
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The impact of the structuring of hydrotropes in water on the mesoscale solubilisation of a third hydrophobic component†
Thomas
Buchecker‡
a,
Sebastian
Krickl‡
b,
Robert
Winkler
b,
Isabelle
Grillo
c,
Pierre
Bauduin
d,
Didier
Touraud
b,
Arno
Pfitzner
 *a and
Werner
Kunz
*b
*a and
Werner
Kunz
*b
aInstitute of Inorganic Chemistry, University of Regensburg, 93040 Regensburg, Germany. E-mail: arno.pfitzner@chemie.uni-regensburg.de
bInstitute of Physical and Theoretical Chemistry, University of Regensburg, 93040 Regensburg, Germany. E-mail: werner.kunz@chemie.uni-regensburg.de
cInstitut Laue-Langevin, 6 rue Jules Horowitz, Grenoble Cedex 9, 38042, France
dInstitut de chimie séparative de Marcoule (ICSM), UMR 5257 (CEA, CNRS, UM, ENSCM), BP 17171, 30207 Bagnols-sur-Cèze, France
First published on 30th November 2016
Abstract
In the present contribution, the pre-structuring of binary mixtures of hydrotropes and H2O is linked to the solubilisation of poorly water miscible compounds. We have chosen a series of short-chain alcohols as hydrotropes and benzyl alcohol, limonene and a hydrophobic azo-dye (Disperse Red 13) as organic compounds to be dissolved. A very weak pre-structuring is found for ethanol/H2O and 2-propanol/H2O mixtures. Pre-structuring is most developed for binary 1-propanol/H2O and tert-butanol/H2O mixtures and supports the bicontinuity model of alcohol-rich and water-rich domains as already postulated by Anisimov et al. Such a pre-structuring leads to a high solubilisation power for poorly water miscible components (limonene and Disperse Red, characterized by high octanol/water partition coefficients, log(P) values of 4.5 and 4.85), whereas a very weak pre-structuring leads to a high solubilisation power for slightly water miscible components (benzyl alcohol). This difference in solubilisation power can be linked to (i) the formation of mesoscale structures in the cases of ethanol and 2-propanol and (ii) the extension of pre-structures in the cases of 1-propanol and tert-butanol. Three different solubilisation mechanisms could be identified: bulk solubilisation, interface solubilisation and a combination of both. These supramolecular structures in binary and ternary systems were investigated by small-and-wide-angle X-ray and neutron scattering, dynamic light scattering and conductivity measurements (in the presence of small amounts of salt).
1 Introduction
Ternary systems comprising a short chain amphiphilic molecule (hydrotrope) and two immiscible liquids, with both of them being fully miscible with the hydrotrope, are considered as powerful solubilisation media1–3 and exhibit anomalies concerning enzymatic reactions4,5 and vapour pressures.6 The existence of well-defined mesoscale inhomogeneities in macroscopically transparent solutions was shown to be responsible for such unexpected behaviour. Such mesoscale inhomogeneities and compartmentation phenomena of oil-rich and water-rich domains are comparable to the micro-domains observed in water/oil/surfactant ternary systems (direct-, bicontinuous- and reverse-microemulsions) as shown using scattering techniques, molecular dynamics simulations (MD simulations) and conductivity measurements.7–9 Only recently, Zemb et al. provided an extended Derjaguin–Landau–Verwey–Overbeek (DLVO) theory as a general explanation for the existence and thermodynamic stability of such surfactant-free microemulsions (SFME). This extended DLVO theory describes the subtle balance between entropy, i.e. homogenous mixing of three components, and enthalpy, i.e. hydrophobic effects between two immiscible fluids causing micellar-like aggregation.10Hydrotropes play a major role in the mesoscale solubilisation phenomenon of SFME. The concept of hydrotropy, defined by C. Neuberg in 1916,11 outlines the capability of short chain amphiphilic molecules to solubilize hydrophobic compounds in a hydrophilic solvent. In SFME showing the compartmentation of hydrophilic and hydrophobic domains, the hydrotrope accumulates mostly at the interface between water-rich and oil-rich domains as deduced from contrast variation experiments with small-angle-neutron-scattering (SANS) and MD simulations. The interface between the oil- and water-rich domains is usually considered as a kind of highly flexible film in dynamic exchange with the surrounding medium.7,12
When using hydrotropes, as in the case of SFME, the solubilisation of hydrophobic compounds in water is only significant at higher hydrotrope concentrations, typically above c(hydrotrope) > 0.2–0.5 M, which may be compared to surfactants for which solubilisation appears above the critical micellar concentration c(surfactant), which is typically >10−5–10−2 M. As a rule of thumb, the solubilisation by hydrotropes starts in the concentration range (c > 1 M), where the average distance between hydrotrope molecules becomes smaller or in the order of the molecular size (∼1 nm), encouraging molecular contact between hydrotrope molecules (cluster formation) and between the hydrotrope and the hydrophobic component to be dissolved.13
In contrast to surfactants, the amphiphilic character of hydrotropes is usually considered to be not enough pronounced to promote micellisation or self-aggregation of the hydrotrope itself in water.13 Nevertheless, some hydrotropes, such as tert-butanol, are known to form clusters in water and thus produce heterogeneities in the sub-nanometer (molecular) or nanometer range.14–17 Only recently, the group of Shimizu et al. developed an approach to describe hydrotropic solubilisation theoretically.18–21 This approach derived from pure statistical thermodynamics uses the exact Kirkwood Buff theory to describe the cooperative phenomena in hydrotropic solubilisation such as (i) the sudden onset of solubilisation of hydrophobic compounds in H2O (commonly referred to as MHC) and (ii) solubility saturation of hydrophobic compounds at high hydrotrope concentrations. In a nutshell, they consider hydrotropic solubilisation to be the result of a subtle balance between solute–hydrotrope interaction and hydrotrope–hydrotrope interaction. In other studies the authors argued that strong hydrotrope–hydrotrope interactions resulting in a pre-structuring of the hydrotrope in H2O diminish the solubilisation efficiency of solutes.22,23 As we will show here, things are even more complex.
