
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemical vapor deposition-prepared sub-nanometer Zr clusters on Pd surfaces: promotion of methane dry reforming†
Lukas
Mayr
ab,
Xue-Rong
Shi
a,
Norbert
Köpfle
a,
Cory A.
Milligan
bc,
Dmitry Y.
Zemlyanov
b,
Axel
Knop-Gericke
d,
Michael
Hävecker
d,
Bernhard
Klötzer
a and
Simon
Penner
*a
aInstitute of Physical Chemistry, University of Innsbruck, Innrain 80-82, Innsbruck, Austria. E-mail: simon.penner@uibk.ac.at; Tel: +43 512 507 58003
bBirck Nanotechnology Center, Purdue University, 1205 West State Street, West Lafayette, IN 47907, USA
cSchool of Chemical Engineering, Purdue University, West Lafayette, Indiana 47907, USA
dDepartment of Inorganic Chemistry, Fritz-Haber-Institute of the Max-Planck-Society, Faradayweg 4–6, D-14195 Berlin, Germany
First published on 8th November 2016
Abstract
An inverse Pd–Zr model catalyst was prepared by chemical vapor deposition (CVD) using zirconium-t-butoxide (ZTB) as an organometallic precursor. Pd–Zr interaction was then investigated with focus on the correlation of reforming performance with the oxidation state of Zr. As test reactions, dry reforming of methane (DRM) and methanol steam reforming (MSR) were chosen. Depending on treatments, either ZrOxHy or ZrO2 overlayers or Zr as sub-nanometer clusters could be obtained. Following the adsorption of ZTB on Pd(111), a partially hydroxylated Zr4+-containing layer was formed, which can be reduced to metallic Zr by thermal annealing in ultrahigh vacuum, leading to redox-active Zr0 sub-nanometer clusters. Complementary density functional theoretical (DFT) calculations showed that a single layer of ZrO2 on Pd(111) can be more easily reduced toward the metallic state than a double- and triple layer. Also, the initial and resulting layer compositions greatly depend on gas environment. The lower the water background partial pressure, the faster and more complete the reduction of Zr4+ species to Zr0 on Pd takes place. Under methanol steam reforming conditions, water activation by hydroxylation of Zr occurs. In excess of methanol, strong coking is induced by the Pd/ZrOxHy interface. In contrast, dry reforming of methane is effectively promoted if these initially metallic Zr species are present in the pre-catalyst, leading to a Pd/ZrOxHy phase boundary by oxidative activation under reaction conditions. These reaction-induced active sites for DRM are stable with respect to carbon blocking or coking. In essence, Zr doping of Pd opens specific CO2 activation channels, which are absent on pure metallic Pd.
1. Introduction
ZrO2 is commonly regarded as a highly stable, “irreducible” refractory oxide. Nevertheless, it is currently under investigation to expand its applicability as a catalyst for selective aldehyde oxidation.1 Regarding CO hydrogenation, the roles of different zirconia polymorphs in the synthesis of higher alcohols were studied both over pure and Pd- or Li-modified ZrO2.2 It has also been shown to promote catalysts for methanol reforming processes.3 In this context, it has been successfully applied as a stabilizing support to prevent CuOx nanoparticles from sintering, and therefore enhances the stability of methanol steam reforming catalysts.4 Zr4+ ions have also been reported to play an important role as dopants in an Au/CeO2 catalyst for CO oxidation5 and in sol–gel-synthesized photocatalysts.6 Interest in the chemical properties of ZrO2 polymorphs also arises from its application as YSZ (yttrium-stabilized zirconium oxide), the standard electrolyte for solid oxide fuel cells, due to its electric insulation properties and permeability for O2− ions.7Atomic layer deposition (ALD), or in its general form chemical vapor deposition (CVD), has been already successfully applied for ZrO2. There are many precursor/oxidative agent pairs known to generate ZrO2 layers,8 with the main application as protection/coating layers. Applications beyond catalysis can also be found in microelectronics, where-prepared ZrO2 was used on SiO2 as an additional insulation layer.9 The synthesis of metallic Zr via the ALD/CVD pathway from metal–organic precursors to form, by sophisticated activation, nanoparticles for catalysis, has – to our knowledge – not been documented sufficiently. Due to the very high formation enthalpy of ZrO2, special preparation techniques for metallic Zr have been developed,10 with the aim to control problems of partial oxidation induced by the strong “getter” effect of Zr0. Therefore, CVD from zirconium-tert-butoxide (ZTB) was chosen in the present study as an easily applicable, reliable and effective way to prepare ZrOxHy overlayers on Pd. The as-prepared CVD layers are not metallic, but contain partially oxidized and partially hydroxylated Zr4+ species, which nevertheless, can be easily reduced on Pd to Zr0 by means of an UHV-based post-treatment. Increased interest in Zr0-based pre-catalysts also arises from the fact, that oxidative segregation of initially bimetallic surfaces under reaction conditions was recently shown to yield active and selective inverse model catalysts.11 Such bimetallic pre-catalysts therefore represent a promising approach to obtain strongly enhanced activities (with respect to surface area) as compared to the related supported powder catalytic systems. The enhancement of surface-specific rates can be assigned to the extremely high number of metal-oxide phase boundary sites induced by localized oxidative segregation starting from an atomically dispersed alloy or intermetallic initial state. One of these examples is the initially bimetallic Cu–Zn system. Starting from a dilute Zn-in-Cu alloy, finely dispersed zinc-(hydr)oxide islands are formed in situ, leading to a maximum of Cu/ZnOxHy phase boundary sites.12 Thus, an extremely active and selective “inverse” MSR catalyst is obtained. The same effect was shown for Cu–Zr, where metallic Zr was sputter-prepared on a Cu substrate and was oxidized/hydroxylated under MSR conditions, yielding phase boundary sites with equally improved CO2-selectivity and activity.11
Two distinct test reactions, methanol steam reforming and dry reforming of methane were chosen to investigate a potential co-catalytic and/or promotional function of surface-near Zr0-dopants on Pd. Although MSR is an efficient way to generate CO-depleted H2, catalyst stability issues and the required minimized CO content of the reformate gas make it still an intensely investigated reaction.4 Cu-based catalysts are known to be excellent to obtain high CO2-selectivity and high reaction rates,12 but also Pd-based intermetallic systems are promising due to the methanol-activating function of Pd.13 As a test reaction, MSR is useful to substantiate and quantify the low-temperature water activation activity of the investigated catalyst systems, because of the fact that CO2 can only be formed via the reaction of activated methanol to formaldehyde, which further reacts with weakly bound –OH species from activated water toward CO2.
Dry reforming of methane (DRM) is a relevant reaction for the one-step conversion of two climate-relevant greenhouse gases, CO2 and CH4, to useful syngas. It is performed at higher temperatures (∼970 K) than MSR and does not so much rely on initial water- but rather on CO2-activation. It is usually initiated by methane decomposition toward different carbon species. If the latter are sufficiently reactive, they can be further converted via the subsequent Boudouard reaction to CO, or else, they remain deposited as unreactive coke. However, also the inverse water-gas-shift reactivity of the catalyst may play a role,14 either as an additional intermediate process to generate water, which may be important for efficient gasification of carbon deposits, or as an unwanted side reaction reducing the H2 yield. Transition metals such as Co, Ni, Ru, Rh, Ir and Pt on alumina and silica supports15,16 have been reported as efficient DRM catalysts. Ceria as a support for Ni and Co was also applied,13 but also mixed ceria and zirconia supports.17 Clean Pd is known for its pronounced methane- but simultaneously quite poor CO2-activation ability. Therefore, DRM was chosen as a reference reaction to investigate the promotion of CO2 activation on a Zr-doped inverse Pd model catalyst, in order to highlight the role of the eventual initial Zr0 nanoclusters for enhanced CO2 activation.
The main goal of this inverse model catalyst study is therefore to highlight the co-catalytic/promotional role of redox-active Zr surface species on Pd beyond the simple function of zirconia as a structurally stabilizing catalyst support. As will be shown in the following, Zr0 clusters of 2–6 atoms adsorbed on Pd can be oxidized, reduced or hydroxylated reversibly at comparably low temperatures around 723 K. Beyond the redox properties, electronic structure nano-effects of small particles and surface islands have also been described for other elements, i.e. gold on TiO2,14,15,18,19 which may alter the catalytic properties substantially. Zr, in its nanostructured highly redox-active surface-adsorbed state, is therefore investigated to be part of a potential redox active cycle and to open new reaction pathways on chemically and electronically altered Pd.
2. Experimental and computational methods
Depending on the experimental requirements, the research was performed using three experimental setups: An Omicron Surface Analysis Cluster (Birck Nanotechnology Center at Purdue University), an UHV-chamber with attached high-pressure recirculating batch reactor, and the Innovative Station for in situ Spectroscopy (Beamline ISISS at BESSY II synchrotron in Berlin).2.1. Omicron setup
The Omicron Surface Analysis Cluster consists of a UHV preparation chamber and an analysis chamber with base pressures of 1 × 10−9 mbar and 5 × 10−11 mbar, respectively. Samples can be transferred between these chambers under UHV. The preparation chamber is equipped with a mass spectrometer, an Ar+ sputtering gun, a gas manifold system, and resistive sample heating. The analysis chamber is equipped with X-ray Photoelectron Spectroscopy (XPS) (electron energy analyzer – Omicron EAC 125 and analyzer controller – Omicron EAC 2000), Low Energy Electron Diffraction (LEED) (Omicron), High Resolution Electron Energy Loss Spectroscopy (HREELS) (ELS5000, LK Technologies), Scanning Tunneling Microscopy (STM) (Omicron ambient temperature UHV STM/AFM), and resistive sample heating. XPS data are acquired using a non-monochromatic Mg Kα X-ray source (hν = 1253.6 eV) at 150 W. High resolution spectra are recorded at the constant pass energy of 20 eV. Photoelectrons are ejected at a 45° angle with respect to the surface normal.The Pd(111) single crystal (orientation accuracy <0.1°, Princeton Scientific) is cleaned by several cycles of Ar+-sputtering (1 kV, 2 μA sample ground current) and annealing to 900–1000 K until no contaminations could be detected by XPS, STM and LEED. Well-ordered monoatomic steps are seen in STM. Due to the potential impact of radiation of the X-ray source on the sample topography/chemistry, STM studies are always done before XPS characterization. Sample heating is performed in the analysis chamber.
Zirconium(IV) tert-butoxide Zr(O-t-C4H9)4 (Strem, purity: 99%) is used as ALD/CVD precursor and filled into an cylinder under Ar. Prior to dosing ZTB, several cycles of freeze–pump–thaw are performed for purification (cooling temperature: 223 K, freezing point ZTB: 269 K). ZTB has a sufficient vapor pressure at room temperature to dose it directly through a leak-valve. Before sample exposure, the preparation chamber is exposed to the precursor at 5 × 10−7 mbar for 5 minutes to passivate the chamber walls against precursor decomposition. The exposure is calculated from uncorrected ion gauge measurements in Langmuir, (1 L = 1 × 10−6 Torr s) involving exposure pressures in the range of 5 × 10−8 to 5 × 10−6 mbar. The adsorbate coverage is determined from XPS data (details can be found in the XPS experimental section or additionally in ref. 20–23).
2.2. UHV-chamber with attached high-pressure recirculating batch reactor
Sample preparation and characterization was performed in a combined preparation/analysis chamber with attached reaction cell, described in more detail elsewhere (base pressure in the low 10−9 mbar range).24 The sample is heated via a home-built e-bombardment setup. Electrons are ejected from a triple-filament emitter (operated with 30 W heating power) set to −500 V, while the sample is set to +300 V. The electron impact heating power is controlled via the filament emission current. For spectroscopic analysis, the chamber is equipped with a hemispherical electron and ion analyzer (Thermo Fisher Electron Alpha 110), a double anode X-ray gun for XPS (Mg/Al, XR 50, Specs), an ion gun sufficient to produce 1 kV He+ ions for ISS (Omicron 100) and an electron beam gun for Auger electron spectroscopy (KPI EGPS-2017B). Additionally, a mass spectrometer (Balzers) for residual gas analysis and an Ar+ ion sputter gun for sample cleaning is attached. A three way gas inlet allows to dose O2 (Messer, 5.0), H2 (Messer 5.0) or O2 cleaned Ar (Messer 5.0) via leak valves into the chamber. All XPS spectra in this chamber are recorded with a non-monochromatic Mg Kα X-ray source (hν = 1253.6 eV) at 250 W and at the magic angle to the analyzer. For XPS the analyzer is operated at a constant pass energy of 20 eV.All catalytic experiments are performed using a 20 × 18 mm ultra-clean polycrystalline Pd foil (Goodfellow, 99.95%) with a thickness of 0.125 mm. For reference experiments on pure ZrO2, a pre-oxidized 0.127 mm Zr foil with the same size (Alfa Aesar, purity: 99.95%) is used. The foils are cleaned before loading to the UHV chamber in a water and an ethanol ultrasonic bath for 20 min, respectively. ZTB (Sigma Aldrich, purity. 99.999%) is filled and mounted to the combined preparation/analysis chamber as described for the Omicron setup above. ZTB exposition is performed as described in the context of the individual experiments discussed below. Sample heating is performed by electron impact heating.
