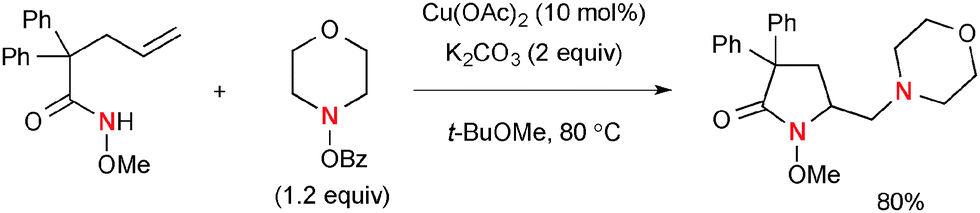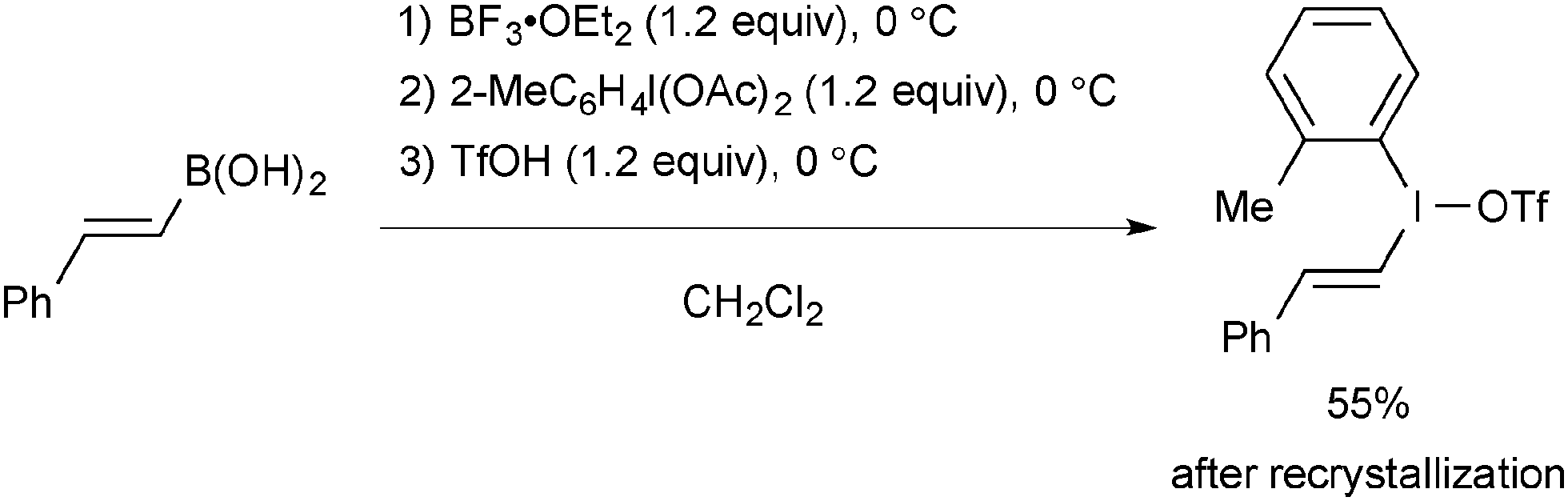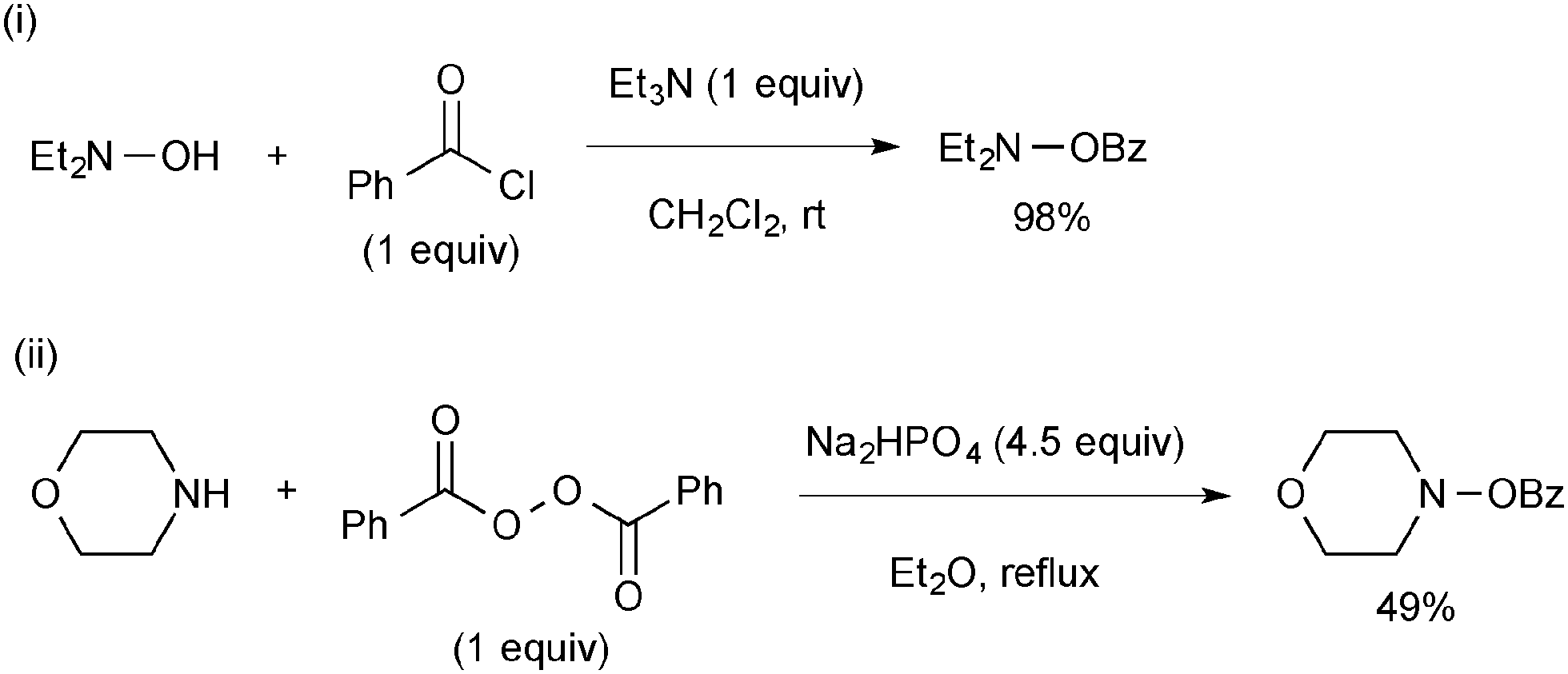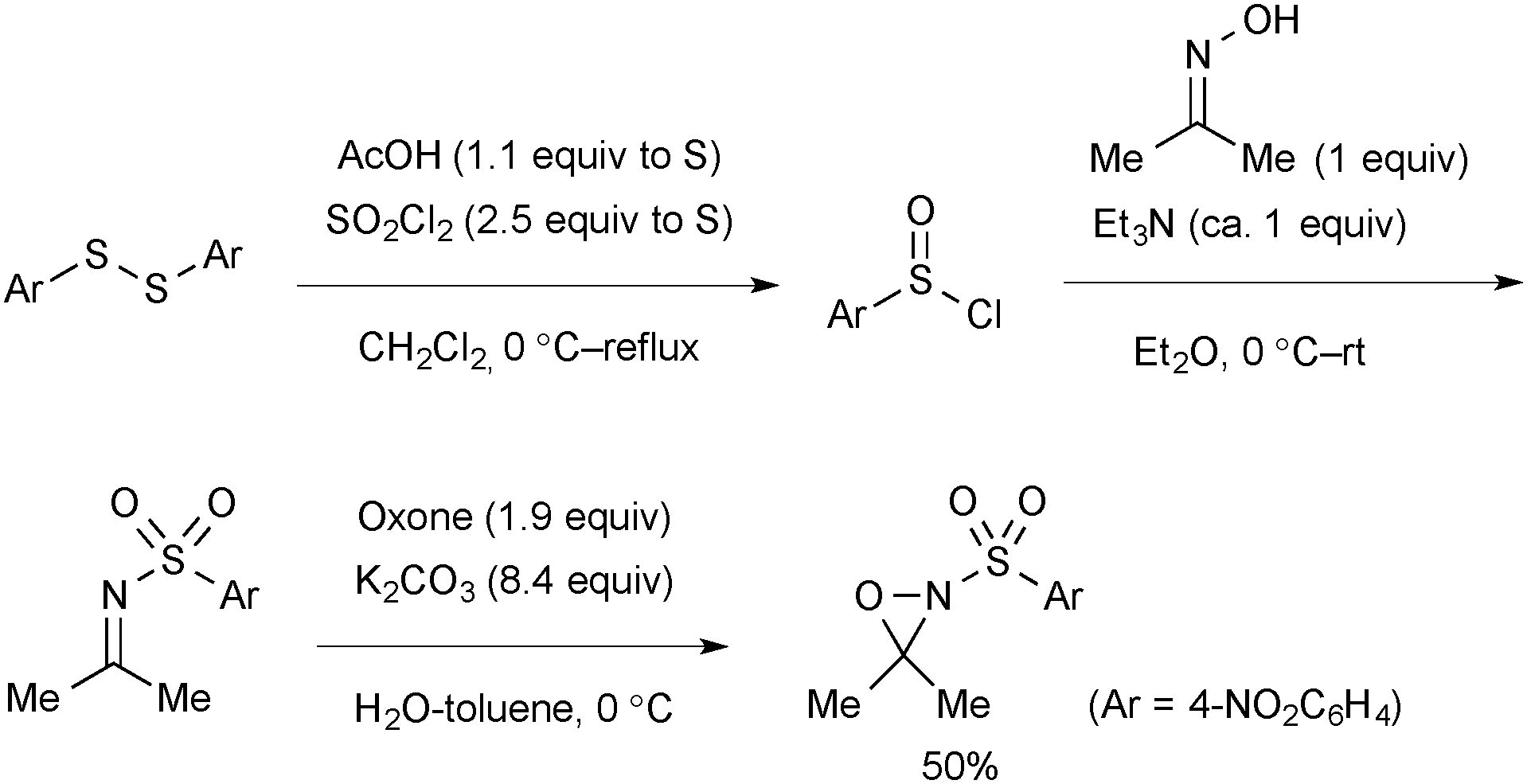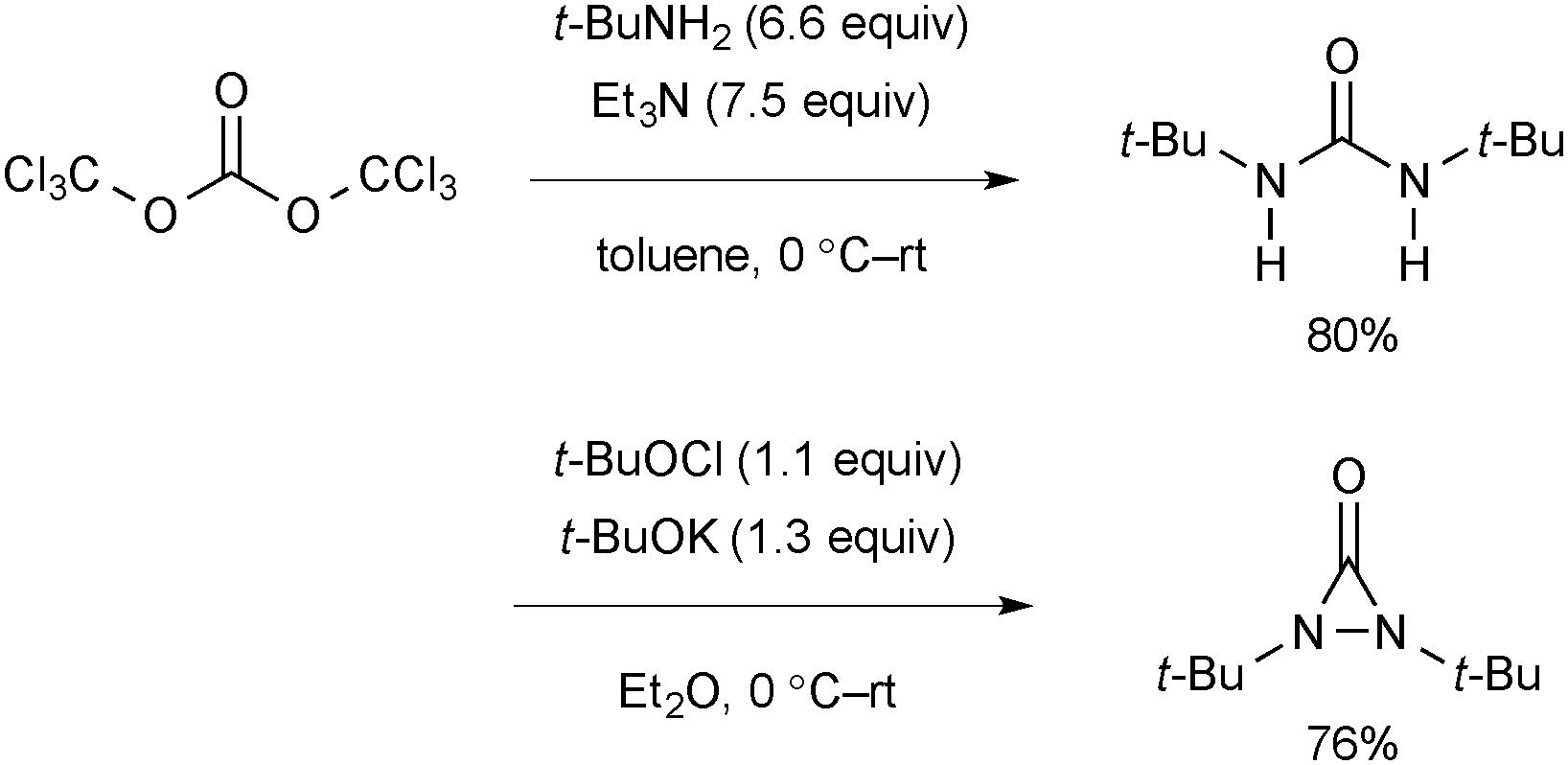DOI:
10.1039/C5CS00882D
(Review Article)
Chem. Soc. Rev., 2016,
45, 4504-4523
Copper-catalyzed oxidative carbon–heteroatom bond formation: a recent update
Received
29th November 2015
First published on 11th February 2016
Abstract
This review updates recent advances in Cu-catalyzed (anaerobic) oxidative carbon–heteroatom bond formation on sp3- and sp2-C–H bonds as well as alkenes, classified according to the types of stoichiometric oxidants.

Xu Zhu
| Xu Zhu received his MSc in chemistry from Soochow University (China) under Prof. Shun-Jun Ji in 2011 and started his PhD studies in the same year. In 2015, he obtained his PhD from Nanyang Technological University under the supervision of Prof. Shunsuke Chiba where he studied methodology development for the synthesis of nitrogen heterocycles. He is currently working as a postdoctoral fellow in the research group of Prof. Corinna S. Schindler at the University of Michigan. |

Shunsuke Chiba
| Shunsuke Chiba earned his PhD in 2006 from the University of Tokyo (Prof. Koichi Narasaka). He was appointed as a research associate at the University of Tokyo in 2005. He began his independent career at Nanyang Technological University (Singapore) as an Assistant Professor in 2007. In 2012, he was promoted to Associate Professor (with tenure) in the same university. His research focus is methodology development in the area of synthetic organic chemistry. |
1. Introduction
Oxidative molecular transformation that incorporates heteroatom units into carbon-based organic scaffolds is one of the most fundamental and important synthetic transformations, enhancing the molecular complexity. Therefore, development of new oxidative synthetic methodologies that convert readily available substrates in lower oxidation states into highly functionalized (oxidized) molecules in a chemo- and stereo-selective manner is a long-standing goal in chemical synthesis. In this context, direct installation of carbon–heteroatom bonds on ubiquitous C–H bonds (both sp3- and sp2-hybridized) is of great significance to streamline the multi-step molecular transformations needed for the synthesis of target functional molecules. However, it is challenging to functionalize such C–H bonds selectively unless otherwise adjacent activating groups are installed because of the inherent inert property and the ubiquitous nature of the C–H bonds. On the other hand, oxidative difunctionalization of alkenes provided another efficient method to address highly oxidized molecular complexity through installing two distinct functional groups in a one-pot fashion. Thus, development of chemo-, regio-, and stereoselective difunctionalization of alkenes is the major concern to be addressed in chemical synthesis.
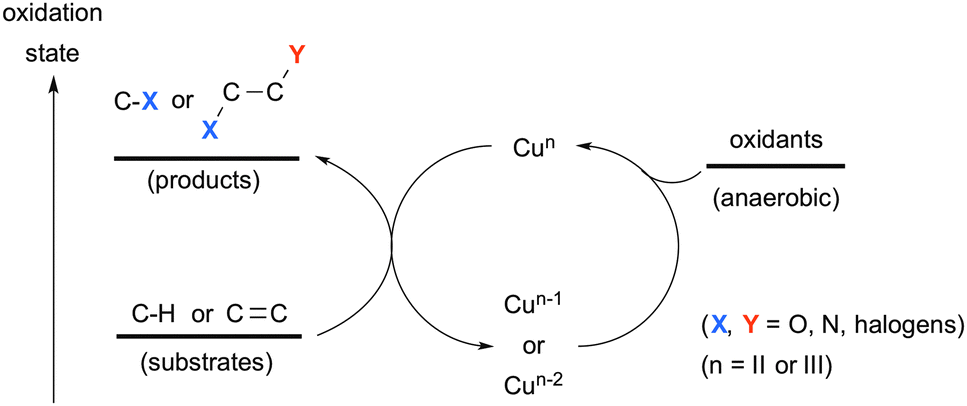
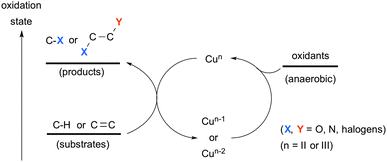
Transition-metals are capable of realizing various state-of-the-art processes for C–H oxidation1 and oxidative difunctionalization of alkenes.2 For the catalysis of choice, ubiquitous first row transition metals have recently attracted much attention not only as alternatives to precious late transition metals, but also for exploration of their unprecedented catalytic processes.3 Among such first row transition metals, copper complexes exhibit unique and versatile reactivity and good functional group tolerance.4 A broad range of oxidation states of copper complexes (mainly from Cu0 to CuIII applied in chemical synthesis)5,6 enables the promotion of redox reactions in either a single-electron or a two-electron-transfer fashion (or both in the sequential processes), depending on the reaction conditions and types of substrates used. A variety of stoichiometric terminal oxidants have been devised and applied for realizing the catalytic turnover in Cu-mediated oxidative molecular transformation and/or serving as the sources of heteroatoms introduced into the products (Scheme 1).
 |
| | Scheme 1 Cu-Catalyzed oxidative carbon–heteroatom bond formation on C–H bonds and alkenes. | |
This review focuses on recent advances in copper-catalyzed oxidative carbon–heteroatom bond forming reactions on C–H bonds as well as alkenes. These reactions are classified according to the different types of stoichiometric terminal oxidants employed in the processes. Among the available oxidants in copper-catalyzed oxidative molecular transformation, molecular oxygen (O2) has been extensively employed as the terminal oxidant for catalytic turnover, enabling a variety of oxidative reactions. As these achievements are reviewed elsewhere in detail,7 this review will exclude copper-catalyzed aerobic reactions.
2. With I(III) reagents
2.1. PhI(OAc)2
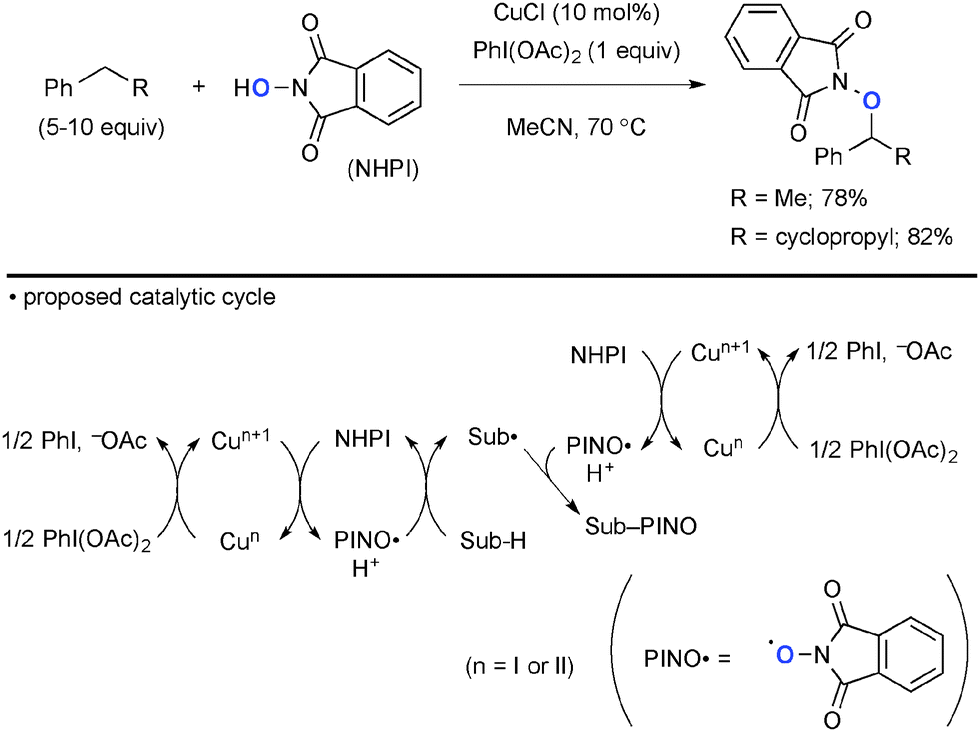
The combination of Cu complexes and PhI(OAc)2 could generate higher oxidation state Cun species (n = II or III), which mediate unprecedented single-electron-oxidation processes. Chang reported benzylic/allylic sp3-C–H oxygenation with N-hydroxyphthalimide (NHPI) under CuCl-catalyzed-PhI(OAc)2-mediated reaction conditions (Scheme 2).8 The radical mechanism is proposed, where the oxidatively formed phthalimide N-oxyl (PINO) radical undergoes H-radical abstraction from the substrates (Sub-H) to generate the corresponding C-radicals (Sub˙). Their subsequent recombination with the PINO radical affords the products. The role of PhI(OAc)2 is to maintain the catalytic cycle by re-oxidizing lower valent Cu species. Interestingly, a radical-clock substrate, cyclopropylmethylbenzene was coupled with the PINO radical keeping the cyclopropyl moiety intact, indicating kinetically faster radical recombination or alternative organometallic mechanism involved.
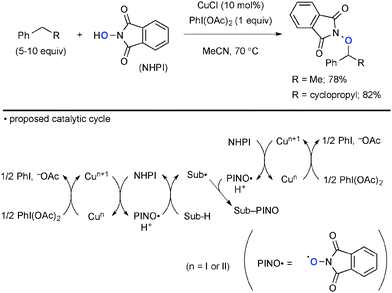 |
| | Scheme 2 Cu-Catalyzed-PhI(OAc)2-mediated sp3-C–H oxygenation with NHPI. | |
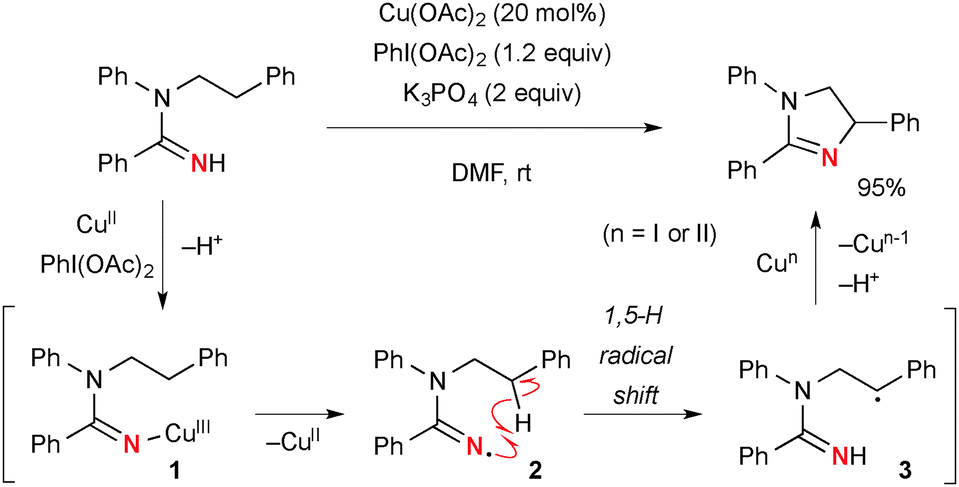
Chiba reported intramolecular aliphatic C–H amination of N-alkylamidines under Cu(OAc)2-catalyzed-PhI(OAc)2-mediated reaction conditions (Scheme 3).9 The reaction is initiated by generation of amidinyl radical 2 probably through formation of Cu(III)-amidine intermediate 1 followed by homolysis of the N–Cu bond. A subsequent 1,5-H radical shift10 of the amidinyl radical affords the corresponding C-radical 3, further single-electron-oxidation of which with Cu(II) or Cu(III) species generates a carbocation and subsequent C–N bond formation to furnish dihydroimidazole. The radical recombination mechanism is not ruled out for C–N bond formation.
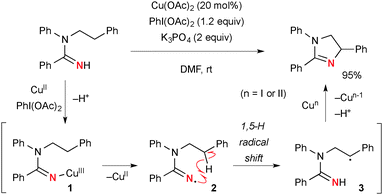 |
| | Scheme 3 Aliphatic C–H amination with amidines under Cu-catalyzed-PhI(OAc)2-mediated reaction conditions. | |
The Cu–PhI(OAc)2 system is capable of oxidizing the aromatic sp2-C–H bond with the assistance of appropriate ortho-directing groups. For example, ortho-C–H amination of aniline derivatives was developed using the picolinamide directing group under Cu-catalyzed-PhI(OAc)2-mediated reaction conditions (Scheme 4).11 The single-electron-oxidation of the benzene ring in chelate complex 4 followed by morpholine transfer to cation radical 5 is proposed for the C–H amination.
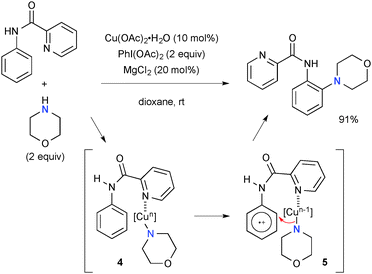 |
| | Scheme 4 Cu-Catalyzed-PhI(OAc)2-mediated directed sp2-C–H amination. | |
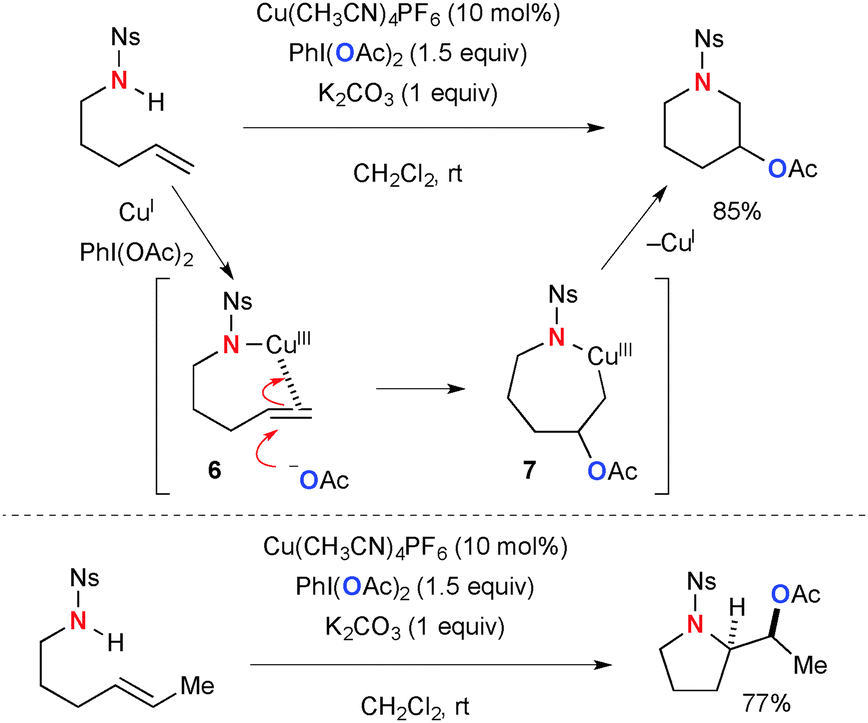
Various modes of oxidative functionalization of alkenes have been realized using Cu-catalyzed-PhI(OAc)2-mediated reaction systems, in which the acetate moiety on PhI(OAc)2 is incorporated during the process. The mechanisms for alkene functionalization vary with the substrates used. Blakey disclosed intramolecular Cu-catalyzed-PhI(OAc)2-mediated aminoacetoxylation of alkenylsulfonamides for synthesis of nitrogen heterocycles (Scheme 5).12 The mode of cyclization (either endo or exo) depends on the alkene substituents. The process is proposed to be initiated by electrophilic activation of alkenes by amide–Cu(III) species 6. Subsequently, acetoxy-cupration of alkenes takes place to afford metallacycle intermediate 7, in which more substituted carbon is preferentially acetoxylated. Reductive elimination of the C–N bond finally furnishes the heterocyclic products.
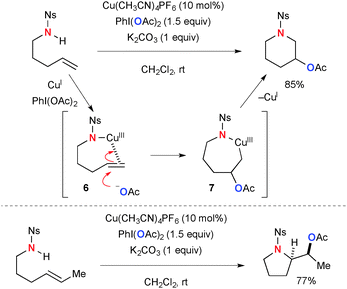 |
| | Scheme 5 Cu-Catalyzed-PhI(OAc)2-mediated aminoacetoxylation of alkenes. | |
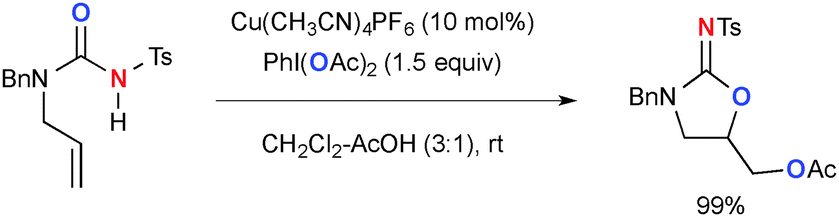
Interestingly, the reactions of alkenylureas under similar reaction conditions give oxyacetoxylation products (Scheme 6).13
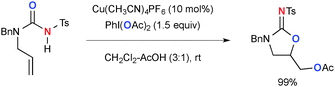 |
| | Scheme 6 Cu-Catalyzed-PhI(OAc)2-mediated oxyacetoxylation of alkenylureas. | |
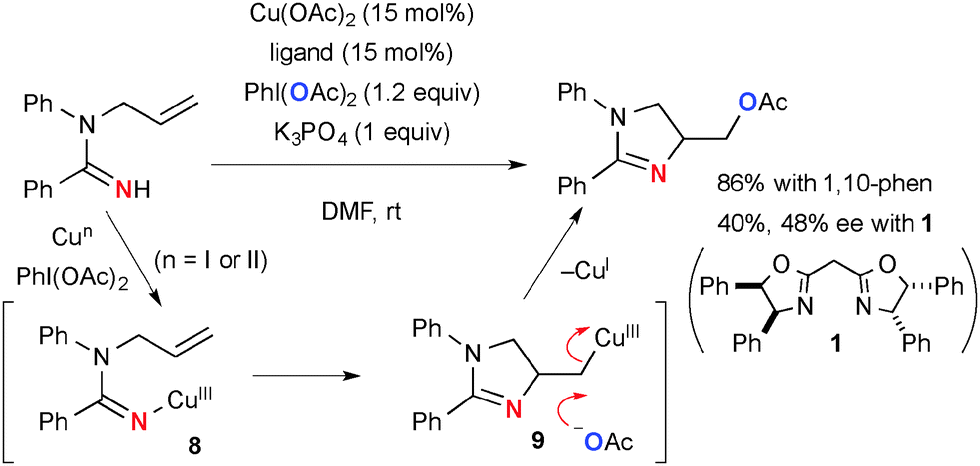
On the other hand, Cu(OAc)2-catalyzed-PhI(OAc)2-mediated reactions of N-allylamidines afford acetoxymethyl dihydroimidazoles via aminoacetoxylation of alkenes (Scheme 7).14 When 2,2′-methylene bis[(4R,5S)-4,5-diphenyl-2-oxazoline] 1 is employed instead of 1,10-phenanthroline, chirality induction of 48% ee is observed in aminoacetoxylation. This observation implicates that the process might involve aminocupration of alkenes by the putative amidinyl copper(III) intermediate 8via an organometallic pathway to form an organocopper(III) intermediate that is unlike the aliphatic CH amination of N-alkylamidines involving free radical intermediates (Scheme 3). Finally, nucleophilic displacement of organocopper(III) moiety 9 with an acetate ion gives the final product.
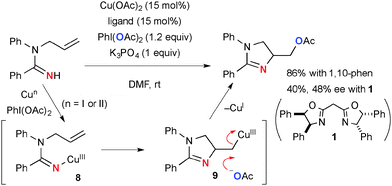 |
| | Scheme 7 Cu-Catalyzed-PhI(OAc)2-mediated aminoacetoxylation of alkenylamidines. | |
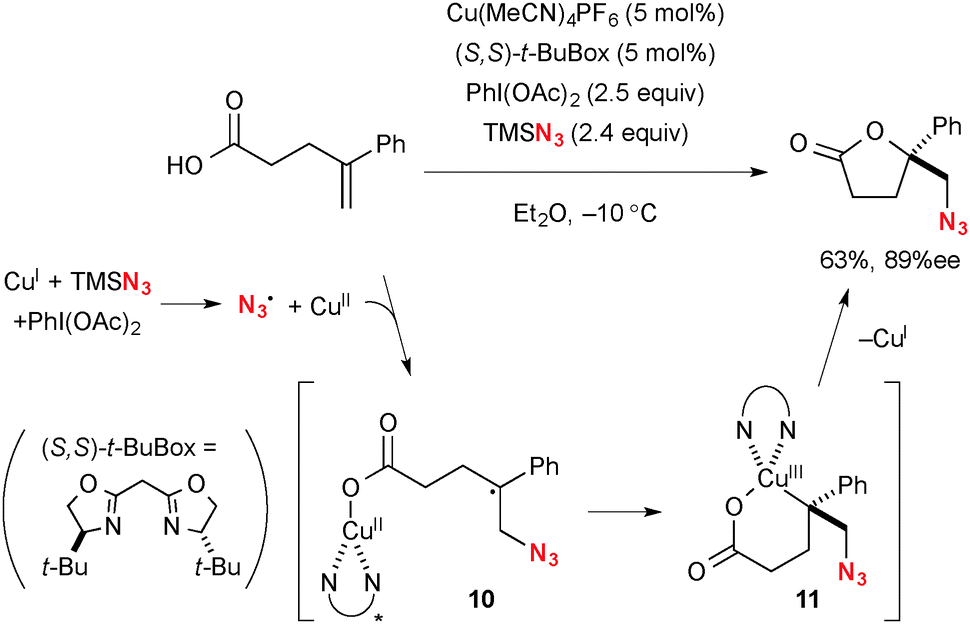
Recently, Buchwald reported Cu-catalyzed enantioselective synthesis of functionalized lactones from alkenylcarboxylic acids through oxyfunctionalization of alkenes as the key step (Scheme 8).15 For example, oxyazidation is enabled using a catalytic amount of Cu(MeCN)4PF6 with TMSN3 and PhI(OAc)2 (Scheme 8). The azido radical and higher valent Cu(II) species are initially formed by the redox reaction between TMSN3, Cu(I) complex, and PhI(OAc)2. The resulting azido radical then adds onto alkenes to generate tertiary radical 10 that recombines with the intramolecular CuII–carboxylate moiety in a stereoselective fashion with the chiral Box ligand. Finally, C–O reductive elimination from metallacycle 11 gives the lactone product with regeneration of the Cu(I) catalyst.
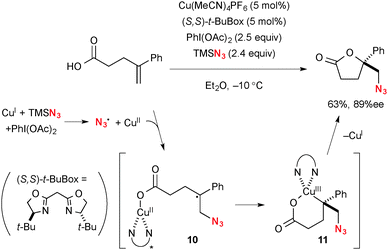 |
| | Scheme 8 Cu-Catalyzed-PhI(OAc)2-mediated asymmetric oxyazidation of alkenes. | |
2.2. Diaryliodonium salts [Ar2IX]
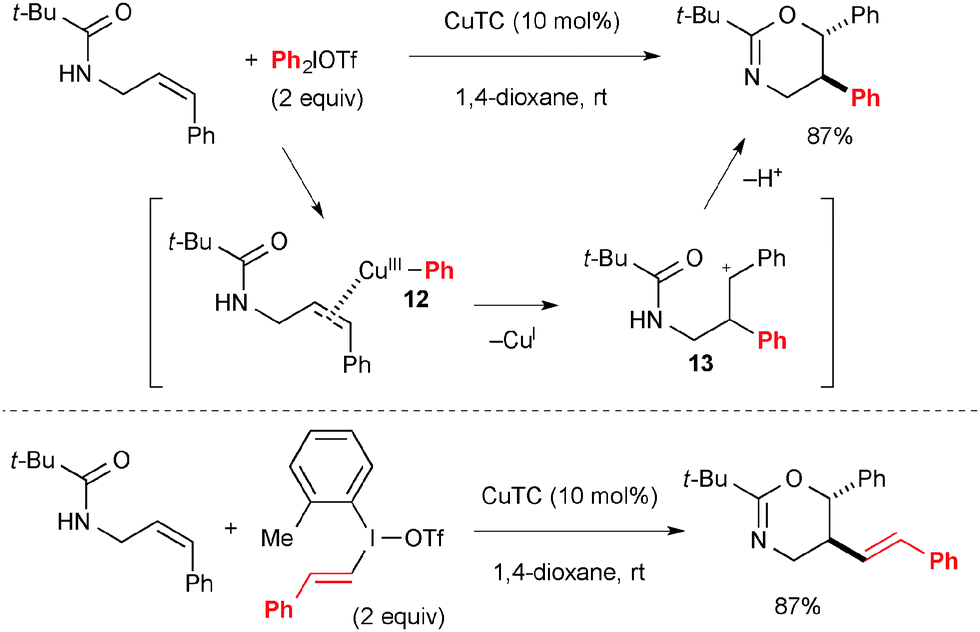
Treatment of Cu(I) complexes with diaryliodonium salts results in formation of aryl-Cu(III) species like 12 in Scheme 9 that could be used for electrophilic activation of alkenes to induce their carbo-functionalization. For example, Gaunt reported that the reaction of N-allylamides could afford oxyarylation products in a diastereoselective fashion through the transient carbocation 13 (Scheme 9).16 Similarly, oxyvinylation was enabled by using vinyl(aryl)iodonium salts. The enantioselective variant was also devised using chiral Cu–bisoxazoline complexes as the catalyst.17
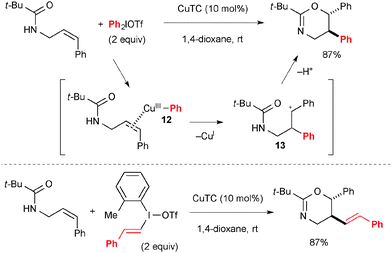 |
| | Scheme 9 Cu-Catalyzed oxy-arylation and -vinylation of allylamides with iodonium salts. | |
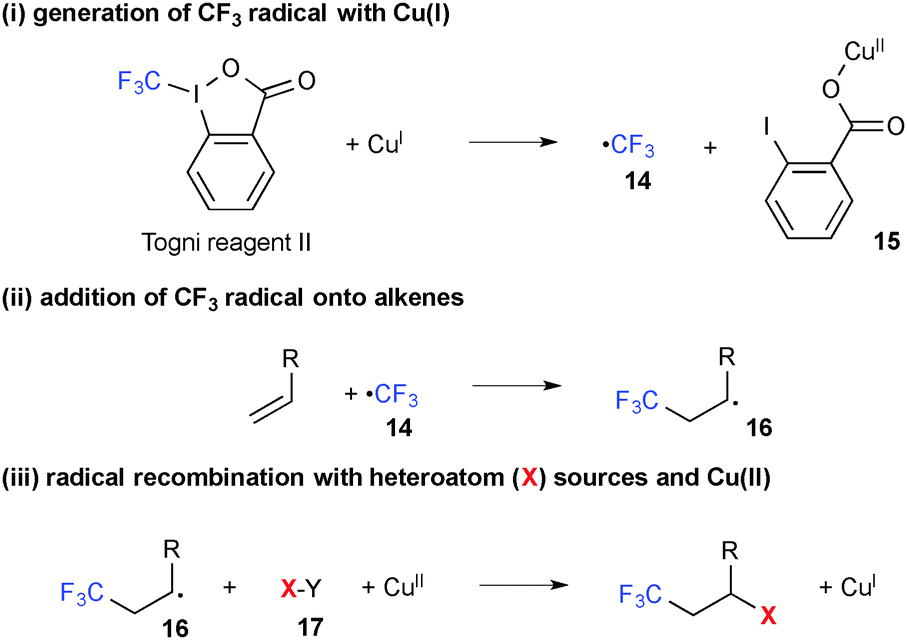
2.3. The Togni reagents
It has been shown that single-electron-reduction of 1-trifluoromethyl-1,2-benziodoxol-3(1H)-one (known as Togni reagent II) by Cu(I) complexes generates CF3 radical 14 along with Cu(II) 2-iodobenzoate 15 (Scheme 10). This reductive process could be combined with the subsequent oxidative alkene difunctionalization through addition of the CF3 radical onto alkenes followed by recombination of the resulting C-radical 16 with external heteroatom sources 17, furnishing the final product and Cu(I) species. The overall process is thus able to have a catalytic turnover.
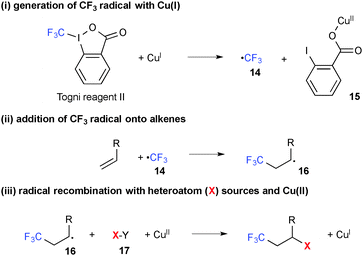 |
| | Scheme 10 Cu-Catalyzed trifluoromethylation of alkenes with the Togni reagent. | |
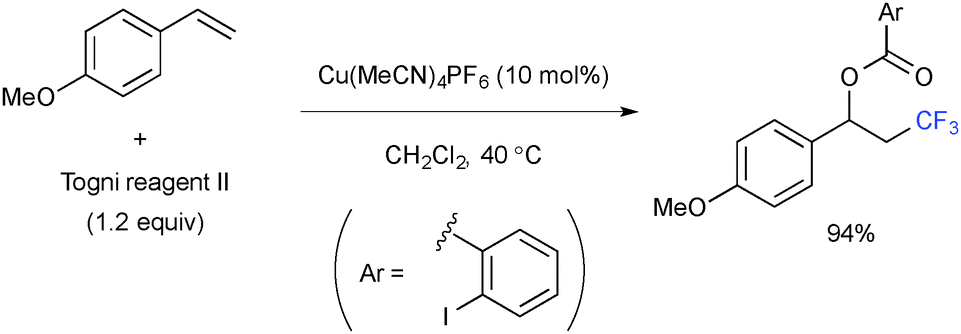
For example, intermolecular oxytrifluoromethylation was realized by radical recombination with the Cu(II) 2-iodobenzoate derived from the Togni reagent (Scheme 11).18
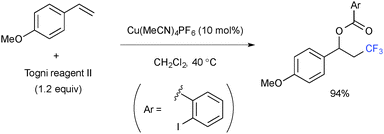 |
| | Scheme 11 Cu-Catalyzed intermolecular oxytrifluoromethylation. | |
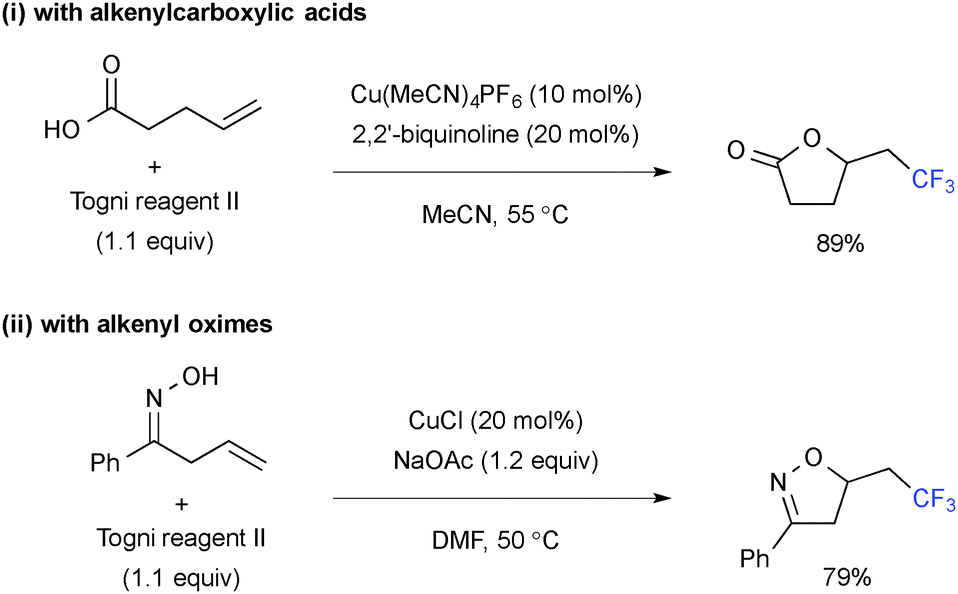
Intramolecular C–O bond formation upon trifluoromethylation was enabled by using alkenylcarboxylic acids (Scheme 12-i)19 and alkenyloximes (Scheme 12-ii), delivering the corresponding trifluoromethylated lactones and isoxazolines, respectively.
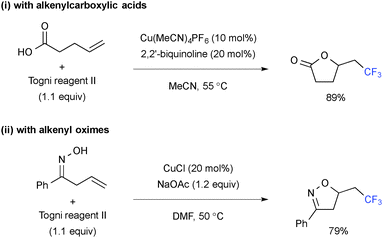 |
| | Scheme 12 Cu-Catalyzed intramolecular oxytrifluoromethylation. | |
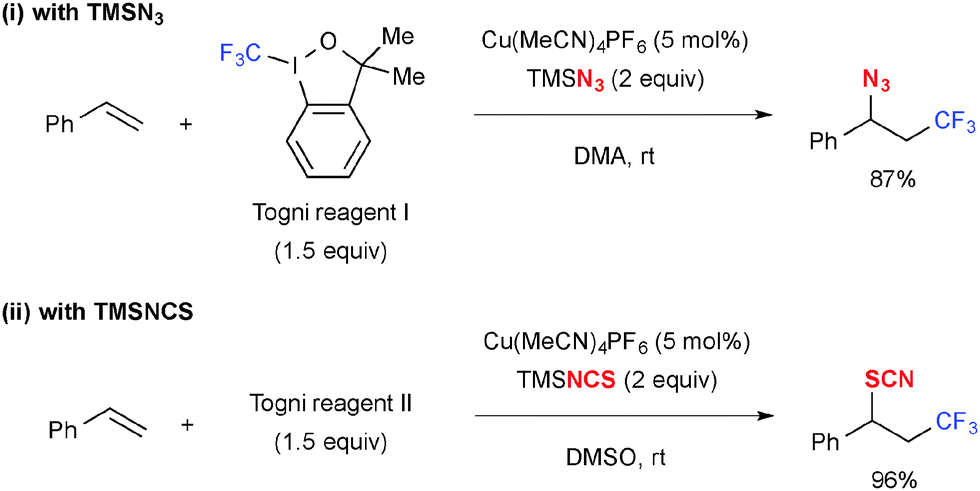
Similarly, trifluoromethylazidation (Scheme 13-i)20 and trifluoromethylthiocyanation (Scheme 13-ii)21 were reported using TMSN3 and TMSNCS, respectively, as the external heteroatom sources. In the case of trifluoromethylazidation, use of 3,3-dimethyl-1,2-benziodoxole (known as Togni reagent I) provided better yields of the desired azidation products as the reaction with 1,2-benziodoxol-3-one generated 2-iodobenzoyloxylaiton product as the side product.
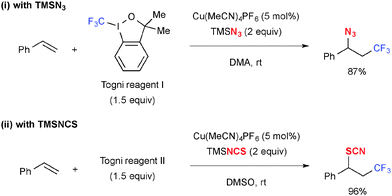 |
| | Scheme 13 Cu-Catalyzed trifluoromethylazidation and trifluoromethylthiocyanation of alkenes. | |
3. With peroxides
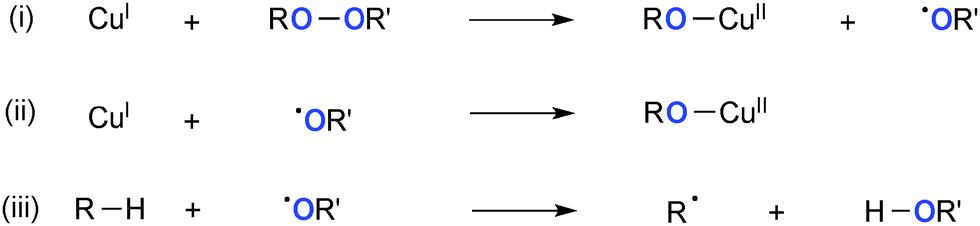
Various types of peroxides have been employed as the stoichiometric oxidants and often as the sources of oxygen functionality in Cu-catalyzed oxidative molecular transformation.4d In principle, reduction of peroxides by lower valent Cu(I) species provides the corresponding higher valent Cu(II) alkoxide and highly reactive alkoxy radical (Scheme 14) that cooperate synergistically to mediate subsequent Kharasch–Sosnovsky22 type oxidative functionalization of the substrates. The resulting alkoxy radical could further oxidize Cu(I) species to give another Cu(II) alkoxide (path-ii) or undergo H-radical abstraction from the substrates to form the C-radical (path-iii).
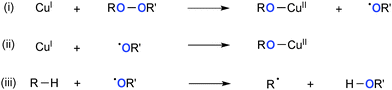 |
| | Scheme 14 The reaction of peroxides with Cu(I) species. | |
3.1. With di-t-butylperoxide (t-BuOOt-Bu)
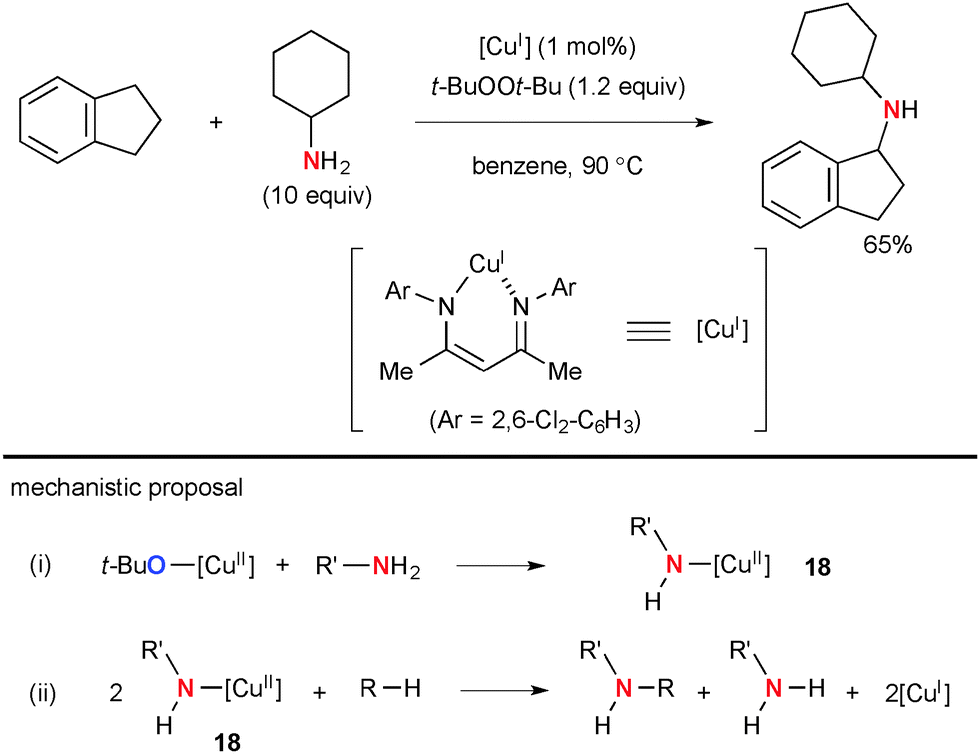
Warren reported seminal studies on aliphatic C–H amination with simple amines using the well-defined Cu(I) β-diketiminate complex and t-BuOOt-Bu as well as their detailed mechanistic studies by kinetic, spectroscopic, and structural analyses of possible intermediates (Scheme 15).1e,23 It is conceivable that Cu(II) amide complex 18 formed by the alkoxy-amide exchange (path-i) undergoes C–H bond amination of alkanes via aliphatic H-radical abstraction and subsequent C–N bond formation with the resulting C-radical (path-ii) along with generation of Cu(I) species that maintains the catalytic cycle with t-BuOOt-Bu.