Already a vast number of publications have focused on the microscopic inhomogeneities of short chain alcohols in H2O.24–39 The microscopic structuring in these binary mixtures is mostly attributed to the hydrophobic hydration of non-polar aliphatic chains of alcohols and a highly dynamic network of hydrogen bonds between alcohols and H2O.15 Anomalies concerning permittivity, surface tension, self-diffusion coefficients, structuring, etc. are most pronounced for tert-butanol–H2O mixtures as tert-butanol provides the largest aliphatic chain among fully water-miscible alcohols.29–38,40,41
The goal of this work is to link such molecular inhomogeneities in binary alcohol/H2O mixtures to the mesoscale solubilisation of a third hydrophobic component, called “third component” hereafter. To establish this link, it is essential to understand the origin of the compartmentation of water- and oil-rich domains in the ternary systems. Three questions are essential in this context:
(i) Do the pre-structuring and microscopic inhomogeneities of the hydrotrope in the binary system alcohol/H2O have an influence on the structuring of the ternary system?
(ii) Is the structuring in SFME caused by the hydrotrope or by a third, (more) hydrophobic component?
(iii) Does the mesoscale solubilisation mechanism of the third, hydrophobic component depend on its hydrophobicity?
Hence, it is necessary to understand the structuring of short chain alcohols in water as a function of the increasing aliphatic moiety of the alcohol. We extend the pioneering studies by M. Anisimov and M. Sédlak on the mesoscale solubilisation of a third component in the pre-structured binary mixture tert-butanol/H2O14,17,42 to the homologous series of alcohols and to other hydrophobic compounds. To this purpose, we investigate the structuring of ethanol (EtOH), 1-propanol (NPA), 2-propanol (IPA) and tert-butanol (TBA) in water using conductivity measurements and scattering techniques. The structuring in the binary alcohol/H2O mixtures is related to its impact on the mesoscale solubilisation of a third hydrophobic component, i.e. benzyl alcohol, limonene and an azo dye, Disperse Red 13 (DR-13). As we will show, the observed differences in solubility are the consequence of different solubilisation mechanisms.
2 Experimental
2.1 Chemicals
Ethanol (purity ≥ 99.8%), acetone (≥99.5%), 2-propanol (≥99.8%), 1-pentanol (≥99%), (R)-(+)-limonene (97%, ee: 98%), dodecane (≥99%) and Disperse Red 13 (DR-13, dye content 95%) were purchased from Sigma-Aldrich (Steinheim, Germany). Sodium bromide (≥99.99%), 1-propanol (≥99.5%), and benzyl alcohol (≥99%) were purchased from Merck (Darmstadt, Germany), and tert-butanol (≥99%) from Carl Roth (Karlsruhe, Germany). Sodium dodecyl sulfate (SDS, ≥99%) was purchased from Applichem (Darmstadt, Germany).All chemicals were used without further purification. Aqueous solutions were prepared using deionized water with a resistivity of 18 MΩ cm.
2.2 Methods and techniques
Small-angle-neutron-scattering (SANS) experiments were performed using a D33 instrument at the ILL, Grenoble, France.44 The wavelength was set at 6 Å and two sample-to-detector distances 2 m and 5 m with collimation at 5.3 m for both configurations were used to cover a q-range from 0.08 to 0.47 Å−1. A 7 × 10 mm2 aperture was placed before the sample. The samples were filled in 1 mm thick Hellma cells and thermostatted at room temperature using a circulating water bath. The raw data were corrected for the electronic background and empty cells, and were normalized on the absolute scale using the attenuated direct beam to calculate the incident flux using the ILL Lamp software.45
The scattering intensity was plotted against the magnitude of q, and the curves were fitted with an Ornstein–Zernike (OZ) function: I(q) = I(0)/(1 + ξ2q2).10
ξ is the correlation length in Å and I(0) is the intensity for q = 0 and takes into account the scattering length density of the different species and volume fraction of each phase. The full fitting of the spectra is beyond the scope of this paper.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 and a RO/TS mass ratio of oil to total surfactant of 21:79. The conductivity curve was determined as described above.
:2 and a RO/TS mass ratio of oil to total surfactant of 21:79. The conductivity curve was determined as described above.
3 Results and discussion
3.1 Binary mixtures
 | ||
| Fig. 1 Self-correlation functions obtained by DLS measurements at 25 °C for the binary systems (a) EtOH/H2O, (b) IPA/H2O, (c) NPA/H2O and (d) TBA/H2O. The symbols refer to different mass ratios of alcohol to H2O. | ||
As the DLS auto-correlation functions are most pronounced for TBA/water mixtures, additional SWAXS and one particular SANS measurement were performed for this system, see Fig. 2 and Fig. S1 in the ESI.†
 | ||
| Fig. 2 SWAXS spectra of the binary mixture TBA/H2O. Symbols indicate the different mass ratios of TBA to H2O. Points (i–iv) denote the different features found in the spectra. | ||
Four features can be observed in SWAXS spectra, assigned to i–iv. A correlation peak (i) is observed at 20 nm−1, originating mainly from the O–O pair correlations in the H-bonding network of H2O. This peak does not significantly shift in position and decreases in intensity as the amount of water decreases.
Another correlation peak (ii) is found at 10.5 nm−1, corresponding to the pair correlations between aliphatic chains. This peak also does not undergo any shift in position and its intensity depends on the TBA concentration.
For TBA concentrations above 70 wt%, a correlation peak (iii) emerges at 5–7 nm−1, which corresponds to average distances between 0.9 and 1.3 nm. This peak can be attributed to the pair-correlations between hydroxyl groups of TBA as its intensity increases with the TBA content. The peak (iii) shifts to lower q-values with increasing water concentrations indicating a swelling of the H-bond network, formed by the TBA hydroxyl groups, by the addition of water. Furthermore (iv), a significant excess of scattering is observed in the low-q range (q <5 nm−1) for 40, 50 and 60 wt% of TBA with a maximum I(0) intensity for 40 wt%. It has been noticed that the low-q scattering is only observed in the absence of a correlation peak of the alcohol (–OH) groups (iii). The existence of low q-scattering in small-angle-X-ray-scattering experiments indicates the existence of a meso-structured system.10,12 Curves exhibiting low q-scattering were fitted with an Ornstein–Zernike formalism to determine the correlation length of different compositions. Correlation lengths of 0.6 nm (60/40 TBA/H2O), 0.7 nm (50/50 TBA/H2O) and 0.8 nm (40/60 TBA/H2O) were found. The scattered intensity is rather constant for q < 1 nm−1 indicating the presence of structures/inhomogeneities with a size of around 6 nm.
SANS measurements on one particular composition, i.e. 40/60 TBA/D2O, confirm the results obtained by SWAXS, see Fig. S1 (ESI†). The existence of low scattering again indicates the presence of a structured system. Fitting the curve with an OZ fit yields a correlation length of 0.7 nm, which is approximately the same as that obtained for SWAXS for this composition (0.8 nm). The slight difference in the correlation lengths obtained from SWAXS and SANS can be ascribed to the different scattering contrasts of SWAXS and SANS.
Therefore, SWAXS and SANS experiments support the results obtained by DLS: the existence of a structured system in the binary mixture TBA/H2O with microscopic inhomogeneities in the order of 4–6 nanometers. The structuring is most pronounced between 30 and 50 wt% of TBA.
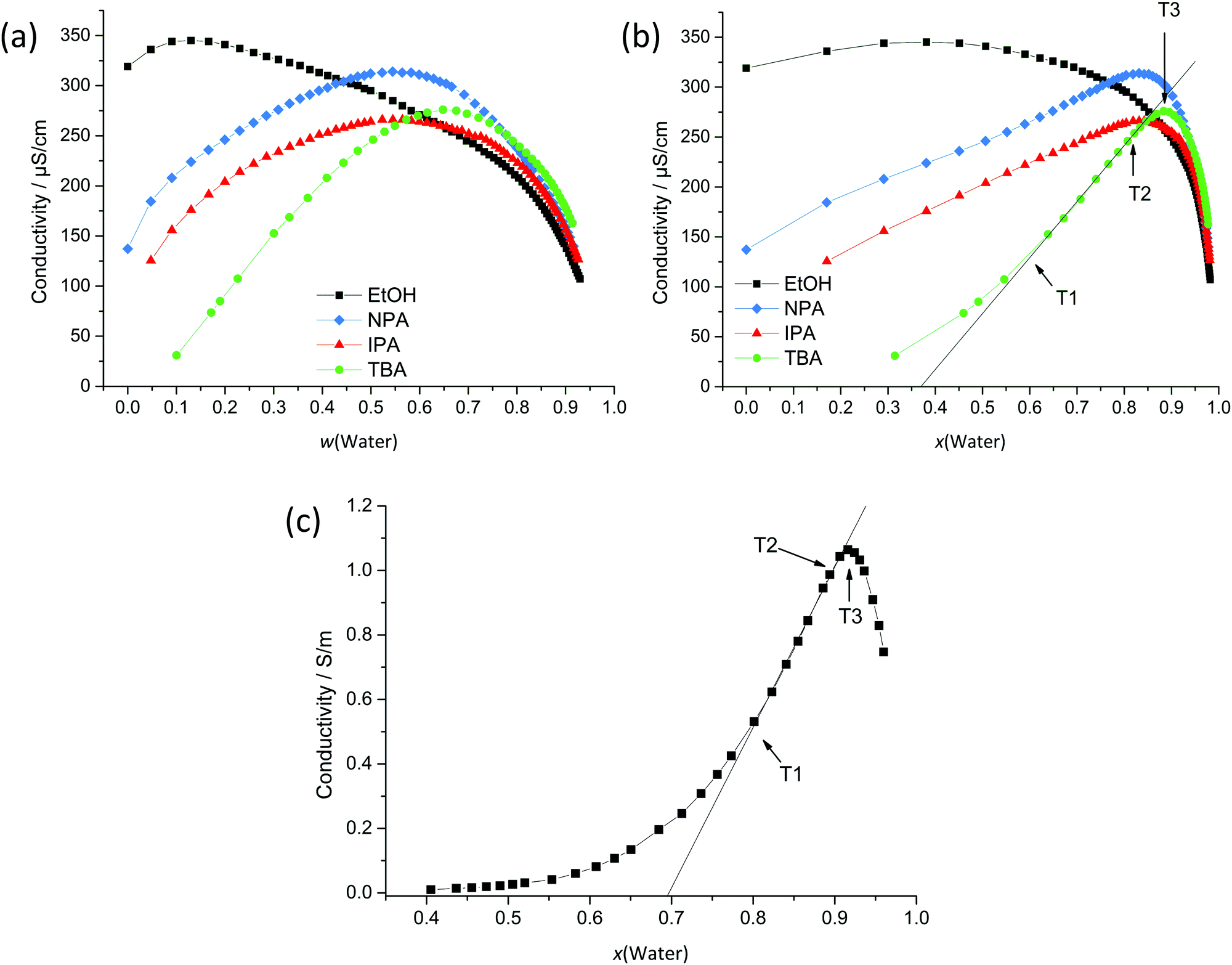 | ||
| Fig. 3 Conductivity measurements at 25 °C for binary mixtures of H2O and EtOH/IPA/NPA/TBA plotted in (a) weight and (b) mole fractions of H2O. In order to ensure measurable conductivities, 0.2 wt% of NaBr was dissolved in the pure alcohol phase for EtOH, NPA and IPA and in a 10 wt% (H2O) mixture for the TBA binary system. (c) Conductivities measured at 25 °C for the system H2O/SDS/1-pentanol/dodecane. Point T1 marks the point of transition to percolative behaviour of w/o microemulsions and points T2 and T3 mark the transitions from w/o to oil-rich and to water-rich bicontinuous microemulsions. | ||
Conductivity was also measured for a classical SDS-based microemulsion, see Fig. 3c.43 Such systems show a flat rise at low water content, followed by a change in the slope passing over into a strong linear increase of conductivity (point T1 in Fig. 3c) with the increasing water content. After this linear increase, the slope of the curve starts to decrease again (point T2) and passes a maximum (point T3). For structured solutions – as is the case for microemulsions – these changes in conductivity are explained by transitions between o/w, bicontinuous, and w/o microemulsions. A w/o microemulsion is present for low water content and causes low conductivity. With the increasing water content, percolation of water droplets is observed, causing the formation of conducting water channels by merging water droplets, which leads to a significant change in the slope (point T1). Further, point T2 marks the transition to an oil-rich bicontinuous microemulsion. A subsequent transition to a water-rich bicontinuous microemulsion is observed upon addition of H2O. The maximum conductivity represents the presence of an extended water-rich bicontinuous phase (point T3). For very high water contents (x > 0.9), an o/w microemulsion is formed and dilution effects prevail, causing conductivities to decrease.