In order to validate the CVD-based catalytic results and to obtain a broader experimental basis of the Pd–Zr0 system with respect to reforming performance, also an intermetallic bulk phase of Pd and Zr0 is prepared. The preparation is performed under HV conditions (base pressure 1 × 10−7 mbar) by heating small pieces of the above-specified pure Pd and Zr foil samples resistively in a Ta crucible in the nominal atomic ratio 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. At a temperature slightly above the melting point of Pd (1828 K), spontaneous reaction between Pd and Zr leads to an intermetallic Pd–Zr melt, which then recrystallizes toward Pd–Zr bulk phases during cooling in vacuum. Thereafter, this sample is transferred to the UHV system with attached batch reactor for analysis and catalytic testing. Note, that this sample, in comparison to the CVD-prepared ones, is exposed to air at room temperature between preparation and characterization. The nominal 2:1 Pd:Zr stoichiometry (i.e. with Pd excess) is chosen with the idea to maintain at least some active Pd metal surface coexisting with oxidatively segregated Zr species under reaction conditions. Moreover, an excess of Pd is necessary because the melting point of Zr is very high and complete intermetallic formation reaction of Pd with Zr is necessary in order to distribute the Zr0 homogeneously in the melt. The corresponding X-ray diffraction patterns of the initial Pd–Zr sample is made up by a complex superposition of at least two different Pd–Zr intermetallic phases, including Pd3Zr and Pd4Zr3, alongside a small amount of oxidized monoclinic ZrO2. Note that this is in striking contrast to the similarly prepared Cu–Zr samples,24 indicating a much less initial homogeneous melt. In due course, as will be discussed below, after the DRM reaction, the patterns are even more complicated, indicating phase transformations and partial (oxidative) decomposition of the intermetallic Pd–Zr compounds.
:1. At a temperature slightly above the melting point of Pd (1828 K), spontaneous reaction between Pd and Zr leads to an intermetallic Pd–Zr melt, which then recrystallizes toward Pd–Zr bulk phases during cooling in vacuum. Thereafter, this sample is transferred to the UHV system with attached batch reactor for analysis and catalytic testing. Note, that this sample, in comparison to the CVD-prepared ones, is exposed to air at room temperature between preparation and characterization. The nominal 2:1 Pd:Zr stoichiometry (i.e. with Pd excess) is chosen with the idea to maintain at least some active Pd metal surface coexisting with oxidatively segregated Zr species under reaction conditions. Moreover, an excess of Pd is necessary because the melting point of Zr is very high and complete intermetallic formation reaction of Pd with Zr is necessary in order to distribute the Zr0 homogeneously in the melt. The corresponding X-ray diffraction patterns of the initial Pd–Zr sample is made up by a complex superposition of at least two different Pd–Zr intermetallic phases, including Pd3Zr and Pd4Zr3, alongside a small amount of oxidized monoclinic ZrO2. Note that this is in striking contrast to the similarly prepared Cu–Zr samples,24 indicating a much less initial homogeneous melt. In due course, as will be discussed below, after the DRM reaction, the patterns are even more complicated, indicating phase transformations and partial (oxidative) decomposition of the intermetallic Pd–Zr compounds.
For catalytic testing in the ambient pressure recirculating batch reaction cell, a long z-transfer rod allows fast and reliable transfer without exposure to air. The all-quartz-glass high-pressure (up to 1 bar) batch reactor is equipped with a gas chromatograph with either intermediate or continuous EID-MS detection to determine the exact gas composition at any point of reaction. Continuous partial pressure detection is performed via a capillary leak to the GC-MS. The quartz-glass reactor with a total circulation volume of 296 ml is designed to measure small reaction rates and selectivity patterns within a temperature range between room temperature and 1300 K. A circulation pump ensures a constant flow and gas intermixing and an attached gas-premixing unit allows to set arbitrary compositions of the attached reactant vapors or introduced gases (methanol, methane (5.0), deionized and degassed water, CO2 (5.0), O2 (5.0), H2 (5.0), Ar (5.0) and He (5.0). The sample holder itself is entirely made of quartz glass to avoid background reactivity from hot metal parts and is designed for 20 mm × 18 mm metal foils.
A partial pressure of 8–30 mbar argon added to all gas mixtures allows to account for the thermal expansion due to the temperature increase and the simultaneous gas loss through the capillary leak for continuous EID-MS detection. For partial pressure calculations, all base-line-corrected MS signals are calibrated using pure substances with quantitative consideration of fragmentation. For all MSR catalytic experiments shown in this work, the following initial conditions are applied: 12 mbar methanol, 24 mbar water, 8 mbar argon, He added to a total pressure of 1 bar. After an equilibration and premixing period of 10 min, a heating routine (rate 10 K min−1) up to 623 K is performed, followed by an isothermal period at 623 K. For all DRM catalytic experiments shown in this work, 50 mbar methane, 50 mbar CO2, 30 mbar Ar, adding He added to a total pressure of 1 bar were chosen. After an equilibration and premixing period of 10 min, a temperature ramp up to 1073 K within 30 min was performed, followed by a corresponding isothermal period at 1073 K. For discussion about mass and heat transport limitations, we refer to a thorough discussion of the catalytic setup in ref. 25.
2.3. In situ XPS setup
Synchrotron-based in situ XPS experiments were performed at the ISISS (Innovative Station for in situ Spectroscopy) beamline at the BESSY II synchrotron in Berlin, Germany. The experimental apparatus consists of a load lock and an in situ cell connected to the XPS spectrometer via differential pumping stages. The experimental apparatus has been described in the literature in detail previously.26 Samples are heated in the in situ cell via a near-infrared semiconductor laser (λ = 808 nm) from the rear. The temperature is measured by a K-type (chromel–alumel) thermocouple positioned between sample holder back plate and Pd foil. All in situ experiments are performed on the same Pd foil sample that is used for the model catalyst preparation in the UHV instrument with attached batch reactor.The same ZTB cylinder/leak valve setup described above is transferred to ISISS beamline. Due to the fact that ZTB only interacts with surfaces hotter than 500 K, it is safe to dose the organometallic precursor into the analysis chamber without any Zr deposition on the components of the vacuum system or on the X-ray window. The growth of ZTB can then be followed directly via XPS. To determine a potential influence of the X-ray beam on the sample structure and chemistry, heating was performed at random checking with and without X-ray beam. As no distinct changes in the spectra between the two runs have been observed, this influence is considered to be marginal.
Monochromator control allows to chose photon energies corresponding to kinetic energies of the ejected photoelectrons of 120 eV for all monitored core-level photoemission peaks in order to extract information from a constant information depth and to yield the same attenuation of the photoelectrons through the gas phase. Due to the fact that 95% of the signal arises from a sample depth up to ∼1 nm, this operation mode is considered to be maximum “surface sensitive”.
Photoelectrons are collected in the direction normal to the surface at constant pass energy of 10 eV. Binding energies were referenced to the Fermi edge, which is measured each time the monochromator moves to a new position (i.e. whenever the incident photon energy was changed). Photoemission peak intensities are corrected for the respective photon flux at a given photon energy. Since the BESSY II synchrotron operates in top-up mode (constant ring current), no additional correction for the ring current was required. Since all photoemission peaks were collected at the same kinetic energy of photoelectrons (120 eV), the attenuation through the gas phase was the same for all-core levels and, thus, cancels out in coverage calculations.
2.4. Analysis of the XPS data
All spectra are analyzed using the CasaXPS software program, version 2.3.16 Pre-rel 1.4 (Casa Software Ltd).27 A Shirley background is applied to all spectra and the associated Scofield relative sensitivity factors are used for quantification. For peak fitting of the Zr 3d peaks a weighted sum of Gaussian and Lorenzian peak shapes (CasaXPS line shape SGL(30)) is assumed, using a doublet separation (Zr 3d5/2vs. Zr 3d3/2) of 2.4 eV for both metallic Zr28 and zirconia29 was used for fitting. The doublet area is kept constant at 3:2 as arising from spin–orbit d-electron coupling. Electron attenuation lengths were taken from the NIST database SR 8230 and the orbital asymmetric parameter from the ELETTRA online database of ref. 31 The quantification of the XPS data is given as atomic percentages or coverage/thickness. The atomic percentage is estimated assuming homogeneously mixed elements. Since an adlayer on the substrate surface is not a homogeneous system, the coverage/thickness gives a better representation. The ZrOxHy surface coverage is calculated assuming a non-attenuating adlayer on a semi-infinite substrate.20–23 As the maximum ZrOxHy layer thicknesses remained in the sub-monolayer regime in this study, the influence of a potential attenuation effect of the photoelectrons by the overlayer remains negligible even for the highest exposures. This is tested by comparing the results on a ∼1 ML ZrOxHy covered sample, using both an attenuating and non-attenuating overlayer model, which eventually shows negligible differences. Details of these calculations are given in ref. 20–23 and in the ESI,† Section A.
2.5. Density functional theory (DFT)
All calculations are performed with plane-wave DFT using the Vienna ab initio simulation package (VASP) using the projector augmented wave (PAW) method.32,33 To elucidate the role of the van der Waals interaction in this system, we perform vdW-DF calculations using the optB88 functional.34,35 The cutoff energy for the plane wave basis set is fixed at 400 eV. Geometry optimization is performed with a conjugate-gradient algorithm and considered to be converged when the forces on each unconstrained atom was <0.01 eV Å−1. A Monkhorst–Pack grid of 2 × 2 × 1 is employed due to the large unit cell. We use a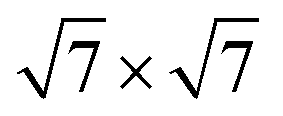 surface unit cell for Pd(111) with five layers thickness. The bottom two layers are fixed at their bulk position. On top of the Pd surface, 1 to 3 layers of a (2 × 2) cubic ZrO2 film are adsorbed and fully optimized. According to the previous experimental STM observations and theoretical work,16,17 we take the (111) surface of cubic ZrO2 as the surface orientation of the ZrO2 film. The calculated lattice mismatch between each other is 1.3%. The reaction energy ΔE of deposited ZrO2 films on the Pd surface is calculated by
surface unit cell for Pd(111) with five layers thickness. The bottom two layers are fixed at their bulk position. On top of the Pd surface, 1 to 3 layers of a (2 × 2) cubic ZrO2 film are adsorbed and fully optimized. According to the previous experimental STM observations and theoretical work,16,17 we take the (111) surface of cubic ZrO2 as the surface orientation of the ZrO2 film. The calculated lattice mismatch between each other is 1.3%. The reaction energy ΔE of deposited ZrO2 films on the Pd surface is calculated by| ΔE = [Etotal(ZrO2–Pd) − Etotal(Pdsurf) − nZrO2Etotal(ZrObulk2)]/Acell | (1) |
The formation energy of ZrO2 films with a different number of layers is defined by
| ΔEfilm = Etotal(ZrO2) − nZrO2Etotal(ZrObulk2) | (2) |
| ΔEads = Etotal(ZrO2–Pd) − Etotal(Pdsurf) − Etotal(ZrO2). | (3) |
The temperature- and pressure-dependent terms of condensed phases are small and tend to be neglected in the free energy calculation. The differences between the chemical potentials can be approximated by the difference between their calculated DFT electronic energies (Etotal). For the gas phase, the thermal contributions from the translational, rotational and vibrational enthalpies and entropies are added.36 By this definition, the reaction energy ΔGdiss for oxygen dissolution into Pd sub-layer and ΔGdiff for oxygen diffusion to the available bare Pd(111) surface are not affected by the reaction temperature and pressure, and will be equal to the reaction energy at 0 K.