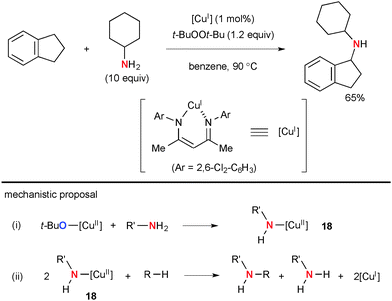 |
| | Scheme 15 Cu-Catalyzed-t-BuOOt-Bu-mediated aliphatic C–H amination. | |
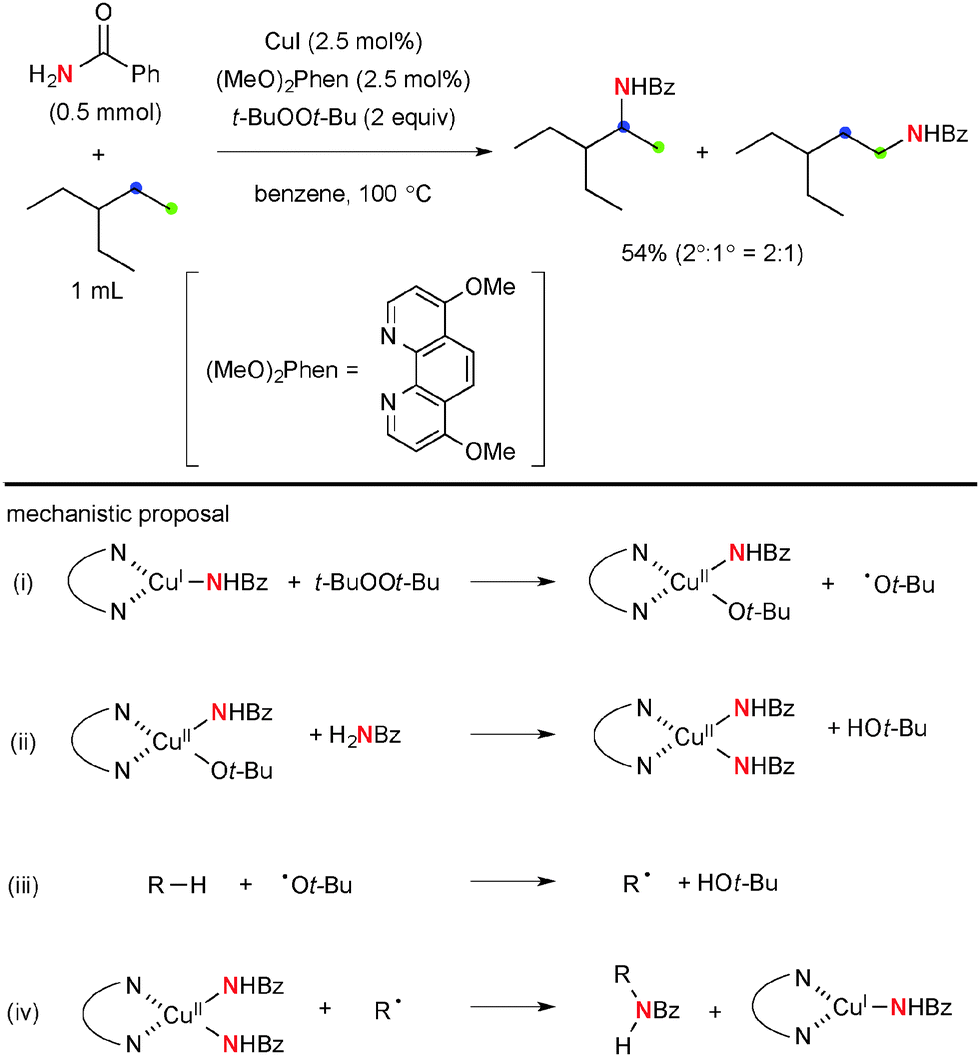
More recently, Hartwig disclosed Cu-catalyzed aliphatic C–H amidation and imidation using t-BuOOt-Bu as the stoichiometric oxidant (Scheme 16).24 The reactions prefer to oxidize secondary C–H bonds than primary ones, while tertiary C–H bonds are interestingly the least reactive. The stoichiometric reaction analyses using the isolated well-defined copper amidate complexes implicated that the C–H amidation is enabled by H-radical abstraction with the t-butoxy radical (path-iii) and radical recombination of the resulting C-radical with transient Cu(II) amidate complexes (path-iv). Analogous ligand-free Cu-catalyzed aliphatic C–H amidation and imidation with t-BuOOt-Bu were developed independently by Huang and Yu/Cheng.25
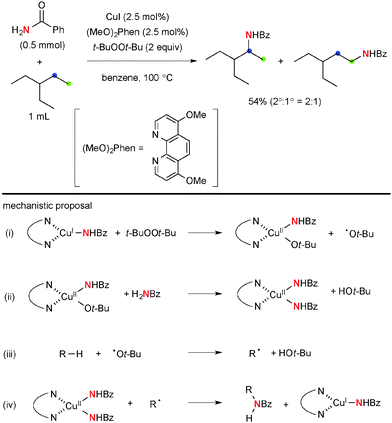 |
| | Scheme 16 Cu-Catalyzed-t-BuOOt-Bu-mediated aliphatic C–H amidation. | |
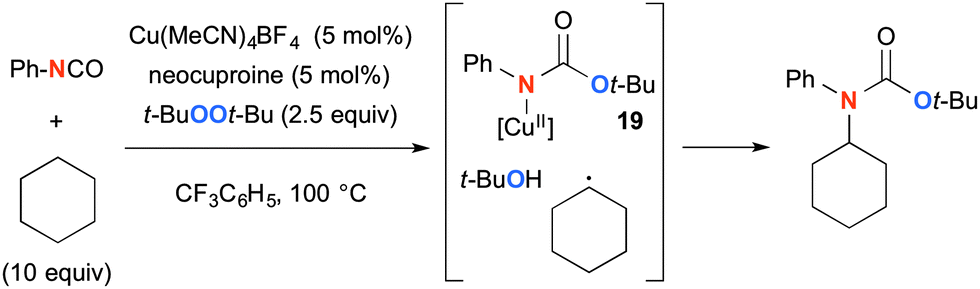
This type of Cu-catalyzed-t-BuOOt-Bu-mediated aliphatic C–H oxidation strategy could be further applied for synthesis of tertiary carbamates using isocyanates as the amide source (Scheme 17).26 The reaction of Cu(I) species with t-BuOOt-Bu and isocyanate generates Cu(II)–amide complex 19, which is coupled with the C-radical derived from alkanes via H-radical abstraction by the transient t-butoxy radical, affording tertiary carbamates.
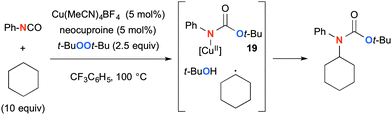 |
| | Scheme 17 Cu-Catalyzed-t-BuOOt-Bu-mediated synthesis of tertiary carbamates with isocyanates as the amide source. | |
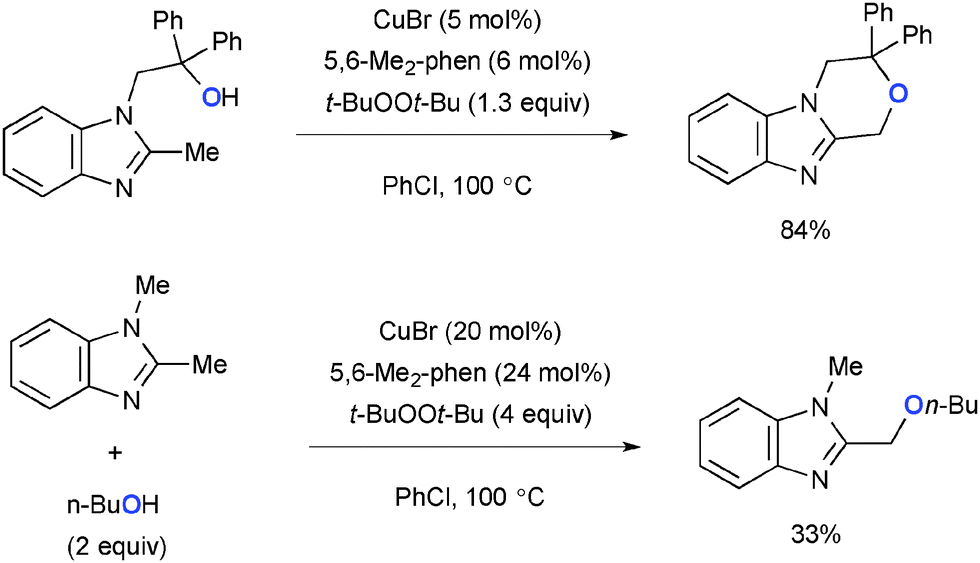
Intramolecular benzylic C–H alkoxylation of aromatic heterocycles having a hydroxyalkyl tether was devised under Cu-catalyzed-t-BuOOt-Bu-mediated reaction conditions (Scheme 18).27 The intermolecular variant also worked in the same system, while the yields of the C–H alkoxylation products were moderate.
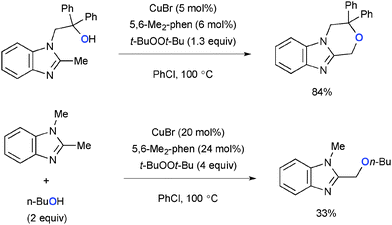 |
| | Scheme 18 Cu-Catalyzed-t-BuOOt-Bu-mediated sp3 C–H oxygenation. | |
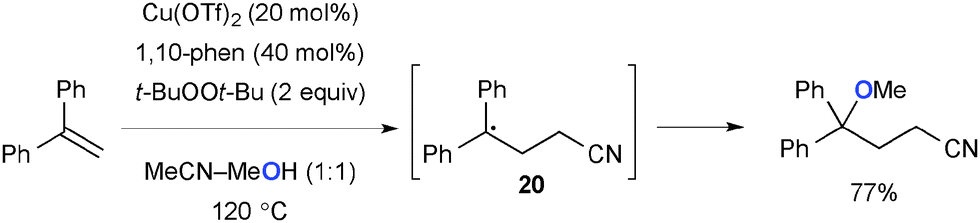
Zhu recently reported carboetherification of aryl alkenes with solvent amounts of acetonitrile and alcohol under Cu-catalyzed-t-BuOOt-Bu-mediated reaction conditions (Scheme 19).28 The radical clock experiment implicated that benzylic radical intermediate 20 is formed by the addition of acetonitrile through either carbocupration followed by homolysis of the resulting C–Cu bond or addition of the α-cyanomethyl radical. The final product is delivered through formation of the C–O bond via radical recombination of the benzylic radical intermediate with alcohol.
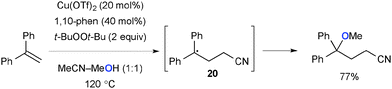 |
| | Scheme 19 Cu-Catalyzed carboetherification of alkenes. | |
3.2. With t-butylhydroperoxide (t-BuOOH: TBHP)
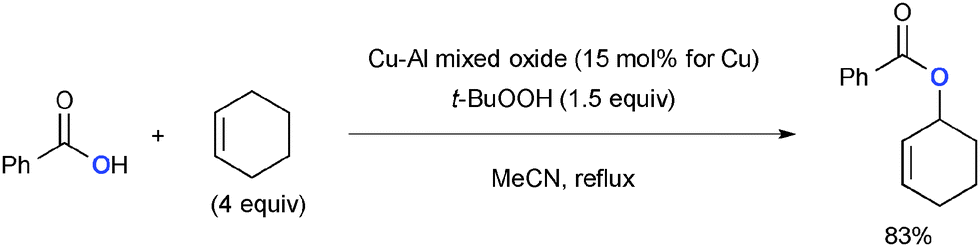
t-Butylhydroperoxide (TBHP) exhibits reactivity analogous with that of di-t-butylperoxide for Cu-catalyzed aliphatic C–H oxidation with various heteroatom sources such as amides via radical intermediates (Scheme 20). For example, Guerra recently reported that Cu–Al mixed oxide could be utilized as a heterogeneous catalyst for Kharasch–Sosnovsky type allylic C–H oxygenation with TBHP as the stoichiometric oxidant.29,30
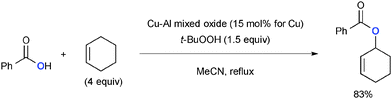 |
| | Scheme 20 Cu-Catalyzed allylic C–H oxygenation. | |
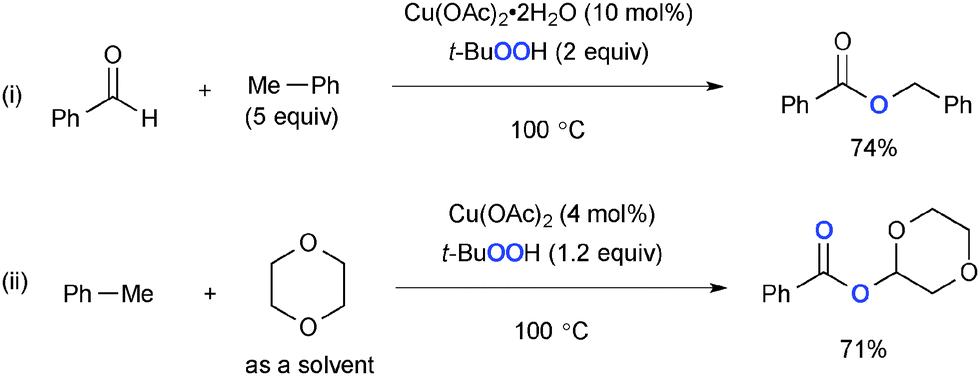
In addition, TBHP uniquely serves as an oxygen source for enhancing the oxidation state of the substrates in aliphatic C–H oxidation. Patel developed Cu-catalyzed-TBHP-mediated synthesis of esters from aldehydes and alkylarenes such as toluene (Scheme 21-i),31 in which Cu–alkoxides formed via benzylic oxygenation with the TBHP couple with aldehydes to deliver esters. Toluene could serve as a benzoate precursor in synthesis of esters via C–H oxygenation of cyclic ethers (Scheme 21-ii).32,33
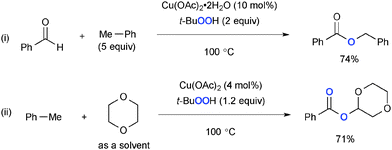 |
| | Scheme 21 Cu-Catalyzed-TBHP-mediated ester synthesis. | |
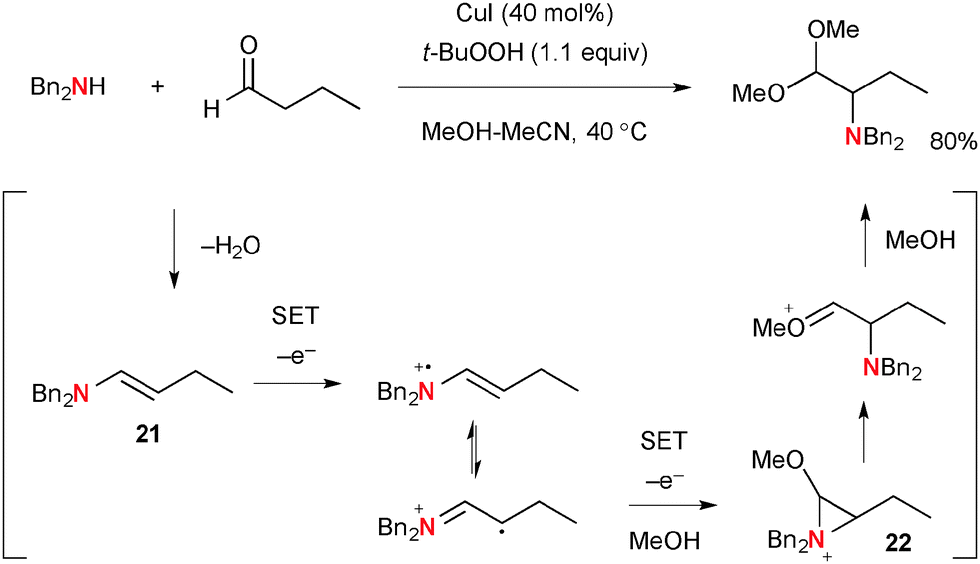
As a mechanistically distinct example of aliphatic C–H oxidation, Loh developed unique α-amination of aldehydes with secondary amines under Cu-catalyzed-TBHP-mediated reaction conditions that provides α-amino acetals as the products (Scheme 22).34 The process is composed of a multi-step sequence including two successive single-electron-oxidations of enamine intermediate 21 derived from condensation of aldehydes and amines. The resulting α-methoxy aziridinium ion 22 undergoes ring-opening with methanolysis, giving α-amino acetal. The detailed roles of CuI and TBHP in the single-electron-transfer processes are not certain.
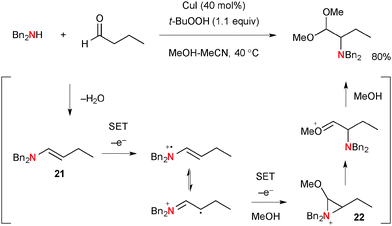 |
| | Scheme 22 Cu-Catalyzed α-amination of aldehydes. | |
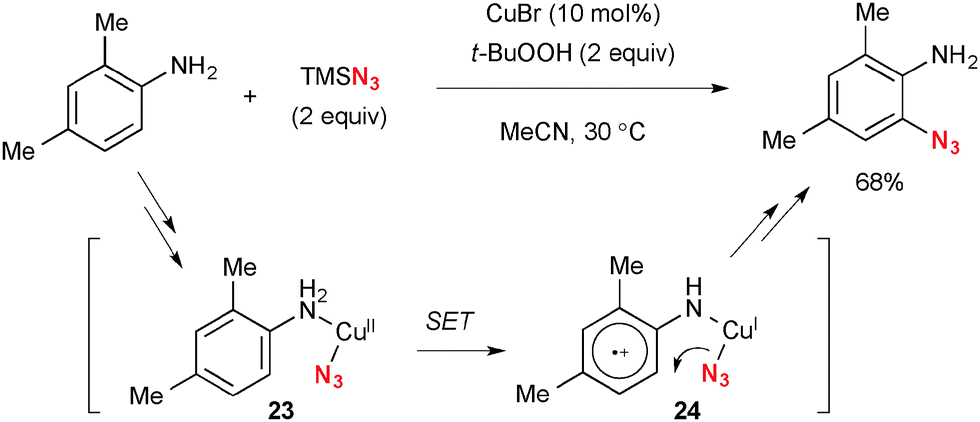
The Cu–TBHP system is also capable of oxidizing sp2-C–H bonds. Jiao disclosed ortho-azidation of anilines with TMSN3 under Cu-catalyzed-TBHP-mediated reaction conditions at ambient temperature (Scheme 23).35 The single-electron-oxidation of the benzene ring by higher valent Cu(II) species 23 adjacent to the primary amine moiety followed by azido ion transfer in cation radical 24 is proposed for the C–H azidation. This is a rare example of the directed ortho C–H functionalization of anilines.
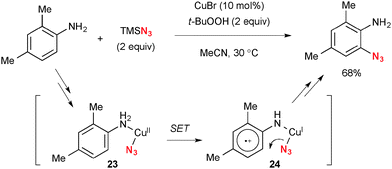 |
| | Scheme 23 Cu-Catalyzed ortho-C–H azidation. | |
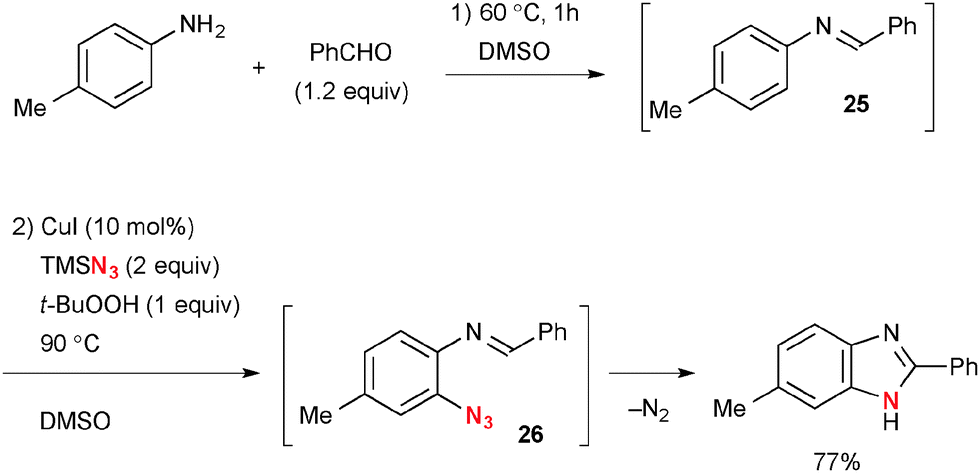
Benzaldimines 25 derived from condensation of anilines and aldehydes could be used for analogous ortho-azidation and the resulting 2-azidoarylimines 26 undergo subsequent denitrogenative cyclization to give benzimidazoles (Scheme 24).36 A 2-pyridyl group has also been utilized as the directing group in aromatic C–H oxidation under Cu-catalyzed-TBHP-mediated reaction conditions.37
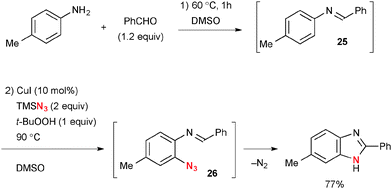 |
| | Scheme 24 Cu-Catalyzed-t-BuOOH-mediated aromatic ortho-azidation. | |
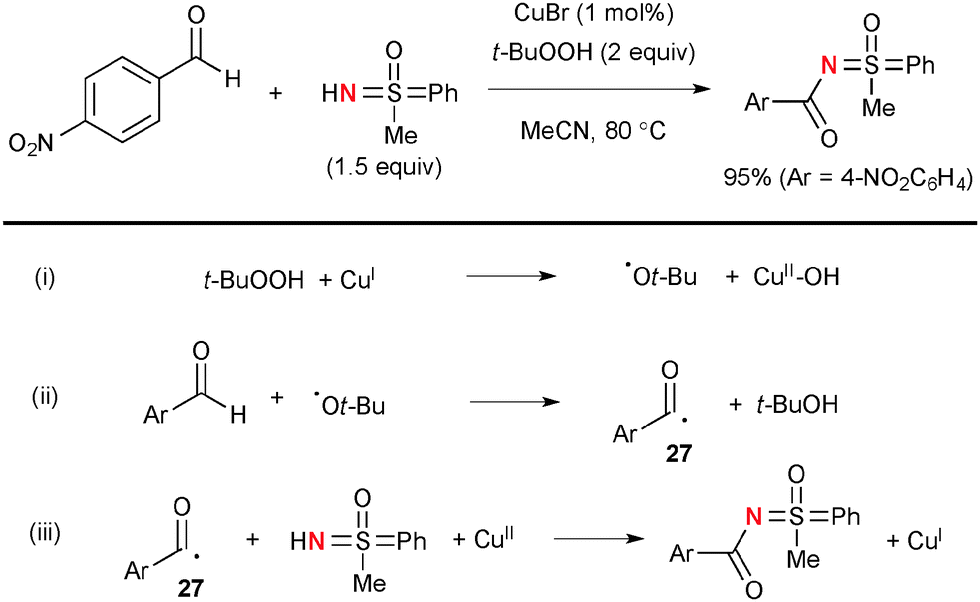
Bolm developed Cu-catalyzed oxidative N-acylation of sulfoximines with aldehydes (Scheme 25).38 The t-butoxy radical derived from decomposition of TBHP by Cu(I) complexes (Scheme 25-i) could abstract the H-radical from aldehydes to generate the corresponding acyl radicals 27 (Scheme 25-ii). The transient acyl radicals 27 undergo radical recombination with sulfoximines mediated by higher valent Cu(II) species to give the products along with re-generation of lower valent Cu(I) species (Scheme 25-iii).
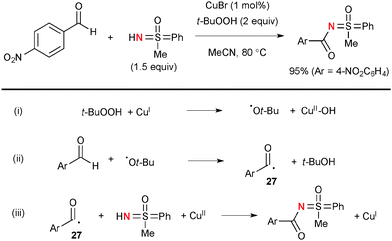 |
| | Scheme 25 Cu-Catalyzed oxidative N-acylation of sulfoximines. | |
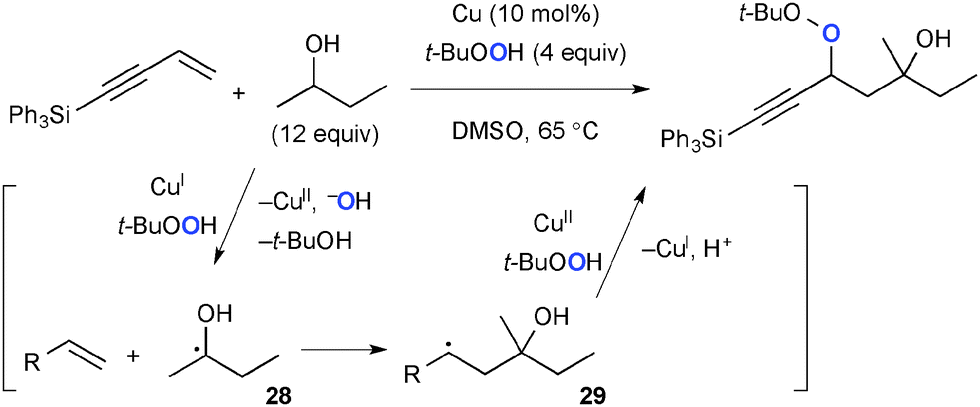
In the presence of Cu(I) complexes, TBHP can serve as the source of the t-butoxy radical that mediates H-radical abstraction to give the C-radical, whereas TBHP itself can also be introduced as the new oxygen functionality via the C–O bond forming process. Loh demonstrated Cu-catalyzed three-component coupling of alkenes, aliphatic alcohols, and TBHP for construction of the corresponding carbooxygenation products. In this process, α-hydroxy radicals 28 generated from aliphatic alcohols add to alkenes to give secondary radicals 29 that recombine with TBHP mediated by Cu(II) species (Scheme 26).39 Patel reported analogous Cu-catalyzed three-component coupling of electron-deficient alkenes, cycloalkanes, and TBHP.40
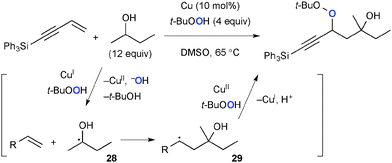 |
| | Scheme 26 Cu-Catalyzed carbooxygenation of alkenes. | |
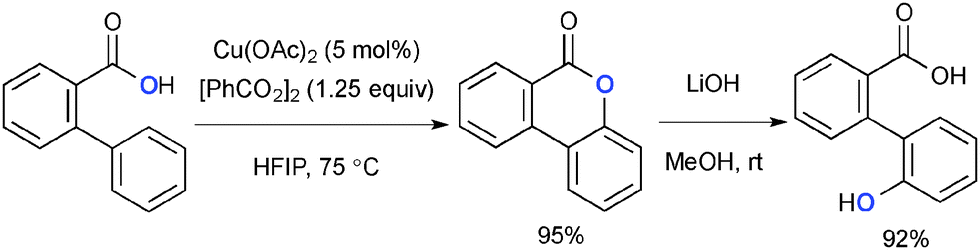
3.3. With benzoyl peroxide (BPO)
While benzoyl peroxide (BPO) has rarely been utilized as the partner with Cu-catalyzed oxidative molecular transformation, Martin recently reported Cu-catalyzed intramolecular aromatic C–H oxygenation of 2-arylbenzoic acids specifically mediated by BPO as the terminal oxidant (Scheme 27).41 The reaction could not be facilitated by TBHP. Together with treatment of the biaryl lactones with LiOH for hydrolysis, the overall process is considered as formal aromatic C–H hydroxylation.