A comparison of the conductivity curve of this SDS system with the curves of TBA and NPA (Fig. 3b and c) shows many similarities regarding the range of increase, the percolation behaviour and the decrease in conductivity for high water contents. The maximum conductivity for NPA/H2O lies in a range of approximately 75–90 mol% water content and for TBA in a range of approximately 85–95 mol% water content. This maximum conductivity coincides with the highest correlation coefficients in DLS and the strongest signal in low q of SWAXS. It is also well known for TBA/H2O that the binary mixture displays two eutectics, one of them being located at 94 mol% H2O, which also coincides with the range of maximum conductivity.48 It was found that thermodynamic anomalies and inhomogeneous mixing are most pronounced for this composition even at temperatures above the deep eutectic temperature. The presence of a percolation point around 40 mol% H2O in the case of TBA and a conductivity behaviour, see points T1–T3 in Fig. 3b, similar to the SDS system lead to the conclusion that the structuring of TBA/H2O is similar to the structuring of H2O/SDS/1-pentanol/dodecane. Therefore, we propose the presence of a bicontinuity of water-rich and TBA-rich domains, as already suggested by the research group of Anisimov et al. for the ternary system H2O/TBA/propylene oxide, based on MD simulations and the observation of mesoscale inhomogeneities.42 However, conductivity measurements here suggest the formation of a bicontinuous phase already in the binary mixture TBA/H2O. Apparently, this bicontinuous structuring behaviour is not only restricted to TBA, but is also observable for aqueous NPA mixtures, as suggested by DLS measurements, although to a much smaller extent.
The conductivity curve of EtOH differs strongly from the conductivity curves of IPA, NPA, TBA and SDS regarding the ranges of increase, decrease and the position of the maximum. The small conductivity increase for low water contents might be explained by an enhanced ion dissociation as well as a higher electrophoretic mobility in aqueous media. For the increasing water content, the conductivity decrease can be explained by the excessive dilution of charge carriers. Therefore, it is assumed that microscopic inhomogeneities in the case of EtOH are very weak and cannot be detected in detail by conductivity measurements.
The conductivity of IPA is in between the conductivities of EtOH and NPA indicating a progressive structuring with the increasing aliphatic chain length. Therefore, EtOH/H2O is a good example of a weakly structured system and TBA/H2O is a representative binary mixture of a system with pronounced structuring.
3.2 Ternary mixtures
In the next step, three different hydrophobic compounds were dissolved in alcohol/H2O binary mixtures, i.e. benzyl alcohol, limonene and DR-13. The difference between these compounds is their miscibility with water. Benzyl alcohol is slightly miscible with H2O. Limonene, as well as DR-13, are poorly water miscible compounds. The realms of solubility in the cases of benzyl alcohol and limonene are shown in the ternary phase diagrams in Fig. 4a and b and the OD as a measure of the solubility of DR-13 in Fig. 4c.49,50 The solubilisation power of the alcohols in the case of benzyl alcohol decreases in the following order: EtOH > IPA > NPA ≈ TBA, while phase diagrams with NPA and TBA are very similar.
 | ||
| Fig. 4 Ternary phase diagrams for the systems (a) H2O/hydrotrope/benzyl alcohol and (b) H2O/hydrotrope/limonene provided in mol%. (c) The results of the OD measurements of DR-13 in H2O/hydrotrope mixtures given in mole fractions of hydrotrope. ODs obtained were divided by the OD value of the corresponding neat hydrotrope (ODmax). T1, T2 and T3 mark the transition points of the binary system TBA/H2O in mole fractions as determined in Fig. 3b. | ||
The solubilisation power of the alcohols in the case of limonene gradually increases in the following order: EtOH < IPA < NPA < TBA. Its miscibility with H2O is very poor (less than in the case of benzyl alcohol) and therefore the two-phase region is larger than in the case of benzyl alcohol. The solubility of limonene in TBA/H2O starts to increase significantly at x(TBA) = 10 mol% (see point T3 in Fig. 4) and at x(TBA) > 40 mol% and the miscibility gap is closed. Furthermore, and most importantly, the hydrotrope efficiency for the solubilisation of limonene is reversed compared to the solubilisation efficiency of the alcohols considered for benzyl alcohol.
In the case of the solubilisation of DR-13, the dissolved amount of DR-13 increases in the following order: EtOH ≪ IPA < NPA < TBA for the hydrotrope content x(hydrotrope) < 0.4. For the hydrotrope content x(hydrotrope) > 0.4 the situation is different. Note that in the case of the solubilisation of DR-13 in TBA/H2O, the solubility starts to increase at point T3 (remember: maximum of the water-rich bicontinuous phase in TBA/H2O). After point T3, the solubility of DR-13 increases linearly up to the point T1, where the slope of the solubility of DR-13 in TBA diminishes. All in all, the solubilisation power of the alcohols for DR-13 follows the same trend as for limonene. However, comparing the results of DR-13 solubilisation to the solubilisation of benzyl alcohol and limonene, there are huge differences regarding the absolute amounts of dissolved hydrophobic compound on the order of ≈106 mol.
 | ||
| Fig. 5 Conductivity measurements at 25 °C for ternary mixtures of H2O/hydrotrope/benzyl alcohol plotted in weight fractions of H2O. The measurements were performed by diluting a binary mixture hydrotrope/benzyl alcohol with H2O. The symbols refer to different mass ratios of hydrotrope to benzyl alcohol (BA) in the binary starting mixture hydrotrope/benzyl alcohol before diluting with H2O. | ||
Taking into account the large difference in the solubilisation power of benzyl alcohol and limonene, different solubilisation mechanisms are proposed.
For EtOH and IPA, no or very weak structuring is observed for their binary mixtures with H2O. The addition of benzyl alcohol and limonene, respectively, induces the formation of a well-ordered system.2,6 Hydrophobic hydration and agglomeration cause the hydrophobic compound to form clusters with accumulation of the hydrotrope mostly at the interface between the benzyl alcohol clusters and the water-rich domain. Consequently, the solubility of the hydrophobic compound is enhanced with increasing hydrotrope concentration. This is in perfect agreement with the ideas forwarded recently.22,23,51
However, in contrast to weakly structured EtOH/H2O and IPA/H2O, the situation is completely different for binary pre-structured TBA/H2O and NPA/H2O systems. As a bicontinuous structure is already given, the structure is not induced by the third component, but by the hydrotrope itself. Due to the formation of a bicontinuous system, an aliphatic-rich domain as well as a water-rich domain and a large area of interface are created.
In such a case of a pre-structured binary hydrotrope–water system, three possible solubilisation pathways are possible: (i) solubilisation in the water-rich bulk phase (appears only for highly polar solutes which are not considered here), (ii) solubilisation in the aliphatic-rich bulk phase and (iii) solubilisation within the interfacial film between the aliphatic and water-rich pseudo-bulk phases, see Fig. 6.
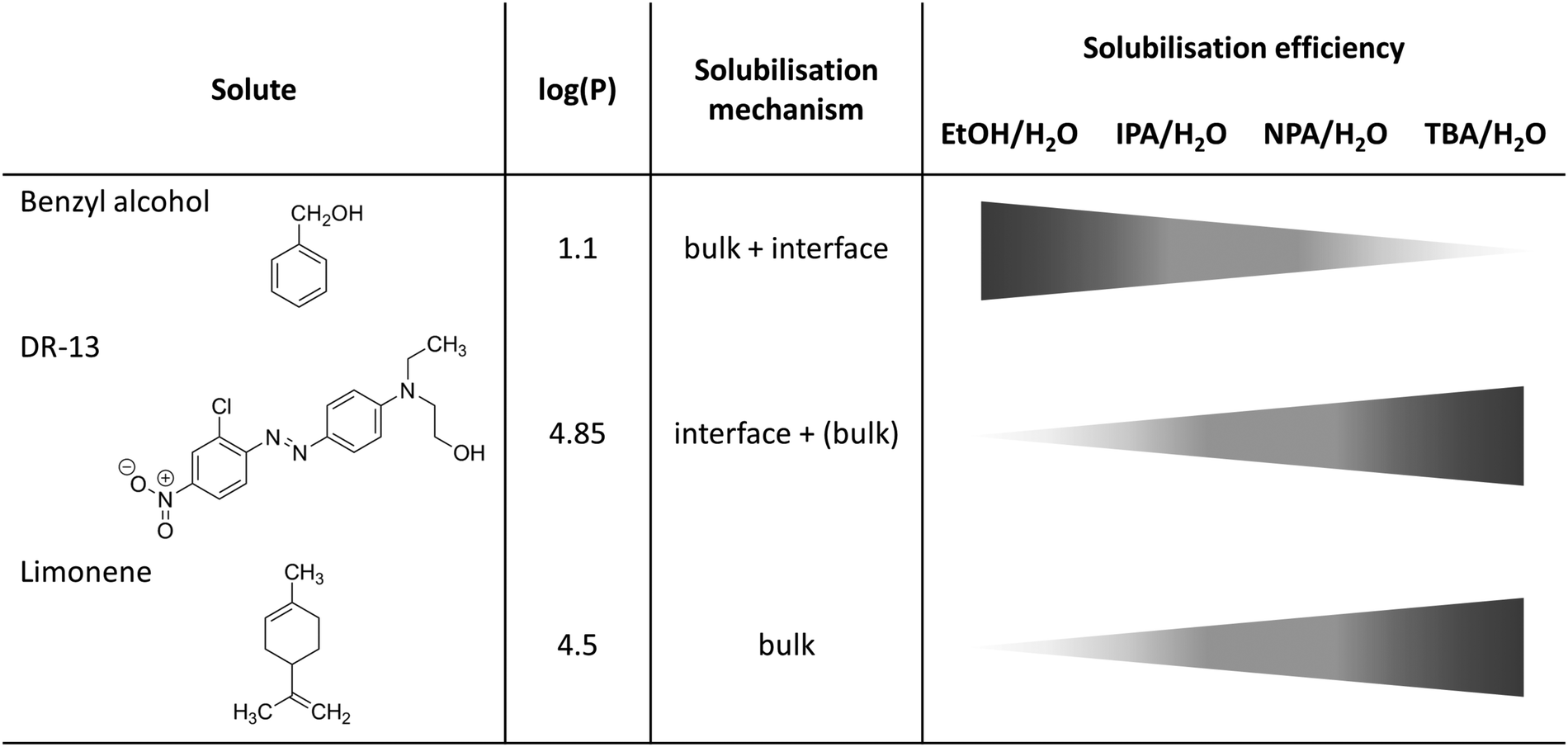 | ||
| Fig. 6 Schematic overview of different solubilisation mechanisms and efficiencies of benzyl alcohol, DR-13 and limonene in binary water/alcohol mixtures. Octanol/water partition coefficients log(P) were taken from ref. 54–56. | ||
The aliphatic-rich bulk-like pseudo-phase permits the incorporation of large amounts of hydrophobic components. This is confirmed by the solubilisation power of the hydrotropes in the case of limonene, where TBA shows the highest solubilisation power and EtOH the lowest solubilisation power. Further indications are conductivity measurements for ternary systems H2O/alcohol/benzyl alcohol, where the absolute conductivity of NPA and TBA is maintained, indicating the retention of the pre-structures.
The SANS spectrum of one exemplary composition of a ternary mixture D2O/TBA/benzyl alcohol (60/32/8 wt%) was compared to a SANS measurement of a binary TBA/H2O mixture (40/60 wt%) to further support this fact, see Fig. S1 (ESI†). The substitution of TBA by benzyl alcohol leads to a large increase of the scattered intensity. Analysis of the correlation length of these curves (0.8 nm for the binary mixture and 2.1 nm for the ternary mixture) indicates an enlargement of the correlation length of the TBA/benzyl alcohol aliphatic rich phase. This implies that benzyl alcohol and certainly also TBA are “driven” towards the aliphatic rich continuum which is “blown up” and causes an increase of the correlation length. Therefore, the structure is reinforced by the addition of benzyl alcohol.
But in the case of benzyl alcohol, the situation is somehow different from the case of limonene, as it contains an alcohol functional group in addition to the aliphatic moiety. This leads us to the assumption that benzyl alcohol is not only dissolved within the aliphatic-rich continuum, due to its low water miscibility, but may also accumulate at the interfacial film, similar to a co-surfactant. Another indicator is the fact that TBA provides the lowest solubilisation power for benzyl alcohol in the series EtOH, IPA and NPA. In EtOH/H2O mixtures, benzyl alcohol induces the formation of mesoscale structures, whereas in the case of TBA/H2O mixtures, it intercalates into the given pre-structure. As soon as the given pre-structure is completely “swollen up” and the interfacial film is saturated, further addition of benzyl alcohol causes the system to collapse and form two phases.
In the case of the solubilisation of DR-13, the situation is again different from both the limonene and benzyl alcohol solubilisation processes, since DR-13 bears polar functional groups and an aromatic backbone much larger than that of benzyl alcohol. The solubilisation power of the hydrotropes in the case of DR-13 follows the same series as for limonene. However, a closer look at the amount of dissolved DR-13 per amount of the hydrotrope reveals a huge difference of the order of several orders of magnitude. Furthermore, DR-13 is assumed to be not capable of inducing micro-structuring, as the overall solubility in H2O and H2O/hydrotrope is too low. Therefore, the solubility in EtOH/H2O is very low. However, as the solubility increases with progressive structuring of the H2O/hydrotrope, we assume that the difference in solubility is based on a completely different solubilisation mechanism as compared to limonene. Instead of a bulk solubilisation within a hydrophobic mesoscale pseudo-phase, we suppose DR-13 to be mostly dissolved within the interfacial film of the two pseudo-bulk phases. This assumption is in good accordance with all results, because an interfacial film provides much less space than a bulky pseudo-phase for the solubilisation of DR-13. The solubility of DR-13 in TBA/H2O mixtures starts to increase at point T3 representing the maximum of the water-rich bicontinuous phase, see Fig. 3b and 4c. Beginning from T1 (representing the percolation threshold) the solubilisation power of TBA/H2O diminishes compared to the region T3–T1. It is assumed that the interface of pre-structured binary alcohol/H2O mixtures is rapidly saturated for low concentrations of DR-13. The further increase of the solubilisation of DR-13 for a higher TBA content (x(TBA) > 0.4) may be ascribed to bulk solubilisation due to saturation of the interface. Hence, DR-13 may preferentially solubilise at the interface in highly structured regimes of TBA 0.1 < x(TBA) < 0.4, passing over to a more bulk-like solubilisation mechanism for a higher TBA content. A more detailed analysis of the curve shapes clarifies these findings even better. A second-order differentiation of the fitted curves reveals huge differences in the solvent character of the hydrotrope (see Fig. S2, ESI†). In the case of EtOH the curvature of the curve stays always positive over the whole miscibility range whereas there is a negative curvature in the case of TBA in the range of approximately 0.23 < x(TBA) < 0.6. This second order differentiation shows that EtOH possesses a more co-solvent character, whereas TBA shows a hydrotropic behaviour when DR-13 is dissolved. The proposed interfacial solubilisation mechanism is further supported by the fact that DR-13 has a very poor solubility both in pure dodecane and pure water, for which both do not provide an interface at all. As a consequence, DR-13 is also scarcely soluble in poorly structured mixtures, such as EtOH/H2O.
4 Conclusion
We investigated the structuring of alcohol/H2O binary mixtures, where the alcohol is miscible with H2O in any proportion, i.e. EtOH, IPA, NPA and TBA. DLS, SWAXS, SANS and conductivity measurements revealed microscopic inhomogeneities in the cases of NPA/H2O and TBA/H2O. As in the case of micellar systems, we infer from conductivity measurements that these microscopic inhomogeneities are similar to direct or reverse micellar solutions (meaning organic aggregates in water or water aggregates in an organic pseudo-phase, respectively) and also, at some concentration ratios, to bicontinuous structuring with rapidly fluctuating water-rich and alcohol-rich domains in equilibrium. In the case of EtOH, no significant structuring could be detected by our methods. IPA marks the transition from a weakly structured system in the case of EtOH to structured systems in the cases of NPA and TBA.It is found that this pre-structuring of a binary alcohol/H2O system can be pivotal to the mesoscale solubilisation of a third component. In the cases of unstructured binary EtOH/H2O and IPA/H2O, a significant structuring is induced upon the addition of a third poorly water miscible component, i.e. limonene and benzyl alcohol. Until now, this solubilisation mechanism is considered to be dominant and fundamental in SFME, as is the case of the well-studied H2O/EtOH/1-octanol system.2,7,12,52,53 It was further argued that this structuring induced by the addition of the third, hydrophobic component, is crucial for a pronounced solubilisation power of the hydrotrope.22,23,51 And indeed, the lower the pre-structuring in the binary hydrotrope–water mixture, the less hydrotrope is required to make benzyl alcohol water-miscible.
However, as we demonstrate here, hydrotropic solubilisation is more complicated. Firstly, it is just the other way around in the case of limonene: the more pronounced the pre-structuring in the binary hydrotrope–water mixture, the less hydrotrope is required to make the much more hydrophobic (compared to benzyl alcohol) limonene water-miscible. Secondly, the more pronounced the pre-structuring in the binary hydrotrope–water mixture, the lower is the minimum hydrotropic concentration (m.h.c.) required to dissolve the dye DR-13, a very hydrophobic compound that is nevertheless nearly insoluble in both water and dodecane.
Two different solubilisation mechanisms can explain the hydrotropic solubilisation of very hydrophobic compounds: (i) pseudo-bulk solubilisation of hydrophobic compounds in the aliphatic-rich moiety of pre-structured hydrotrope–water mixtures and (ii) interface solubilisation of hydrophobic, but still slightly amphiphilic compounds in the interfacial film.
This leads to the conclusion that for solubilising a hydrophobic compound in an aqueous medium, the choice of the alcohol (hydrotrope) depends primarily on the nature of the hydrophobic substance. For more hydrophilic compounds bearing polar functional groups, short-chained alcohols are more favourable, which is in agreement with ref. 22, 23 and 51. In order to solubilise hydrophobic compounds without polar groups, structure-forming hydrotropes like TBA and NPA should be used instead.
At first glance, our conclusions may appear contradictory to recent statistical thermodynamic considerations of hydrotropy.19–21 These state that the predominant driving forces for hydrotropic solubilisation are (i) solute induced interactions between the solute and hydrotrope molecules leading to a solute–hydrotrope association. In contrast (ii) pre-structuring of hydrotropes (hydrotrope–hydrotrope interactions) in water is considered to be rather obstructive for good solubilisation of the solute. Indeed, this holds true for solutes, which are able to interact with the hydrotrope due to the presence of polar functional groups, as is the case for benzyl alcohol. For this case, our results are in total agreement with the results obtained by thermodynamic calculations. However, these statistic thermodynamic studies by Shimizu et al. consider almost exclusively hydrotropes with polar functional groups, e.g. ester functional groups. Yet, for very hydrophobic substances (as is the case for limonene), the situation becomes subtler. In this case, the solute–hydrotrope interactions are expected to be weaker and no longer play the dominant role since there is no functional group to interact with the hydrotrope. Instead, it becomes more important that the highly hydrophobic solute molecules can intercalate into a significantly hydrophobic, aliphatic pseudo-phase (as is the case for TBA in H2O), originated by the pre-structuring of the hydrotrope in water. For DR-13, we found a similar trend for its solubility as for the solubilisation of limonene, but based on a different solubilisation mechanism, due to its slightly amphiphilic character. Thus, the solubility depends on the chemical nature of both the solute and the hydrotrope. Furthermore, we would like to emphasise that in addition to a different choice of solutes, the aforementioned studies by Shimizu et al. focused in most of the cases on completely different concentration ranges of the solute and hydrotrope. They considered a very low amount of solute in aqueous hydrotrope solutions. (This is important for the validity of Kirkwood–Buff theory used for statistical thermodynamic calculations in these studies.) Thus, our studies provide a more general and molecular-based model concept of hydrotrope solubilisation.
All in all, taking into account the different ways in which structuring can be induced and how they influence the involved solubilisation mechanisms, our picture of mesoscale solubilisation in ternary systems becomes more general and more complete now. Microscopic inhomogeneities and compartmentation phenomena in SFME can not only be induced by the addition of a third hydrophobic component to a H2O/hydrotrope binary mixture, but can also have their origin in the pre-structuring of alcohol/H2O binary mixtures.
Acknowledgements
T. B. thanks the BFHZ (Bayerisch-französisches Hochschulzentrum) for the grant number FK41_15 for covering travel expenses and the DFG project GRK1626 chemical photocatalysis for financial support. S. K. thanks the Fonds der chemischen Industrie (FCI) for a scholarship. The authors thank Olivier Diat and Bruno Corso for assistance with SWAXS measurements.Notes and references
- M. L. Klossek, D. Touraud, T. Zemb and W. Kunz, ChemPhysChem, 2012, 13, 4116–4119 CrossRef CAS PubMed.
- M. L. Klossek, D. Touraud and W. Kunz, Phys. Chem. Chem. Phys., 2013, 15, 10971–10977 RSC.
- V. Fischer, J. Marcus, D. Touraud, O. Diat and W. Kunz, J. Colloid Interface Sci., 2015, 453, 186–193 CrossRef CAS PubMed.
- M. Zoumpanioti, H. Stamatis, V. Papadimitriou and A. Xenakis, Colloids Surf., B, 2006, 47, 1–9 CrossRef CAS PubMed.
- Y. L. Khmelnitsky, R. Hilhorst and C. Veeger, Eur. J. Biochem., 1988, 176, 265–271 CrossRef CAS PubMed.
- J. Marcus, M. L. Klossek, D. Touraud and W. Kunz, Flavour Fragrance J., 2013, 28, 294–299 CrossRef CAS.
- S. Schöttl, J. Marcus, O. Diat, D. Touraud, W. Kunz, T. Zemb and D. Horinek, Chem. Sci., 2014, 5, 2909–3340 RSC.
- P. Bošković, V. Sokol, T. Zemb, D. Touraud and W. Kunz, J. Phys. Chem. B, 2015, 119, 9933–9939 CrossRef PubMed.
- Z. Li, X. Liu, Y. Lian, J. Xie, X. Gao and T. Chang, World J. Eng., 2016, 13, 142–148 Search PubMed.
- T. Zemb, M. L. Klossek, T. Lopian, J. Marcus, S. Schöttl, D. Horinek, S. Prevost, D. Touraud, O. Diat, S. Mar
![[c with combining breve]](https://www.rsc.org/images/entities/char_0063_0306.gif) elja and W. Kunz, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 4260–4265 CrossRef CAS PubMed.
elja and W. Kunz, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 4260–4265 CrossRef CAS PubMed. - C. Neuberg, Biochem. Z., 1916, 76, 107–108 CAS.
- O. Diat, M. L. Klossek, D. Touraud, B. Deme, I. Grillo, W. Kunz and T. Zemb, J. Appl. Crystallogr., 2013, 46, 1665–1669 CAS.
- P. Bauduin, A. Renoncourt, A. Kopf, D. Touraud and W. Kunz, Langmuir, 2005, 21, 6769–6775 CrossRef CAS PubMed.
- D. Subramanian and M. Anisimov, J. Phys. Chem. B, 2011, 115, 9179–9183 CrossRef CAS PubMed.
- D. Subramanian, C. T. Boughter, J. B. Klauda, B. Hammouda and M. Anisimov, Faraday Discuss., 2013, 167, 217–238 RSC.
- M. Sédlak, J. Phys. Chem. B, 2006, 110, 13976–13984 CrossRef PubMed.
- M. Sedlák and D. Rak, J. Phys. Chem. B, 2014, 118, 2726–2737 CrossRef PubMed.
- S. Shimizu and N. Matubayasi, J. Phys. Chem. B, 2014, 118, 10515–10524 CrossRef CAS PubMed.
- S. Shimizu and N. Matubayasi, J. Phys. Chem. B, 2014, 118, 3922–3930 CrossRef CAS PubMed.
- S. Shimizu and N. Matubayasi, Phys. Chem. Chem. Phys., 2016, 18, 25621–25628 RSC.
- T. W. J. Nicol, N. Matubayasi and S. Shimizu, Phys. Chem. Chem. Phys., 2016, 18, 15205–15217 RSC.
- J. J. Booth, S. Abbott and S. Shimizu, J. Phys. Chem. B, 2012, 116, 14915–14921 CrossRef CAS PubMed.
- J. J. Booth, M. Omar, S. Abbott and S. Shimizu, Phys. Chem. Chem. Phys., 2015, 17, 8028–8037 RSC.
- S. Dixit, J. Crain, W. C. K. Poon, J. L. Finney and A. K. Soper, Nature, 2002, 416, 829–832 CrossRef CAS PubMed.
- O. Gereben and L. Pusztai, J. Phys. Chem. B, 2015, 119, 3070–3084 CrossRef CAS PubMed.
- A. Ghoufi, F. Artzner and P. Malfreyt, J. Phys. Chem. B, 2016, 120, 793–802 CrossRef CAS PubMed.
- M. L. Tan, B. T. Miller, J. Te, J. R. Cendagorta, B. R. Brooks and T. Ichiye, J. Chem. Phys., 2015, 142, 0645011 Search PubMed.
- L. Almásy, G. Jancsó and L. Cser, Appl. Phys. A: Mater. Sci. Process., 2002, 74, 1376–1378 CrossRef.
- K. Nishikawa, H. Hayashi and T. Iijima, J. Phys. Chem., 1989, 93, 6559–6565 CrossRef CAS.
- D. T. Bowron, J. L. Finney and A. K. Soper, J. Phys. Chem. B, 1998, 102, 3551–3563 CrossRef CAS.
- K. Yoshida, T. Yamaguchi, A. Kovalenko and F. Hirata, J. Phys. Chem. B, 2002, 106, 5042–5049 CrossRef CAS.
- T. Fukasawa, Y. Tominaga and A. Wakisaka, J. Phys. Chem. A, 2004, 108, 59–63 CrossRef CAS.
- A. A. Bakulin, M. S. Pshenichnikov, H. J. Bakker and C. Petersen, J. Phys. Chem. A, 2011, 115, 1821–1829 CrossRef CAS PubMed.
- H. A. R. Gazi and R. Biswas, J. Phys. Chem. A, 2011, 115, 2447–2455 CrossRef CAS PubMed.
- B. Kezic and A. Perera, J. Chem. Phys., 2012, 137, 0145011 Search PubMed.
- M. D. Hands and L. V. Slipchenko, J. Phys. Chem. B, 2012, 116, 2775–2786 CrossRef CAS PubMed.
- L. Comez, M. Paolantoni, L. Lupi, P. Sassi, S. Corezzi, A. Morresi and D. Fioretto, J. Phys. Chem. B, 2014, 119, 9236–9243 CrossRef PubMed.
- D. Banik, A. Roy, N. Kundu and N. Sarkar, J. Phys. Chem. B, 2015, 119, 9905–9919 CrossRef CAS.
- S. K. Allison, J. P. Fox, R. Hargreaves and S. P. Bates, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 1–5 CrossRef.
- P. G. Kusalik, A. P. Lyubartsev, D. L. Bergman and A. Laaksonen, J. Phys. Chem. B, 2000, 104, 9526–9532 CrossRef CAS.
- M. Tomsic, A. Jamnik, G. Fritz-Popovski, O. Glatter and L. Vladek, J. Phys. Chem. B, 2007, 111, 1738–1751 CrossRef CAS PubMed.
- D. Subramanian, J. B. Klauda, P. J. Collings and M. Anisimov, J. Phys. Chem. B, 2014, 118, 5994–6006 CrossRef CAS PubMed.
- M. L. Dekker, M. Clausse, H. L. Rosano and A. Zradba, Microemulsion Systems, Surfactant Science Ser., New York, 24th edn, 1987 Search PubMed.
- C. D. Dewhurst, I. Grillo, D. Honecker, M. Bonnaud, M. Jacques, C. Amrouni, A. Perillo-Marcone, G. Manzin and R. Cubitt, J. Appl. Crystallogr., 2016, 49, 1–14 CrossRef CAS.
- http://https://www.ill.eu/instruments-support/computing-for-science/cs-software/all-software/lamp/ .
- B. Lagourette, J. Peyrelasse, C. Boned and M. Clausse, Nature, 1979, 281, 60–62 CrossRef.
- M. Clausse, J. Peyrelasse, J. Heil, C. Boned and B. Lagourette, Nature, 1981, 293, 636–638 CrossRef CAS.
- D. Subramanian and M. A. Anisimov, Fluid Phase Equilib., 2014, 362, 170–176 CrossRef CAS.
- A. Arce, A. Marchiaro and A. Soto, J. Solution Chem., 2004, 33, 561–569 CrossRef CAS.
- H. Li and K. Tamura, Fluid Phase Equilib., 2008, 263, 223–230 CrossRef CAS.
- W. Kunz, K. Holmberg and T. Zemb, Curr. Opin. Colloid Interface Sci., 2016, 22, 99–107 CrossRef CAS.
- J. Marcus, D. Touraud, S. Prévost, O. Diat, T. Zemb and W. Kunz, Phys. Chem. Chem. Phys., 2015, 17, 32528–32538 RSC.
- T. Lopian, S. Schöttl, S. Prévost, S. Pellet-Rostaing, D. Horinek, W. Kunz and T. Zemb, ACS Cent. Sci., 2016, 2, 467–475 CrossRef CAS PubMed.
- L. O. Copolovici and Ü. Niinemets, Chemosphere, 2005, 61, 1390–1400 CrossRef CAS PubMed.
- Stephen B. Roscoe, Neal A. Rakow, Michael L. Husberg and Lester H. McIntosh, US Pat., 2005/0065062, 2005 Search PubMed.
- G. G. Briggs, J. Agric. Food Chem., 1981, 29, 1050–1059 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp06696h |
| ‡ Authors contributed equally to the experimental work and to writing the paper. |
| This journal is © the Owner Societies 2017 |