The reaction energy ΔGdiss for oxygen dissolution into Pd sub-layer is calculated by
| ΔGdiss = Etotal(ZrO2–O–Pd) − Etotal(ZrO2–Pd). | (4) |
E total(ZrO2–O–Pd) is the total energy of ZrO2–Pd system with one oxygen dissolving into the Pd sub-layer. The reaction energy ΔGdiff for oxygen diffusion to the available neighboring bare Pd(111) surface is then calculated by
| ΔGdiff = Etotal[(ZrO2)Ovacancy–Pd] + Etotal(O–Pd) − Etotal(ZrO2–Pd) − Etotal(Pdsurf). | (5) |
| ΔGO2 = ΔE0 + 0.5G(T) | (6) |
| ΔE0 = Etotal[(ZrO2)Ovacancy–Pd] + 0.5Etotal(O2) − Etotal(ZrO2–Pd). | (7) |
3. Results
3.1. Synthesis of the starting catalyst structure and redox chemistry of Zr
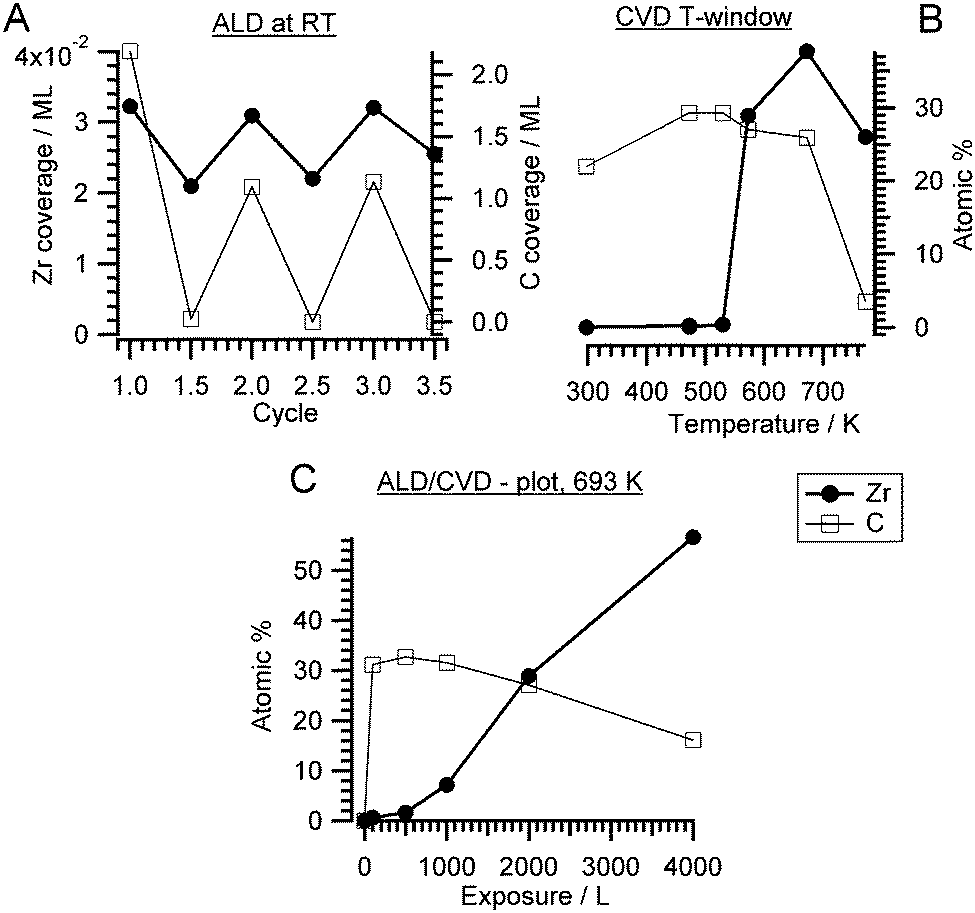 | ||
| Fig. 1 (A) Zr and C coverage as a function of number of ALD-like cycles: 1st half cycle – 2000 L ZTB adsorption at RT and 2nd half cycle – annealing at 673 K in vacuum. Prior to the first ALD cycle, the clean Pd(111) surface was exposed to 2000 L O2 at 298 K; (B) constant exposure of 2000 L at selected temperatures, indicating the CVD window. (C) Langmuir plot of Zr and C atomic% derived from Zr 3d, C 1s and Pd 3d versus ZTB exposure at 693 K (isothermal conditions). | ||
Typically, an ALD/CVD process is characterized by a specific temperature window. For instance, the exposure temperature is recommended to be around 650 K for Si.9 The ALD window for the decomposition of ZTB on a SiO2 wafer was between 573 K and 673 K.37,38 The activation barrier was explained by a hydride elimination step, which was considered as a rate limiting step, and the formation of isobutene.37 A pure ALD mechanism was supposed to occur below 573 K. We investigated the dependence of the zirconium amount from the ZTB exposure temperature. Fig. 1B shows the amount of zirconium and carbon (in atomic percent for a rough quantitative estimation) calculated from the Zr 3d and C 1s XPS peaks as a function of ZTB exposure temperature. Carbon can be mainly found on the surface following exposure at temperatures below a sharp threshold of ∼573 K. Above 573 K, the Zr/Pd ratio increases rapidly and the amount of carbon decreases. A remarkably similar behavior was observed during ZrO2 growth from ZTB on Si(100) by Cameron and George,38 where the CVD rate of ZrO2 increased sharply above 573 K and then started to drop above 773 K. At high temperatures, ZrO2 deposition was supposed to be partially poisoned by carbon. Fourier transform infrared spectroscopy (FTIR) demonstrated hydroxyl (ZrOH*) and butoxide (ZrOC(CH3)3*) species on ZrO2, whose concentration decreased roughly linearly with temperature from 300–800 K.33 In our case, we do not detect much carbon deposition at high temperatures. However, a much lower ZTB pressure is used in our study (∼10−5 Torr vs. 0.05 Torr in ref. 38) and Pd exhibits a high carbon dissolution capability.39 Therefore, on Pd the amount of deposited Zr decreases above 673 K most likely due to kinetic limitations: fast desorption of ZTB effectively competes with dissociation. The temperatures between 550–773 K are most efficient for zirconium deposition, more effective than multiple ALD-like cycles at room temperature. The temperature of 693 K is usually used for the CVD experiments described in this study because of the empirically determined trade-off between reducing the unwanted carbon deposition and optimizing the Zr amount.
A Langmuir-type plot (deposited amount of Zr and C versus ZTB exposure in Langmuir) obtained at 673 K is shown in Fig. 1C. No zirconium saturation was observed even at 4000 L, while the carbon amount was decreasing with exposure. Increasing with exposure, the Zr amount is more typical for CVD than for ALD. The CVD was also reported for ZTB adsorption on Si(100).38 Interestingly, carbon is displaced by zirconium compounds, which might reflect a competition for adsorption/dissociation sites between ZTB and its decomposition products (i.e. hydrocarbons). ZTB dissociation products could hinder ZTB adsorption and block further ZTB dissociation. A similar situation was observed during trimethylaluminum and ferrocene adsorption on Pd(111) and Pt(111).21,22 The CVD growth of ZrOxHy on Cu is also monitored in situ. The corresponding XP spectra are shown in the ESI† in Fig. S1.
In order to identify adsorption species and to understand the low zirconium uptake below 550 K, ZTB adsorption is investigated by HREELS (Fig. 2). Molecularly adsorbed ZTB was prepared by 2000 L exposure on Pd(111) at 180 K and followed by warming up to 293 K (spectrum (a) in Fig. 2). Despite the rather complex spectrum, all detected vibrations are very similar to those detected from FTIR in gas phase for the tert-butyl group and ZTB.40–43 In XPS (ESI,† Fig. S2), two separate C 1s peaks can be seen at 286.9 eV and 284.9 eV following ZTB exposure, which can be attributed to carbon bound to oxygen or carbon, respectively. By comparison with the Zr 3d5/2 peak area, the C/Zr ratios for the oxygenated and non-oxygenated species are 4.9 and 17.4, respectively. This allows us to conclude that adsorption of ZTB is mostly molecular under these conditions. The characteristic frequencies of ZTB on Pd(111) are summarized in the ESI† in Table S2 and compared with literature values. The HREELS spectrum obtained following ZTB adsorption on Pd(111) at 293 K (spectrum (b) in Fig. 2) is slightly different. The main difference is a new peak at 1685 cm−1 (marked dark-green). This peak can be assigned to the stretching of the double C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond,44–46 which most likely is due to adsorbed isobutylene species. Indeed, thermal desorption of isobutylene correlated with the ZrO2 growth rate,38 and this observation along with FT-IR-detected butoxide species (ZrOC(CH3)3,ads) allowed Cameron and George38 to conclude that the butoxide species decomposition via β-hydride elimination is the rate-limiting step for ZrO2 film growth. This conclusion is consistent with our HREELS data showing dissociation of ZTB and the appearance of the adsorbed isobutylene species. The intensity enhancement of a skeletal vibration of tert-butyl group, ν(C–C), at 780 cm−1 infers that the surface is covered with ZTB dissociation products. The C/Zr ratio increased compared to the molecular adsorption as well. The ratio for the room temperature-exposure roughly is 30:1 atomic% C:Zr. Thus, hydrocarbon products of ZTB dissociation “passivate” the Pd(111) surface against further ZTB adsorption.
C bond,44–46 which most likely is due to adsorbed isobutylene species. Indeed, thermal desorption of isobutylene correlated with the ZrO2 growth rate,38 and this observation along with FT-IR-detected butoxide species (ZrOC(CH3)3,ads) allowed Cameron and George38 to conclude that the butoxide species decomposition via β-hydride elimination is the rate-limiting step for ZrO2 film growth. This conclusion is consistent with our HREELS data showing dissociation of ZTB and the appearance of the adsorbed isobutylene species. The intensity enhancement of a skeletal vibration of tert-butyl group, ν(C–C), at 780 cm−1 infers that the surface is covered with ZTB dissociation products. The C/Zr ratio increased compared to the molecular adsorption as well. The ratio for the room temperature-exposure roughly is 30:1 atomic% C:Zr. Thus, hydrocarbon products of ZTB dissociation “passivate” the Pd(111) surface against further ZTB adsorption.
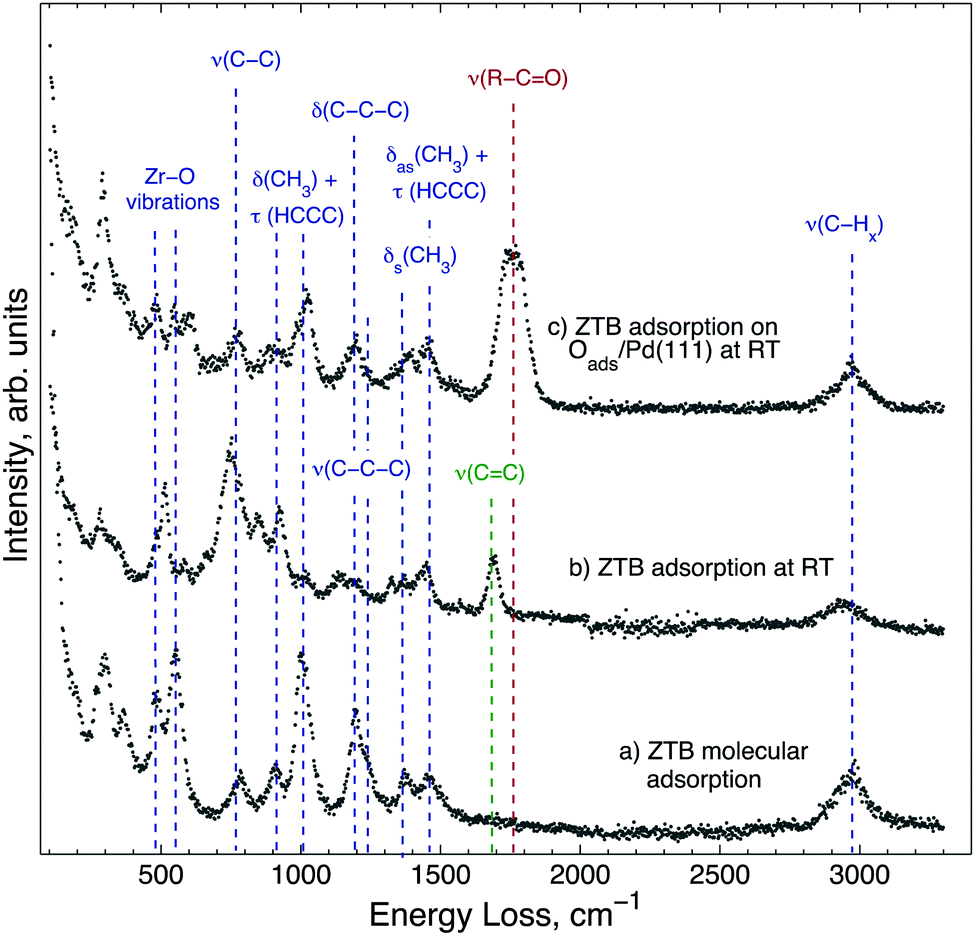 | ||
| Fig. 2 HREEL spectra obtained following 2000 L ZTB exposure of the Pd(111) surface (a) at 180 K and heated to 293 K in UHV, (b) and (c) at 293 K; for (c), Pd(111) was pre-exposed to 2000 L O2 at 673 K. HREELS spectra were collected at 293 K. The energy losses, which are characteristic of ZTB, are marked in blue. | ||
Spectrum (c) in Fig. 2 was obtained following 2000 L ZTB exposure of Pd(111) covered by Oads. Interestingly, the spectrum is close to the one for the molecular adsorption of ZTB (spectrum (a), Fig. 2): it is likely that the surface contains less ZTB dissociation products, as no adsorbed isobutylene species are found on the surface. On the other hand, a new peak appeared at 1745–1775 cm−1 (dark-red), which can be due to a C–O stretching vibration in aldehyde or ketones.44–46 Likely, an isobutyl fragment splitting off ZTB could attach to a surface oxygen center without β-hydride elimination. No OH vibration following ZTB adsorption in all three cases is observed.
According to the HREELS data, even slight heating of ZTB adlayers in UHV resulted in decomposition of hydrocarbon species (ESI,† Fig. S3). Structure-wise, following adsorption of 100 L ZTB at 673 K, the surface is covered with a layer of rather disordered ZrOx mixed with carbon as determined from STM images (Fig. 3A). This layer is not uniform and contains holes and cracks (Fig. 3B). The surface carbon species seen in this stage are most likely CxHy fragments and/or graphitic deposits, as deduced from the main C 1s components around 284 eV (not shown). A relatively broad height distribution within the terraces (not shown) indicates the limited uniformity of the overlayer.
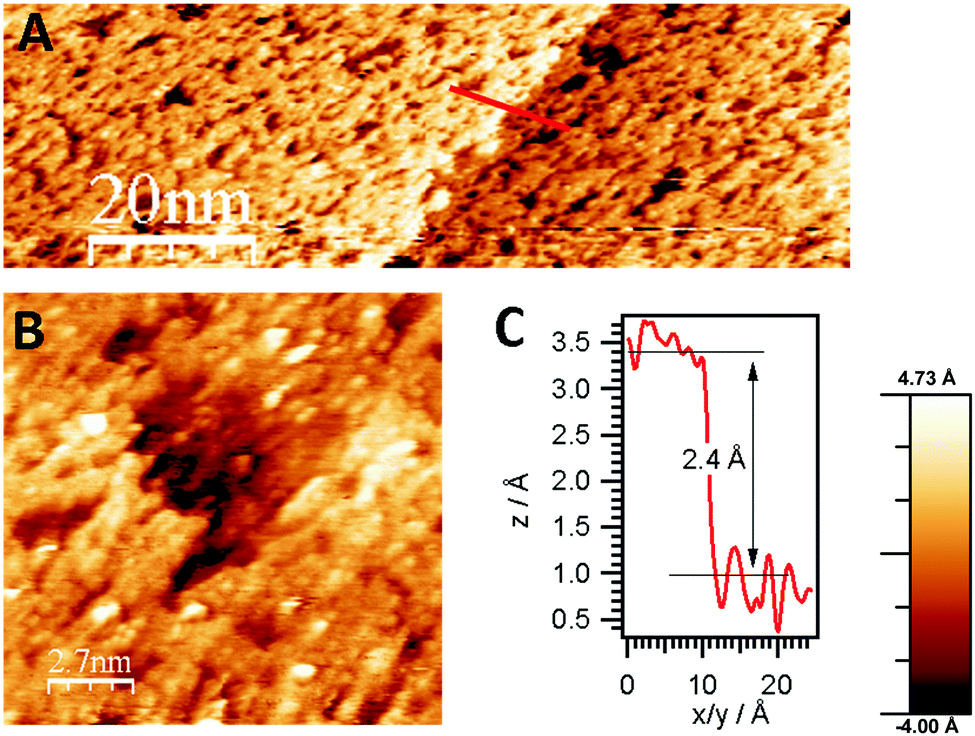 | ||
| Fig. 3 (A) STM images of the CVD-grown ZrOx overlayer on Pd(111) (100 L ZTB at 673 K, ZrOxHy–carbon layer coverage estimated to ∼1 ML, bias voltage: −0.8 V, tunneling current: 0.4 nA), including monoatomic step edge profile in the Pd(111) surface, (B) crack in the non-uniform mixed ZrOxHy + carbon overlayer, C) height profile of step edge along the line shown in A. | ||
Based on the data discussed above, we propose the following mechanism of ZTB–Pd surface interaction.
On clean Pd(111), ZTB dissociates via β-hydride elimination, resulting in adsorbed isobutylene and butoxide (ZrOC(CH3)3,ads) species. Isobutylene species either desorb as isobutylene or decompose and hydrogenate leaving carbon on the surface. The adsorbed butoxide species dissociates further, resulting in ZrOx deposition (* denoting an adsorption site, n and x are stoichiometric factors):
| Zr(OC4H9)4 + n* → Zr(OC(CH3)3)x,ads + (4−x)CH2CH–(CH3)2,ads + (4−x)Oads (x = 1, 2 and n = 7, 5) | (R1) |
| Zr(OC(CH3)3)x,ads + x* → ZrOx + xCH2CH–(CH3)2,ads | (R2) |
| CH2CH–(CH3)2,ads → CH2CH–(CH3)2↑ + * | (R3) |
| CH2CH–(CH3)2,ads + 3* → 4H2↑ + 4C | (R4) |
| ZrO + Oads → ZrO2 + *. | (R5) |
As dissolution of carbon atoms in Pd is fast above 573 K on Pd,39 we assume this process to dominate the ALD vacuum annealing step. Some oxygen could be consumed for hydrogen and carbon oxidation. The further fate of ZrO2 is discussed separately. We can conclude that at temperatures below the threshold of ∼550 K, the surface is poisoned by carbon compounds and above 773 K, the deposition rate starts to decline due to kinetic limitations (too fast desorption of ZTB).
On Oads/Pd(111), the main difference is the appearance of alkoxide-like species showing CO stretching vibrations. The exact configuration of this species is out of this work's scope and would require further investigation.
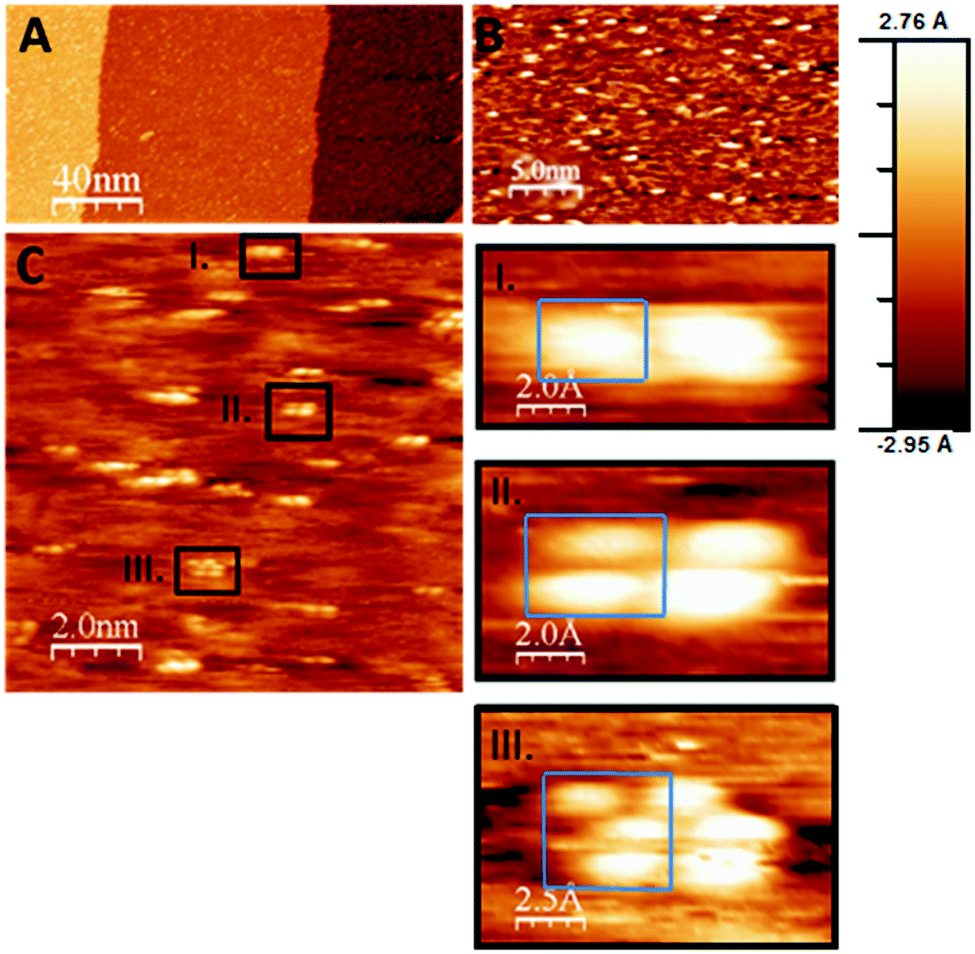 | ||
| Fig. 4 (A–C) STM images obtained after CVD growth of the ZrOxHy + C overlayer (100 L ZTB at 673 K) on Pd(111) followed by UHV-annealing to 723 K in 5 × 10−11 mbar. The resulting Zr0 coverage was ∼0.1 ML, and no more carbon was detected after annealing. The formed Zr-nanoclusters are shown in panel C and sub-panels I–III. (gap voltage: 0.2 V, constant current mode, feedback setting: 0.2 nA). Clusters containing 1–3 Zr atoms (blue frames) are seen (note that the structure most likely is imaged using a double tip. However, as all structural features seem to be doubled along the scan direction, the clusters containing 1–3 Zr atoms seen along the direction almost perpendicular to it are true Zr clusters being present on the surface). | ||
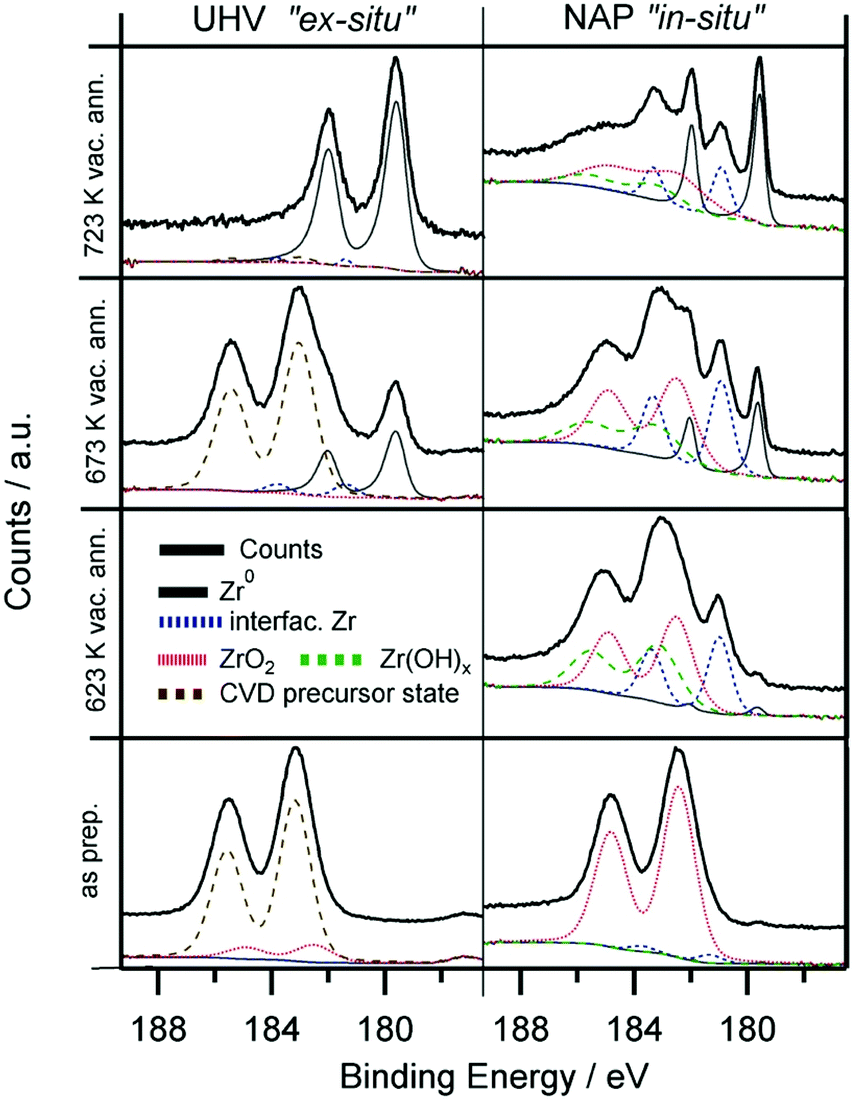 | ||
| Fig. 5 Zr 3d spectra of the CVD-grown ZrOxHy overlayer (673 K, 2000 L ZTB) at room temperature (“as prepared”) and after annealing in vacuum to 623 K, 673 K and 723 K. Left panels: experiments on Pd(111), base pressure of the UHV XPS chamber 5 × 10−11 mbar. Right panels: experiments on a Pd foil at the NAP-XPS setup of ISISS beamline at HZB/BESSY II, base pressure 5 × 10−8 mbar. The initial Zr coverage for both experiments is approximately 0.6 ML. | ||
The unusual behavior on Pd under vacuum conditions, leading to the formation of nanostructured Zr0 islands, is explained by efficient removal of oxygen atoms and an extremely low re-oxidation rate. As deduced from STM images and DFT calculations shown later, metal support interaction effects are also highly beneficial. Even though a number of Pd–Zr intermetallic bulk phases and/or compounds of varying stoichiometry are known,47,48 no such Pd–Zr phases can be formed at temperatures as low as 673 K. The Zr0 metallic surface state was confirmed on the Pd(111) surface by (i) almost no shift of the Pd 3d5/2 peak (peak position: 335.2 eV) and (ii) the fact that the Zr 3d intensity does not change following annealing (neither dissolution nor evaporation) and (iii) the shift of the Zr 3d binding energy toward 179.4 eV typical for metallic Zr species.49,50 On the other hand, the interface between ZrOx and the noble metal (Pd or Pt) likely plays an important role since this reduction is only working on Pd and Pt, but e.g. not on Cu, which is a less active material.
In order to extend our understanding of ZTB CVD under UHV to more realistic conditions, we compare the UHV results to the in situ XPS data obtained at the synchrotron radiation facility BESSY-II. Fig. 5 shows the Zr 3d spectra obtained during the in vacuo heating of the CVD grown ZrOx film (∼2000 L ZTB at 673 K, deposited amount ∼0.6 ML) on Pd(111) and on polycrystalline Pd foil by the UHV-XPS (Omicron) and in situ by the NAP-XPS at BESSY II, respectively. The better vacuum conditions in the UHV-XPS set-up (5 × 10−11 mbar, water free) compared with the 5 × 10−8 mbar water-containing background in the NAP-XPS chamber allowed to obtain completely reduced zirconium. In in situ NAP-XPS, the highly reactive ZTB precursor was converted to mainly ZrO2 right after preparation (Zr 3d5/2 at 182.4 eV). Also the amount of carbon is significantly lower in UHV-XPS, which we assign to enhanced precursor decomposition due to the intense synchrotron beam of the in situ experiment and partially to the high base pressure. In both experiments, heating in vacuum results in reduction: only metallic Zr (179.4 eV) was detected in UHV-XPS, whereas a larger fraction of “partially reduced” zirconium (interfacial species) is additionally observed in the in situ NAP-XPS experiment, as a Zr 3d5/2 component is found at ca. 181.0 eV. This state is assigned to an ultrathin layer of ZrO2.49 We tentatively assign this peak to oxygen-deficient and not fully metallic interfacial ZrOxHy, e.g. with metal–metal bonds to the Pd substrate and still remaining Zr–O–Zr entities to the outermost ZrO2 layer(s). This may explain the suggested oxidation state between Zr4+ and Zr0, but rather if e.g. a bi- or multilayered precursor would be affected by loss of interfacial oxygen between Zr and Pd.
The reduction of Zr4+ is more difficult in the NAP-XPS-System, as not all Zr can be fully reduced to the metallic state (Fig. 5). In the ex situ UHV experiment, where the reduction is fast and complete, no such partially reduced intermediates could be detected. This difference most likely arises from the much poorer vacuum conditions of the in situ NAP-XPS chamber as discussed above. The background pressure of any oxygen containing molecules was, thus, found to be crucial for the eventual efficiency of Zr reduction. Also, the growth mode of the Zr4+ precursor species (i.e. layer thickness or island formation) may be already pre-determined by the residual gas conditions during deposition and/or annealing.
Upon further annealing (T > 780 K), dissolution of Zr into the Pd bulk occurs, leading to a significant loss of the Zr 3d intensity and the formation of a near-surface Pd–Zr alloy, as indicated by a slightly higher BE shift of the Pd 3d peaks (not shown herein). The BE of the dissolving Zr is found to be 179.6 eV, which is shifted about 0.2–0.4 eV towards higher BE from the position for bulk Zr0.50 The most likely reason is the enhanced Pd–Zr coordination in the subsurface regions, but charge-transfer processes and final state effects might also play a role.
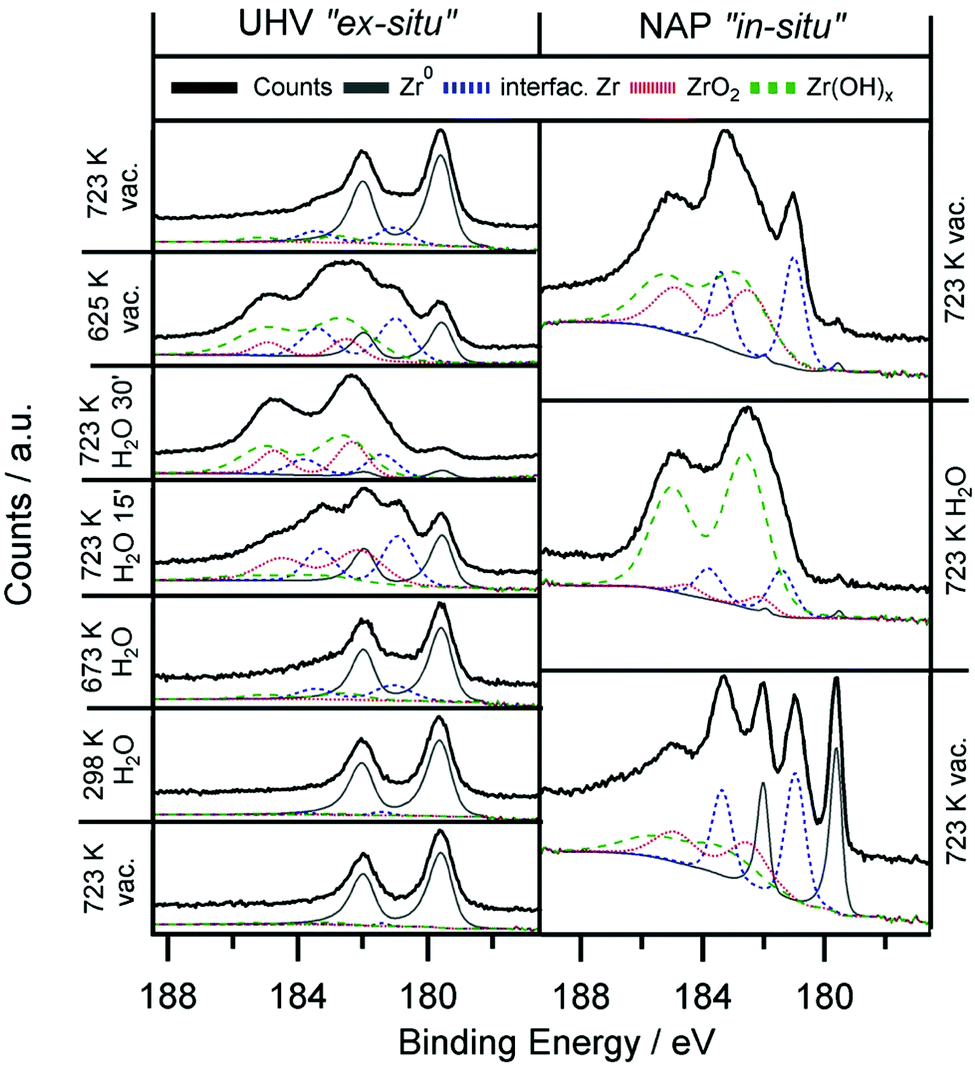 | ||
| Fig. 6 Zr 3d spectra obtained ex situ and in situ during heating of 0.2 ML Zr0/ZrOxHy in water. Zr0/ZrOxHy is prepared by annealing of CVD-grown ZrOxHy (at 673 K, 2000 L ZTB) in 5 × 10−11/5 × 10−8 mbar for the ex situ and in situ experiment, respectively. Left panels: ex situ UHV XPS experiments on Pd(111), water pressure 5 × 10−7 mbar. Right panels: in situ NAP-XPS experiments on a Pd foil, water pressure 0.3 mbar for 20 min. | ||
Only in the UHV ex situ experiment the hydroxylation process is fully reversible: annealing at 723 K fully re-establishes the metallic state of Zr. Most likely, the degree of reversibility of the reduction process is highly dependent on the background pressure of oxygen containing molecules such as H2O and O2 in the residual gas.
Even though a catalytic reforming cycle should rather involve reversibly formed hydroxyl groups at Zr sites, an analogous experiment can also be performed with O2 instead of H2O. Oxidative treatments in O2 (ex situ) and 0.3 mbar O2 (in situ) leads in both cases to fully oxidized ZrO2. Oxidation in 5 × 10−7 mbar O2 for the ex situ experiments is fast, after 10 min no more metallic Zr is found. Again, full reversibility to the Zr metal state is only achieved in the UHV chamber. Water-analogous results are also obtained for the in situ experiments with 0.3 mbar O2 pressure. Here, again only partially reversible reduction is possible, highlighting the aforementioned limitations by the poorer background pressure.
We then investigated three possible competing reactions for oxygen atom removal: to release 0.5O2 into the gas phase directly, to dissolve an O atom into the Pd sub-layer and to diffuse the O atom to the available neighboring bare Pd(111) surface. The free reaction energy at 725 K, calculated for an equilibrium pressure above the surface of PO2 = 10−12 mbar, is summarized in Table 1. Among the three possible competing reactions the interface oxygen diffusion to the neighboring bare Pd(111) surface is in any case the most favorable, followed by direct desorption into the gas phase as 0.5O2 and dissolution into the Pd sub-layer, which is the least favorable. The corresponding reaction energies for the interface-to-Pd oxygen diffusion are 0.74, 1.43, and 0.94 eV for the 1 ML, 2 ML and 3 ML thick ZrO2 films, respectively, while the direct desorption of 0.5O2 into the gas phase is ∼0.20 eV higher than the diffusion. The corresponding reaction energies for oxygen dissolution in the Pd sub-layer amount to 2.00, 3.09 and 2.48 eV, which is in all cases much higher than the diffusion to the neighboring Pd(111) surface. We also found that the surface oxygen of the topmost layer and the interface oxygen of the lowest layer show the same reactivity for the direct O2 desorption for the 1 ML and 2 ML ZrO2 films. However, for the 3 ML ZrO2 film, the interface oxygen is more active than the surface oxygen, indicating that the oxygen vacancy is more stable at the interface than at the outer surface. The corresponding structures for oxygen diffusion to the neighboring Pd(111) surface and dissolution into the sub-layer of the Pd substrate are shown in Fig. 7. As shown therein, the surface Pd atom moves up significantly by ∼0.40–0.50 Å in order to adapt the dissolved oxygen atom. In Table 1, we also included the movement of an interface Zr atom. As expected, atomic Zr dissolution from the interface to the Pd sub-layer is less favorable than atomic oxygen dissolution due to the large size of atomic Zr. The corresponding reaction energies are 2.78, 3.45 and 3.29 eV for the 1 ML, 2 ML, and 3 ML ZrO2 films. Comparing all the reaction energies, it is noted that to move either O or Zr from the 2 ML ZrO2 film is more difficult than that from the 1 ML and 3 ML films, suggesting that the 2 ML arrangement is the most stable one.
| ZrO2 | 1 layer | 2 layer | 3 layer |
|---|---|---|---|
| ΔGdiss(Ointerface → Osub-layer/Pd) | 2.00 | 3.09 | 2.48 |
| ΔGO2(Ointerface → 0.5O2) | 0.94 | 1.63 | 1.14 |
| ΔGO2(Osurface → 0.5O2) | 0.94 | 1.68 | 1.46 |
| ΔGdiff(Ointerface → OPd) | 0.74 | 1.43 | 0.94 |
| ΔGdiss(Zrinterface → Zrsub-layer/Pd) | 2.78 | 3.45 | 3.29 |
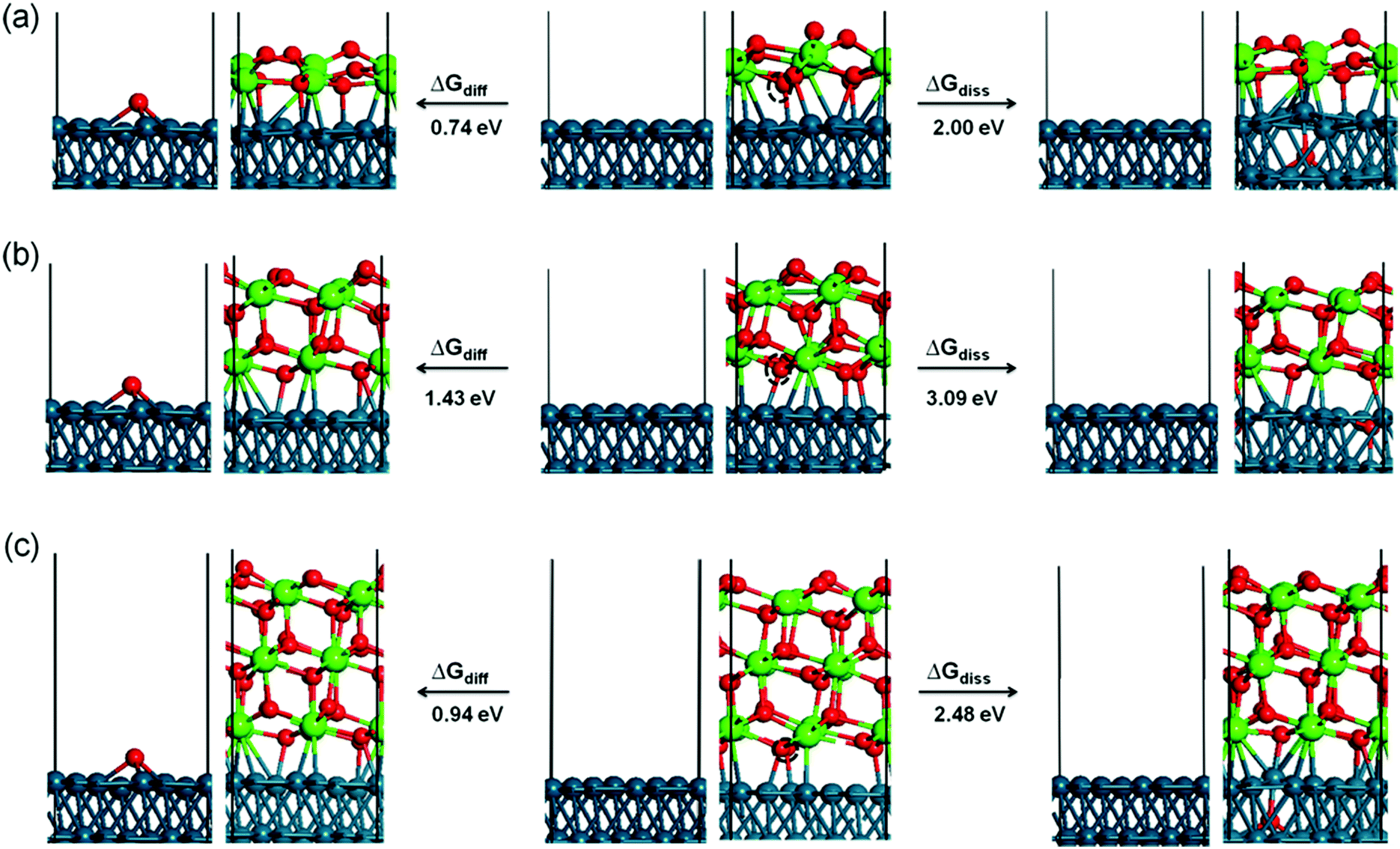 | ||
| Fig. 7 Structural models of interface oxygen dissolution to the sub-layer of Pd(111) and diffusion to the available neighboring bare Pd(111) surface. The reaction energies are labeled. Left: the Pd–O diffusion scenario; right: dissolution scenario; middle: 1 (a), 2 (b), and 3 (c) ML ZrO2 on the Pd(111) substrate. The moving O atom is marked by a dashed black circle. Small red spheres: O; large green spheres: Zr; blue spheres: Pd. The boxes indicate the computational supercell. | ||
As discussed above, among the three possible competing reactions the interface oxygen rather prefers to diffuse to the available neighboring bare Pd(111) surface. Following this process, atomic oxygen may recombine to desorb as O2 from the Pd surface. A number of previous experimental and theoretical studies have investigated O2 adsorption/desorption on the Pd(111) surface.53–57 Using thermal desorption spectrometry, one of our previous studies showed that the O2 desorption rate from a chemisorbed O(ads) layer on Pd(111) reached its maximum at ∼750 K, which implies that associative O2 desorption from bare Pd is both possible and irreversible under the UHV-reduction conditions chosen in the present study.58
The conclusions from this section regarding the optimum growth conditions for subsequent full reducibility are: (i) preferential single layer growth of ZrO2 and (ii) a large phase boundary of the sub-monolayer ZrO2 islands toward the surrounding bare Pd surface should be realized already during the deposition process.
3.2. Catalytic results
To correlate the redox chemistry of Zr on Pd with catalytic properties, the previously discussed Pd–Zr systems have been tested in two model reactions: methanol steam reforming and dry reforming of methane. While for the former no catalytically promoting role has been found (for an overview of the catalytic results, see ESI,† Section F, Fig. S4–S6), the latter is clearly promoted by Zr doping of Pd. These experiments, which show the opening of new CO2 activation channels on Pd, are now discussed in Section 3.2.1.:Zr = 2:1 intermetallic “IM” reference catalyst described in the experimental section, are tested for methane dry reforming (DRM, CH4 + CO2 → 2CO + 2H2). For this reaction, no initial water activation is required, but active sites both for CO2 and CH4 activation are mandatory. Water can in principle play a “co-catalytic” role as an intermediate or spoil the reaction as a side product, if CH4 is not stoichiometrically converted and the inverse WGS reaction is active: | Water-free | Water-involving |
| CH4 → C + 2H2 | CH4 → C + 2H2 |
| C + CO2 → 2CO | CO2 + H2 → CO + H2O |
| C + H2O → CO + H2 |
The results of the DRM experiments are shown in Fig. 8. In none of the catalytic experiments shown therein, H2O was detected, thus, at least the side reaction to water associated with the inverse WGS reaction is of minor importance. Carbon was detected with XPS on the surface after the reaction, but in too low amounts (relative to the gaseous reactant/product amounts) to affect the overall stoichiometry. Fig. 8 displays the DRM selectivity/activity pattern for a choice of “monofunctional” (pure Pd or Zr) and “bifunctional” (Pd–Zr) model catalysts. Clean Pd is completely inactive, as well as clean ZrO2. Fig. 9 shows the corresponding XP spectra before and after selected DRM experiments of Fig. 8. A pure Zr metal foil was oxidized in 1 bar O2 at 673 K to form a ZrO2 layer that is thicker than the XPS analysis depth (95% of signal from topmost 5 nm for ZrO2 with Mg Kα radiation). After the DRM reaction, the ZrO2 film is reduced within the accessible information depth and a single state of Zr is detected in the XPS 3d region at a binding energy of 179.4 eV, which agrees well both with literature BE values of Zr050 and zirconium carbide ZrC.59 Destructive (sputter) depth profiling of this surface after catalysis is shown in the ESI† in Fig. S7, revealing a thickness of the carburized ZrC layer on top of Zr of at least 20 nm.
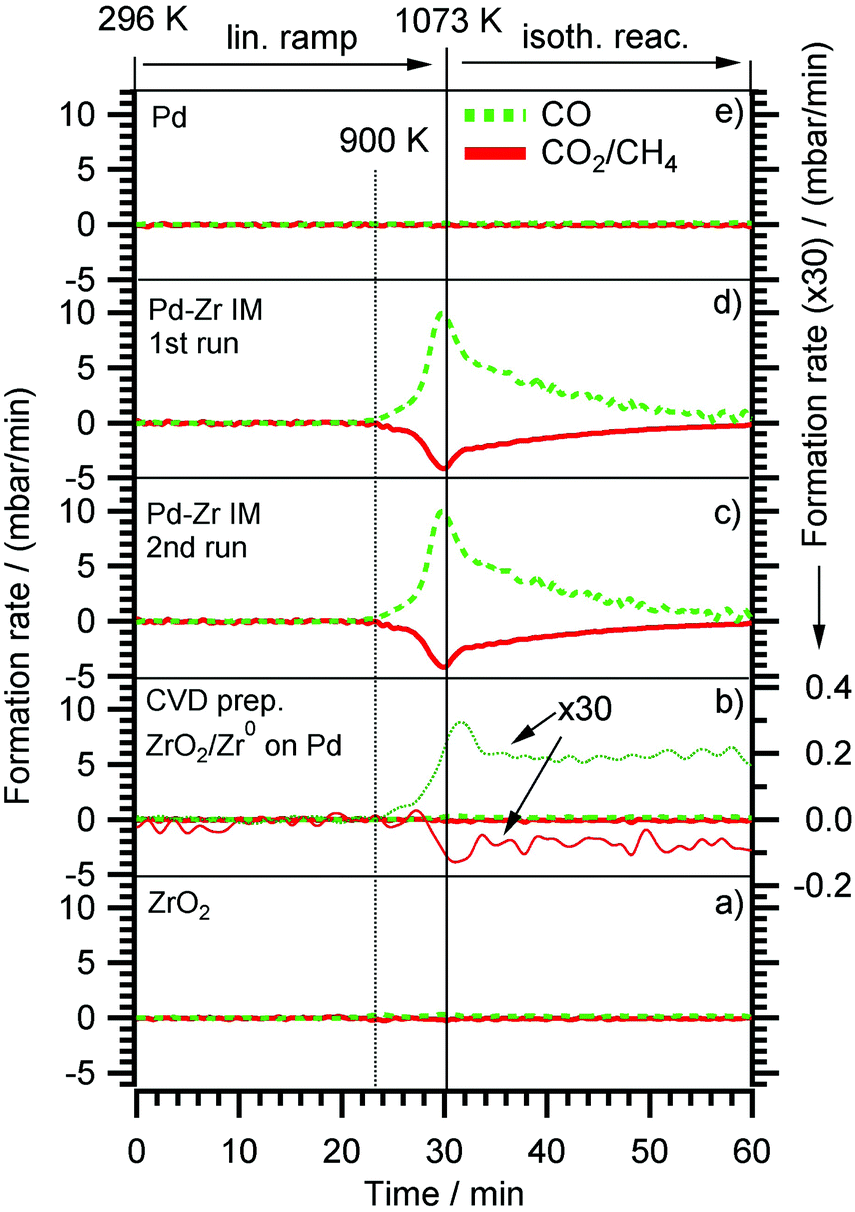 | ||
| Fig. 8 Methane dry reforming (DRM) on (a) Clean oxidized Zr foil (identical with pattern for 0.3 ML ZrO2 on Pd, prepared by post annealing of CVD grown ZrOxHy in 5 × 10−7 mbar O2 at 700 K). (b) 0.3 ML Zr0/ZrOxHy, prepared by annealing of CVD grown ZrOxHy in 5 × 10−9 mbar vacuum (the reactant consumption- and CO product formation rates were multiplied by a factor of 30 and plotted in the Y-scale range of the “IM” experiments (c and d) two consequent DRM runs of an intermetallic Pd:Zr = 2:1 “IM” bulk phase, (e) pure Pd. Initial DRM conditions: CO2:CH4 = 1:1, total reactant pressure 100 mbar). | ||
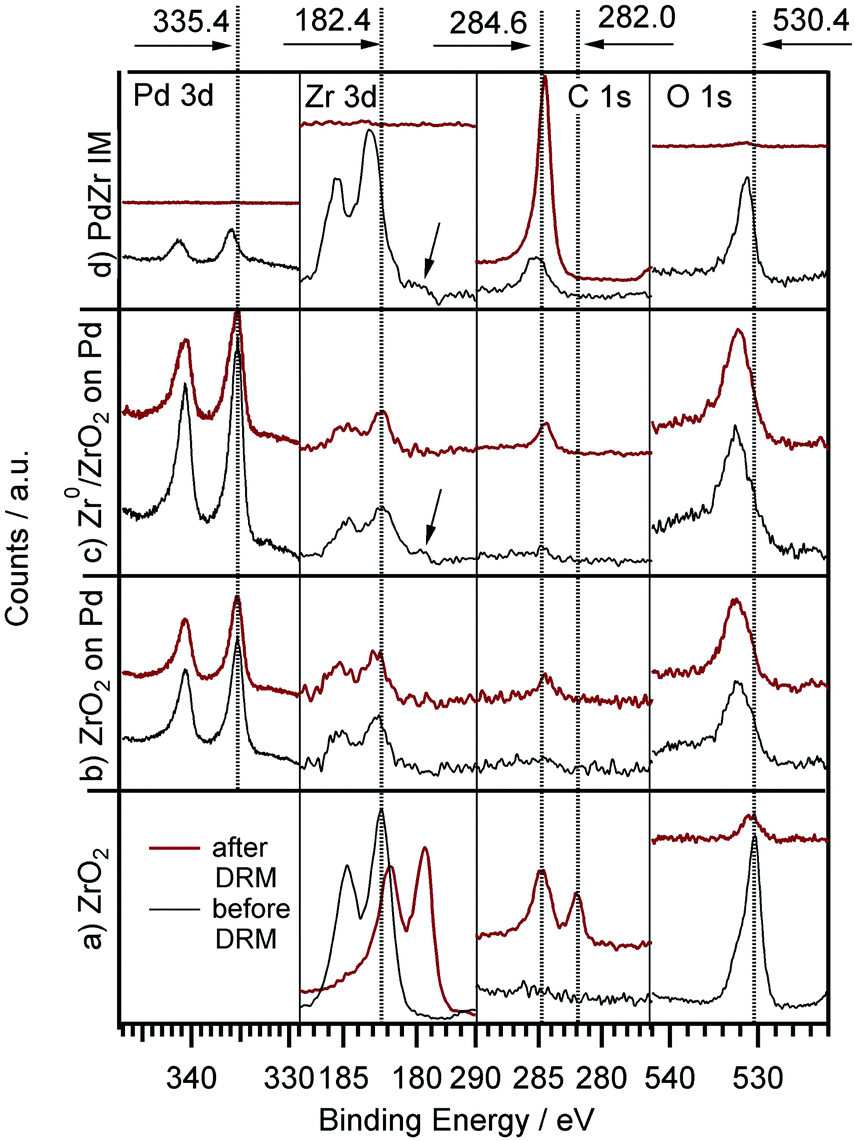 | ||
| Fig. 9 XPS (Pd 3d, Zr 3d, C 1s and O 1s (overlapping with Pd 3p) region) before and after DRM experiments for: (a) clean Zr foil, pre-oxidized in 5 × 10−7 mbar O2 at 750 K for 15 min; (b) 0.3 ML ZrO2 on Pd, prepared by thermal annealing of CVD grown ZrOxHy in 5 × 10−7 mbar O2 at 700 K; (c) 0.3 ML Zr0/ZrOxHy, prepared by annealing of a CVD grown ZrOxHy in 5 × 10−9 mbar vacuum at 700 K; (d) intermetallic Pd/Zr (2:1) “IM” bulk phase before first and after second DRM run. The arrow indicates the Zr0 component. | ||
For the CVD-prepared and then vacuum- or oxygen-annealed Pd/Zr0/ZrOxHy or Pd/ZrO2 samples, the activity in DRM depends on the initial oxidation state of Zr. As soon as there is a small fraction of Zr0 available in the pre-catalyst (see arrow in Fig. 9), a measurable CO formation rate could be detected. In Fig. 8, the reaction rate is multiplied by a factor of 30 to show that there is some CO2 consumed and CO formed at the stoichiometric ratio 1:2, but at a 30× lower rate as compared to the bulk-intermetallic “IM” catalyst described below. Again, no water side product formation could be detected. The temperature-programmed reaction profile for fully oxidized ZrO2 on Pd (generated by annealing of the “as-grown” CVD layer in O2 at 750 K) is not shown since no reactivity at all was detected, but the XP spectra for both ZrO2 and ZrOxHy/Zr0 on Pd are shown in Fig. 9 before and after DRM. On this basis, the fundamental activating function of Zr0, which becomes in situ oxidized during DRM, could be shown qualitatively in spite of the very small initial Zr0 amount. In order to substantiate this result, we decided to test an all-intermetallic bulk phase of Pd and Zr in the stoichiometry of 2:1 under otherwise identical DRM conditions (“IM” sample prepared by strongly exothermic co-melting, see chapter 2). The sample gets (surface) oxidized at room temperature over time, leading to a loss of Zr0 in XPS (analysis depth ∼2–3 nm). This oxidation was not found to be crucial for the catalyst performance. It does not matter whether Pd–Zr intermetallic is surface pre-oxidized in air at room temperature almost within the XPS analysis depth (Fig. 9). The “before DRM” – IM spectra in Fig. 9 shows the most air-oxidized state right before the catalytic experiment. Nevertheless, it shows residual Zr0.
On this sample, the formation rate of CO is ∼30 times higher than on the vacuum annealed CVD Pd/Zr0/ZrOxHy sample. This substantiates our interpretation that the (in situ) oxidative segregation from an initially intermetallic phase and/or surface leads to an enhanced number of reforming-active interfacial Pd/ZrOxHy sites. Again, we suggest the idea that the (in situ) oxidation of an atomically homogeneous intermetallic phase leads to the most disperse Zr4+OxHy surface-near distribution possible and therefore, to a maximum number of Pd/ Zr4+OxHy interface sites, i.e. to a quasi “atomically dispersed” phase boundary. The validity of this concept has previously been proven for Cu/Zn12, Pd/Zn13 and Cu/Zr11 and is now also confirmed for Pd/Zr. The enhanced DRM reactivity on the Pd/Zr0/ZrOxHy pre-catalyst is therefore mainly assigned to the synergistic action of very small Zr0 clusters, which become partially oxidized and/or hydroxylated under reaction conditions and exhibit a high number of direct Pd neighbours. Obviously, the number of these desirable interfacial sites is even higher on the in situ oxidized IM bulk phase pre-catalyst, eventually leading to a 30 times higher reaction rate.
Note that no deactivation of the IM catalyst was found on the time scale of our experiments (2 × 60 min each, 1st and 2nd run). In both cases, the decrease of the reaction rate in the isothermal reaction section is due to complete consumption of CO2 and CH4, as the reactor is operated in re-circulating batch mode. This reproducibility and stability with respect to catalytic performance holds despite a huge amount of carbon deposition (or, possibly, surface segregation) after cooling in the reaction mixture (see C 1s region of uppermost panel in Fig. 9). The binding energy of ∼284.5 eV and the rather large peak width (as compared to pure graphite) suggest a mix of sp3- or sp2-hybridized carbon species. The carbon layer is thick enough for complete shielding of both the Pd and Zr signals. This renders the absent deactivation of the catalyst after the first run even more surprising. Apparently, the catalytically active sites become again accessible during heating, e.g. if the carbon layer is reacted off by CO2 and/or becomes re-dissolved in the bulk. Since the reaction sets in at ∼900 K, the onset of the Boudouard reaction might explain this phenomenon. A final note on the bulk structure of the DRM-tested intermetallic Pd–Zr sample should be provided at this point. As already outlined in Section 2 and further elaborated upon in the discussion of Fig. S8 (ESI†), highlighting the X-ray diffractograms before and after the DRM reaction, despite the nominal 2:1 composition, the initial patterns reveal the presence of at least two intermetallic compounds with Pd excess, namely Pd3Zr and Pd4Zr3. Upon performing the DRM reaction, the patterns get even more complex, including the complete removal of the Pd3Zr phase and the formation of a new Pd9Zr phase. Elementary, graphite-like carbon and partially oxidized ZrO2 in the monoclinic and tetragonal modifications, formed by partial (oxidative) decomposition of the Pd–Zr intermetallic compounds, are equally observed. This is in striking contrast to Cu–Zr, where only one Cu51Zr14 compound was observed after preparation.24 A common behavior (despite the different reactions) are the massive operando observed bulk structural changes accompanying the surface chemical alterations. However, due to the initially less homogeneous melt in the Pd–Zr case, the structural complexity is significantly more pronounced.
4. Conclusions
The CVD process of zirconium-t-butoxide ZTB was studied on Pd(111) single crystal and on polycrystalline Pd foil samples. A substrate temperature of ∼700 K was found to be ideal to create a partially hydroxylated, carbon-depleted and vacuum-reducible ZrOxHy overlayer. Depending on the quality of the vacuum conditions, reversible partial or full reduction of the ZrOxHy overlayer (i.e. Zr4+) to surface Zr0 nanoclusters could then be accomplished. This very dynamic redox behaviour of ZrOxHy overlayers on Pd manifests itself in repeated oxidation (in water or oxygen) and vacuum-reduction cycles to the adsorbed Zr0 metallic state without alloying up to 770 K. Nanoclusters of 2–6 Zr0 atoms were found by STM if Pd/ZrOxHy was UHV-reduced at 723 K. DFT calculations show that the interfacial oxygen, especially that of a single-layer cubic ZrO2 film, energetically prefers to diffuse to the available neighboring bare Pd(111) surface, rather than to desorb as O2 or to become dissolved in the Pd subsurface region.CVD-prepared and vacuum-reduced Pd/Zr0/ZrOxHy model catalysts are not MSR active up to 623 K, due to the formation of an interlayer of graphitic carbon between Zr and Pd, resulting in the loss of active Zr/Pd phase boundary sites. In contrast to the related Cu/ZrOxHy system, MSR on Pd/Zr0/ZrOxHy does, therefore, not proceed via the low (<623 K) temperature/partial dehydrogenation route with intermediate formaldehyde, which is thereafter totally oxidized by activated water to CO2. On Zr-doped Pd, rather, the expected full dehydrogenation of methanol toward CO takes place on the bare Pd surface patches, followed by the (also water-activation dependent) water gas shift route toward CO2 at higher temperatures above ∼650 K. The latter process is only possible in the presence of an active Pd/ZrOxHy phase boundary, which is re-established above ∼650 K by reactive removal of carbon deposits.
Dry reforming activity with almost 100% CO-selectivity is only observed if Zr0 species either in a CVD-prepared/vacuum-reduced or melt-prepared intermetallic Pd–Zr “pre-catalyst” are initially present. With an intermetallic pre-catalyst bulk phase with a nominal 2:1 composition consisting of Pd0 and Zr0, the highest CO formation rates were obtained. At the surface of both systems, Zr0 is not stable under DRM conditions, and Zr4+OxHy species are formed by in situ reactive oxidation/hydroxylation. This process is likely to lead to a particularly high number of active phase boundary sites. As discussed above, further studies will most likely be focused on identifying sophisticated synthesis routines, allowing to prepare single-phase Pd–Zr intermetallic compounds, whose intrinsic (catalytic) properties can, thus, be studied.
DFT calculations to describe the Pd/ZrOxHy induced CO2 activation and the reaction mechanism to CO and H2 are planned. Besides enhanced CO2 activation, also formation of surface adsorbed carbon from CH4 may be positively influenced by Pd/ZrOxHy. The present study provides phenomenological evidence for an enhanced phase boundary synergism, including a beneficial carbon chemistry. Thus, theoretical studies of the microscopic reaction mechanism at the active sites are imperative, since Zr doping of Pd obviously opens up special CO2 activation pathways on chemically and electronically Pd, which are not available on clean undoped metallic Pd.
Acknowledgements
L. Mayr and Z. Shi acknowledge financial support via FWF SFB “FOXSI” project part F4503-N16, L. Mayr via a scholarship of the Carinthian Confederation of Industry (Industriellenvereinigung Kärnten). The computational results presented have been achieved (in part) using the Vienna Scientific Cluster (VSC). Support from the Department of Energy, Office of Basic Energy Sciences, Chemical Sciences, under Grant DE-FG02-03ER15408 is gratefully acknowledged. The authors acknowledge the Purdue Catalysis Center, We are grateful to Michael Detwiler, Amir Gharachorlou, Ronald G. Reifenberger and Fabio H. Ribeiro for many helpful suggestions and motivating discussions. We also thank the Birck Nanotechnology Center staff for their assistance and cooperation throughout. We gratefully acknowledge the Helmholtz-Zentrum-Berlin (HZB) for providing beamtime at the ISISS beamline.References
- K.-I. Fujimoto, F. H. Ribeiro, M. Avalos-Borja and E. Iglesia, J. Catal., 1998, 179, 431–442 CrossRef CAS.
- S. H. Liu, G. K. Chuah and S. Jaenicke, J. Mol. Catal. A: Chem., 2004, 220, 267–274 CrossRef CAS.
- D. He, Y. Ding, H. Luo and C. Li, J. Mol. Catal. A: Chem., 2004, 208, 267–271 CrossRef CAS.
- M. Behrens and M. Arnbrüster, Methanol Steam Reforming, in Catalysis for Alternative Energy Generation, ed. L. Guczi and A. Erdohelyi, Springer, New York, 2012, pp. 175–235 Search PubMed.
- P. Sudarsanam, B. Mallesham, P. S. Reddy, D. Großmann, W. Grünert and B. M. Reddy, Appl. Catal., B, 2014, 144, 900–908 CrossRef CAS.
- W. Du, G. Zhao, H. Chang, F. Shi, Z. Zhu and X. Qian, Mater. Charact., 2013, 83, 178–186 CrossRef CAS.
- A. Atkinson, S. Barnett, R. J. Gorte, J. T. S. Irvine, A. J. McEvoy, M. Mogensen, S. C. Singhal and J. Vohs, Nat. Mater., 2004, 3, 17–27 CrossRef CAS PubMed.
- R. L. Puurunen, J. Appl. Phys., 2005, 97, 121301 CrossRef.
- J. P. Chang, Y.-S. Lin, S. Berger, A. Kepten, R. Bloom and S. Levy, J. Vac. Sci. Technol., B, 2001, 19, 2137–2143 CAS.
- L. Mayr, N. Köpfle, A. Auer, B. Klötzer and S. Penner, Rev. Sci. Instrum., 2013, 84, 094103 CrossRef PubMed.
- L. Mayr, B. Klötzer, D. Zemlyanov and S. Penner, J. Catal., 2015, 321, 123–132 CrossRef CAS.
- C. Rameshan, W. Stadlmayr, S. Penner, H. Lorenz, N. Memmel, M. Hävecker, R. Blume, D. Teschner, T. Rocha, D. Zemlyanov, A. Knop-Gericke, R. Schlögl and B. Klötzer, Angew. Chem., 2012, 124, 3057–3061 CrossRef.
- C. Rameshan, W. Stadlmayr, C. Weilach, S. Penner, H. Lorenz, M. Hävecker, R. Blume, T. Rocha, D. Teschner, A. Knop-Gericke, R. Schlögl, N. Memmel, D. Zemlyanov, G. Rupprechter and B. Klötzer, Angew. Chem., Int. Ed., 2010, 49, 3224–3227 CrossRef CAS PubMed.
- S. Wang, G. Q. Lu and G. J. Millar, Energy Fuels, 1996, 10, 896–904 CrossRef CAS.
- P. Ferreira-Aparicio, A. Guerrero-Ruiz and I. Rodríguez-Ramos, Appl. Catal., A, 1998, 170, 177–187 CrossRef CAS.
- H. Ay and D. Üner, Appl. Catal., B, 2015, 179, 128–138 CrossRef CAS.
- N. H. Elsayed, N. R. M. Roberts, B. Joseph and J. N. Kuhn, Appl. Catal., B, 2015, 179, 213–219 CrossRef CAS.
- F. Kertis, J. Snyder, L. Govada, S. Khurshid, N. Chayen and J. Erlebacher, JOM, 2010, 62, 50–56 CrossRef CAS.
- M. Valden, X. Lai and D. W. Goodman, Science, 1998, 281, 1647–1650 CrossRef CAS PubMed.
- A. Gharachorlou, M. D. Detwiler, X. K. Gu, L. Mayr, B. Klötzer, J. Greeley, R. G. Reifenberger, W. N. Delgass, F. H. Ribeiro and D. Y. Zemlyanov, ACS Appl. Mater. Interfaces, 2015, 7, 16428–16439 CAS.
- A. Gharachorlou, M. D. Detwiler, L. Mayr, X. K. Gu, J. Greeley, R. G. Reifenberger, W. N. Delgass, F. H. Ribeiro and D. Y. Zemlyanov, J. Phys. Chem. C, 2015, 119, 19059–19072 CAS.
- A. Gharachorlou, M. D. Detwiler, A. V. Nartova, Y. Lei, J. Lu, J. W. Elam, W. N. Delgass, F. H. Ribeiro and D. Y. Zemlyanov, ACS Appl. Mater. Interfaces, 2014, 6, 14702–14711 CAS.
- R. Paul, R. G. Reifenberger, T. S. Fisher and D. Y. Zemlyanov, Chem. Mater., 2015, 27, 5915–5924 CrossRef CAS.
- L. Mayr, D. Schmidmair, M. Armbrüster, N. Köpfle, J. Bernardi, S. Schwarz, B. Klötzer and S. Penner, ChemCatChem, 2016, 8, 1778–1781 CrossRef CAS.
- L. Mayr, R. Rameshan, B. Klötzer, S. Penner and C. Rameshan, Rev. Sci. Instrum., 2014, 85, 055104 CrossRef PubMed.
- A. Knop-Gericke, E. V. Kleimenov, M. Hävecker, R. Blume, D. Teschner, S. Zafeiratos and R. Schlögl, Adv. Catal., 2009, 52, 213–272 CAS.
- CasaXPS Version 2.3.16 Pre-rel 1.4, Casa Software Ltd, 2011.
- C. D. Wagner, W. M. Riggs, L. E. Davis, J. F. Moulder and G. E. Muilenberg, Handbook of X-Ray Photoelectron Spectroscopy, Perkin-Elmer Corporation, Physical Electronics Division, Eden Prairie, Minnesota, 1979, vol. 55344 Search PubMed.
- D. Majumdar and D. Chatterjee, J. Appl. Phys., 1991, 70, 988–992 CrossRef CAS.
- C. J. Powell and A. Jablonski, NIST Electron Effective-Attenuation-Length Database SRD 82, Version 1.3, Instititue of Standards and Technology, Gaithersburg, 2011 Search PubMed.
- J. J. Yeh, Atomic Calculation of Photoionization Cross-Sections and Asymmetry Parameters, Gordon and Breach Science Publishers, Langhorne, PE, USA, 1993 Search PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. Klimes, D. R. Bowler and A. Michaelides, J. Phys.: Condens. Matter, 2010, 22, 022201 CrossRef PubMed.
- J. Klimeš, D. R. Bowler and A. Michaelides, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 195131 CrossRef.
- X.-R. Shi, S.-G. Wang, J. Hu, Z. Qin and J. Wang, Surf. Sci., 2012, 606, 1187–1194 CrossRef CAS.
- J. P. Chang, Y.-S. Lin and K. Chu, J. Vac. Sci. Technol., B, 2001, 19, 1782–1787 CAS.
- M. A. Cameron and S. M. George, Thin Solid Films, 1999, 348, 90–98 CrossRef CAS.
- H. Gabasch, E. Kleimenov, D. Teschner, S. Zafeiratos, M. Hävecker, A. Knop-Gericke, R. Schlögl, D. Zemlyanov, B. Aszalos-Kiss, K. Hayek and B. Klötzer, J. Catal., 2006, 242, 340–348 CrossRef CAS.
- C. T. Lynch, K. S. Mazdiyasni, W. J. Crawford and J. S. Smith, Anal. Chem., 1964, 36, 2332–2336 CrossRef CAS.
- I. Martinko, R. Verhoef, Y. Engelmann, J. Cornil, S. Salah, R. Snyders, O. Antonin and P. Raynaud, 22nd International Symposium on Plasma Chemistry, 2015, vol. III, pp. 6–35 Search PubMed.
- R. Verhoef, P. Raynaud, S. Ligot, R. Snyders, T. Nelis, R. V. Gonzalez and S. Vitale, 21nd International Symposium on Plasma Chemistry, 2013, p. OR 214 Search PubMed.
- Y. Sert, L. M. Singer, M. Findlater, H. Dogan and G. Cirak, Spectrochim. Acta, Part A, 2014, 128, 46–53 CrossRef CAS PubMed.
- H. J. Oelichmann, D. Bougeard and B. Schrader, J. Mol. Struct., 1981, 77, 179–194 CrossRef CAS.
- H. J. Oelichmann, D. Bougeard and B. Schrader, J. Mol. Struct., 1981, 77, 149–163 CrossRef CAS.
- N. F. Brown and M. A. Barteau, J. Am. Chem. Soc., 1992, 114, 4258–4265 CrossRef CAS.
- M. W. Chase, J. Phys. Chem. Ref. Data, Monogr., 1998, 9, 1–1951 Search PubMed.
- J.-Q. Hu, M. Xie, Y. Pan, Y.-C. Yang, M.-M. Liu and J.-M. Zhang, Comput. Mater. Sci., 2012, 51, 1–6 CrossRef CAS.
- H. Li, J.-I. J. Choi, W. Mayr-Schmölzer, C. Weilach, C. Rameshan, F. Mittendorfer, J. Redinger, M. Schmid and G. Rupprechter, J. Phys. Chem. C, 2015, 119, 2462–2470 CAS.
- S. Sinha, S. Badrinarayanan and A. P. B. Sinha, J. Less-Common Met., 1987, 134, 229–236 CrossRef CAS.
- C. Huang, Z. Tang and Z. Zhang, J. Am. Chem. Soc., 2001, 84, 1637–1638 CAS.
- K. Meinel, A. Eichler, S. Förster, K. M. Schindler, H. Neddermeyer and W. Widdra, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 235444 CrossRef.
- F. P. Leisenberger, G. Koller, M. Sock, S. Surnev, M. G. Ramsey, F. P. Netzer, B. Klötzer and K. Hayek, Surf. Sci., 2000, 445, 380–393 CrossRef CAS.
- Y. Cao and Z.-X. Chen, Phys. Chem. Chem. Phys., 2007, 9, 739–746 RSC.
- M. Ji, C. Hao, Z. Xie, S. Liu and J. Qiu, J. Comput. Theor. Nanosci., 2012, 9, 394–400 CrossRef CAS.
- J. Klikovits, E. Napetschnig, M. Schmid, N. Seriani, O. Dubay, G. Kresse and P. Varga, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 045405 CrossRef.
- H. Gabasch, W. Unterberger, K. Hayek, B. Klötzer, G. Kresse, C. Klein, M. Schmid and P. Varga, Surf. Sci., 2006, 600, 205–218 CrossRef CAS.
- B. Klötzer, K. Hayek, C. Konvicka, E. Lundgren and P. Varga, Surf. Sci., 2001, 482–485, 237–242 CrossRef.
- R. Kaufmann, H. Klewe-Nebenius, H. Moers, G. Pfennig, H. Jenett and H. J. Ache, Surf. Interface Anal., 1988, 11, 502–509 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Overview of XPS Overlayer model, Zr 3d core level region showing in situ CVD growth monitored at 673 K, C 1s and Zr 3d core level regions of molecularly adsorbed ZTB, summary of ZTB HREELS peak assignments, HREELS spectra of ZTB decomposition, X-ray diffractograms, MSR reaction profiles. See DOI: 10.1039/c6cp07197j |
| This journal is © the Owner Societies 2016 |