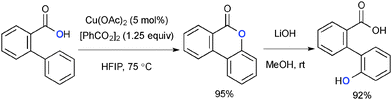 |
| | Scheme 27 Cu-Catalyzed aromatic C–H oxygenation. | |
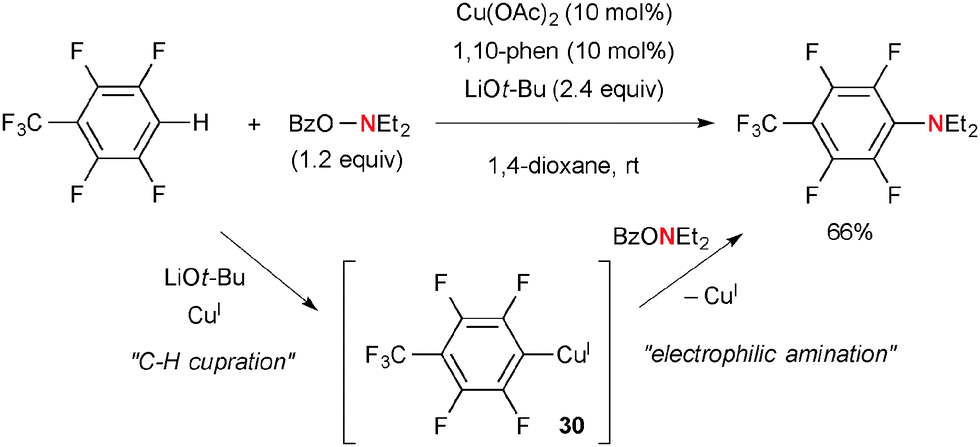
4. With O-benzoyl-N,N-dialkylhydroxylamines
Among various types of hydroxyl amine derivatives, O-benzoyl-N,N-dialkylhydroxylamines (BzO–NR2) have been of particular use for electrophilic amination of organocopper(I) species. For example, Hirano/Miura reported direct aromatic C–H amination of electron-deficient arenes with BzO–NEt2 by the Cu(OAc)2-1,10-phen catalytic system in the presence of LiOt-Bu (Scheme 28).42 The process is composed of aromatic C–H cupration for the formation of aryl-Cu(I) species 30 and subsequent electrophilic amination with BzO–NEt2. The mechanism of electrophilic amination was previously investigated by Johnson and suggested as the SN2 mechanism.43 This strategy was applied for C2-amination of quinoline-N-oxides by Li/Wu.44
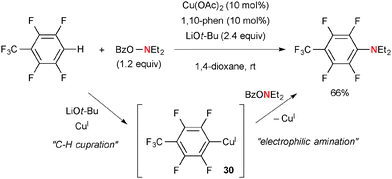 |
| | Scheme 28 Cu-Catalyzed aromatic C–H amination with hydroxylamines. | |
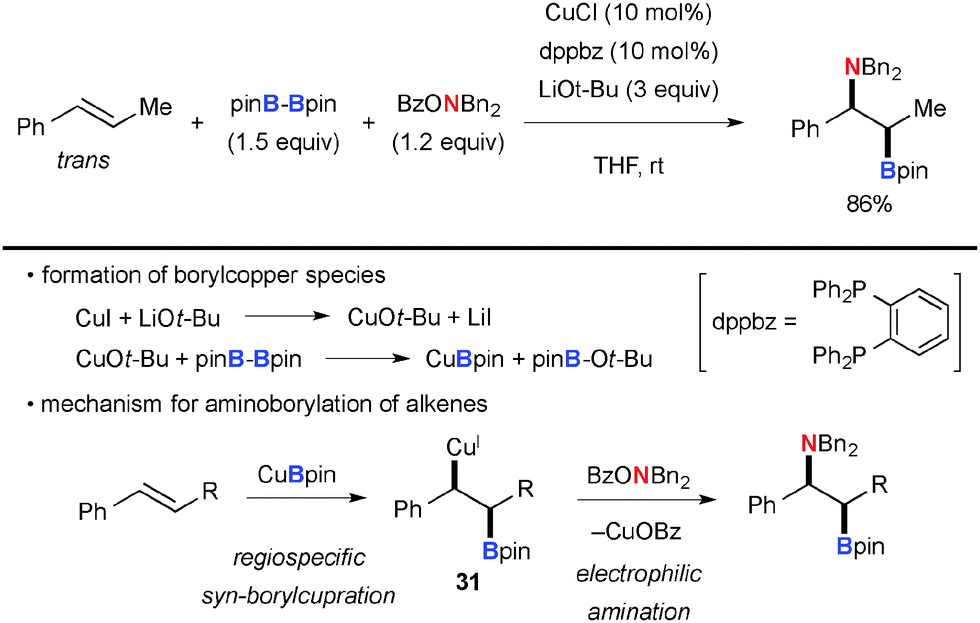
Hirano/Miura demonstrated the combination of boryl-cupration of alkenes with subsequent electrophilic amination of the resulting alkyl-Cu species with BzO–NR2, offering elegant aminoboration of alkenes in stereo- and regio-selective manners. For example, the reactions of arylalkenes such as trans-β-methylstyrene with bis(pinacolato)diboron (pinB-Bpin) and BzO–NBn2 under the CuCl-dppbz catalytic system in the presence of LiOt-Bu resulted in the formation of aminoboration products (Scheme 29).45 As boryl-cupration of alkenes takes place in syn-selective and regiospecific fashions to form organocopper intermediate 31 and the subsequent electrophilic amination proceeds with retention of the configuration of the organocopper moiety, the overall stereochemical outcome of the process is regioselective syn-aminoboration of alkenes.
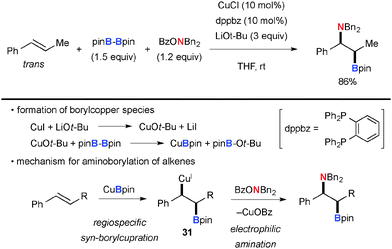 |
| | Scheme 29 Cu-Catalyzed aminoboration of alkenes. | |
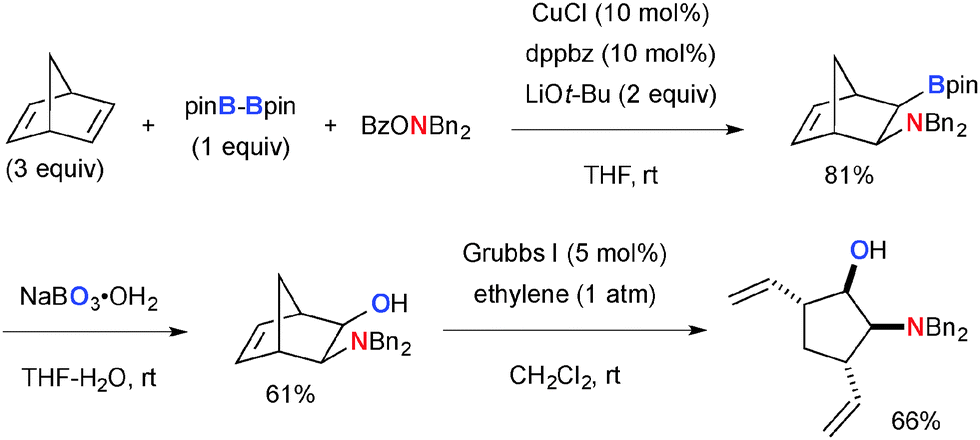
This Cu-catalyzed aminoboration of alkenes was capable of functionalizing bicyclic alkenes (Scheme 30).46 The 1,2-aminoborane product from norbornadiene could be further converted into the corresponding diastereomerically pure cyclopentane derivative via hydroxylation of the C–B bond by sodium perborate followed by ring-opening cross metathesis with ethylene.
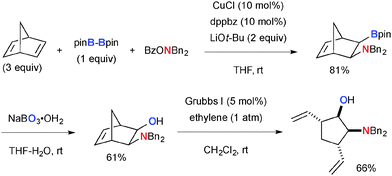 |
| | Scheme 30 Cu-Catalyzed aminoboration of norbornadiene and further molecular transformation. | |
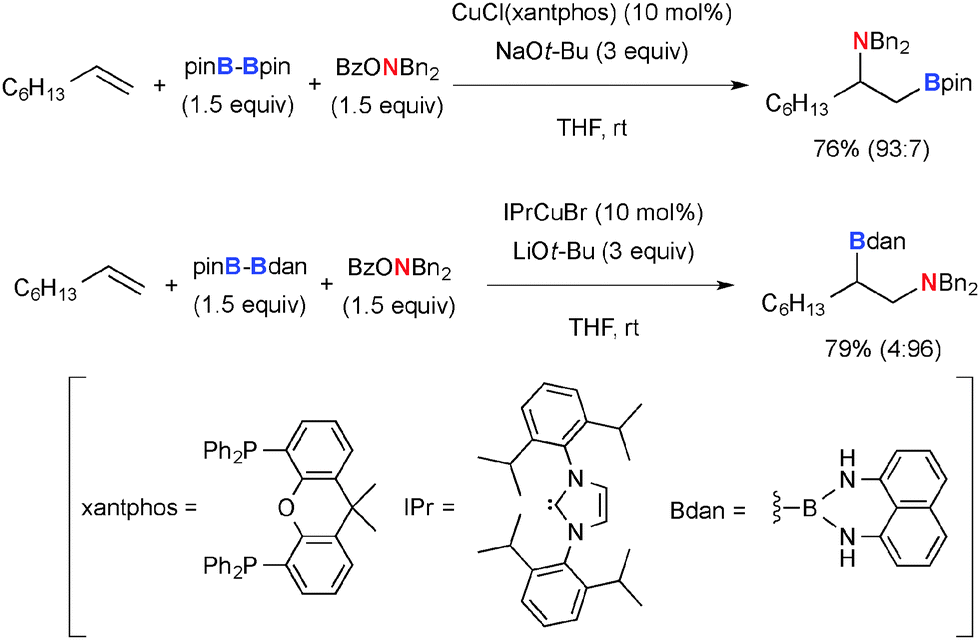
As for the aminoboration of non-activated terminal alkenes, its regioselectivity could be controlled by switching the ligands on the Cu(I) catalysts (Scheme 31).47 Namely, the CuCl–xantphos system installs the amine moiety at the internal carbon, while the CuBr-N-heterocyclic carbene (IPrCuBr) complex induces the amination at the terminal carbon.
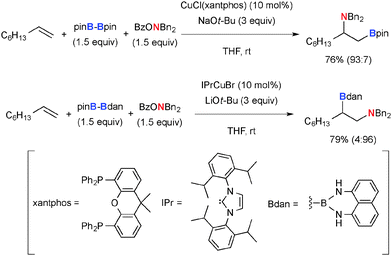 |
| | Scheme 31 Cu-Catalyzed regiodivergent aminoboration of non-activated terminal alkenes. | |
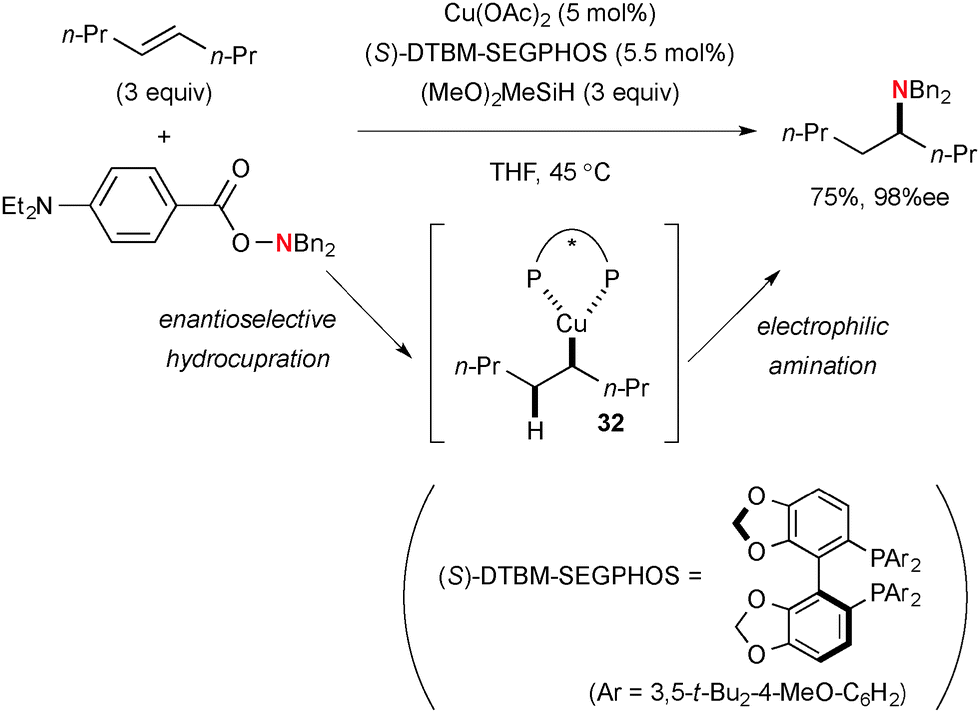
Buchwald, Hirano/Miura, and Hartwig independently reported formal hydroamination of alkenes by the sequence of Cu-catalyzed hydrocupration of alkenes with hydrosilanes and electrophilic amination of the resulting alkylcopper species 32 with BzO–NR2. Various types of alkenes such as aryl alkenes,48 unactivated 1,1-disubstituted alkenes,49 unactivated internal alkenes (Scheme 32),50 oxa/aza-bicyclic alkenes,51 and alkenylsilanes52 have been employed for regio- and enantioselective hydroamination, in which choice of ligands on copper catalysts is crucial to control the reactions (i.e. to prevent the side reactions such as hydride reduction of hydroxyl amines).
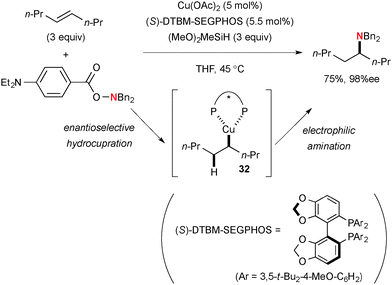 |
| | Scheme 32 Cu-Catalyzed enantioselective formal hydroamination of non-activated internal alkenes. | |
Wang recently developed Cu-catalyzed diamination of alkenes using alkenyl O-Me-hydroxamic acids and BzO–NR2 that afforded functionalized nitrogen heterocycles (Scheme 33).53 The process is composed of intramolecular aminocupration of alkenes and subsequent electrophilic amination. As a deuterium-labeling experiment on the terminal alkenyl carbon revealed that the reaction does not retain the original stereochemistry of alkenes, the radical intermediates are supposed to be involved prior to the electrophilic amination.
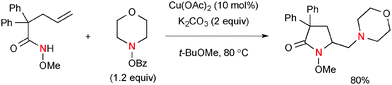 |
| | Scheme 33 Cu-Catalyzed diamination of alkenyl O-Me hydroxamic acids with O-benzoyl hydroxylamines. | |
5. With oxaziridines
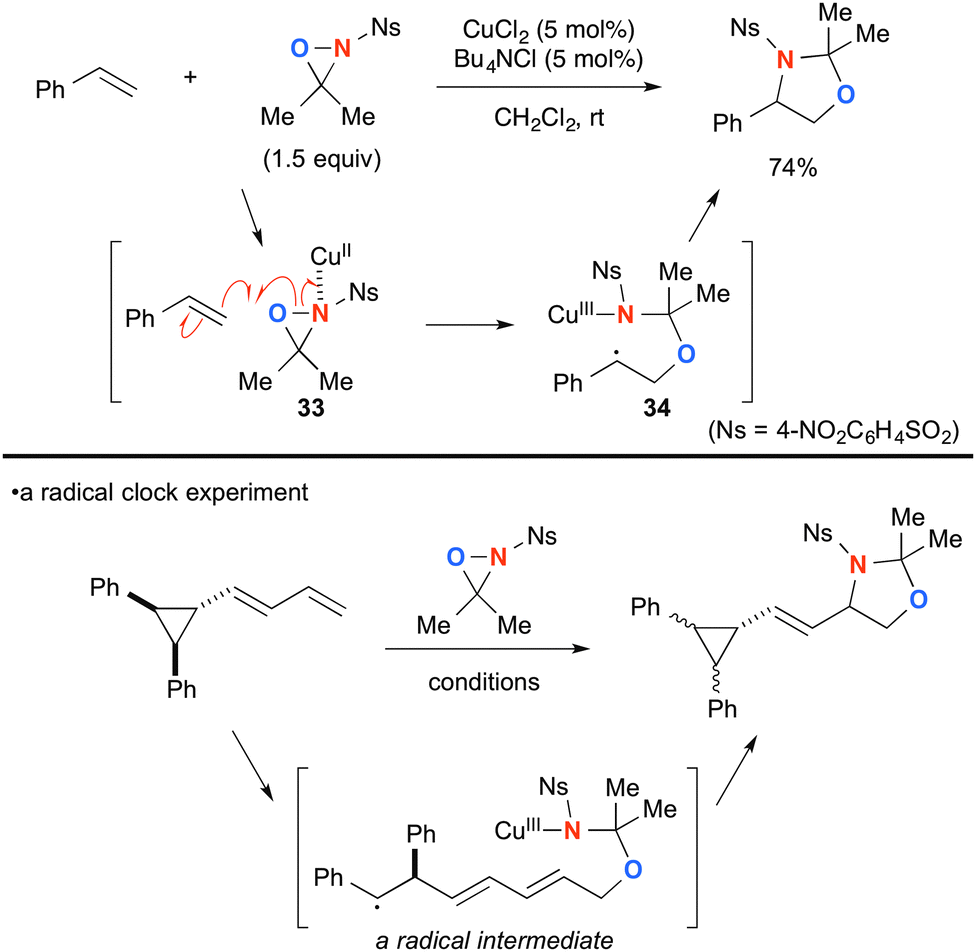
Highly strained three-membered ring oxaziridines work with copper complexes to facilitate aminooxygenation of alkenes. Yoon developed CuCl2-catalyzed aminooxygenation of alkenes with N-sulfonyloxaziridines that could be dramatically facilitated by chloride additives such as Bu4NCl (Scheme 34).54 The detailed mechanistic investigation elucidated that Cu(II)–oxaziridine complex 33 undergoes C–O bond forming radical addition onto alkenes to generate C-radical intermediate 34 tethered with a Cu(III) sulfonamide moiety. Subsequent radical recombination forms the C–N bond and regenerates Cu(II) species that can maintain catalytic turnover further. The presence of the radical intermediate was proved by a radical clock experiment.
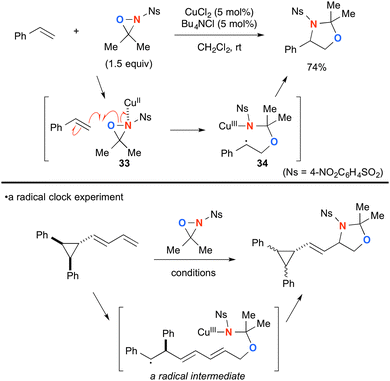 |
| | Scheme 34 CuCl2-catalyzed aminooxygenation of alkenes with N-sulfonyloxaziridines. | |
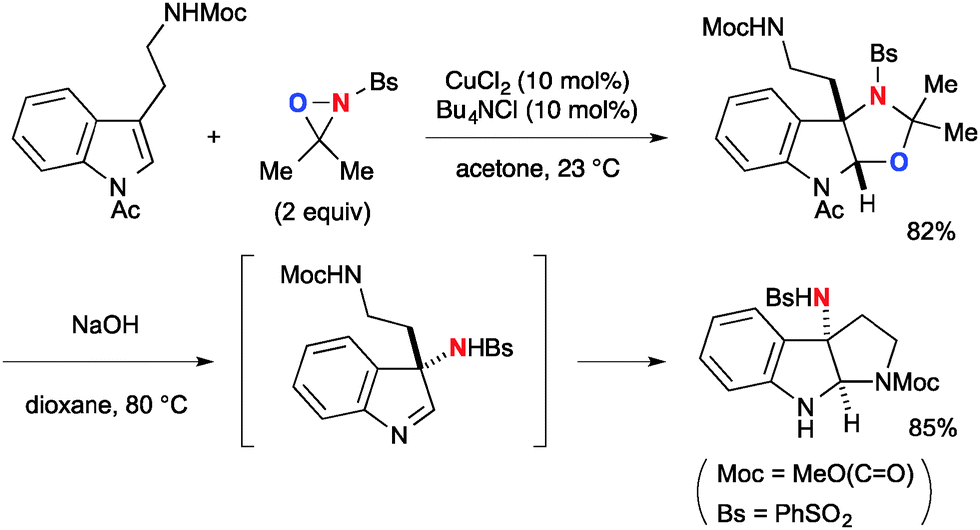
This aminooxygenation strategy with N-sulfonyl oxaziridines was capable of functionalizing indoles (Scheme 35).55 It is worthy to note that the resulting aminal product derived from N-acyltryptamine was readily transformed to 3-aminopyrroloindoline by base treatment.
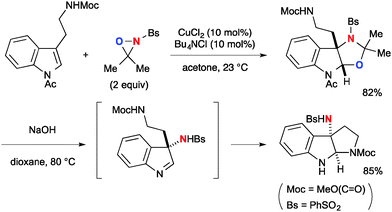 |
| | Scheme 35 Cu-Catalyzed aminooxygenation of indoles with N-sulfonyloxaziridines. | |
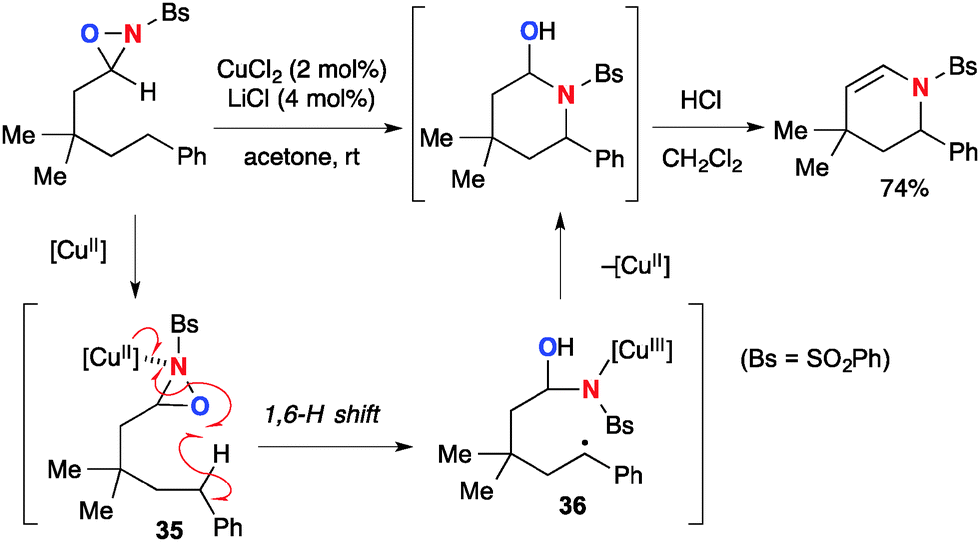
The Cu(II)–oxaziridine complexes also undergo the remote H-radical abstraction to enable aliphatic C–H amination (Scheme 36).56 The reaction of N-sulfonyl oxaziridines having an alkyl tether under the CuCl2–LiCl catalytic system provides intramolecular C–H amination products via 1,6-H-radical abstraction by Cu(II)–oxaziridine complex 35 followed by subsequent radical recombination of the resulting C-radical 36 to form the C–N bond. The resulting hemiaminal product could be converted into cyclic enamide by acid treatment.
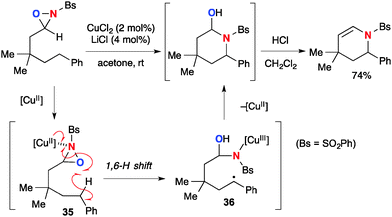 |
| | Scheme 36 Cu(II)-catalyzed C–H amination with N-sulfonyloxaziridines. | |
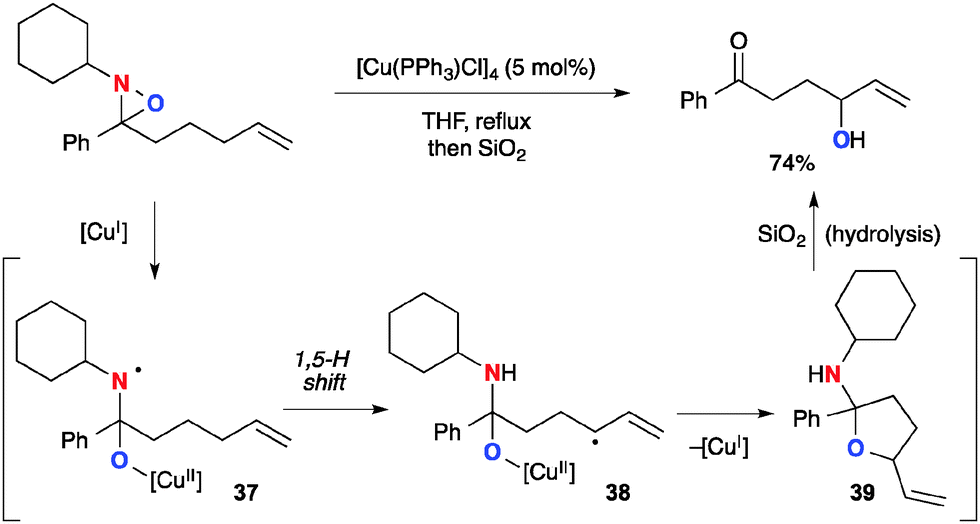
On the other hand, Aubé recently reported Cu(I)-catalyzed allylic sp3 C–H oxygenation with N-alkyl oxaziridines (Scheme 37).57 This method could oxidize the allylic position via the sequence of (1) formation of aminyl radical 37 having a Cu(II)–alkoxide tether through single-electron-reduction of N-alkyl oxaziridines by the Cu(I) complex; (2) generation of the allylic radical by a 1,5-H radical shift to form C-radical 38 and subsequent radical recombination with the Cu(II)–alkoxide moiety to form cyclic hemiaminal 39 with regeneration of Cu(I) species; (3) hydrolysis to form the final product, γ-hydroxy ketone.
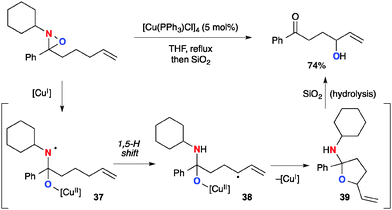 |
| | Scheme 37 Cu(I)-catalyzed C–H oxygenation with N-alkyloxaziridines. | |
6. With diaziridinone derivatives
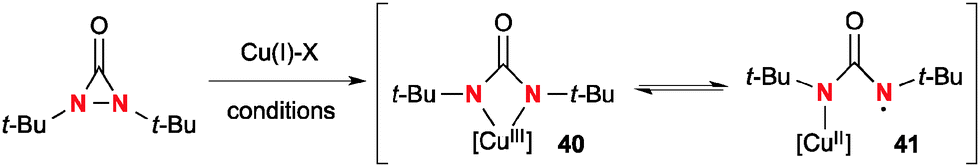
Similar to oxaziridines, N,N-di-t-butyldiaziridinone shows strain-driven oxidative reactivity towards Cu(I) complexes, enabling catalytic diamination of various types of alkenes.4b Shi revealed that N,N-di-t-butyldiaziridinone oxidizes Cu(I) complexes to form an equilibrium mixture of four-membered Cu(III) species 40 and Cu(II))–N radical species 41 (Scheme 38).
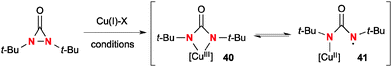 |
| | Scheme 38 The reaction of di-t-butyldiaziridinone with Cu(I) complexes. | |
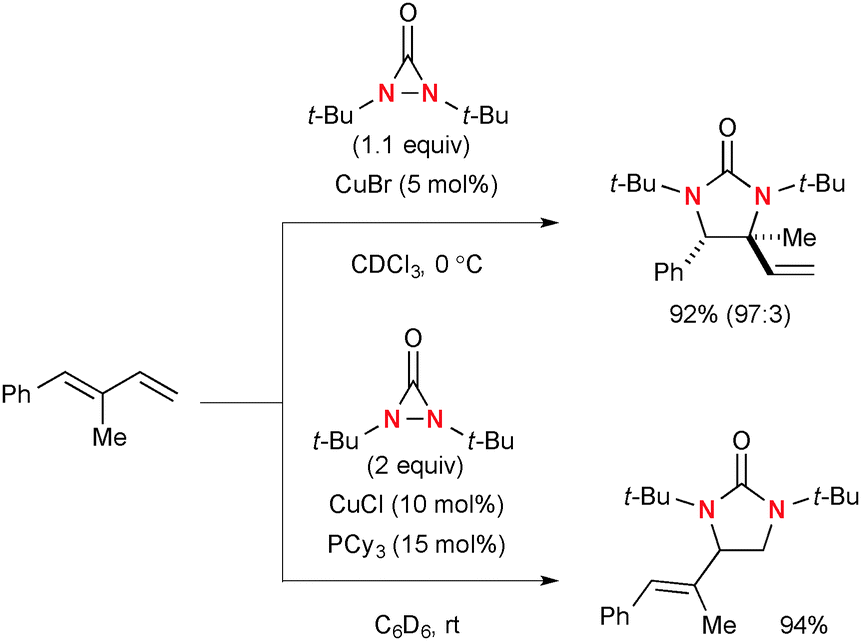
Of particular interest is that the regioselectivity in diamination of conjugated dienes could be switched by the choice of Cu(I) catalyst systems and electronic nature of dienes with the different reaction mechanisms.58 Namely, conjugated dienes and N,N-di-t-butyldiaziridinone in the presence of a catalytic amount of CuBr generally undergo diamination of internal alkenes, whereas terminal alkenes could be functionalized under the CuCl–phosphine ligand catalytic system (Scheme 39).
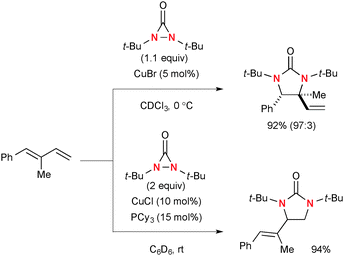 |
| | Scheme 39 Regioselectivity in Cu(I)-catalyzed diamination of conjugated dienes with N,N-di-t-butyldiaziridinone. | |
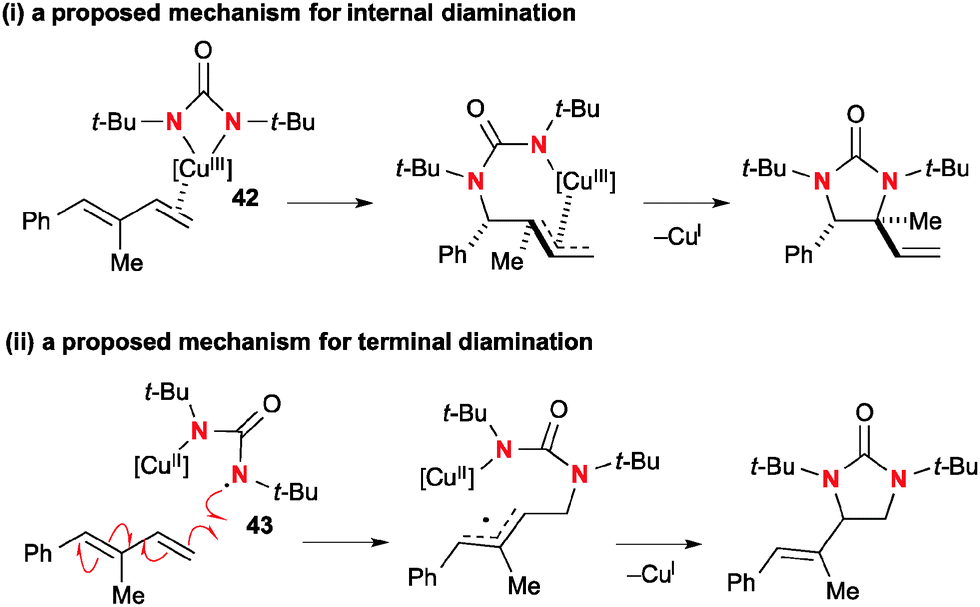
Detailed mechanistic investigation suggested that internal diamination proceeds with the four-membered Cu(III) species 42via (1) coordination and migratory insertion to dienes; (2) C–N reductive elimination that renders the overall process cis-diamination (Scheme 40-i). On the other hand, diamination of terminal alkenes involves Cu(II)–N radical species 43 that initiates radical C–N bond formation to the sterically less hindered terminal carbon to generate an allyl radical. The second C–N bond formation is enabled by the radical recombination of the allyl radical with an N–Cu(II) moiety to afford the diamination product along with regeneration of the Cu(I) catalyst (Scheme 40-ii).
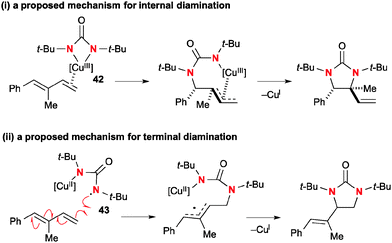 |
| | Scheme 40 The proposed reaction mechanisms for diamination. | |
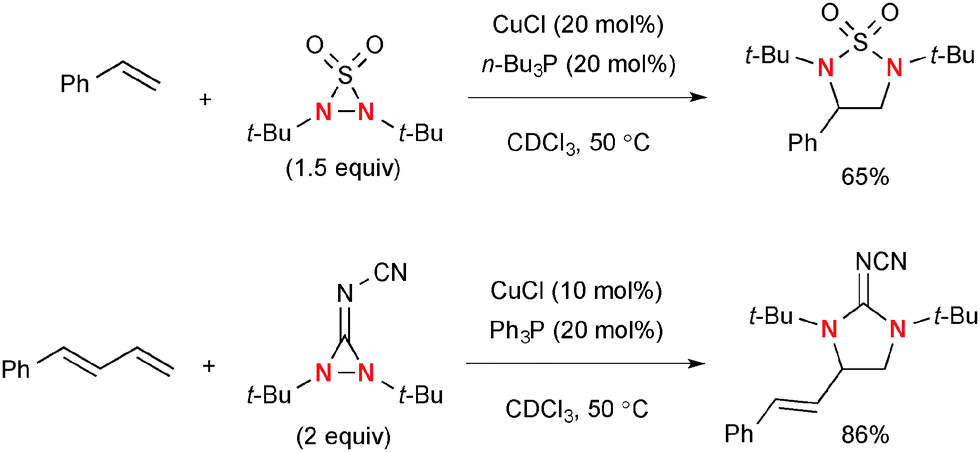
This stepwise radical-mediated diamination of alkenes with N,N-di-t-butyldiaziridinone by the Cu(I)–phosphine ligand catalytic system is amenable to functionalize not only conjugated dienes but also 1,1-disubstituted alkenes.59 Asymmetric terminal diamination of conjugated dienes was also developed by the CuCl–chiral phosphine60 and Cu(I)–chiral phosphate systems.61 Analogous to N,N-di-t-butyldiaziridinone, N,N-di-t-butylthiadiaziridine 1,1-dioxide62 and N,N-di-t-butyl-3-(cyanimino)-diaziridine63 could be utilized for catalytic radical diamination of conjugated alkenes under the CuCl–phosphine ligand systems (Scheme 41).
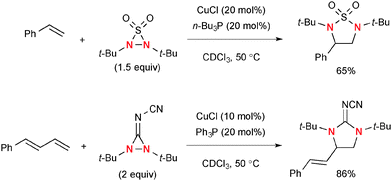 |
| | Scheme 41 The reaction of di-t-butyldiaziridinone with Cu(I) complexes. | |
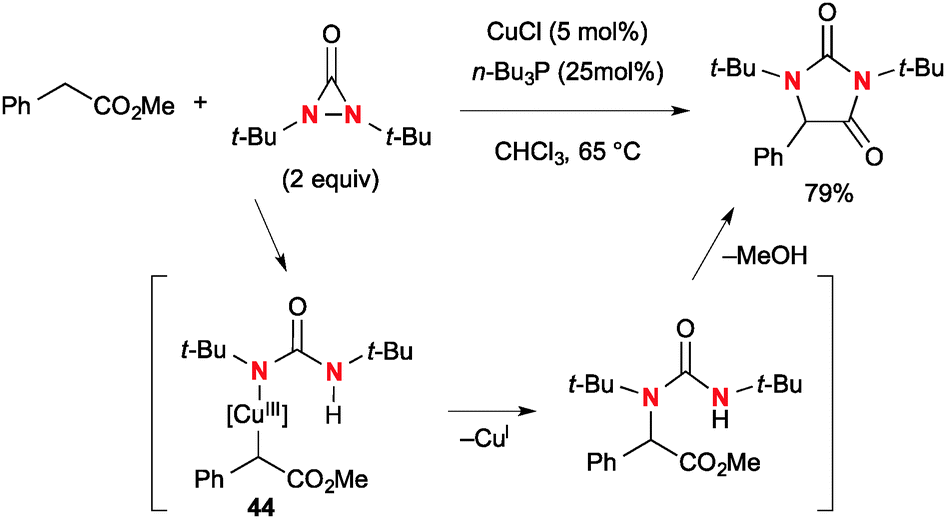
In addition to diamination of alkenes, N,N-di-t-butyldiaziridinone could be utilized for Cu(I)-catalyzed α-amination of esters (Scheme 42).64 The reaction of esters with N,N-di-t-butyldiaziridinone under the CuCl-n-Bu3P catalytic system provides the corresponding hydantoins. The proposed mechanism involves α-cupration of esters by the transient Cu(II)–N radical species or four-membered Cu(III) species derived from N,N-di-t-butyldiaziridinone and CuCl. The resulting α-cupro(III)-esters 44 undergo C–N reductive elimination that is followed by cyclization to afford hydantoins.
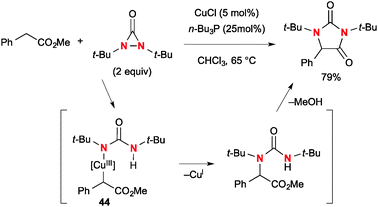 |
| | Scheme 42 Synthesis of hydantoins from esters and N,N-di-t-butyldiaziridinone via α-amination. | |
7. With fluoroamine derivatives
7.1. With N-fluorobenzenesulfonimide (NFSI)
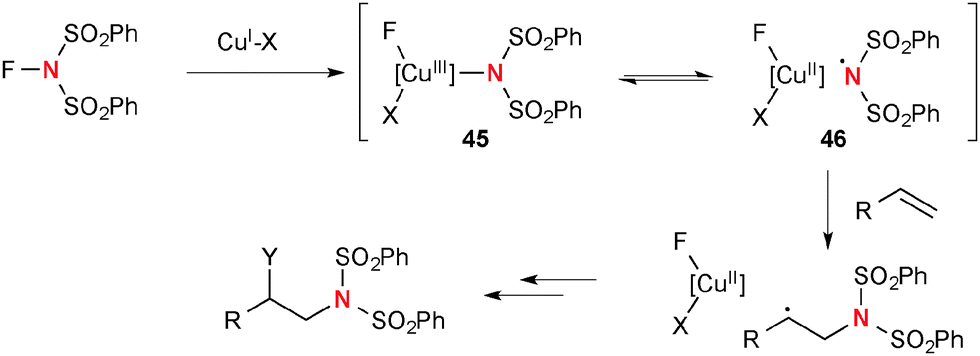
A highly reactive oxidant, N-fluorobenzenesulfonimide (NFSI) reacts readily with Cu(I) complexes to afford Cu(III)-imide species 45 that is in equilibrium with Cu(II)-stabilized sulfonimide radical 46 (Scheme 43). Thus, this nitrogen-centered radical derived from NFSI and Cu(I) complexes could initiate aminofunctionalization of alkenes.
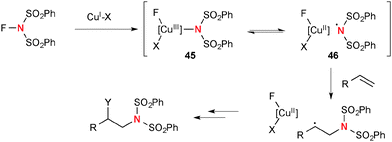 |
| | Scheme 43 The reaction of NFSI with Cu(I) complexes for aminofunctionalization of alkenes. | |
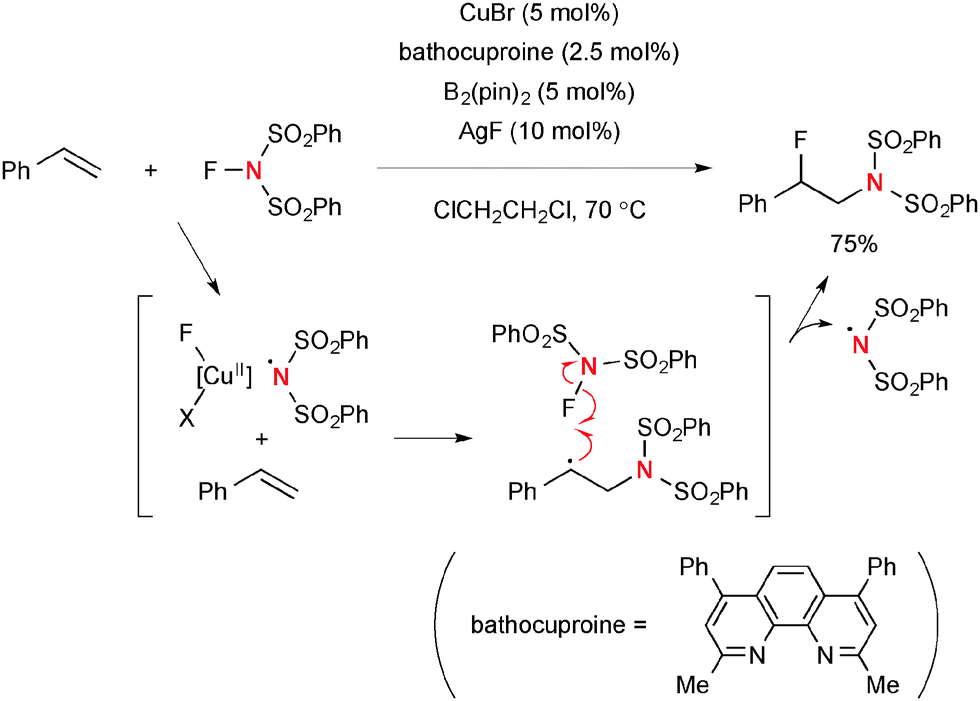
For example, Zhang reported regioselective aminofluorination of styrenes with NFSI under CuBr/bathocuproine-catalyzed reaction conditions (Scheme 44).65 The use of bis(pinacolato)diborone (B2pin2) and AgF as additives was crucial to facilitate the aminofluorination. The DFT calculations suggested that the C–F bond formation is likely enabled by F-radical abstraction from NFSI. This process concurrently generates the sulfonimide radical that can maintain the radical chain for aminofluorination.
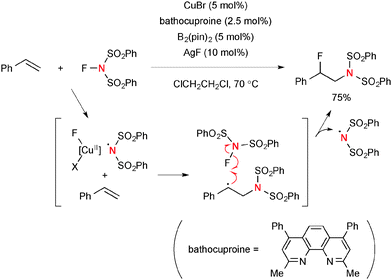 |
| | Scheme 44 Cu(I)-Catalyzed aminofluorination of styrene with NFSI. | |
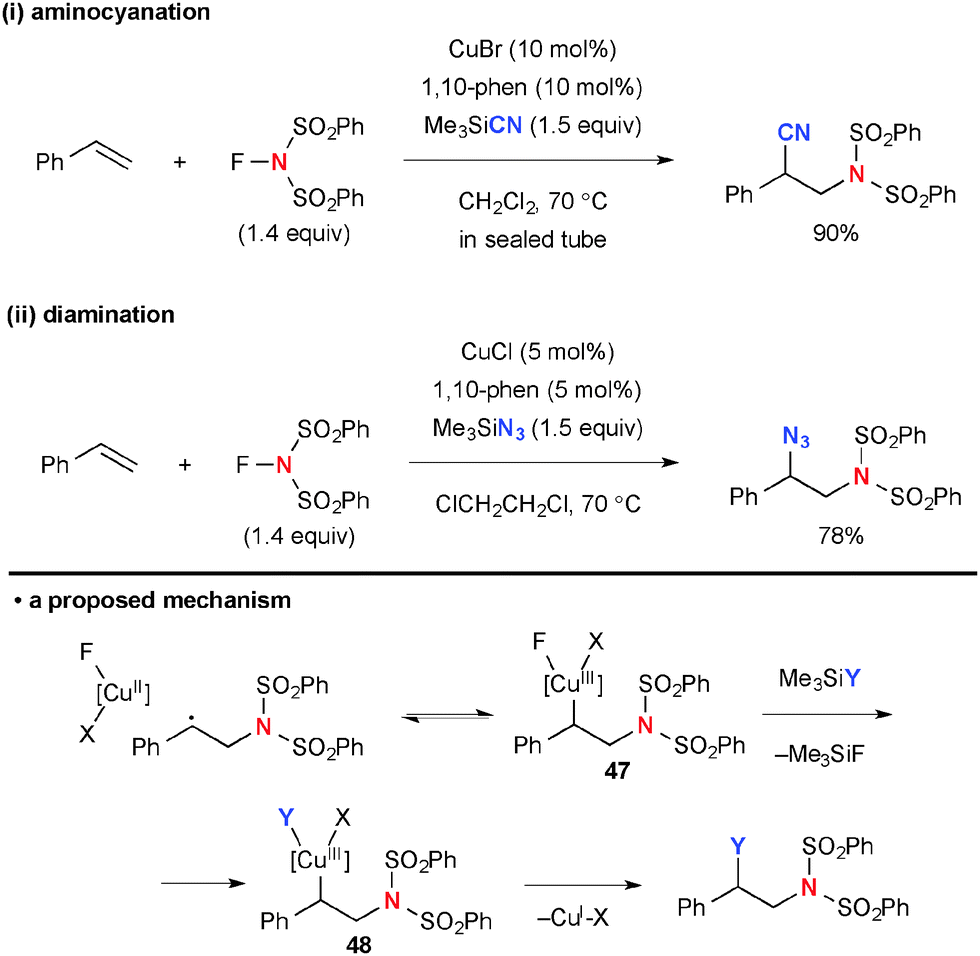
In place of fluorine incorporation, cyano, amido, and azido moieties could be installed by Cu-catalyzed radical aminofunctionalization with NFSI. Xiong/Li/Zhang revealed that the reaction of styrene with NFSI and TMSCN under the CuBr-1,10-phen catalytic system gives an aminocyanation product (Scheme 45-i).66 Similarly, aminoazidation was developed by Studer using TMSN3 (Scheme 45-ii).67 The C–CN and C–N3 bond formation is mediated via radical recombination presumably through formation of organo-Cu(III) intermediate 47 followed by ligand exchange with TMSCN or TMSN3 to afford another organo-Cu(III) species 48 and subsequent C–CN or C–N3 reductive elimination. The driving force of the ligand exchange could be preferential elimination of TMSF due to the strong affinity between Si and F atoms.
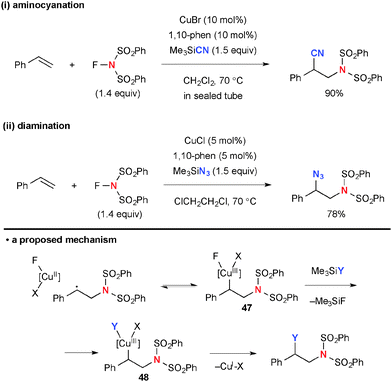 |
| | Scheme 45 Cu(I)-catalyzed aminocyanation and diamination of styrene with NFSI. | |
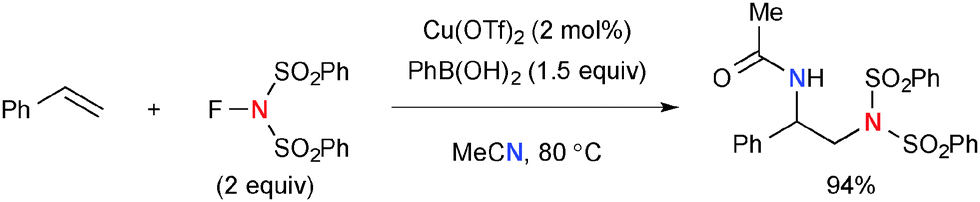
Interestingly, when the Cu-catalyzed reaction of styrene with NFSI was conducted in the presence of PhB(OH)2 in acetonitrile, a diamination product was formed through incorporation of acetonitrile by the Ritter-type reaction (Scheme 46).67
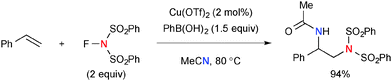 |
| | Scheme 46 Cu(OTf)2-catalyzed diamination of styrene with NFSI and acetonitrile. | |
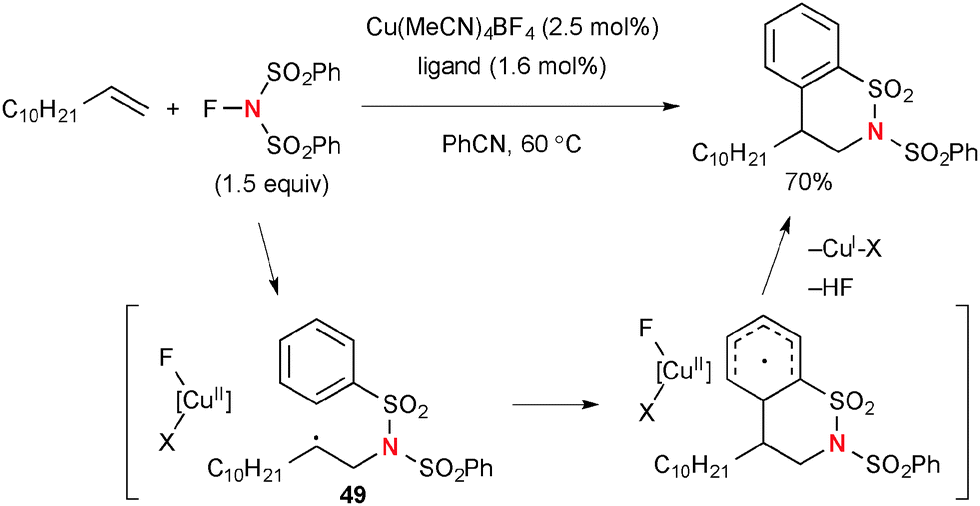
On the other hand, the Cu(I)-catalyzed reactions of aliphatic alkenes with NFSI provide non-stabilized secondary radicals 49, which undergo intramolecular radical addition to the phenylsulfonyl moiety to afford sultams (Scheme 47).68
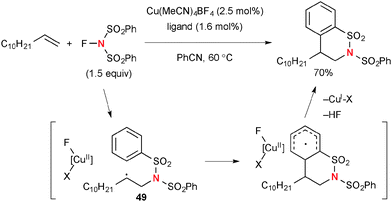 |
| | Scheme 47 Synthesis of sultams by the Cu(I)-catalyzed reaction of aliphatic alkenes with NFSI. | |
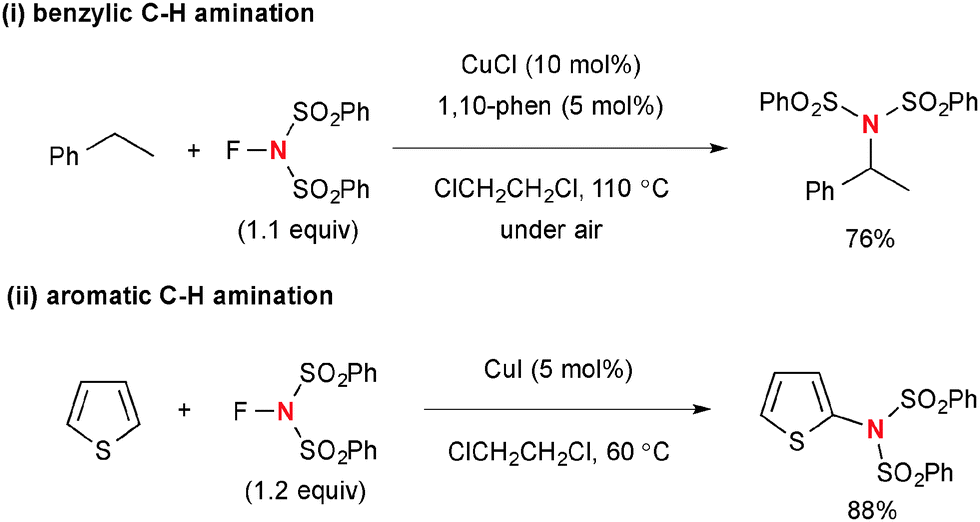
The combination of the Cu(I)-catalyst and NFSI is also capable of functionalizing benzylic sp3 C–H bonds (Scheme 48-i)69 as well as sp2 C–H bonds (Scheme 48-ii)70 on 5-membered-aromatic heterocycles such as thiophene and furan through the radical mechanism with the transient sulfonimide radical.
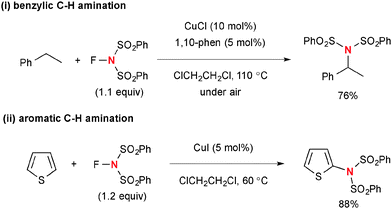 |
| | Scheme 48 Cu(I)-catalyzed C–H amination with NFSI. | |
7.2. With Selectfluor® and its derivatives (F-TEDA-X)
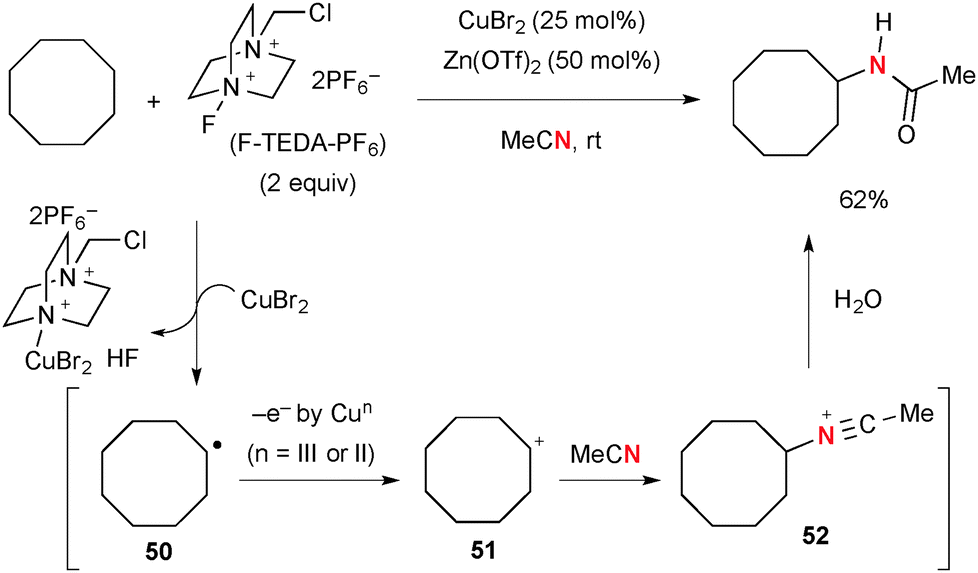
Selectfluor® [1-chloromethyl-4-fluoro-diazoniabicyclo[2.2.2]octane bis(tetrafluoroborate] and its derivatives (known as F-TEDA-X reagents, where X stands for their counter anions) have been used as versatile fluorination reagents in organic synthesis,71 while these reagents could be utilized as strong oxidants of Cu(II) and Cu(I) complexes to generate highly reactive Cu(III) species that can abstract the H-radical from sp3 hybridized carbons. The combination of Cu catalysts and F-TEDA-X reagents is thus capable of oxidizing unactivated aliphatic C–H bonds. For example, Baran developed Cu-catalyzed Ritter-type aliphatic C–H amination with acetonitrile in the presence of F-TEDA-PF6 (Scheme 49).72,73 The C–H amination is likely enabled by a stepwise sequence involving (1) H-radical abstraction; (2) SET oxidation of the resulting C-radical 50 to carbocation 51; (3) Ritter-type amination by solvent acetonitrile. Nitrilium ion 52 is finally hydrolyzed to give acetamide products.
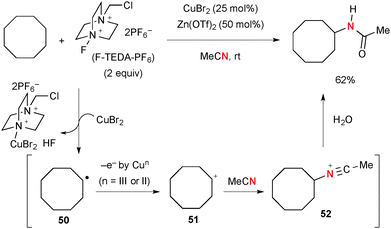 |
| | Scheme 49 CuBr2-catalyzed C–H amination of cyclooctane with F-TEDA-PF6. | |
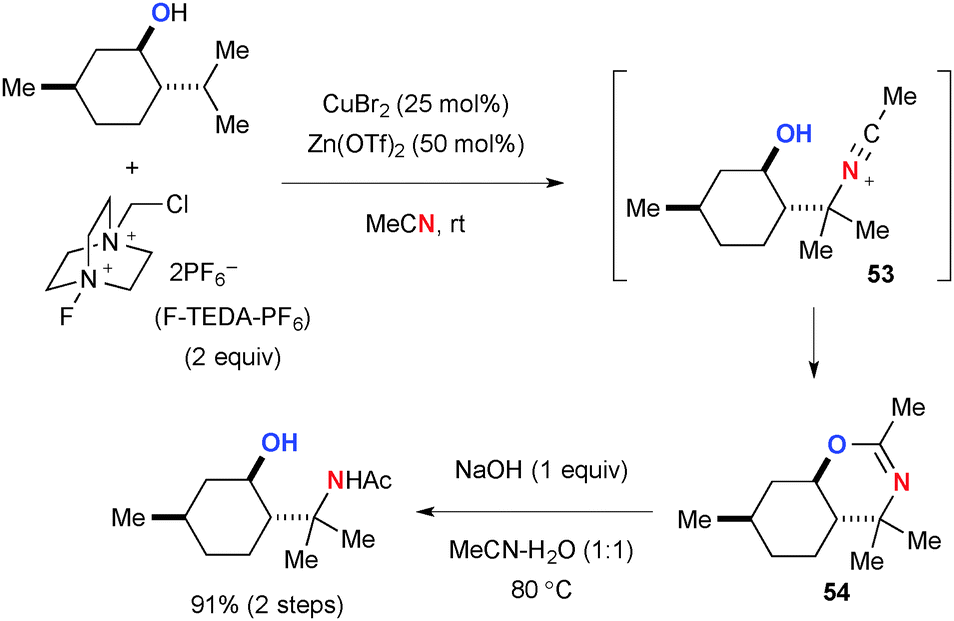
Interestingly, substrates having hydroxyl or carbonyl groups rendered the C–H amination process more chemo-selective and efficient presumably by their chelation effect. For example, the CuBr2-catalyzed reaction of (−)-menthol with F-TEDA-PF6 afforded the chemo-selective C–H amination product dihydrooxazine in very high yield through intramolecular trap of the transient nitrilium ion 53 by the hydroxyl group (Scheme 50). Dihydrooxazine moiety 54 could be easily hydrolyzed into the corresponding 1,3-aminoalcohol.
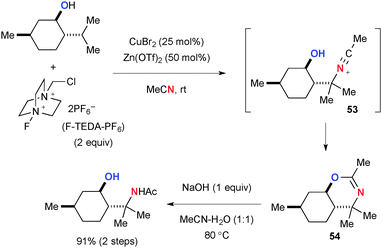 |
| | Scheme 50 CuBr2-catalyzed C–H amination of menthol with F-TEDA-PF6. | |
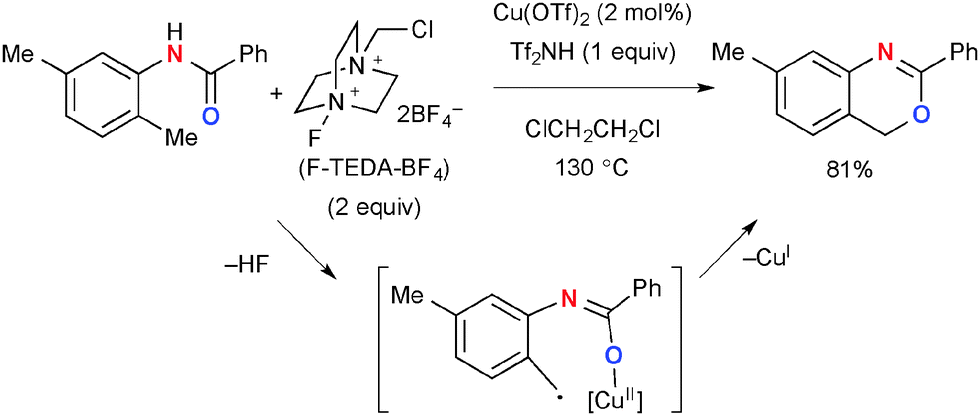
A similar directing effect in chemo-selective aliphatic C–H oxygenation was observed in the reactions of N-(2-alkylphenyl)benzamides in the presence of Cu(OTf)2 as the catalyst and F-TEDA-BF4 (Selectfluor®) for synthesis of 4H-3,1-benzoxazines through ortho-aliphatic C–H oxygenation (Scheme 51).74 The reactions selectively functionalize the ortho-alkyl group presumably via H-radical abstraction by the amide–Cu chelate intermediate, whereby aliphatic C–H bonds in the other positions (e.g. meta-methyl group) are kept intact.
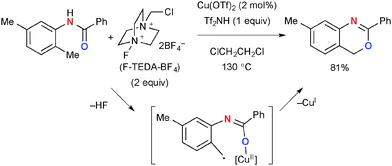 |
| | Scheme 51 Cu(OTf)2-catalyzed C–H oxygenation with F-TEDA-BF4. | |
8. With 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO)
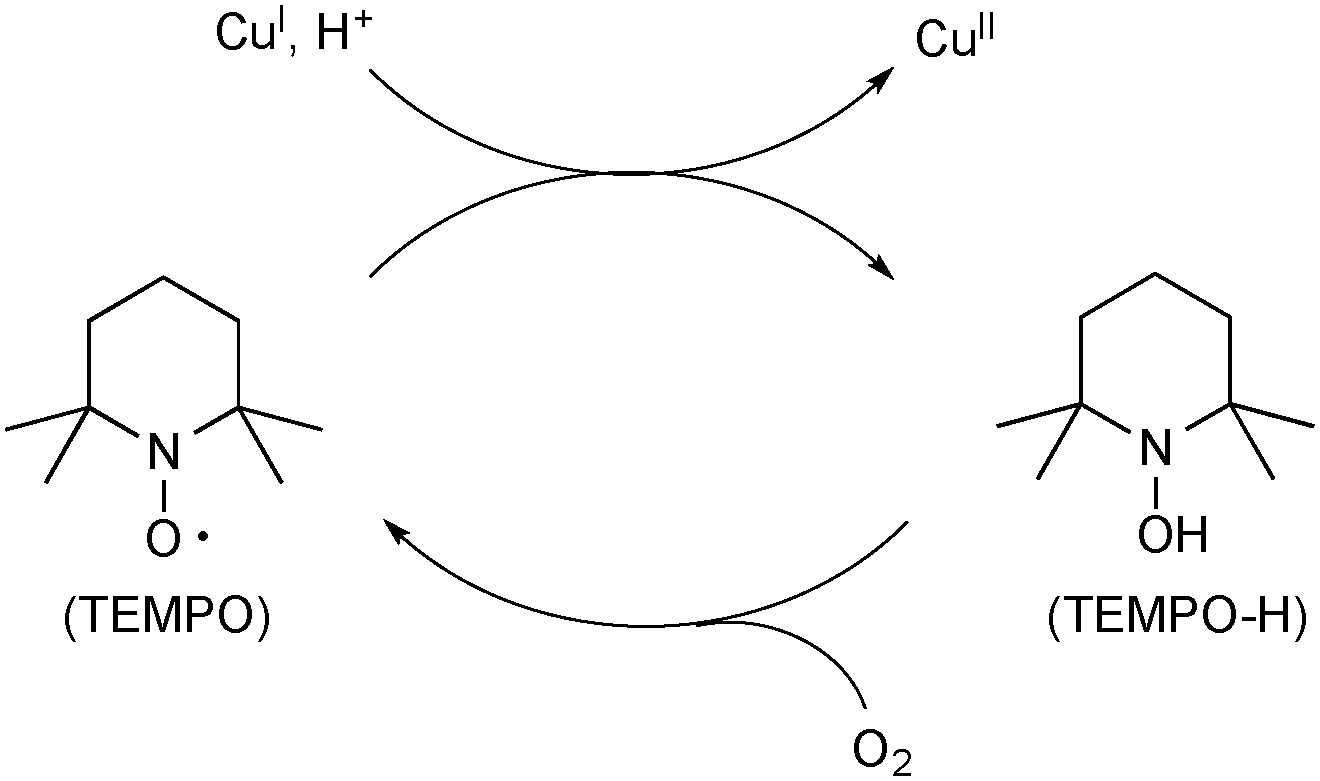
A persistent radical, TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxy), has been utilized for Cu-catalyzed oxidative C–O bond forming reactions. The unique feature of TEMPO is that it works as an oxidant of Cu(I) species to generate Cu(II) species and its reduced form TEMPO-H could be reoxidized by molecular O2 to regenerate TEMPO (Scheme 52).75
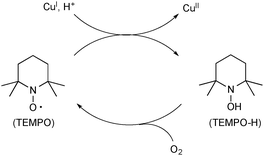 |
| | Scheme 52 Oxidation of Cu(I) species to Cu(II) species by TEMPO. | |
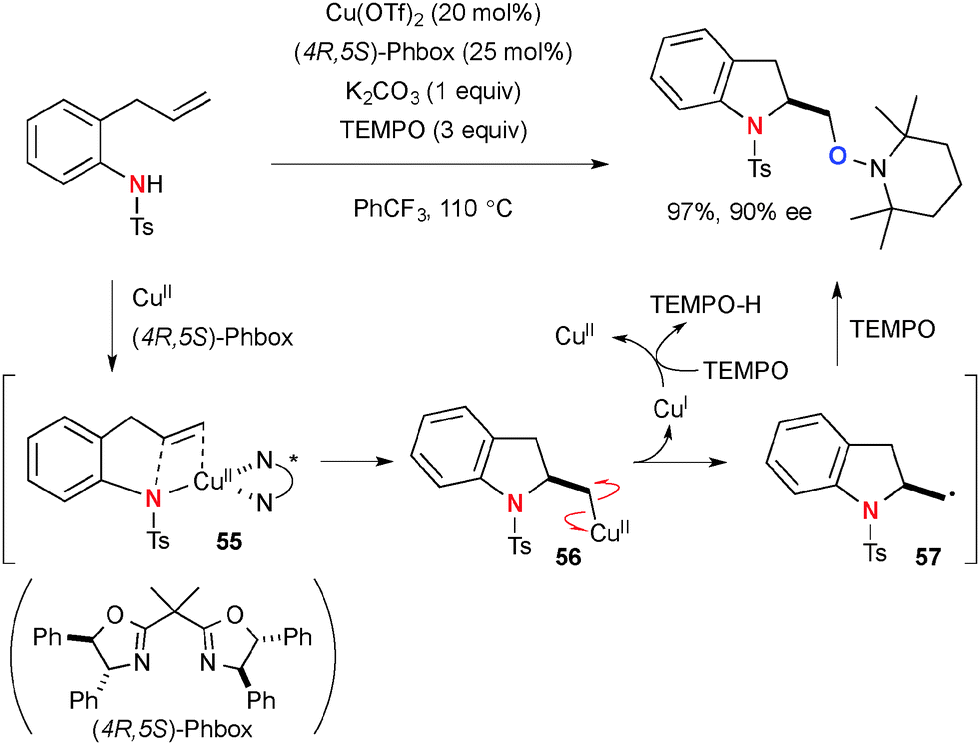
Chemler developed Cu-catalyzed enantioselective intra/intermolecular aminooxygenation of alkenyl N-sulfonamides with TEMPO for synthesis of indoline and pyrrolidine derivatives (Scheme 53), in which TEMPO plays two roles as the external oxygen source and stoichiometric oxidant to realize catalytic turnover.76 The detailed mechanistic analyses in experimental and theoretical manners revealed that the process is initiated by concerted syn-aminocupration of alkenes by Cu(II)-amido species 55 to construct heterocyclic frameworks having organocopper(II) moiety 56.77 Subsequent C–Cu(II) bond homolysis results in formation of C-radical 57 that is trapped by TEMPO to afford the aminooxygenation product. The resulting lower valent Cu(I) species is reoxidized to the Cu(II) complex by TEMPO.
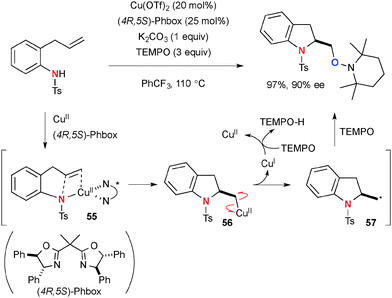 |
| | Scheme 53 Cu-Catalyzed aminooxygenation of alkenes with TEMPO. | |
In some cases especially when N-pentenylsulfonamides were employed, the use of molecular oxygen as an atmosphere could make the aminooxygenation process more efficient (Scheme 54). The TEMPO loading could be reduced to 1.5 equiv. as molecular oxygen serves as an oxidant to reoxidize TEMPO-H to TEMPO.
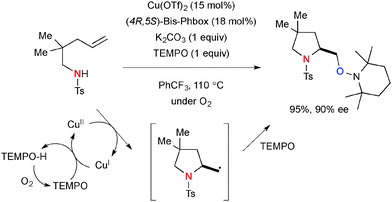 |
| | Scheme 54 Cu-Catalyzed aminooxygenation of alkenes with TEMPO under an oxygen atmosphere. | |
9. With metallic oxidants
Mild and cost-economical metallic oxidants have been employed as the terminal stoichiometric oxidants to regenerate higher valent active Cu species to realize catalytic turnover for the Cu-mediated oxidative molecular transformation.
9.1. MnO2
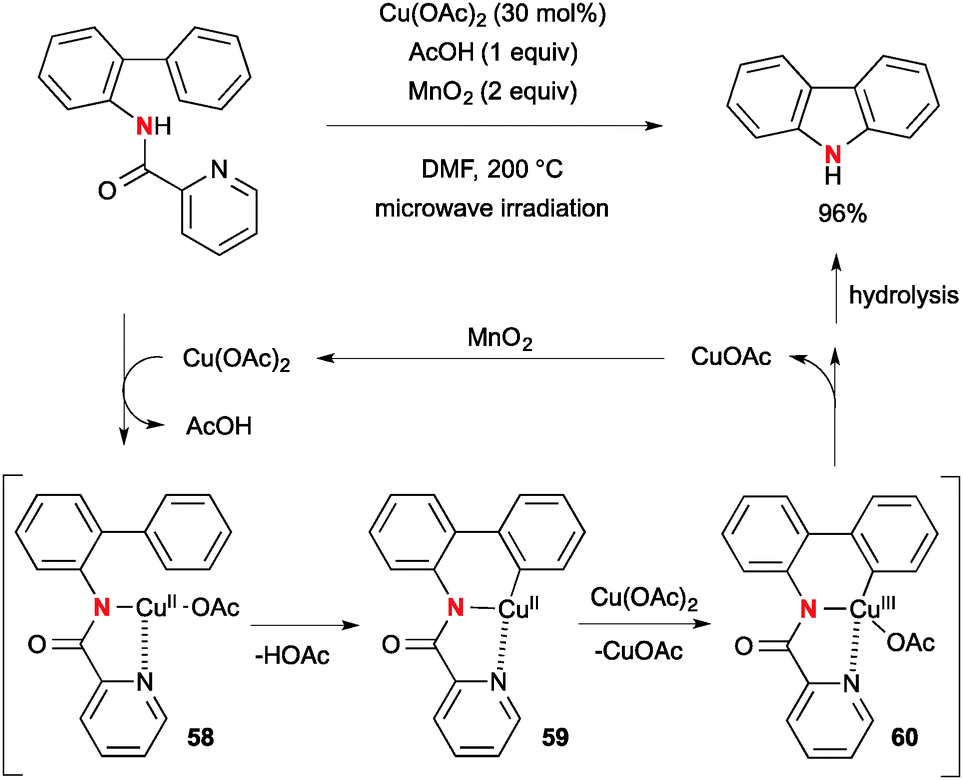
Hirano/Miura reported Cu(OAc)2-catalyzed intramolecular aromatic C–H amination of biaryl-2-picolinamide for synthesis of carbazoles with MnO2 as the stoichiometric terminal oxidant to realize the catalytic turnover (Scheme 55).78 The reaction is initiated by aromatic C–H cupration by copper(II)–picolinamide chelate complex 58 to afford organo-Cu(II) intermediate 59. Further redox disproportionation with Cu(OAc)2 forms copper(III) intermediate 60 and subsequent C–N bond reductive elimination establishes formation of the carbazole product along with generation of a lower valent Cu(I) species that is oxidized by MnO2 to regenerate the Cu(II) catalyst.
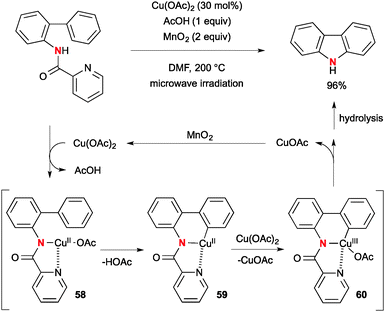 |
| | Scheme 55 Cu-Catalyzed-MnO2-mediated aromatic C–H amination for synthesis of carbazoles. | |
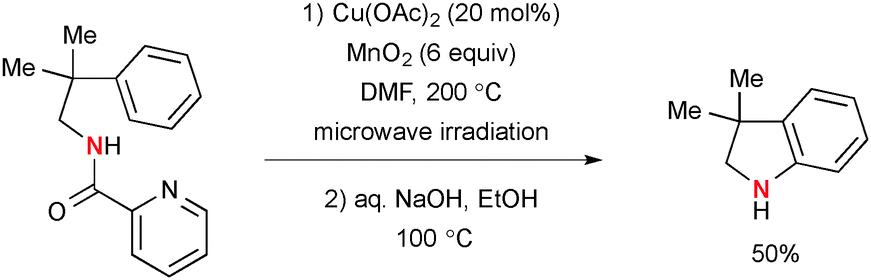
The analogous Cu-catalyzed-MnO2-mediated C–H amination strategy with picolinamides was applied for synthesis of indolines (Scheme 56).79
 |
| | Scheme 56 Cu-Catalyzed-MnO2-mediated aromatic C–H amination for synthesis of indolines. | |
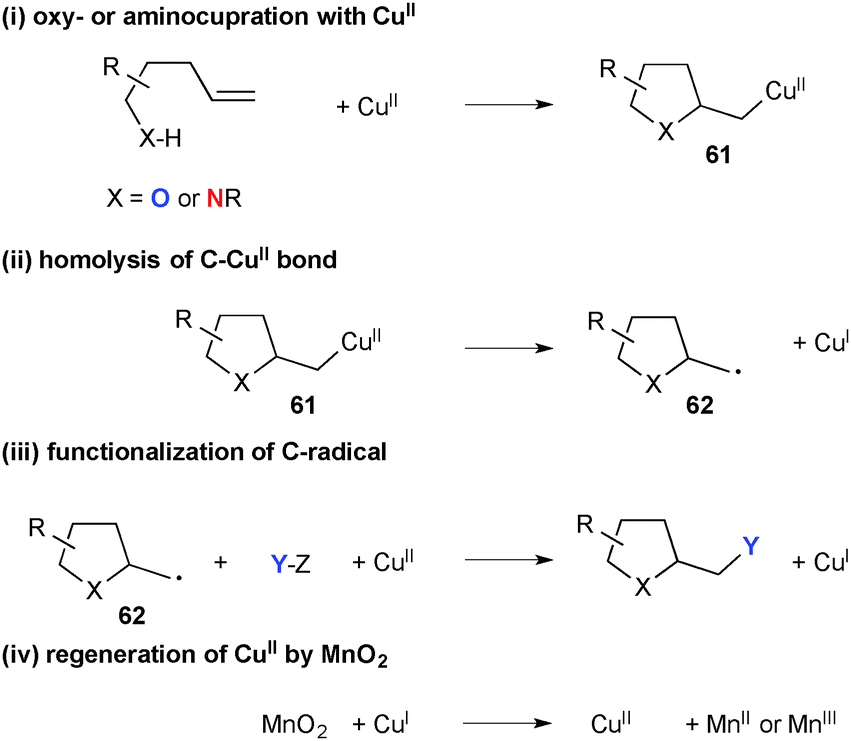
Using MnO2 as the terminal oxidant, Chemler developed a series of Cu(II)-catalyzed amino- and oxy-functionalization of alkenes with alkenylsulfonamides, -anilines, and -alcohols for synthesis of the corresponding heterocycles. As shown in Scheme 57, the process is composed of a multi-step sequence involving (i) amino- or oxy-cupration of alkenes to form five-membered ring organocopper(II) intermediates 61; (ii) C–Cu(II) bond homolysis to generate C-radical 62 and Cu(I) species; (iii) radical recombination with various internal/external carbon or heteroatom sources to provide difunctionalized final products; (iv) regeneration of the higher valent Cu(II) species by oxidation of the lower valent Cu(I) species by MnO2.
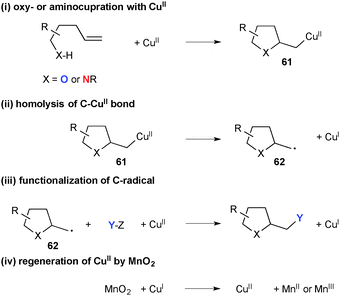 |
| | Scheme 57 Cu-Catalyzed-MnO2-mediated oxy- and amino-functionalization of alkenes. | |
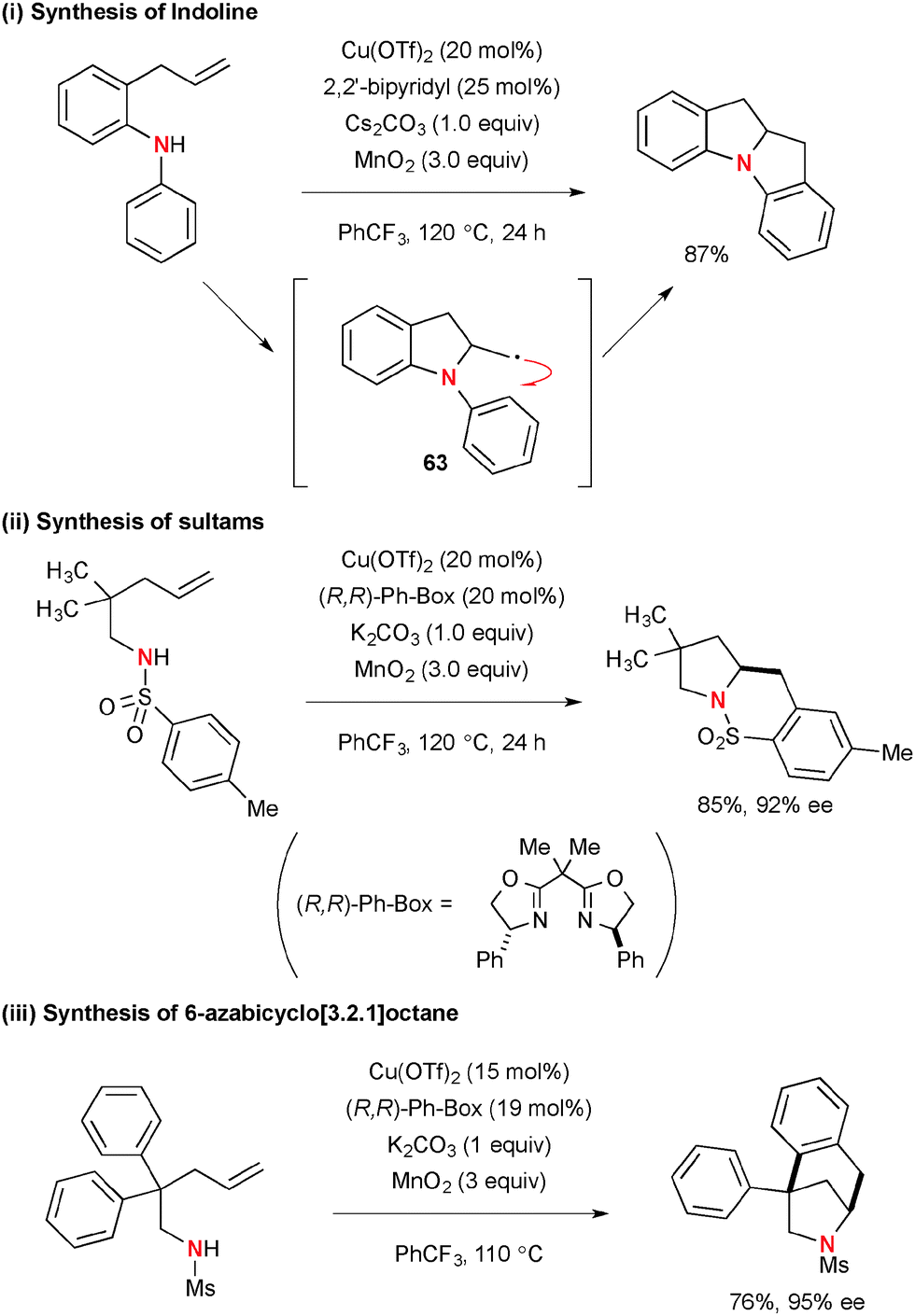
For example, treatment of N-aryl-2-allylaniline with 20 mol% of Cu(OTf)2 in the presence of Cs2CO3 and MnO2 afforded indoline derivative through intramolecular carboamination of alkenes (Scheme 58).80 In this process, the resulting C-radical 63 was added directly to the intramolecular benzene ring to construct the new C–C bond (Scheme 58-i). Enantioselective carboamination of N-pentenyl(p-tolyl)sulfonamides was enabled by using the (R,R)-Ph-Box ligand for the Cu(OTf)2 catalyst, delivering optically active bicyclic sultams (Scheme 58-ii). On the other hand, the reactions of N-mesyl-4-pentenylamines having the geminal diaryl moiety at the C2 position under the Cu(OTf)2-(R,R)-Ph-Box catalytic system in the presence of MnO2 provided 6-azabicyclo[3.2.1]octane in high enantioselectivity through carboamination of alkenes (Scheme 58-iii).81
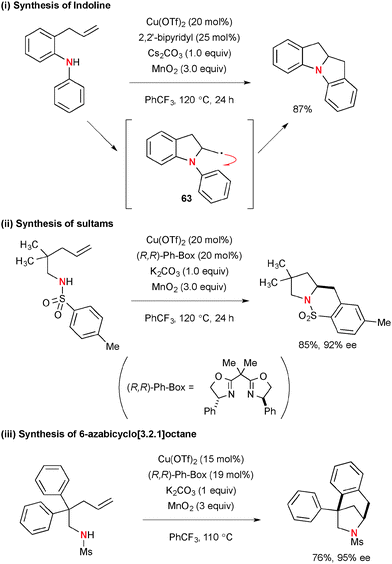 |
| | Scheme 58 Cu-Catalyzed-MnO2-mediated carboamination of alkenes. | |
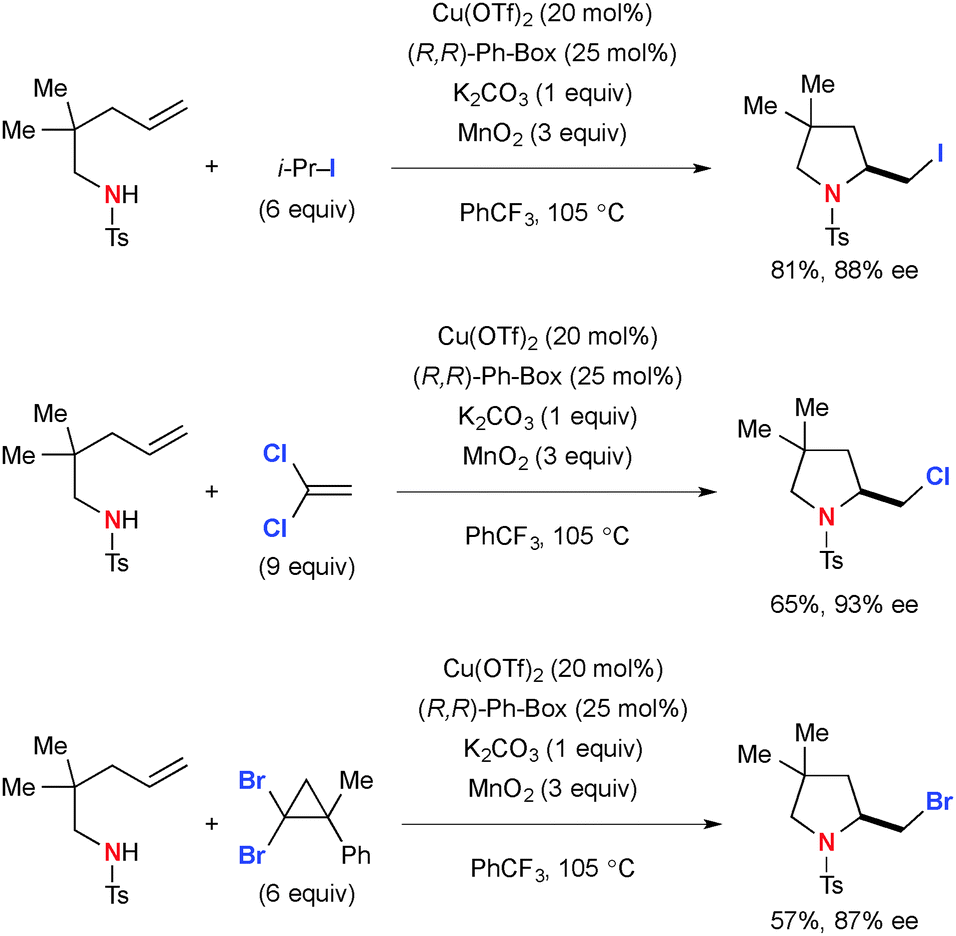
The transient C-radicals generated via aminocupration of N-sulfonyl alkenylamines could undergo an iodine transfer reaction with isopropyl iodide to form the corresponding 2-iodomethyl indolines and pyrrolidines (Scheme 59).82 Similarly, chlorination and bromination reactions were achieved in moderate yields using 1,1-dichloroethylene and (2,2-dibromo-1-methylcyclopropyl)benzene.
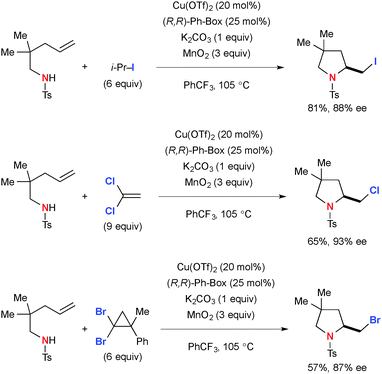 |
| | Scheme 59 Cu-Catalyzed-MnO2-mediated-aminohalogenation of alkenes. | |
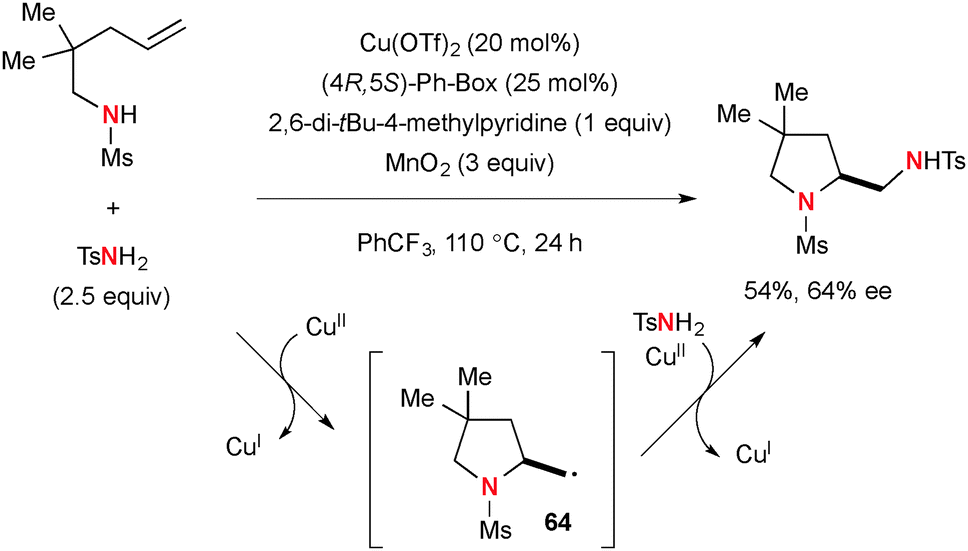
Cu-Catalyzed diamination of N-sulfonyl-alkenylamines was developed using tosylamide (TsNH2) as the external nitrogen source (Scheme 60).83 In this case, the second C–N bond formation was enabled by radical recombination of C-radical 64 with TsNH2 and Cu(II) species.
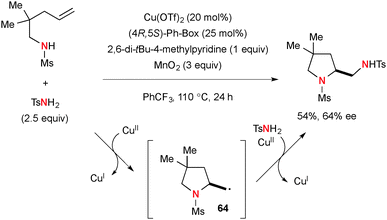 |
| | Scheme 60 Cu-Catalyzed-MnO2-mediated diamination of alkenes. | |
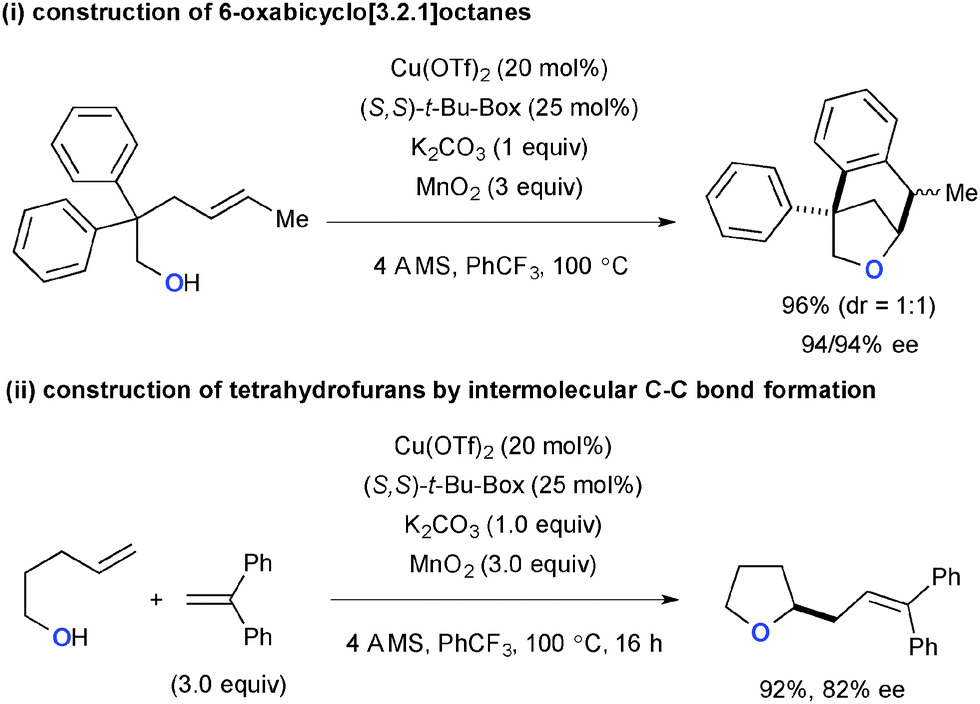
The Cu(OTf)2-catalyzed-MnO2-mediated reaction conditions were amenable to carboetherification of alkenyl alcohols, in which the second C–C bond formation was possible both in intra- and intermolecular fashions (Scheme 61).84 Construction of 6-oxabicyclo[3.2.1]octanes was carried out using 4-pentenylalcohol with a geminal diaryl moiety at the C2 position via oxycupration of alkenes followed by intramolecular radical cyclization on the aryl group (Scheme 61-i). Intermolecular C–C bond formation was also realized with aryl alkenes, in which the transient C-radical underwent the oxidative Heck-type coupling with aryl alkenes to deliver 2-allyltetrahydrofurans (Scheme 61-ii).
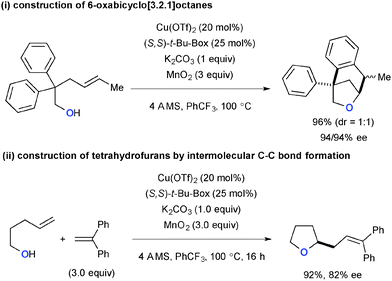 |
| | Scheme 61 Cu-Catalyzed-MnO2-mediated carboetherification of alkenes. | |
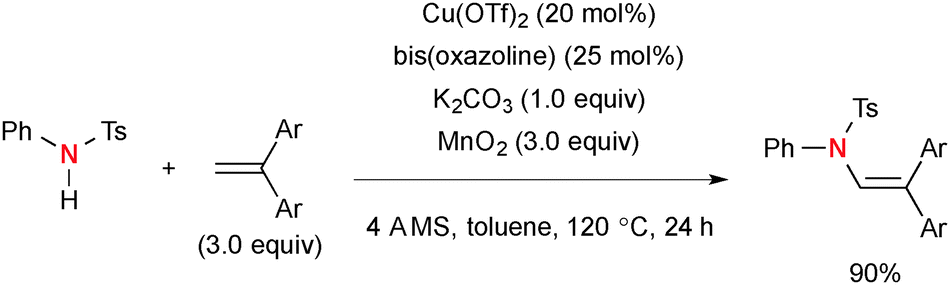
Chemler also reported Cu-catalyzed intermolecular amination of 1,1-disubstited alkenes with N-arylsulfonamides in the presence of MnO2 as a terminal oxidant (Scheme 62).85 The reactions uniquely afforded N-aryl enamide products in an anti-Markovnikov fashion.
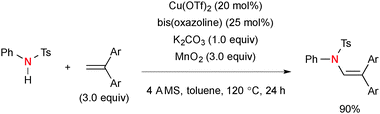 |
| | Scheme 62 Cu-Catalyzed-MnO2-mediated intermolecular amination of 1,1-disubstituted alkenes. | |
9.2. Ag2CO3
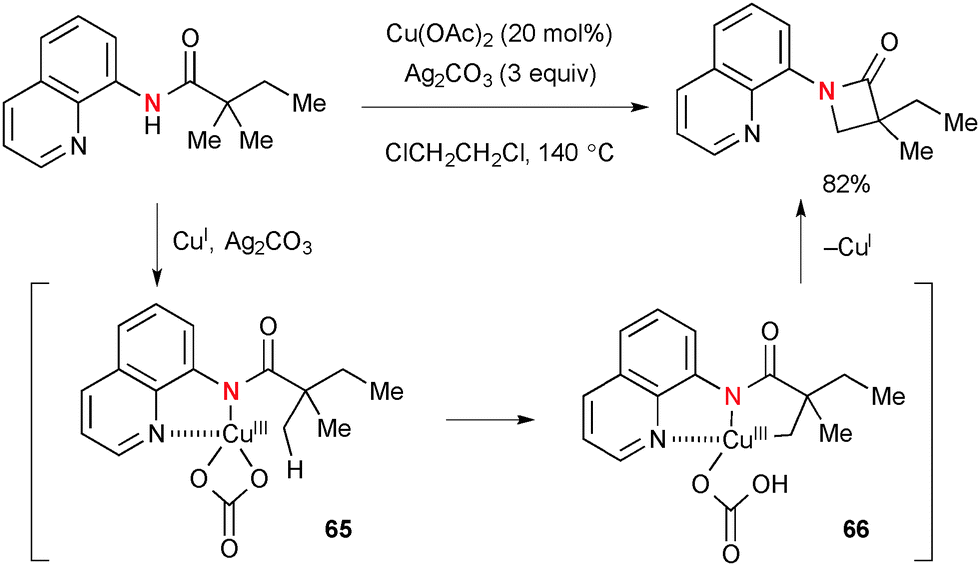
Ag2CO3 has been specifically employed as the stoichiometric oxidant for Cu-catalyzed aliphatic and aromatic C–H oxidation. Kuninobu/Kanai reported synthesis of β-lactams by Cu-catalyzed intramolecular sp3 C–H amidation of N-(8-quinolinyl)amides in the presence of Ag2CO3 (Scheme 63).86,87 Installation of the N-8-quinolinyl moiety is crucial on the amide substituent to allow it to serve as the bidentate directing group. The C–H functionalization is mediated by the transient amide–Cu(III) complex 65 through concerted metalation–deprotonation on sp3-C–H bonds with the acetate or carbonate counter ions (the reaction with the carbonate counter ion is shown below) on the copper to provide metallacycle intermediate 66. Finally, C–N bond forming reductive elimination affords β-lactam along with a lower valent CuI species that reacts further with amide and Ag2CO3 to regenerate the amide–Cu(III) complex to maintain the catalytic turnover.
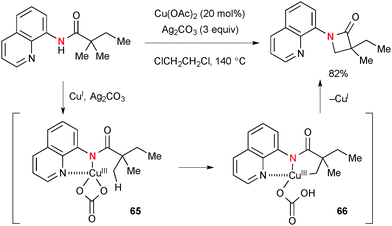 |
| | Scheme 63 Cu-Catalyzed-Ag2CO3-mediated directed sp3 C–H amidation. | |
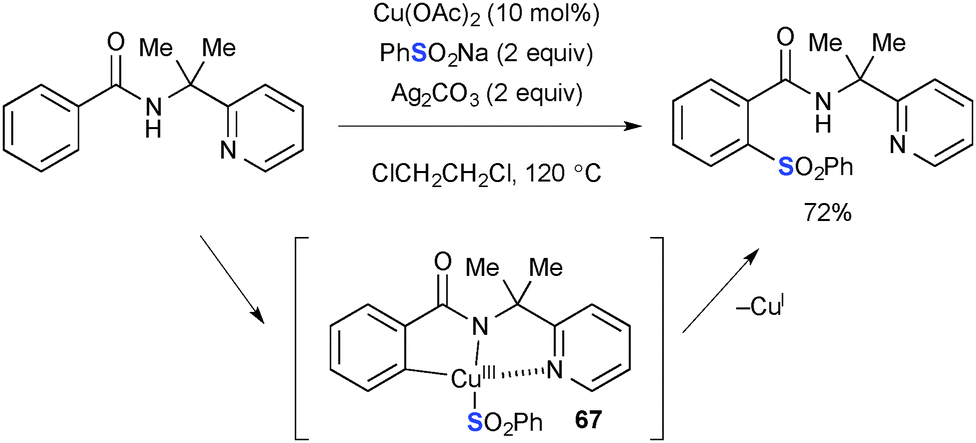
The Cu(OAc)2–Ag2CO3 reaction system was also utilized for aromatic C–H functionalization of benzamides having a 2-pyridylmethyl moiety on the amide nitrogen through directed concerted metalation–deprotonation. Shi demonstrated Cu-catalyzed ortho-aromatic C–H sulfonylation of benzamides using sodium sulfinate as the sulfonylation reagent (Scheme 64).88 The C–S bond forming reductive elimination from the transient Cu(III) metallacycle 67 furnishes the sulfonylation product.
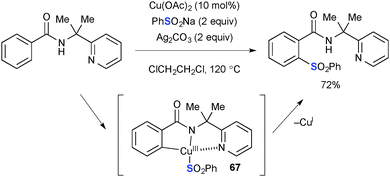 |
| | Scheme 64 Cu-Catalyzed-Ag2CO3-mediated directed sp2 C–H sulfonylation. | |
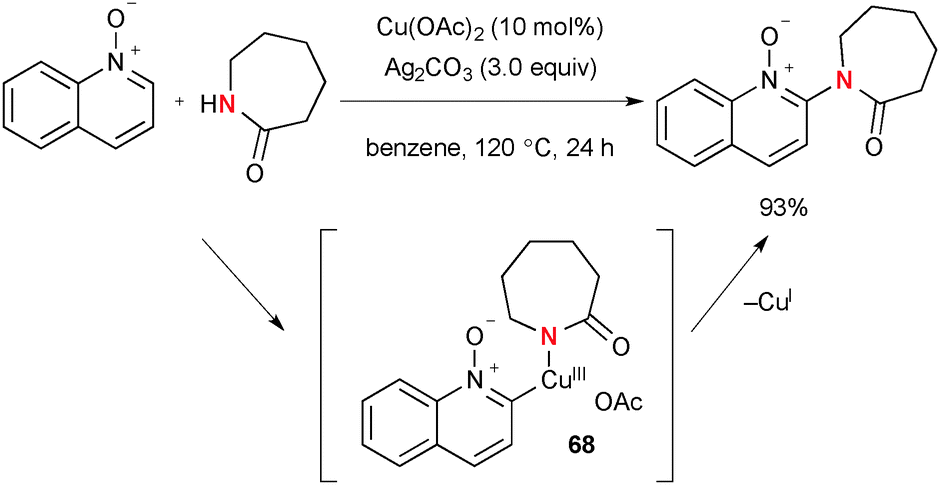
Intermolecular sp2 C–H amidation/amination of quinoline N-oxides was also reported under the Cu(OAc)2–Ag2CO3 reaction system (Scheme 65).89 Various lactams/cyclic amines are incorporated into the key organocopper(III) intermediate 68 prior to its C–N bond reductive elimination to deliver the final products.
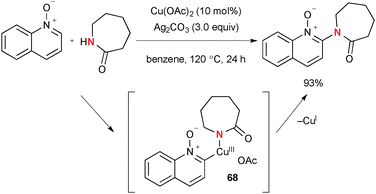 |
| | Scheme 65 Cu-Catalyzed-Ag2CO3-mediated sp2 C–H amidation. | |
10. Information of the oxidants: their commercial availability and preparation methods
Among the terminal oxidants for the Cu-catalyzed oxidative carbon–heteroatom bond formation discussed in this review, the price of the commercially available ones from Sigma-Aldrich is summarized in Table 1. Togni reagent II90 (entry 4) and peroxides (entries 5–7) are potentially explosive, so the reactions with these reagents should need special care with proper protecting shields.
Table 1 A price list of commercially available oxidants
| Entry |
Oxidant |
Price (US$)a |
CAS no. |
|
http://www.sigmaaldrich.com/united-states.html.
|
| 1 |
PhI(OAc)2 |
21.6/5 g |
3240-34-4 |
| 2 |
Ph2IOTf |
114/1 g |
66003-76-7 |
| 3 |
Togni reagent I |
66.4/250 mg |
887144-97-0 |
| 4 |
Togni reagent II |
50.6/250 mg |
887144-94-7 |
| 5 |
t-BuOOt-Bu |
48.1/250 mL |
110-05-4 |
| 6 |
t-BuOOH (5–6 M in decane) |
151/100 mL |
75-91-2 |
| 7 |
(PhCO2)2 |
43.5/50 g |
94-36-0 |
| 8 |
(PhSO2)2NF (NFSI) |
77.2/5 g |
133745-75-2 |
| 9 |
Selectfluor® |
34.1/5 g |
140681-55-6 |
| 10 |
TEMPO |
44.9/5 g |
2564-83-2 |
| 11 |
MnO2 (>99%) |
49.4/100 g |
1313-13-9 |
| 12 |
Ag2CO3 (>99%) |
34.9/5 g |
534-16-7 |
The typical preparation methods of non-commercialized oxidants such as vinyl(aryl)iodonium triflate (Section 2.2.), O-benzoyl-N,N-dialkylhydroxylamines (Section 4), oxaziridines (Section 5), and diaziridinones (Section 6) are illustrated in Schemes 66–69, respectively.
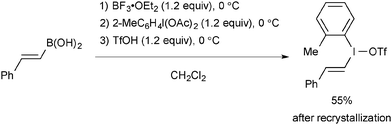 |
| | Scheme 66 Preparation of N-noxyl-3,3-dimethyloxaziridine. | |
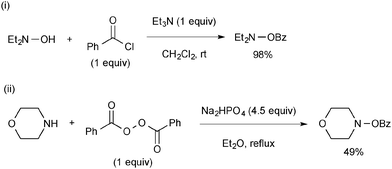 |
| | Scheme 67 Preparation of O-benzoyl-N,N-dialkylhydroxylamines. | |
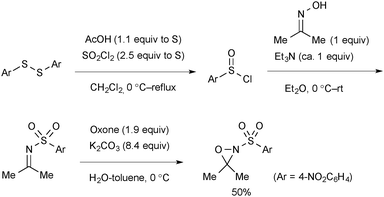 |
| | Scheme 68 Preparation of N-noxyl-3,3-dimethyloxaziridine. | |
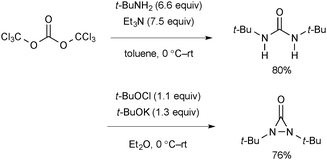 |
| | Scheme 69 Preparation of N,N-di-t-butyldiaziridinone. | |
Vinyl(aryl)iodonium triflate is readily prepared from the sequential treatment of the corresponding alkenylboronic acids with BF3·OEt2, 2-iodotoluene diacetate, and TfOH (Scheme 66).91
Preparation of O-benzoyl-N,N-dialkylhydroxylamines is conducted either by benzoylation of N,N-dialkylhydroxylamines with benzoyl chloride (Scheme 67-i) or by nucleophilic substitution reactions of dibenzoyl peroxide with the corresponding secondary amines (Scheme 67-ii).92O-Benzoyl-N,N-dialkylhydroxylamines should be stored in the freezer.
Oxaziridines are synthesized by oxidation of the corresponding imines with Oxone®. The scalable procedure for synthesis of N-nosyl-3,3-dimethyloxaziridine reported by Yoon is shown in Scheme 68.54,93
The synthetic procedure of N,N-di-t-butyldiaziridinone includes a sequence of preparation of di-t-butylurea and oxidative intramolecular N–N bond formation (Scheme 69).94N,N-di-t-butyldiaziridinone should be stored in the dark.
11. Conclusions
This review highlighted up-to-date developments in copper-catalyzed (anaerobic) oxidative formation of carbon–heteroatom bonds on C–H bonds and alkenes. Various combinations of copper catalysts and readily available stoichiometric oxidants have been devised to enable unique and unprecedented oxidative molecular transformations. Unlike other transition metals, the reaction modes enabled by the copper species are multifarious. For example, many of the carbon–heteroatom bond-forming processes in Cu-catalyzed oxidative molecular transformations include organocopper (C–Cu) species as the key intermediates. The chemical reactivity of the organocopper species is uniquely diverse depending on the oxidation state of the copper moiety. Higher valent C–CuIII species undergo substitution reactions with heteroatom nucleophiles or reductive elimination of the C–heteroatom bond, whereas lower valent C–CuI species exhibit a nucleophilic character to react with heteroatom electrophiles such as O-benzoyl-N,N-dialkylhydroxylamines. On the other hand, C–CuII species undergo homolysis to generate C-radicals that could be further functionalized with radical trapping reagents such as TEMPO or heteroatom nucleophiles through radical recombination pathways with the aid of CuII species. More challenges and opportunities still remain for elucidation of the detailed reaction mechanisms and identification of active Cu-species that control the course of the reaction and improve catalytic turnovers and overall process efficiency. We anticipate that copper complexes have inexhaustible potential as catalysts to enhance our synthetic capability further.
Acknowledgements
Our co-workers whose names appear in the references are gratefully acknowledged for their intellectual and experimental contributions. This work was supported by funding from Nanyang Technological University and Singapore Ministry of Education (Academic Research Fund Tier 2: MOE2010-T2-1-009 and MOE2012-T2-1-014).
Notes and references
- For recent selected reviews on transition-metal-catalyzed C–H oxidation, see:
(a) J. L. Jeffrey and R. Sarpong, Chem. Sci., 2013, 4, 4092 RSC;
(b) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS PubMed;
(c) D. Y.-K. Chen and S. W. Youn, Chem. – Eur. J., 2012, 18, 9452 CrossRef CAS PubMed;
(d) M. C. White, Science, 2012, 335, 807 CrossRef CAS PubMed;
(e) R. T. Gephart III and T. H. Warren, Organometallics, 2012, 31, 7728 CrossRef;
(f) J. L. Roizen, M. E. Harvey and J. Du Bois, Acc. Chem. Res., 2012, 45, 911 CrossRef CAS PubMed;
(g) T. A. Ramirez, B. Zhao and Y. Shi, Chem. Soc. Rev., 2012, 41, 931 RSC;
(h) H. M. L. Davies, J. Du Bois and J.-Q. Yu, Chem. Soc. Rev., 2011, 40, 1855 RSC;
(i) T. Newhouse and P. S. Baran, Angew. Chem., Int. Ed., 2011, 50, 3362 CrossRef CAS PubMed;
(j) J. Du Bois, Org. Process Res. Dev., 2011, 15, 758 CrossRef CAS PubMed;
(k) F. Collet, C. Lescot and P. Dauban, Chem. Soc. Rev., 2011, 40, 1926 RSC;
(l) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed;
(m) F. Collet, R. H. Dodd and P. Dauban, Chem. Commun., 2009, 5061 RSC;
(n) M. M. Díaz-Requejo and P. J. Pérez, Chem. Rev., 2008, 108, 3379 CrossRef PubMed.
- For recent selected reviews on transition-metal-catalyzed difunctionalization of alkenes, see:
(a) X. Zeng, Chem. Rev., 2013, 113, 6864 CrossRef CAS PubMed;
(b) S. R. Chemler and M. T. Bovino, ACS Catal., 2013, 3, 1076 CrossRef CAS PubMed;
(c) K. Muñiz and C. Martínez, J. Org. Chem., 2013, 78, 2168 CrossRef PubMed;
(d) W. Wu and H. Jiang, Acc. Chem. Res., 2012, 45, 1736 CrossRef CAS PubMed;
(e) S. E. Denmark, W. E. Kuester and M. T. Burk, Angew. Chem., Int. Ed., 2012, 51, 10938 CrossRef CAS PubMed;
(f) T. J. Donohoe, C. K. A. Callens, A. R. Lacy and C. Winter, Eur. J. Org. Chem., 2012, 655 CrossRef CAS;
(g) D. M. Schultz and J. P. Wolfe, Synthesis, 2012, 351 CAS;
(h) R. I. McDonald, G. Liu and S. S. Stahl, Chem. Rev., 2011, 111, 2981 CrossRef CAS PubMed;
(i) R. I. McDonald, G. Liu and S. S. Stahl, J. Am. Chem. Soc., 2011, 111, 2981 CAS;
(j) K. H. Jensen and M. S. Sigman, Org. Biomol. Chem., 2008, 6, 4083 RSC;
(k) J. P. Wolfe, Synlett, 2008, 2913 CrossRef CAS;
(l) K. H. Jensen and M. S. Sigman, Org. Biomol. Chem., 2008, 6, 1153 RSC;
(m) A. Minatti and K. Muñiz, Chem. Soc. Rev., 2007, 36, 1142 RSC;
(n) E. M. Beccalli, G. Broggini, M. Martinelli and S. Sottocomola, Chem. Rev., 2007, 107, 5318 CrossRef CAS PubMed;
(o) S. R. Chemler and P. H. Fuller, Chem. Soc. Rev., 2007, 36, 1153 RSC;
(p) A. Minatti and K. Muñiz, Chem. Soc. Rev., 2007, 36, 1142 RSC.
- For reviews on molecular transformations catalyzed by front row transition metals, see:
(a) B. Su, Z.-C. Cao and Z.-J. Shi, Acc. Chem. Res., 2015, 48, 886 CrossRef CAS PubMed;
(b) J. Miao and H. Ge, Eur. J. Org. Chem., 2015, 7859 CrossRef CAS . For Fe catalysis, see: ;
(c) I. Bauer and H.-J. Knölker, Chem. Rev., 2015, 115, 3170 CrossRef CAS PubMed;
(d) J. Cornil, L. Gonnard, C. Bensoussan, A. Serra-Muns, C. Gnamm, C. Commandeur, M. Commandeur, S. Reymond, A. Guérinot and J. Cossy, Acc. Chem. Res., 2015, 48, 761 CrossRef CAS PubMed;
(e) F. Jia and Z. Li, Org. Chem. Front., 2014, 1, 194 RSC;
(f) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Rev., 2011, 111, 1293 CrossRef CAS PubMed;
(g) A. A. O. Sarhan and C. Bolm, Chem. Soc. Rev., 2009, 38, 2730 RSC;
(h) A. Correa, O. G. Mancheño and C. Bolm, Chem. Soc. Rev., 2008, 37, 1108 RSC . For Co catalysis, see: ;
(i) M. Moselage, J. Li and L. Ackermann, ACS Catal., 2016, 6, 498 CrossRef CAS;
(j) K. Gao and N. Yoshikai, Acc. Chem. Res., 2014, 47, 1208 CrossRef CAS PubMed . For Mn catalysis, see: ;
(k) W. Liu and J. T. Groves, Acc. Chem. Res., 2015, 48, 1727 CrossRef CAS PubMed.
- For recent selected reviews on Cu-catalyzed molecular transformation, see:
(a) C. U. Maheswari, G. S. Kumar and K. R. Reddy, Curr. Org. Chem., 2016, 20, 512 CrossRef CAS;
(b) X. X. Guo, D. W. Gu, Z. Wu and W. Zhang, Chem. Rev., 2015, 115, 1622 CrossRef CAS PubMed;
(c) Y. Zhu, R. G. Cornwall, H. Du, B. Zhao and Y. Shi, Acc. Chem. Res., 2014, 47, 3665 CrossRef CAS PubMed;
(d) S. A. Girard, T. Knauber and C.-J. Li, Angew. Chem., Int. Ed., 2014, 53, 74 CrossRef CAS;
(e) X. Yan, X. Yang and C. Xi, Catal. Sci. Technol., 2014, 4, 4169 RSC;
(f) Y. Shimizu and M. Kanai, Tetrahedron Lett., 2014, 55, 3727 CrossRef CAS;
(g) J. X. Qiao and P. Y. S. Lam, Synthesis, 2011, 829 CrossRef CAS;
(h) H. Rao and H. Fu, Synlett, 2011, 745 CAS;
(i) J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302 RSC;
(j) F. Monnier and M. Taillefer, Angew. Chem., Int. Ed., 2009, 48, 6954 CrossRef CAS PubMed;
(k) G. Evano, N. Blanchard and M. Toumi, Chem. Rev., 2008, 108, 3054 CrossRef CAS PubMed;
(l) D. Ma and Q. Cai, Acc. Chem. Res., 2008, 41, 1450 CrossRef CAS PubMed;
(m) S. R. Chemler and P. H. Fuller, Chem. Soc. Rev., 2007, 36, 1153 RSC;
(n) I. P. Beletshkaya and A. V. Cheprakov, Coord. Chem. Rev., 2004, 248, 2337 CrossRef;
(o) S. V. Ley and A. W. Thomas, Angew. Chem., Int. Ed., 2003, 42, 5400 CrossRef CAS PubMed.
- The oxidation state of Cu species in the catalytic cycle should be carefully considered. Even if the reaction is initiated by Cu(II) salts and oxidants, the active higher valent Cu species in the catalytic cycle is not necessarily Cu(III) as Cu(II) species could be readily reduced to Cu(I) by various factors such as nucleophiles and solvents. For several relevant literature precedents, see:
(a) S. Chiba, Chem. Lett., 2012, 41, 1554 CrossRef CAS;
(b) Y.-F. Wang, K. K. Toh, J.-Y. Lee and S. Chiba, Angew. Chem., Int. Ed., 2011, 50, 5927 CrossRef CAS PubMed;
(c) J. Kim and S. Chang, J. Am. Chem. Soc., 2010, 132, 10272 CrossRef CAS PubMed;
(d) R. J. Phipps, N. P. Grimster and M. J. Gaunt, J. Am. Chem. Soc., 2008, 130, 8172 CrossRef CAS PubMed;
(e) S. Liu, Y. Tu and L. S. Liebeskind, Org. Lett., 2007, 9, 1947 CrossRef CAS PubMed;
(f) A. Y. S. Malkhasian, M. E. Finch, B. Nikolovski, A. Menon, B. E. Kucera and F. A. Chavez, Inorg. Chem., 2007, 46, 2950 CrossRef CAS PubMed;
(g) J. J. Teo, Y. Chang and H. C. Zeng, Langmuir, 2006, 22, 7369 CrossRef CAS PubMed.
- Cu(II) species could undergo disproportionation to generate higher valent Cu(III) and lower valent Cu(I) species to facilitate the catalytic turnover. For a relevant review, see: A. Casitas and X. Ribas, Chem. Sci., 2013, 4, 2301 RSC.
- For recent reviews on copper-catalyzed aerobic oxidation of organic molecules, see:
(a) S. D. McCann and S. S. Stahl, Acc. Chem. Res., 2015, 48, 1756 CrossRef CAS PubMed;
(b) S. Chiba, Bull. Chem. Soc. Jpn., 2013, 86, 1400 CrossRef CAS;
(c) S. E. Allen, R. R. Walvoord, R. Padilla-Salinas and M. C. Kozlowski, Chem. Rev., 2013, 113, 6234 CrossRef CAS PubMed;
(d) A. N. Campbell and S. S. Stahl, Acc. Chem. Res., 2012, 45, 851 CrossRef CAS PubMed;
(e) Z. Shi, C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3381 RSC;
(f) C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3464 RSC;
(g) A. E. Wendlandt, A. M. Suess and S. S. Stahl, Angew. Chem., Int. Ed., 2011, 50, 11062 CrossRef CAS PubMed.
- J. M. Lee, E. J. Park, S. H. Cho and S. Chang, J. Am. Chem. Soc., 2008, 130, 7824 CrossRef CAS PubMed.
- H. Chen, S. Sanjaya, Y.-F. Wang and S. Chiba, Org. Lett., 2013, 15, 212 CrossRef CAS PubMed.
- For a recent review, see: S. Chiba and H. Chen, Org. Biomol. Chem., 2014, 12, 4051 CAS.
-
(a) Q. Li, S.-Y. Zhang, G. He, Z. Ai, W. A. Nack and G. Chen, Org. Lett., 2014, 16, 1764 CrossRef CAS PubMed;
(b) Á. M. Martínez, N. Rodríguez, R. G. Arrayás and J. C. Carretero, Chem. Commun., 2014, 50, 2801 RSC.
- D. E. Manncheno, A. R. Thornton, A. H. Stoll, A. Kong and S. B. Blakey, Org. Lett., 2010, 12, 4110 CrossRef PubMed.
- W.-H. Rao, X.-S. Yin and B.-F. Shi, Org. Lett., 2015, 17, 3758 CrossRef CAS PubMed.
- S. Sanjaya and S. Chiba, Org. Lett., 2012, 14, 5342 CrossRef CAS PubMed.
- R. Zhu and S. L. Buchwald, J. Am. Chem. Soc., 2015, 137, 8069 CrossRef CAS PubMed.
- E. Cahard, N. Bremeyer and M. J. Gaunt, Angew. Chem., Int. Ed., 2013, 52, 9284 CrossRef CAS PubMed.
- E. Cahard, H. P. J. Male, M. Tissot and M. J. Gaunt, J. Am. Chem. Soc., 2015, 137, 7986 CrossRef CAS PubMed.
-
(a) H. Egami, R. Shimizu and M. Sodeoka, Tetrahedron Lett., 2012, 53, 5503 CrossRef CAS;
(b) P. G. Janson, I. Ghoneim, N. O. Ilchenko and K. J. Szabó, Org. Lett., 2012, 14, 2882 CrossRef CAS PubMed.
-
(a) R. Zhu and S. L. Buchwald, Angew. Chem., Int. Ed., 2013, 52, 12655 CrossRef CAS PubMed;
(b) R. Zhu and S. L. Buchwald, J. Am. Chem. Soc., 2012, 134, 12462 CrossRef CAS PubMed.
-
(a) F. Wang, X. Qi, Z. Liang, P. Chen and G. Liu, Angew. Chem., Int. Ed., 2014, 53, 1881 CrossRef CAS PubMed;
(b) M. Yang, W. Wang, Y. Liu, L. Feng and X. Ju, Chin. J. Chem., 2014, 32, 833 CrossRef CAS.
- Z. Liang, F. Wang, P. Chen and G. Liu, Org. Lett., 2015, 17, 2438 CrossRef CAS PubMed.
- For a review, see: M. B. Andrus and J. C. Lashley, Tetrahedron, 2002, 58, 845 CrossRef CAS.
-
(a) R. T. Gephart III, C. L. McMullin, N. G. Sapiezynski, E. S. Jang, M. J. B. Aguila, T. R. Cundari and T. H. Warren, J. Am. Chem. Soc., 2012, 134, 17350 CrossRef PubMed;
(b) R. T. Gephart III, D. L. Huang, M. J. B. Aguila, G. Schmidt, A. Shahu and T. H. Warren, Angew. Chem., Int. Ed., 2012, 51, 6488 CrossRef PubMed;
(c) S. Wiese, Y. M. Badiei, R. T. Gephart, S. Mossin, M. S. Varonka, M. M. Melzer, K. Meyer, T. R. Cundari and T. H. Warren, Angew. Chem., Int. Ed., 2010, 49, 8850 CrossRef CAS PubMed.
- B. L. Tran, B. Li, M. Driess and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 2555 CrossRef CAS PubMed.
-
(a) H.-T. Zeng and J.-M. Huang, Org. Lett., 2015, 17, 4276 CrossRef CAS PubMed;
(b) F. Teng, S. Sun, Y. Jiang, J.-T. Yu and J. Cheng, Chem. Commun., 2015, 51, 5902 RSC.
- P. K. Chikkade, Y. Kuninobu and M. Kanai, Chem. Sci., 2015, 6, 3195 RSC.
- N. Takemura, Y. Kuninobu and M. Kanai, Org. Biomol. Chem., 2014, 12, 2528 CAS.
- C. Chatalova-Sazepin, Q. Wang, G. M. Sammis and J. Zhu, Angew. Chem., Int. Ed., 2015, 54, 5443 CrossRef CAS PubMed.
- A. L. García-Cabeza, R. Martín-Barrios, F. J. Moreno-Dorado, M. J. Ortega, G. M. Massanet and F. M. Guerra, Org. Lett., 2014, 16, 1598 CrossRef PubMed.
- For other recent reports on Cu-catalyzed aliphatic C–H oxidation with TBHP, see:
(a) D. Talukdar, S. Borah and M. K. Chaudhuri, Tetrahedron Lett., 2015, 56, 2555 CrossRef CAS;
(b) G. S. Kumar, B. Pieber, K. R. Reddy and C. O. Kappe, Chem. – Eur. J., 2012, 18, 6124 CrossRef CAS PubMed;
(c) Y. Zhang, H. Fu, Y. Jiang and Y. Zhao, Org. Lett., 2007, 9, 3813 CrossRef CAS PubMed.
- S. K. Rout, S. Guin, K. K. ghara, A. Banerjee and B. K. Patel, Org. Lett., 2012, 14, 3982 CrossRef CAS PubMed.
- S. K. Rout, S. Guin, W. Ali, A. Gogoi and B. K. Patel, Org. Lett., 2014, 16, 3086 CrossRef CAS PubMed.
- For an analogous ester synthesis from benzyl alcohols and cyclic ethers, see: K. B. Raju, B. N. Kumar and K. Nagaiah, RSC Adv., 2014, 4, 50795 RSC.
- J.-S. Tian and T.-P. Loh, Chem. Commun., 2011, 47, 5458 RSC.
- C. Tang and N. Jiao, J. Am. Chem. Soc., 2012, 134, 18924 CrossRef CAS PubMed.
- D. Mahesh, P. Sadhu and T. Punniyamurthy, J. Org. Chem., 2015, 80, 1644 CrossRef CAS PubMed.
-
(a) S. K. Rout, S. Guin, A. Gogoi, G. Majji and B. K. Patel, Org. Lett., 2014, 16, 1614 CrossRef CAS PubMed;
(b) A. B. Khemnar and B. M. Bhanage, Org. Biomol. Chem., 2014, 12, 9631 RSC;
(c) A. Behera, S. K. Rout, S. Guin and B. K. Patel, RSC Adv., 2014, 4, 55115 RSC;
(d) Y.-J. Bian, C.-B. Xiang, Z.-M. Chen and Z.-Z. Huang, Synlett, 2011, 2407 CAS.
- L. Wong, D. L. Priebbenow, L.-H. Zou and C. Bolm, Adv. Synth. Catal., 2013, 355, 1490 CrossRef.
- J.-K. Cheng and T.-P. Loh, J. Am. Chem. Soc., 2015, 137, 42 CrossRef CAS PubMed.
- A. Banerjee, S. K. Santra, A. Mishra, N. Khatun and B. K. Patel, Org. Biomol. Chem., 2015, 13, 1307 CAS.
- J. Gallardo-Donaire and R. Martin, J. Am. Chem. Soc., 2013, 135, 9350 CrossRef CAS PubMed.
- N. Matsuda, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2011, 13, 2860 CrossRef CAS PubMed.
- M. J. Campbell and J. S. Johnson, Org. Lett., 2007, 9, 1521 CrossRef CAS PubMed.
- G. Li, C. Jia, K. Sun, Y. Lv, F. Zhao, K. Zhou and H. Wu, Org. Biomol. Chem., 2015, 13, 3207 CAS.
- N. Matsuda, K. Hirano, T. Satoh and M. Miura, J. Am. Chem. Soc., 2013, 135, 4934 CrossRef CAS PubMed.
- R. Sakae, K. Hirano, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2015, 54, 613 CAS.
- R. Sakae, K. Hirano and M. Miura, J. Am. Chem. Soc., 2015, 137, 6460 CrossRef CAS PubMed.
-
(a) J. S. Bandar, M. T. Pirnot and S. L. Buchwald, J. Am. Chem. Soc., 2015, 137, 14812 CrossRef CAS PubMed;
(b) S. Zhu, N. Niljianskul and S. L. Buchwald, J. Am. Chem. Soc., 2013, 135, 15746 CrossRef CAS PubMed;
(c) Y. Miki, K. Hirano, T. Sato and M. Miura, Angew. Chem., Int. Ed., 2013, 52, 10830 CrossRef CAS PubMed.
- S. Zhu and S. L. Buchwald, J. Am. Chem. Soc., 2014, 136, 15913 CrossRef CAS PubMed.
-
(a) Y. Yang, S.-L. Shi, D. Niu, P. Liu and S. L. Buchwald, Science, 2015, 340, 62 CrossRef PubMed;
(b) Y. Xi, T. W. Butcher, J. Zhang and J. F. Hartwig, Angew. Chem., Int. Ed., 2016, 55, 776 CrossRef CAS PubMed.
- Y. Miki, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2014, 16, 1498 CrossRef CAS PubMed.
- N. Niljianskul, S. Zhu and S. L. Buchwald, Angew. Chem., Int. Ed., 2015, 54, 1638 CrossRef CAS PubMed.
- K. Shen and Q. Wang, Chem. Sci., 2015, 6, 4279 RSC.
-
(a) T. Benkovics, J. Du, I. A. Guzei and T. P. Yoon, J. Org. Chem., 2009, 74, 5545 CrossRef CAS PubMed;
(b) D. J. Michaelis, K. S. Williamson and T. P. Yoon, Tetrahedron, 2009, 65, 5118 CrossRef CAS PubMed;
(c) D. J. Michaelis, M. A. Ischay and T. P. Yoon, J. Am. Chem. Soc., 2008, 130, 6610 CrossRef CAS PubMed;
(d) D. J. Michaelis, C. J. Shaffer and T. P. Yoon, J. Am. Chem. Soc., 2007, 129, 1866 CrossRef CAS PubMed.
- T. Benkovics, I. A. Guzei and T. P. Yoon, Angew. Chem., Int. Ed., 2010, 49, 9153 CrossRef CAS PubMed.
- C. P. Allen, T. Benkovics, A. K. Turek and T. P. Yoon, J. Am. Chem. Soc., 2009, 131, 12560 CrossRef CAS PubMed.
- H. F. Motiwala, B. Gülgeze and J. Aubé, J. Org. Chem., 2012, 77, 7005 CrossRef CAS PubMed.
-
(a) B. Zhao, X. Peng, Y. Zhu, T. A. Ramirez, R. G. Cornwall and Y. Shi, J. Am. Chem. Soc., 2011, 133, 20890 CrossRef CAS PubMed;
(b) B. Zhao, X. Peng, S. Cui and Y. Shi, J. Am. Chem. Soc., 2010, 132, 11009 CrossRef CAS PubMed;
(c) W. Yuan, H. Du, B. Zhao and Y. Shi, Org. Lett., 2007, 9, 2589 CrossRef CAS PubMed.
- Y. Wen, B. Zhao and Y. Shi, Org. Lett., 2009, 11, 2365 CrossRef CAS PubMed.
- H. Du, B. Zhao, W. Yuan and Y. Shi, Org. Lett., 2008, 10, 4231 CrossRef CAS PubMed.
- B. Zhao, H. Du and Y. Shi, J. Org. Chem., 2009, 74, 8392 CrossRef CAS PubMed.
- B. Zhao, W. Yuan, H. Du and Y. Shi, Org. Lett., 2007, 9, 4943 CrossRef CAS PubMed.
- B. Zhao, H. Du and Y. Shi, Org. Lett., 2008, 10, 1087 CrossRef CAS PubMed.
- B. Zhao, H. Du and Y. Shi, J. Am. Chem. Soc., 2008, 130, 7220 CrossRef CAS PubMed.
- H. Zhang, Y. Song, J. Zhao, J. Zhang and Q. Zhang, Angew. Chem., Int. Ed., 2014, 53, 11079 CrossRef CAS PubMed.
- H. Zhang, W. Pu, T. Xiong, Y. Li, X. Zhou, K. Sun, Q. Liu and Q. Zhang, Angew. Chem., Int. Ed., 2013, 52, 2529 CrossRef CAS PubMed.
- B. Zhang and A. Studer, Org. Lett., 2014, 16, 1790 CrossRef CAS PubMed.
- K. Kaneko, T. Yoshino, S. Matsunaga and M. Kanai, Org. Lett., 2013, 15, 2502 CrossRef CAS PubMed.
- Z. Ni, Q. Zhang, T. Xiong, Y. Zheng, Y. Li, H. Zhang, J. Zhang and Q. Liu, Angew. Chem., Int. Ed., 2012, 51, 1244 CrossRef CAS PubMed.
- S. Wang, Z. Ni, X. Huang, J. Wang and Y. Pan, Org. Lett., 2014, 16, 5648 CrossRef CAS PubMed.
- For a review, see: P. T. Nyffeler, S. G. Durón, M. D. Burkart, S. P. Vincent and C.-H. Wong, Angew. Chem., Int. Ed., 2005, 44, 192 CrossRef CAS PubMed.
- Q. Michaudel, D. Thevenet and P. S. Baran, J. Am. Chem. Soc., 2012, 134, 2547 CrossRef CAS PubMed.
- For aliphatic C–H oxygenation with carboxylic acids under substantially the same reaction conditions as those in ref. 71, see: J. Zhou, C. Jin, X. Li and W. Su, RSC Adv., 2015, 5, 7232 RSC.
- Y. Li, Z. Li, T. Xiong, Q. Zhang and X. Zhang, Org. Lett., 2012, 14, 3522 CrossRef CAS PubMed.
- T. Vogler and A. Studer, Synthesis, 2008, 1979 CAS.
- P. H. Fuller, J. W. Kim and S. R. Chemler, J. Am. Chem. Soc., 2008, 130, 17638 CrossRef CAS PubMed.
- For the mechanistic and computational studies, see:
(a) M. C. Paderes, J. B. Keister and S. R. Chemler, J. Org. Chem., 2013, 78, 506 CrossRef CAS PubMed;
(b) L. Belding, S. R. Chemler and T. Dudding, J. Org. Chem., 2013, 78, 10288 CrossRef CAS PubMed.
- K. Takamatsu, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2014, 16, 2892 CrossRef CAS PubMed.
- K. Takamatsu, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2015, 80, 3242 CrossRef CAS PubMed.
-
(a) W. Zeng and S. R. Chemler, J. Am. Chem. Soc., 2007, 129, 12948 CrossRef CAS PubMed;
(b) E. S. Sherman and S. R. Chemler, Adv. Synth. Catal., 2009, 351, 467 CrossRef CAS PubMed.
- B. J. Casavant, A. Z. Hosseini and S. R. Chemler, Adv. Synth. Catal., 2014, 356, 2697 CrossRef CAS PubMed.
- M. T. Bovino and S. R. Chemler, Angew. Chem., Int. Ed., 2012, 51, 3923 CrossRef CAS PubMed.
- B. W. Turnpenny and S. R. Chemler, Chem. Sci., 2014, 5, 1786 RSC . For a recent updated report of diamination from alkenylureas, see: S. Fu, H. Yang, G. Li, Y. Deng, H. Jiang and W. Zeng, Org. Lett., 2015, 17, 1018 CrossRef CAS PubMed.
-
(a) Y. Miller, L. Miao, A. S. Hosseini and S. R. Chemler, J. Am. Chem. Soc., 2012, 134, 12149 CrossRef CAS PubMed;
(b) M. T. Bovino, T. W. Liwosz, N. E. Kendel, Y. Miller, N. Tyminska, E. Zurek and S. R. Chemler, Angew. Chem., Int. Ed., 2014, 53, 6383 CrossRef CAS PubMed.
- T. W. Liwosz and S. R. Chemler, Chem. – Eur. J., 2013, 19, 12771 CrossRef CAS PubMed.
- Z. Wang, J. Ni, Y. Kuninobu and M. Kanai, Angew. Chem., Int. Ed., 2014, 53, 3496 CrossRef CAS PubMed.
- Ge recently utilized duroquinone as the stoichiometric oxidant in Cu-catalyzed aliphatic C–H amidation of N-(quinolin-8-yl)alkylamides for synthesis of β-lactams, see: X. Wu, Yan. Zhao, G. Zhang and H. Ge, Angew. Chem., Int. Ed., 2014, 53, 3706 CrossRef CAS PubMed.
- W.-H. Rao and B.-F. Shi, Org. Lett., 2015, 17, 2784 CrossRef CAS PubMed.
- G. Li, C. Jia and K. Sun, Org. Lett., 2013, 15, 5198 CrossRef CAS PubMed.
- N. Fiederling, J. Happer and H. Schramm, Org. Process Res. Dev, 2013, 17, 318 CrossRef CAS.
- M. G. Suero, E. D. Bayle, B. S. L. Collins and M. J. Gaunt, J. Am. Chem. Soc., 2013, 135, 5332 CrossRef CAS PubMed.
-
(a) A. M. Berman and J. S. Johnson, J. Am. Chem. Soc., 2004, 126, 5680 CrossRef CAS PubMed;
(b) A. J. Biloski and B. Ganem, Synthesis, 1983, 537 CrossRef CAS.
- W. B. Jennings, S. P. Watson and D. R. Boyd, J. Chem. Soc., Chem. Commun., 1988, 931 RSC.
- H. Du, B. Zhao and Y. Shi, Org. Synth., 2009, 86, 315 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence