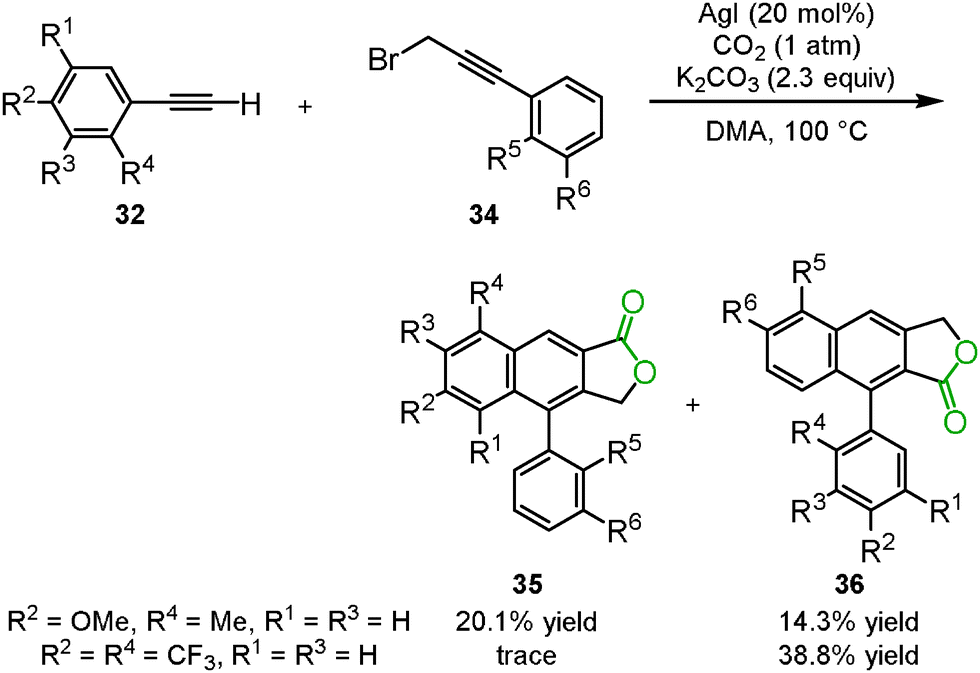
Anastas's group reported that the three-component reaction between phenyl acetylene 32, carbon dioxide and 3-bromo-1-phenyl-propyne 34 successfully proceeded in the presence of silver iodide and potassium carbonate in DMA to produce two isomers of the arylnaphthalene lactones 35 and 36 (Scheme 22).39 During the first stage of the reaction, propiolic acid was produced by the carboxylation of the terminal alkyne. Propiolic acid then reacted with 3-bromo-1-phenyl-propyne to afford the corresponding 1,6-diyne compound. Finally, the [2+2+2] cyclization of the 1,6-diyne occurred to provide the two isomers of the arylnaphthalene lactones. The selectivity depended on the electronic properties of the aromatics of the terminal alkyne. Interestingly, in the case of gold iodide, no arylnaphthalene lactone was detected, but a 1,6-diyne compound was obtained. Copper iodide was ineffective in this reaction, though the 1,4-diyne compound was obtained from phenyl acetylene and 3-bromo-1-phenyl-propyne.
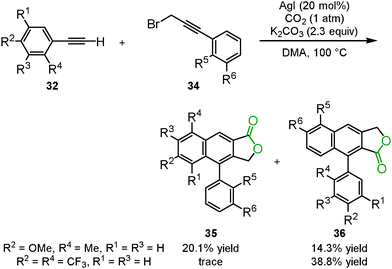 |
| | Scheme 22 Silver-catalyzed one-pot synthesis of arylnaphthalene lactones. | |
4.2 Carboxylation of arylboronic esters
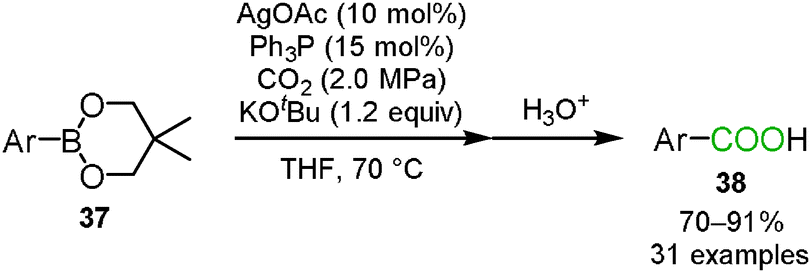
Lu, Zhang and co-workers reported the silver-catalyzed carboxylation of the arylboronic ester 37 with carbon dioxide (Scheme 23).40 They hypothesized that based on the reaction mechanism of the rhodium- or copper-catalyzed carboxylation of organoboronic esters, carbon dioxide could insert into the silver–carbon bond which was generated by the transmetallation of the silver catalyst with the arylboronic ester. The Ph3P ligand played a key role in increasing the yield of the corresponding benzoic acids 38.
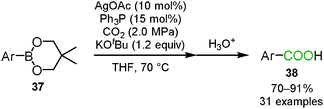 |
| | Scheme 23 Carboxylation of arylboronic esters by silver catalysts. | |
Conclusions
Recent developments in silver-catalyzed carboxylation chemistry using carbon dioxide were described. In some cases, silver catalysts showed an effective reactivity unlike other transition metals, including the coinage metals. The sequential carboxylation and cyclization of propargyl alcohols and amines were promoted by a simple silver salt or a highly efficient silver catalyst. Silver-catalyzed C–C bond forming reactions with carbon dioxide were also described.
Notes and references
- J.-M. Weibel, A. Blanc and P. Pale, Chem. Rev., 2008, 108, 3149–3173 CrossRef CAS PubMed
 .
.
- M. Álvarez-Corral, M. Munoz-Dorado and I. Rodríguez-García, Chem. Rev., 2008, 108, 3174–3198 CrossRef PubMed .
- M. Naodovic and H. Yamamoto, Chem. Rev., 2008, 108, 3132–3148 CrossRef CAS PubMed .
- Y. Yamamoto, Chem. Rev., 2008, 108, 3199–3222 CrossRef CAS PubMed .
- G. Fang and X. Bi, Chem. Soc. Rev., 2015, 44, 8124–8173 RSC .
-
Silver in Organic Chemistry, ed. M. Harmata, Wiley-VHC, 2010 Search PubMed .
-
Carbon Dioxide as Chemical Feedstock, ed. M. Aresta, Wiley-VCH, 2010 Search PubMed .
- L. Zhang and Z. Hou, Chem. Sci., 2013, 4, 3395–3403 RSC .
- Y. Tsuji and T. Fujihara, Chem. Commun., 2012, 48, 9956–9964 RSC .
- W. Yamada, Y. Sugawara, H.-M. Cheng, T. Ikeno and T. Yamada, Eur. J. Org. Chem., 2007, 2604–2607 CrossRef CAS .
- S. Kikuchi, S. Yoshida, W. Yamada, H.-M. Cheng, K. Fukui, K. Sekine, I. Iwakura, T. Ikeno and T. Yamada, Bull. Chem. Soc. Jpn., 2011, 84, 698–717 CrossRef CAS .
- A. S. K. Hashmi, J. P. Weyrauch, W. Frey and J. W. Bats, Org. Lett., 2004, 6, 4391–4394 CrossRef CAS PubMed .
- A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896–7936 CrossRef PubMed .
- R. Ugajin, S. Kikuchi and T. Yamada, Synlett, 2014, 1178–1180 Search PubMed .
- Y. Sugawara, S. Yoshida, S. Kikuchi and T. Yamada, J. Am. Chem. Soc., 2007, 129, 12902–12903 CrossRef CAS PubMed .
- S. Yoshida, K. Fukui, S. Kikuchi and T. Yamada, J. Am. Chem. Soc., 2010, 132, 4072–4073 CrossRef CAS PubMed .
- X. Tang, C. Qi, H. He, H. Jiang, Y. Yuan and G. Ren, Adv. Synth. Catal., 2013, 355, 2019–2028 CrossRef CAS .
- M. Cui, Q. Qian, Z. He, J. Ma, X. Kang, J. Hu, Z. Liu and B. Han, Chem. – Eur. J., 2015, 21, 15924–15928 CrossRef CAS PubMed .
- Q.-W. Song, B. Yu, X.-D. Li, R. Ma, Z.-F. Diao, R.-G. Li, W. Li and L.-N. He, Green Chem., 2014, 16, 1633–1638 RSC .
- Q.-W. Song, W.-Q. Chen, R. Ma, A. Yu, Q. Y. Li, Y. Chang and L. N. He, ChemSusChem, 2015, 8, 821–827 CrossRef CAS PubMed .
- C. Qi, L. Huang and H. Jiang, Synthesis, 2010, 1433–1440 CAS .
- S. Yoshida, K. Fukui, S. Kikuchi and T. Yamada, Chem. Lett., 2009, 38, 786–787 CrossRef CAS .
- M. Yoshida, T. Mizuguchi and K. Shishido, Chem. – Eur. J., 2012, 18, 15578–15581 CrossRef CAS PubMed .
- K. Sekine, R. Kobayashi and T. Yamada, Chem. Lett., 2015, 42, 1407–1409 CrossRef .
- T. Ishida, S. Kikuchi, T. Tsubo and T. Yamada, Org. Lett., 2013, 15, 848–851 CrossRef CAS PubMed .
- C. Ruan, L. Yang, Y. Yuan, Y. Ju and H. Wang, Comput. Theor. Chem., 2015, 1058, 34–40 CrossRef CAS .
- K. Yamashita, S. Hase, Y. Kayaki and T. Ikariya, Org. Lett., 2015, 17, 2334–2337 CrossRef CAS PubMed .
- T. Ishida, S. Kikuchi and T. Yamada, Org. Lett., 2013, 15, 3710–3713 CrossRef CAS PubMed .
- T. Ishida, R. Kobayashi and T. Yamada, Org. Lett., 2014, 16, 2430–2433 CrossRef CAS PubMed .
- S. Kikuchi, K. Sekine, T. Ishida and T. Yamada, Angew. Chem., Int. Ed., 2012, 51, 6989–6992 CrossRef CAS PubMed .
- K. Sekine, A. Takayanagi, S. Kikuchi and T. Yamada, Chem. Commun., 2013, 49, 11320–11322 RSC .
- W.-Z. Zhang, L.-L. Shi, C. Liu, X.-T. Yang, Y.-B. Wang, Y. Luo and X.-B. Lu, Org. Chem. Front., 2014, 1, 275–283 RSC .
- K. Sekine, Y. Sadamitsu and T. Yamada, Org. Lett., 2015, 17, 5706–5709 CrossRef CAS PubMed .
- T. Tsuda, K. Ueda and T. Saegusa, J. Chem. Soc., Chem. Commun., 1974, 380–381 RSC .
- X. Zhang, W.-Z. Zhang, X. Ren, L.-L. Zhang and X.-B. Lu, Org. Lett., 2011, 13, 2402–2405 CrossRef CAS PubMed .
- F. Manjolinho, M. Arndt, K. Gooßen and L. J. Gooßen, ACS Catal., 2012, 2, 2014–2021 CrossRef CAS .
- M. Arndt, E. Risto, T. Krause and L. J. Gooßen, ChemCatChem, 2012, 4, 484–487 CrossRef CAS .
- D. Yu, M. X. Tan and Y. Zhang, Adv. Synth. Catal., 2012, 354, 969–974 CrossRef CAS .
- N. Eghbali, J. Eddy and P. T. Anastas, J. Org. Chem., 2008, 73, 6932–6935 CrossRef CAS PubMed .
- X. Zhang, W.-Z. Zhang, L.-L. Shi, C.-X. Guo, L.-L. Zhang and X.-B. Lu, Chem. Commun., 2012, 48, 6292–6294 RSC .
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence