
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExtensive H-atom abstraction from benzoate by OH-radicals at the air–water interface†
Shinichi
Enami
*a,
Michael R.
Hoffmann
b and
Agustín J.
Colussi
*b
aNational Institute for Environmental Studies, 16-2 Onogawa, Tsukuba, Ibaraki 305-8506, Japan. E-mail: enami.shinichi@nies.go.jp; Tel: +81-29-850-2770
bLinde Center for Global Environmental Science, California Institute of Technology, California 91125, USA. E-mail: ajcoluss@caltech.edu
First published on 2nd November 2016
Abstract
Much is known about OH-radical chemistry in the gas-phase and bulk water. Important atmospheric and biological processes, however, involve little investigated OH-radical reactions at aqueous interfaces with hydrophobic media. Here, we report the online mass-specific identification of the products and intermediates generated on the surface of aqueous (H2O, D2O) benzoate-h5 and -d5 microjets by ∼8 ns ˙OH(g) pulses in air at 1 atm. Isotopic labeling lets us unambiguously identify the phenylperoxyl radicals that ensue H-abstraction from the aromatic ring and establish a lower bound (>26%) to this process as it takes place in the interfacial water nanolayers probed by our experiments. The significant extent of H-abstraction vs. its negligible contribution both in the gas-phase and bulk water underscores the unique properties of the air–water interface as a reaction medium. The enhancement of H-atom abstraction in interfacial water is ascribed, in part, to the relative destabilization of a more polar transition state for OH-radical addition vs. H-abstraction due to incomplete hydration at the low water densities prevalent therein.
Introduction
Benzoic acid (BA) is one of the most abundant carboxylic acid in the particulate matter (PM) found over most polluted urban areas. It has been recently reported that BA concentrations in PM2.5 collected over Beijing (average 1496 ng m−3 in Pekin University, 1278 ng m−3 in Yufa) exceed the concentrations of total diacids (∼1010 ng m−3), fatty acids (∼600 ng m−3) and ketocarboxylic acids (∼120 ng m−3).1 Low-volatility BA, which is produced both from direct traffic emissions and in the atmospheric oxidation of anthropogenic aromatic compounds, largely partitions to the aqueous phase where it reacts further with atmospheric oxidants.1 BA (pKa = 4.2) is largely present as benzoate (BzO) in atmospheric aqueous media. Since BzO is amphiphilic and relatively inert toward O3 (k = 1.2 M−1 s−1),2 the heterogeneous (interfacial) oxidation of BzO(aq) by ˙OH(g) is deemed to control its fate.3–8 Molecular dynamics (MD) calculations and surface-tension data confirm the affinity of BzO for aqueous surfaces.9 It has been recently realized that the photochemical aging of particulate organic matter is not only degradative but generates volatile organic compound (VOC) emissions and reactive species, such as hydroperoxides.10–12 The identification of products and labile intermediates from the heterogeneous oxidation of organic matter in condensed phases has thus emerged as a major issue in the atmospheric chemistry of polluted urban air.6,13–15Here we address this issue and report direct, online mass-specific identification of the products of the oxidation of BzO(aq) by ˙OH(g) pulses on the surface of aqueous microjets (see Methods and Fig. S1, ESI†).11 In such events, ˙OH(g) first sticks to the surface of water and is converted into hydrated ˙OH(H2O)n species,6,11,16 which react with BzO via(R1), or recombine into H2O2 (reaction (R9), Scheme 1) within interfacial layers:17,18
| ˙OH + BzO → products | (R1) |
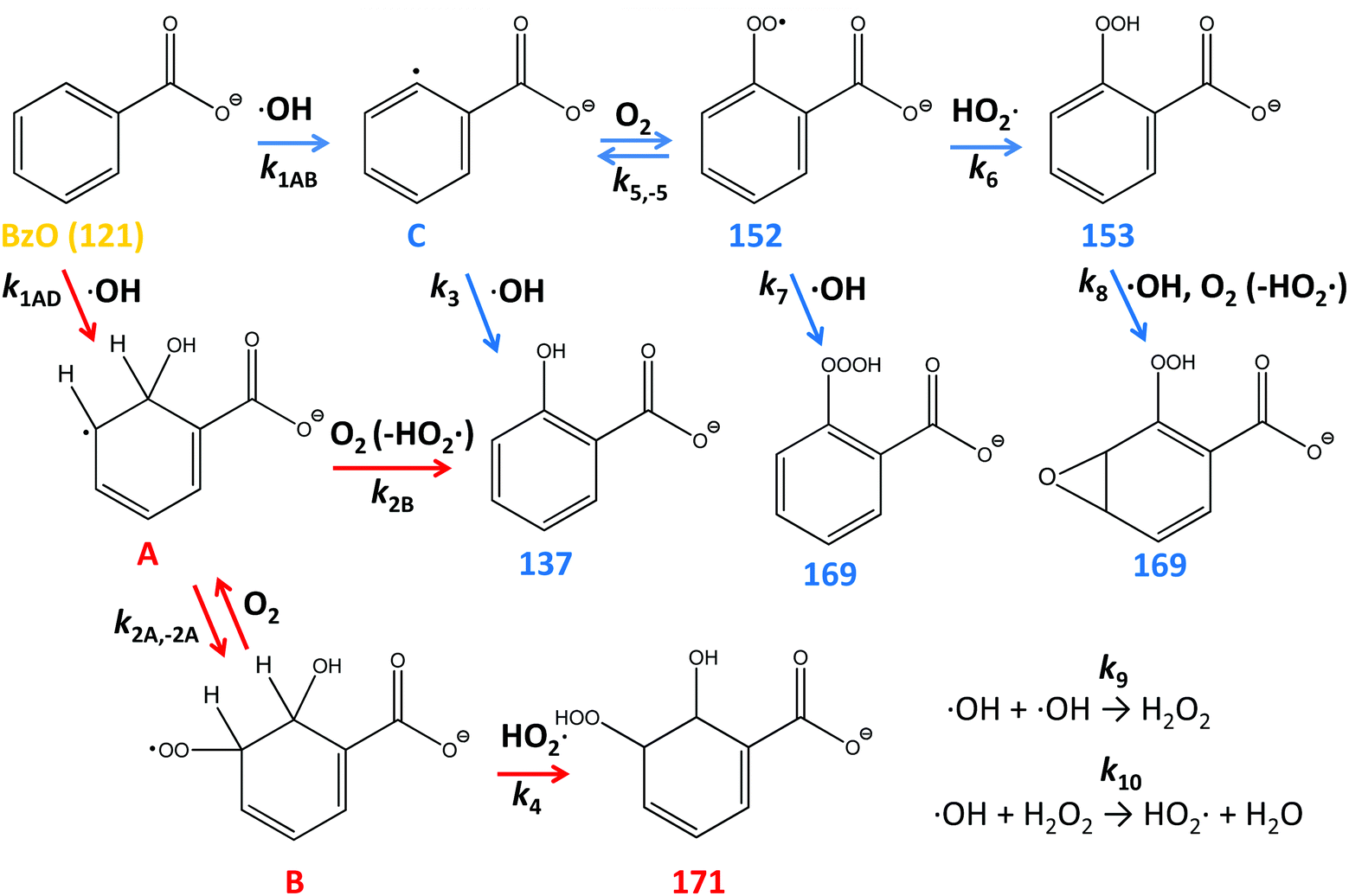 | ||
| Scheme 1 Mechanism of formation and proposed structures (among the various positional and/or functional isomers in each case) of the species generated in the ˙OH-initiated oxidation of benzoate at the air–water interface. | ||
Experimental
The experimental setup has been described in previous publications.11,20 The prompt (within the ∼10 μs lifetime of the intact microjets) formation of anionic products at the air–water interfaces of microjets from the reaction of aqueous reactants with gaseous OH-radicals at 1 atm at 298 K are monitored in situ by an electrospray ionization mass spectrometry (ES-MS, Agilent 6130 Quadrupole LC/MS Electrospray System, see Fig. S1, ESI†).11 Aqueous solutions are pumped (100 μL min−1) into the spraying chamber of the mass spectrometer through a grounded stainless steel needle (100 μm bore) coaxial with a sheath issuing nebulizer N2(g) at high gas velocity (vg ∼ 160 m s−1).20 The surface specificity of our experiments had been previously demonstrated.20–26 The depth (or thickness) of the sampled interfacial layers can be controlled by varying the nebulizer gas velocity vg, as evidenced by the fact that both ion signal intensities and relative anion surface affinities increase with higher gas velocities vg and extrapolate to zero as vg → 0.20 The ions detected by our mass spectrometer are ions that: (1) were already present or produced by chemical reactions in the interfacial layers of microjets (see previous publications for further details),20,23,27,28 (2) become incorporated into charged microdroplets produced during the stripping of interfacial layers by the nebulizing gas, and (3) finally ejected to the gas-phase and admitted into the mass spectrometer section via a polarized inlet port positively biased at 3.5 kV relative to ground.The dissociation of O3(g) by unfocused 266 nm laser pulses (laser beam diameter 10 mm, beam divergence ≤1.5 mrad, pulse duration ∼8 ns) into O(1D), followed by the reaction of O(1D) with H2O(g), in competition with its deactivation by N2(g) and O2(g) into O(3P), yields ˙OH(g) within ∼6 ns. Order of magnitude ˙OH(g) concentrations were estimated as described in previous publication.11
Results and discussion
Fig. 1 shows a typical negative ion electrospray mass spectrum obtained from 1.0 mM BzO(aq) microjets under O3(g)/O2(g)/H2O(g)/N2(g) mixtures as such or after being irradiated with 266 nm laser pulses.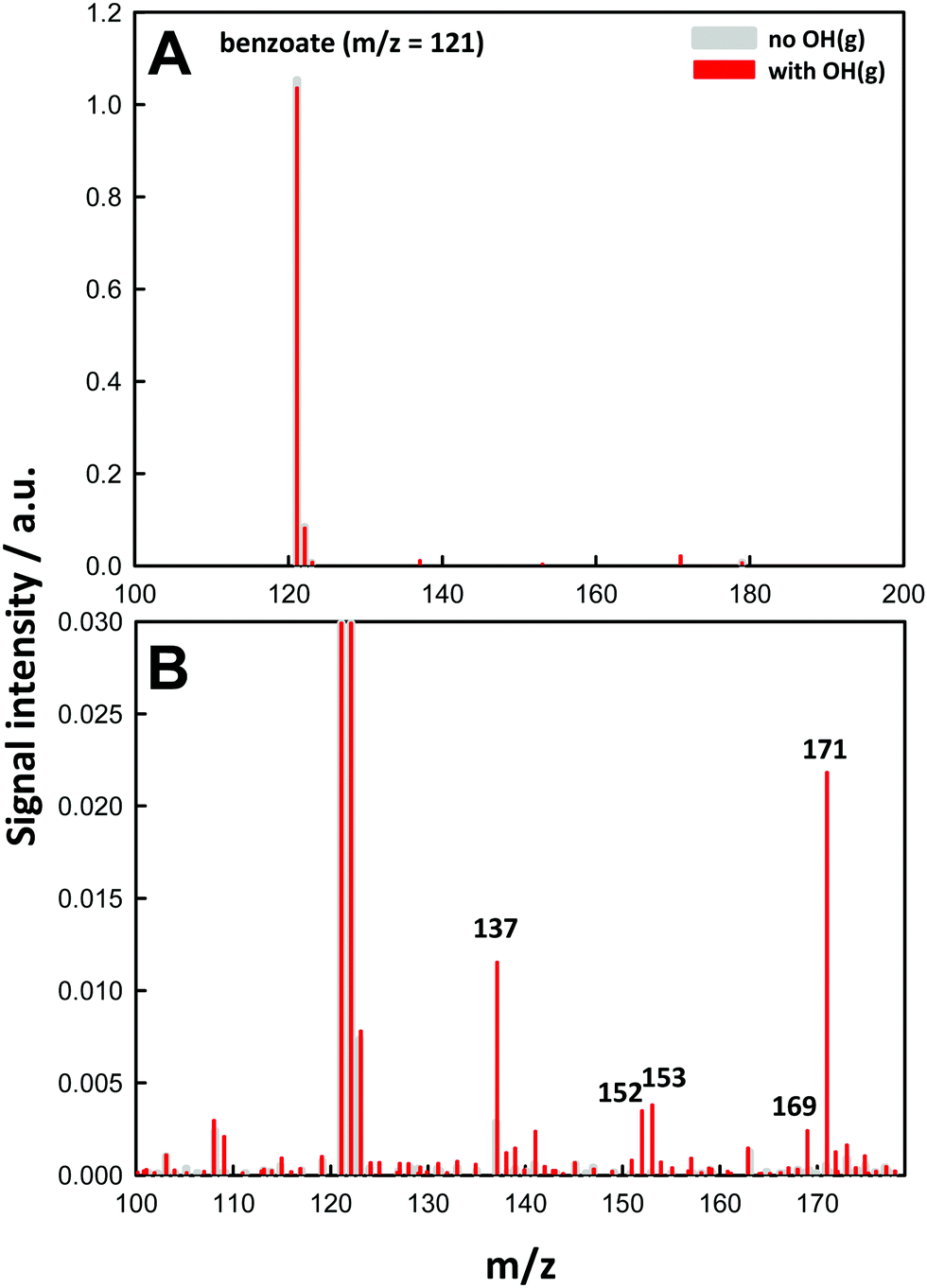 | ||
| Fig. 1 (A) Negative ion electrospray mass spectra of 1.0 mM (pH 4.0) benzoic acid microjets in the presence of 2 × 1015 molecules cm−3 O3(g) in O2(g)/H2O(g)/N2(g) mixtures with the 266 nm laser pulses (40 mJ, ∼8 ns, 10 Hz) off (gray) or on (red). (B) Zooming in the ˙OH-oxidation products. See text for details. | ||
At pH 4.0, ∼40% BA (pKa = 4.2) is dissociated into detectable benzoate C6H5–COO− (BzO) at m/z = 121 (m/z in Thompson units throughout). Recall that neutral species are transparent to mass spectrometry (see above). We verified that in the absence of light, O3(g) does not generate new product signals (Fig. S2, ESI†), in line with the inertness of BzO toward O3 (kBzO+O3 = 1.2 M−1 s−1 in bulk water).2 Upon 266 nm pulse irradiation of the inflowing O3(g)/O2(g)/H2O(g)/N2(g) mixtures, which generates ˙OH(g) in situ within 8 ns, we observe the partial depletion of BzO and the simultaneous appearance of new signals, which we therefore ascribe to products of ˙OH reactions with BzO. The same products, albeit in different proportions, were observed over the [BzO] = 0.01–10 mM range. We confirmed that reactant depletion and product formation require both the participation of O3(g) and actinic 266 nm photons (Fig. S2 and S3, ESI†), that is, the chemistry we observe is neither due to benzoate ozonation or benzoate photolysis, but involves reactions of gas-phase OH-radicals with interfacial benzoate.
For the experiments shown in Fig. 1, we estimate [˙OH(g)]0 ∼ 2 × 1014 molecules cm−3 at 40 mJ pulse−1 (the highest laser pulse energy used in our experiments) within the laser-irradiated volume.11 [˙OH(g)] on the surface of microjets ≤1 mm apart are estimated to be a factor of 10 lower. It might be argued that they are larger than atmospheric concentrations, but we note that reactant conversions during the τ ∼ 10 μs reaction times of our experiments are comparable to those in which aerosol droplets exposed to typical tropospheric [˙OH] ≈ 106 molecules cm−3 concentrations for 40 min. Product distributions, however, are expected to be different. In our experiments, yields of secondary products resulting from organic radical + OH-radical reactions will be enhanced relative to those produced under atmospheric conditions.
The molecular formulas of the species generated in our experiments could be inferred from their mass-to-charge ratios. Thus, the m/z = 137 signal in Fig. 1B is readily assigned to hydroxy-benzoates C6H4(OH)–COO− (BzO-OH): 137 = 121 + 16 = BzO − H + ˙OH. It is important to note that BzO-OH can be produced via two reaction channels: one initiated by ˙OH-addition to BzO: 137 = 121 (BzO) + 17 (˙OH) + 32 (O2) − 33 (HO2˙), and another one initiated by H-abstraction from BzO: 137 = 121 (BzO) − 1 (H) + 17 (˙OH) (Scheme 1). Note that one phenylic H-atom is removed in both cases. Similarly, the m/z = 153 signal corresponds to structures formally derived from O-addition to BzO-OH: 153 = 137 + 16, such as benzoate hydroperoxides C6H4(OOH)–COO− (BzO-OOH) or di-hydroxy-benzoates C6H3(OH)2–COO− (BzO-(OH)2). The difference is that the hydroperoxides will retain 4 phenylic H-atoms but the di-hydroxy species only 3. The m/z = 169 signal corresponds to species formally derived from the addition of a third O-atom to BzO-OOH: 169 = 153 + 16.29,30 The m/z = 171 signal corresponds to the only species produced by successive ˙OH and HO2˙ additions: 171 = 121 (BzO) + 17 (˙OH) + 33 (HO2˙), via processes that retain all 5 phenylic H-atoms. In addition to the above closed-shell species, we detected even mass m/z = 152 signals, which correspond to isomeric peroxyl radicals C6H4(OO˙)–COO− (BzO–O2˙) derived from O2-addition to the phenylic radicals generated by H-abstraction from BzO: 152 = 121 (BzO) − 1 (H) + 32 (O2).31 To our knowledge, this is the first report on the detection of early, labile intermediates, such as peroxyl radicals and hydroperoxides, in the oxidation of aromatics by ˙OH at the air–water interface. Since mass spectrometry reports molecular mass, the structures shown in Scheme 1 stand for all possible positional isomers in each case.32–34
Experiments in D2O provided additional evidence on molecular assignments (Fig. 2). Thus, hydroxy-benzoates and hydroxy-hydroperoxides, which possess one and two exchangeable (O-)H-atoms, generate (M + 1) and (M + 2) species, respectively. We note that the H-containing nascent phenol and hydroperoxide groups are able to exchange with D2O prior to detection ∼1 ms later.10–12 The finding that m/z = 152 does not shift in D2O solvent is clearly consistent with BzO–O2˙ peroxyl radical structural isomers.
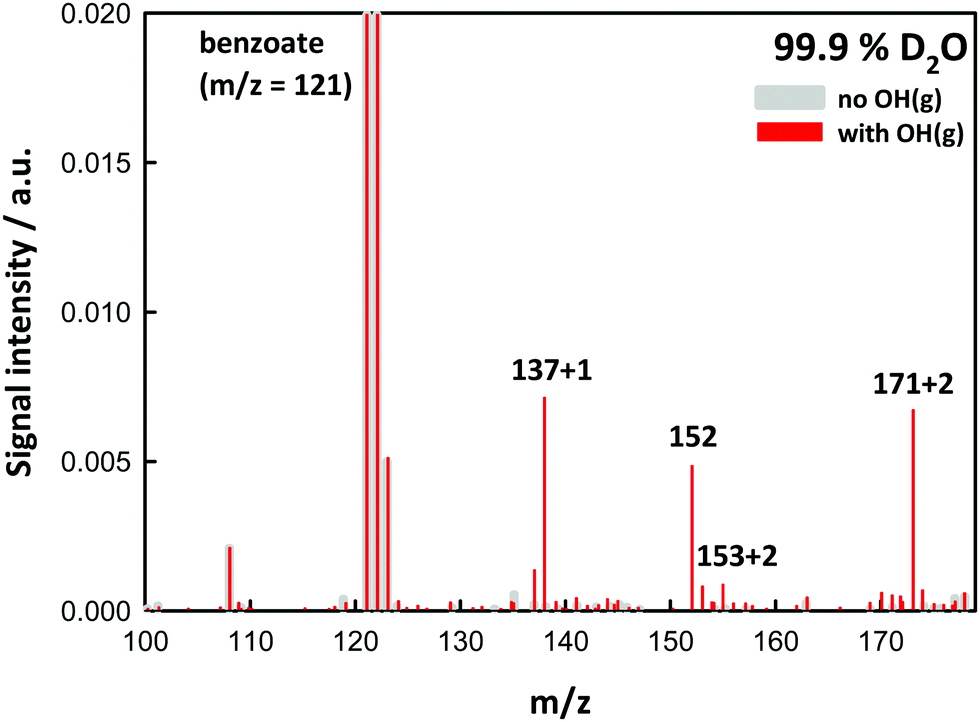 | ||
| Fig. 2 Negative ion electrospray mass spectra of 0.5 mM benzoic acid in D2O (99.9 atom % D) microjets exposed to 6.9 × 1015 molecules cm−3 O3(g) in O2(g)/H2O(g)/N2(g) mixtures at 1 atm and 298 K. Gray: laser off. Red: under 40 mJ, ∼8 ns pulses (at 10 Hz) of 266 nm radiation. | ||
Our assignments were validated in experiments involving isotopically labeled BzO-d5 (benzoate-2, 3, 4, 5, 6-d5) in H2O and D2O as solvents. Fig. 3A shows the mass spectra for the ˙OH oxidation of BzO-d5 (m/z = 126) in H2O. In this case, we detected products at m/z = 141, 156, 157, 173 and 176, which are consistent with the structures proposed in Scheme 1. For example, the hydroxy-benzoates signal shifts from m/z = 137 in BzO-h5 to 141 in BzO-d5, i.e., as expected from species derived from the abstraction of one D-atom from BzO-d5. Similarly, D-abstraction precedes the formation of the benzoate peroxyl radicals (BzO–O2˙) and benzoate hydroperoxides (BzO-OOH), whose signals, originally at m/z = 152 and 153, shift to 156 and 157, respectively. The only species retaining all 5 D-atoms are those corresponding to hydroxy-hydroperoxides (Scheme 1), which shift from m/z = 171 to 176 as proof that they result from OH-addition to the ring. The shift of the m/z = 169 signal in BzO-h5 to m/z = 173 in BzO-d5 excludes a tri-hydroxy-benzoate (BzO-(OH)3) (which would have led to a m/z = 169 to 171 shift) but is consistent with trioxides or epoxide hydroperoxides (Scheme 1).
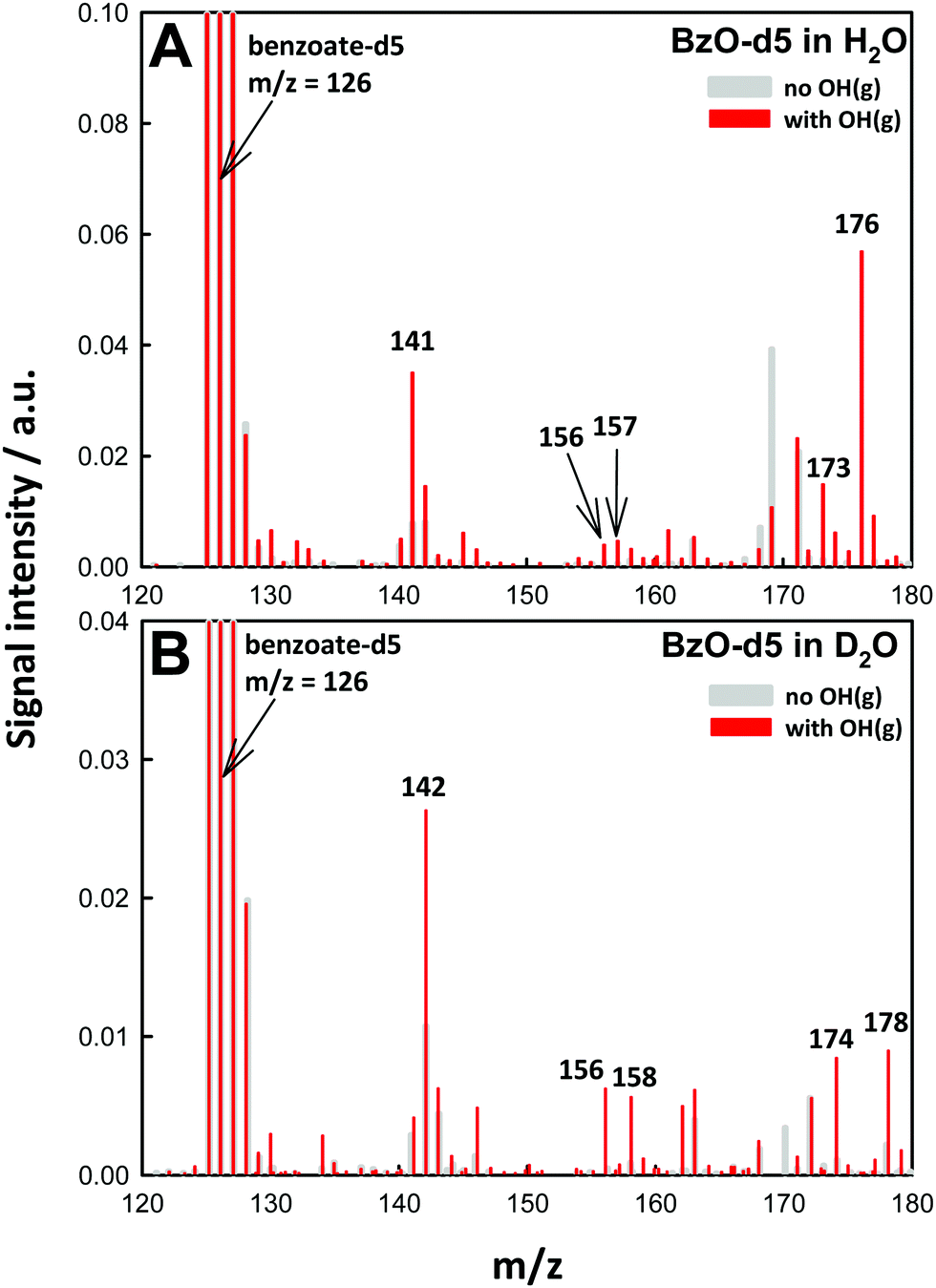 | ||
| Fig. 3 (A) Negative ion electrospray mass spectra (background subtracted) of 10 mM C6D5COOH benzoic acid-d5 microjets in H2O. (B) in D2O, in the presence of 2 × 1016 molecules cm−3 O3(g) at 1 atm and 298 K. Gray: laser off. Red: under 14 mJ, ∼8 ns (at 10 Hz) 266 nm pulses. | ||
The ˙OH oxidation of BzO-d5 in D2O gives rise to m/z = 142, 156, 158, 174 and 178 products (Fig. 3B). The shift of the m/z = 141 signal in H2O (Fig. 3A) to m/z = 142 in D2O (Fig. 3B) is consistent with hydroxy-benzoates containing the exchangeable (O-)H-atom brought by ˙OH. Also, according to the proposed structures, the signals assigned to the hydroxy-hydroperoxides, which contain two (O-)H-atoms, shift by two mass units from m/z = 176 to 178. As expected, signals assigned to benzoate peroxyl radicals (m/z = 156 in Fig. 3A and B) do not shift as D2O replaces H2O as solvent. The m/z = 173 signals in Fig. 3A shift to m/z = 174 in D2O, as expected from the presence of only one exchangeable H-atom in the proposed structures (Scheme 1) rather than to 176 should they correspond to tri-hydroxy isomers (BzO-(OH)3). A benzoate trioxide (m/z = 169) could be formed by the radical–radical reaction (BzO–O2˙ + ˙OH). The known negative temperature dependence of the (CH3O2˙ + ˙OH = CH3O˙ + HO2˙) reaction in the gas-phase35 is indicative of an associative reaction proceeding via a chemically activated trioxide CH3OOOH* intermediate. Thus, it is conceivable that a similar BzO-OOOH trioxide could be formed and stabilized from (BzO–O2˙ + ˙OH) in aqueous media. Alternatively, hydroperoxy epoxides (also m/z = 169) could be formed from the reaction of a hydroperoxide with ˙OH, followed by O2 addition and elimination of HO2˙ (see Scheme 1). The formation of epoxides has been previously proposed in reactions of aromatics with ˙OH in the presence of O2.29
We consider that our key finding is the detection of substantial yields of BzO–O2˙ radicals, which quantify the extent of H-abstraction from the aromatic ring at the air–water interface. Our result is in marked contrast with previous studies in bulk water, in which addition was the exclusive channel in OH-radical reactions with aromatics.31,33,34 Our detection of labile intermediates and products, such as BzO–O2˙ and the hydroperoxides vis-à-vis reports on the exclusive formation of hydroxybenzoates (BzO-OH) reported in the oxidation of BzO by ˙OH in bulk water,33,34 therefore implies that (1) all intermediates we detect within ∼1 ms are ultimately converted to hydroxybenzoates, (2) they may have been missed in the other studies, or (3) the reaction proceeds by different mechanisms in bulk water vs. at the air–water interface. It should be emphasized that, at variance with the oxidation of alkyl-carboxylic acids by ˙OH under similar conditions,10–12 we found no evidence of putative products of BzO–O2˙ self-reactions, such as alkoxyl radicals and alcohol/carbonyls. Thus, self-reactions of the bulky secondary peroxyl radicals BzO–O2˙ are relatively slow in the timeframe of our experiments.36
The dependences of signal intensities as functions of laser energy and benzoate concentration provide valuable mechanistic clues. Fig. 4 shows mass signals as functions of laser energy per pulse. We assume that ˙OH doses increase linearly with pulse energy at low energies before plateauing at high pulse energies. Note that these are not kinetic plots, i.e., mass signals as functions of time. The fact that all species, i.e., primary and secondary, appear at the lowest laser pulse energies, i.e., at the lowest OH-radical concentrations, means that all reactions are very fast (and therefore nearly independent of temperature) and not limited by reactants under present conditions.
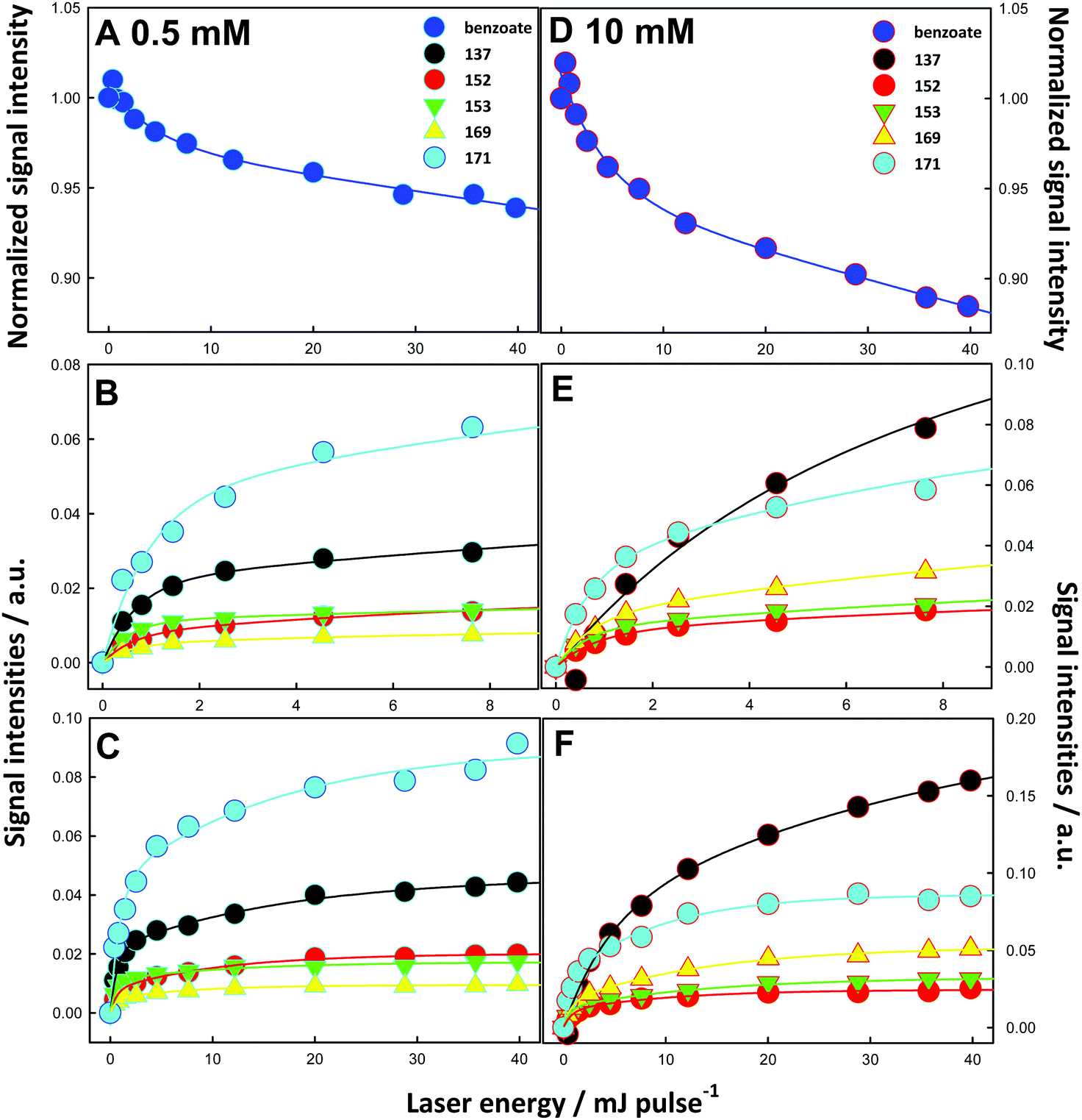 | ||
| Fig. 4 (A) Reactant, (B and C) intermediates and products mass spectral signal intensities from aqueous 0.5 mM benzoic acid at pH 4.2 microjets, (D–F) from 10 mM benzoic acid at pH 3.4 microjets exposed to O3(g)/O2(g)/H2O(g)/N2(g) mixtures, [O3(g)] ∼ 4 × 1015 molecules cm−3, as functions of 266 nm laser energy (in mJ pulse−1). Connecting lines are guides to the eye. See text for details. | ||
Interestingly, we note that the main products in the more dilute 0.5 mM BzO solutions are the m/z = 171 hydroxy-hydroperoxides (Fig. 4C), which, as we have seen, provide a true measure of the addition channel, whereas in 10 mM BzO the main products are the m/z = 137 hydroxybenzoates (Fig. 4F). Rough estimates OH-radical concentrations help to rationalize these findings. We estimate that a 40 mJ pulse generates [˙OH(g)]0 ≈ 3 × 1014 molecules cm−3, which translate (on the basis of the kinetic theory of gases37 and a mean ˙OH speed c = 6.4 × 104 cm s−1 at 298 K) into ∼5 × 1018 molecules cm−2 s−1 flux on the surface of the microjets.38 From the reported thermal accommodation coefficient of ˙OH on water: S ∼ 0.95,39 we estimate that the maximum dose of ˙OH incorporated into the surface of aqueous microjets during ∼8 ns pulses is: N˙OH ≤ 4 × 1010 radicals cm−2.
The remarkable fact that BzO signals decay by ∼7% in 0.5 mM solutions, and by ∼14% in the 20 times more concentrated 10 mM solutions at the same ˙OH doses (Fig. 4A and D) has mechanistic implications. Also note that BzO signals do not decay as single exponentials but bottom out at large pulse energies (i.e., at large ˙OH doses).10–12,40 The rate coefficients of (˙OH + BzO) via (R1AB + R1AD) and (˙OH +˙OH) via R9 (Scheme 1) in bulk water are within a factor of two and correspond to diffusionally controlled reactions: k1AB + k1AD = 2.5 × 109 M−1 s−1, k9 = 5.5 × 109 M−1 s−1.3,41,42 Since they are also expected to be fast and commensurate at the air–water interface, the above observations imply that: (1) ˙OH recombination into relatively inert H2O2 (via R9, Scheme 1) is competitive and more extensive than its reaction with BzO in the more dilute 0.5 mM BzO solutions, and (2) BzO must diffuse back from the bulk solution to replenish the depleted outermost layers. The first observation in turn requires that ˙OH and BzO should have comparable concentrations in the layers where these processes take place. This condition defines the average thickness δ of such layers. At the maximum dose of OH-radicals: N˙OH = 4 × 1010 radicals cm−2, the resulting [˙OH] in layers of thickness δ is given by: [˙OH] = N˙OH ≤ 4 × 1010 radicals cm−2/δ. By equating [˙OH] to [BzO] = 3 × 1017 molecules cm−3 (in 0.5 mM solutions), we derive a δ ≤ 1.3 nm value. It is apparent that our experiments effectively probe reactive events occurring in interfacial nanolayers.
An approximate but realistic estimate of the rates of the competing processes involved in BzO depletion reveals that reactions R1AB + R1AD and R9 take place in ≤1 μs (see Kinetic Model Calculations in ESI† and Fig. S4 and S5). By assuming a typical value of diffusion coefficients in water: DBzO = 2 × 10−5 cm2 s−1, we infer that BzO diffusion from the bulk into depleted layers takes place after chemical reactions are over. In fact, the slower time scale associated with replenishing BzO-depleted layers via diffusion sets an upper limit of ∼10 μs to the lifetimes of intact microjets (i.e., before they break into charged microdroplets). Longer lifetimes would have led to negligible BzO conversions because diffusion would have fully refilled interfacial layers. Shorter lifetimes, in contrast, would have preempted diffusion and led to exponential decays of BzO as a function of pulse energy. It should be emphasized that the much longer microjet lifetimes: ∼10 μs vs. the ∼8 ns laser pulses (which are shot every 100 ms) imply that our experiments correspond to processes taking place in fresh air–water interfaces after being exposed once to strong, short ˙OH pulses.
The 137/171 and 137/152 ratios of signal intensities as a function of laser energy, i.e. ˙OH dose, are shown in Fig. 5A and B. The dependences of product yields, defined as the ratio of the individual signal intensities to the sum of product signal intensities, and the ratios of 137/171 and (152 + 153 + 169)/(137 + 171) as functions of BzO concentration in experiments under 40 mJ pulses are shown in Fig. 6 and 7, respectively.
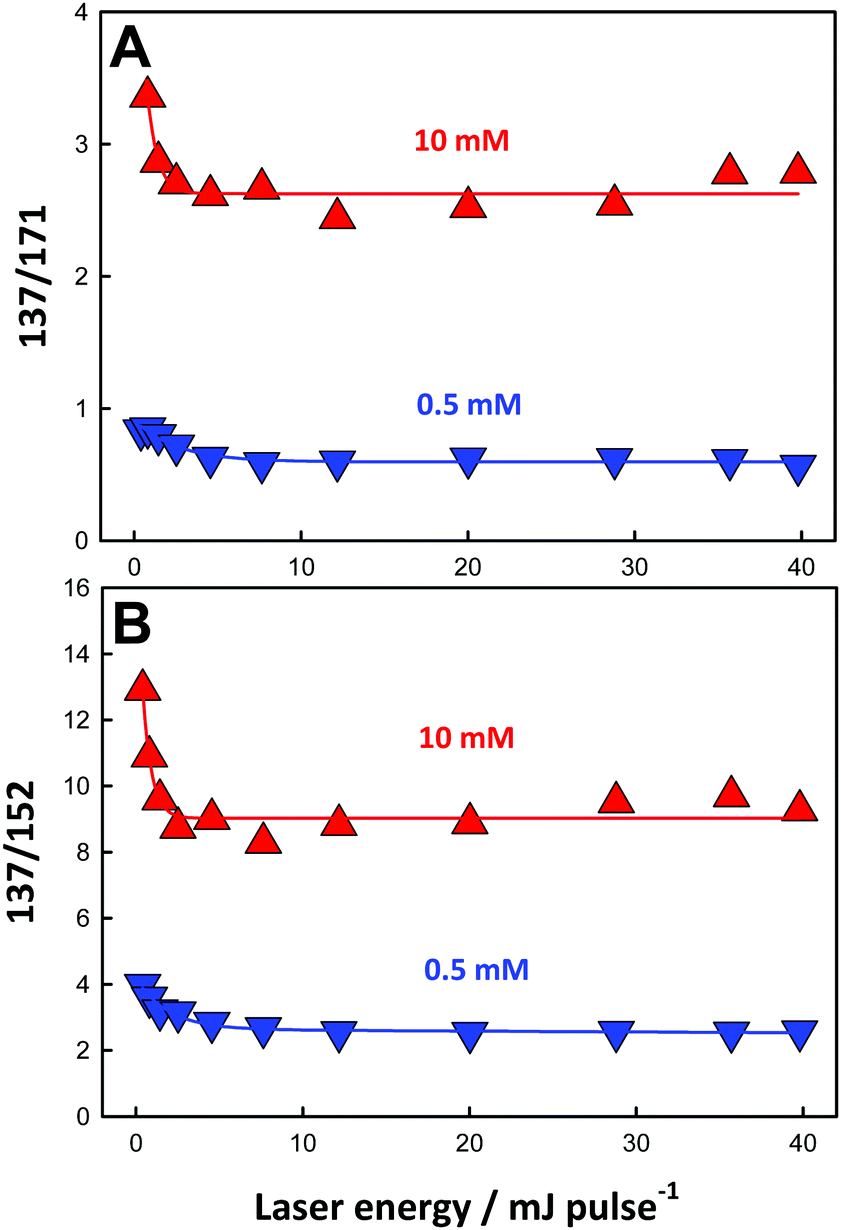 | ||
| Fig. 5 (A) The 137/171 and (B) 137/152 ratios of signal intensities from aqueous 0.5 mM (blue triangles) and 10 mM (red triangles) benzoic acid microjets exposed to O3(g)/O2(g)/H2O(g)/N2(g) mixtures as functions of 266 nm laser energy (in mJ pulse−1). Connecting lines are guides to the eye. See text for details. | ||
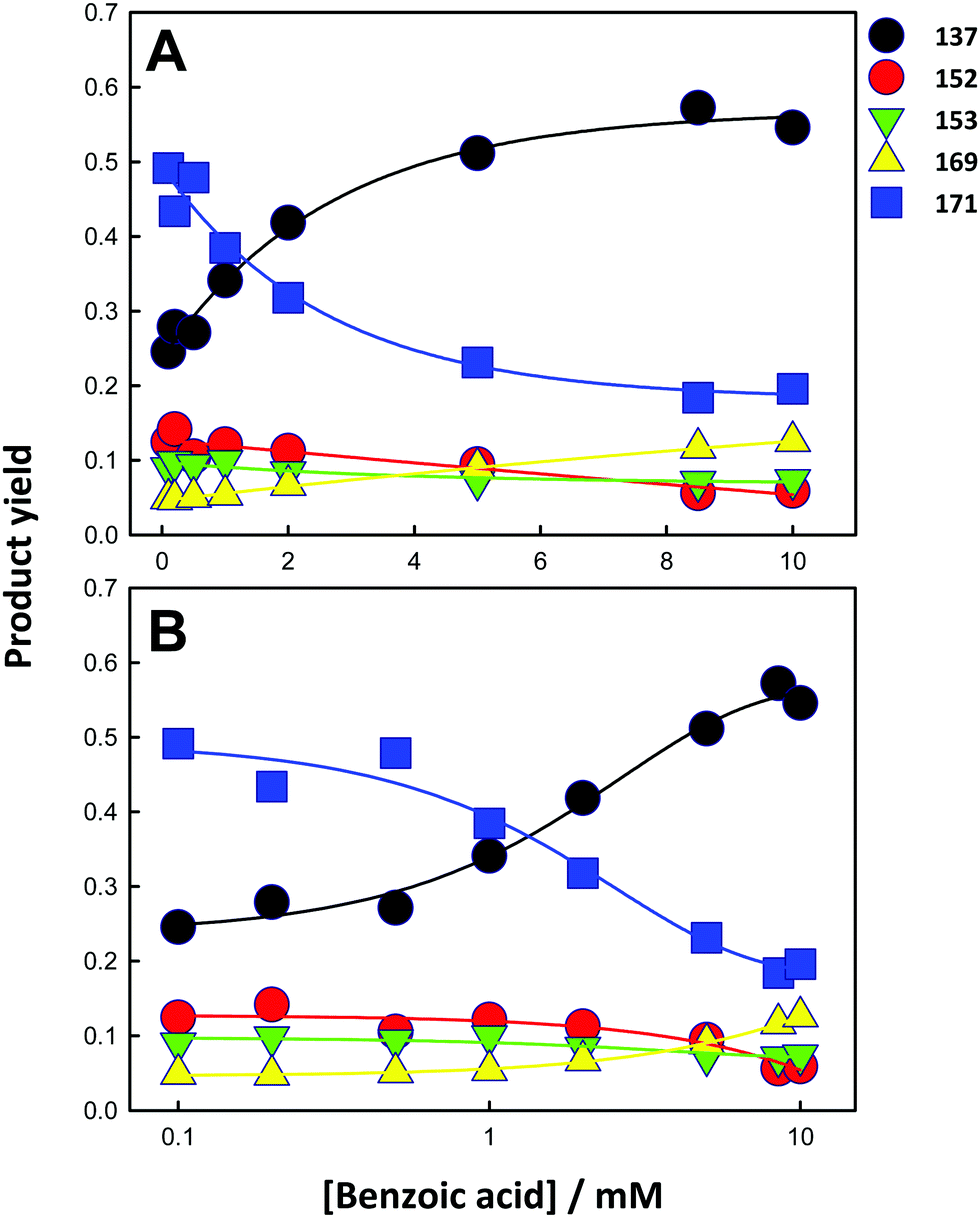 | ||
| Fig. 6 (A) Fractional contributions of individual signal intensities to the mass spectra as functions of benzoic acid concentration in experiments under 40 mJ pulse−1. (B) Semi-log plots. Connecting lines are guides to the eye. | ||
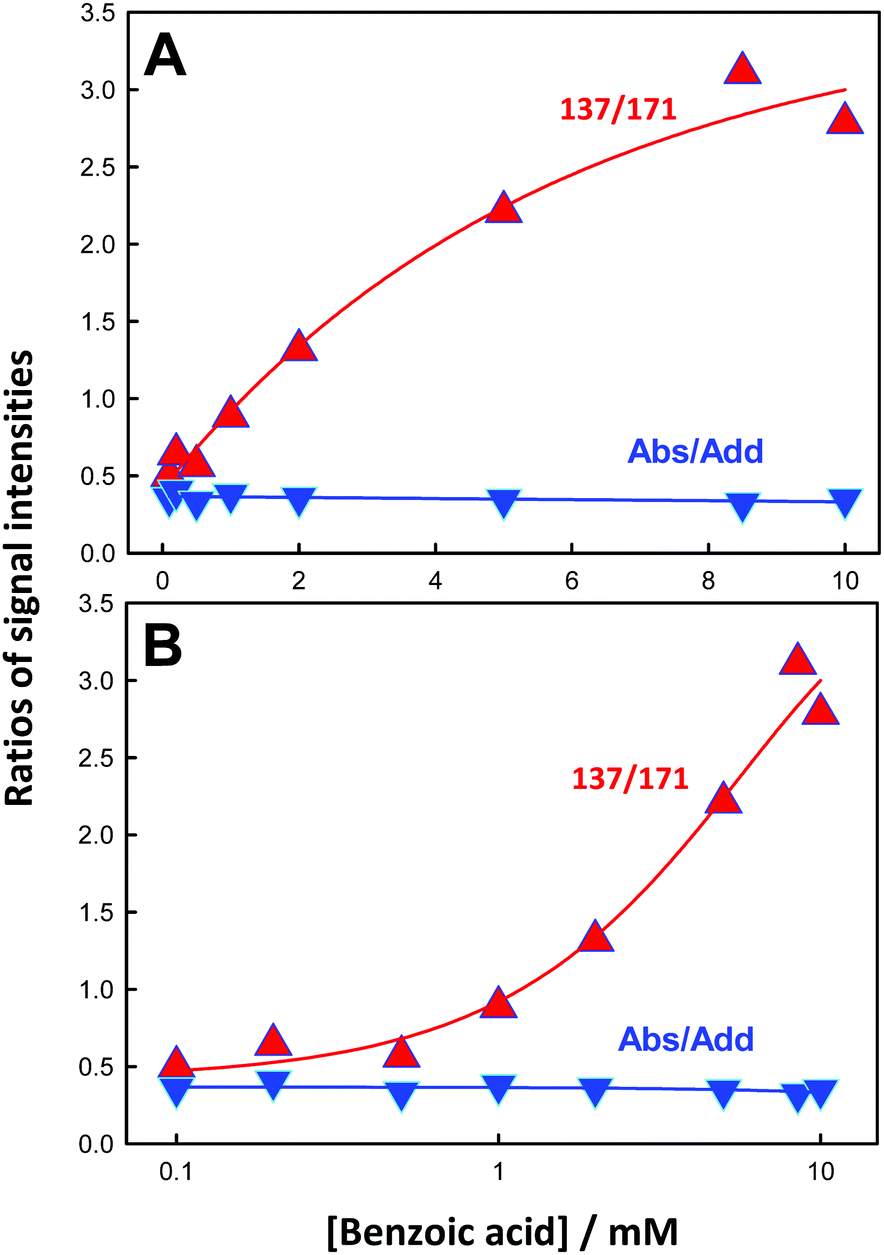 | ||
| Fig. 7 (A) The 137/171 (red triangles) and (152 + 153 + 169)/(137 + 171) (blue triangles) product signal ratios as functions of benzoic acid concentration in experiments under 40 mJ pulse−1. (B) Semi-log plots. Connecting lines are guides to the eye. See text for details. | ||
These trends are consistent with the mechanism outlined in Scheme 1. It should be realized that: (1) m/z = 137 species can be produced both via OH-radical addition and abstraction reactions in the sequences (1AD + 2B) and (1AB + 3), (2) whereas the m/z = 171 species can only be produced from OH-radical addition along the sequence (1AD + 2A + 4), via the hydroxycyclohexadienyls A (undetected) from reaction 1AD in equilibrium with peroxyl radicals B (undetected).43,44 Since little H2O2, the precursor of HO2˙, will be produced at high [BzO] and low ˙OH doses, the low [HO2˙]/[O2] ratios prevailing under such conditions will largely convert the addition intermediate A into the m/z = 137 hydroxy-benzoates rather that into the m/z = 171 hydroxy-hydroperoxide (Scheme 1). Note that neutral HO2˙ is MS-silent. It is apparent that under such conditions the m/z = 137 hydroxybenzoates will be favored over the m/z = 171 hydroxy-hydroperoxide. Thus, the competition between ˙OH reactions with BzO and recombination accounts for the higher 137/171 and 137/152 ratios observed at higher [BzO] and lower ˙OH doses (Fig. 5A and B), as well as the significant increase of the 137/171 ratio at larger [BzO] in Fig. 7.
On the basis of the preceding consideration, we estimate a lower limit to the branching between H-abstraction and addition from the ratio Abs/Add ≥ (152 + 153 + 169)/(137 + 171) = 0.35. This is a lower limit to the extent of abstraction because the numerator is the sum of the signal intensities of the products derived from H-abstraction over those arising from addition by assuming that all 137 ensues from addition. It is apparent that at least 26% [0.35/(1 + 0.35) = 0.26] of the ˙OH reacting with BzO will H-abstract from the aromatic ring at the air–water interface, independent of [BzO]. The ratio Abs/Add ≥ 0.35 is consistent with a difference of Δ = EAbs − EAdd ≤ 0.6 kcal mol−1 (from exp(−Δ/RT) ≥ 0.35) between the activation energies for abstraction and addition at the air–water interface. We note that our Δ ≤ 0.6 kcal mol−1 value is significantly smaller than the Δ = 3 kcal mol−1 reported for such competition in the presence of 2 water molecules,45 and much smaller than the Δ = 6.5 kcal mol−1 in the gas-phase, where H-abstraction is negligible.46
The sensitivity of OH-radical reactions with aromatics to H-bonding with hydrophilic solvents is well established. An experimental and computational study of ˙OH reactions with aromatics in water and in polar, non-hydrophilic acetonitrile,45 had reported rate coefficients ∼65 times larger in water than in acetonitrile, as a clear indication of both the electrophilicity of the OH-radical and the polar nature of the transition states involved. Calculations also showed that the effects of binding one and two water molecules to reactants and transition states on free energies of activation were sensitive to relative orientation.47–50 In view of such information, we propose that H-abstraction is enhanced over addition in the anisotropic hydration environment provided by the air–water interface, where water density drops precipitously over 3 Å.47–52 Molecular dynamics calculations based on many-body potentials that reliably simulate interfacial phenomena could shed further light on these processes.51,52
The atmospheric implications of present findings are briefly discussed below. Our experiments simulate the heterogeneous oxidation of organic aerosol matter by gas-phase OH-radicals that stick to the surface to subsequently react at nearly diffusion controlled rates with aromatics therein. Significantly, they reveal that the oxidation of aqueous benzoate not only leads to hydroxylation but, via a sizable contribution of H-abstraction, to the formation of more reactive species, such as peroxyl radicals and hydroperoxides, that can propagate radical chemistry via solar photolysis,53,54 or metal-catalyzed decomposition55,56 in the condensed aerosol phase. Given the hydrophobic character of benzoate, the most abundant organic species found in the PM2.5 in polluted urban areas,1 our experiments suggest that its oxidation by ˙OH(g) at the air–water interface may be an important process in the photochemical aging of secondary organic aerosols.
Conclusion
Summing up, we report the first direct detection of peroxyl radicals in significant yields during the oxidation of benzoate by OH-radicals at the air–water interface. They originate from significant (>26%) H-abstraction from the aromatic ring, a pathway deemed to be absent in OH-radical reactions with aromatics in water. We also detected hydroperoxides and other hitherto unidentified products in addition to hydroxybenzoates. The significant extent of H-abstraction from the aromatic ring is tentatively ascribed to the relative destabilization of the more polar transition state for the OH-radical addition channel at the low water density prevalent in the outermost interfacial layers.Acknowledgements
S. E. is grateful to the Hakubi Project of Kyoto University, the Iwatani Naoji Foundation's Research Grant and JSPS KAKENHI grant number 15H05328. M. R. H. and A. J. C. acknowledge support from the National Science Foundation (U.S.A.) Grant AC-1238977.References
- K. F. Ho, R. J. Huang, K. Kawamura, E. Tachibana, S. C. Lee, S. S. H. Ho, T. Zhu and L. Tian, Atmos. Chem. Phys., 2015, 15, 3111–3123 CrossRef CAS.
- J. Hoigne and H. Bader, Water Res., 1983, 17, 185–194 CrossRef CAS.
- H. Herrmann, D. Hoffmann, T. Schaefer, P. Brauer and A. Tilgner, ChemPhysChem, 2010, 11, 3796–3822 CrossRef CAS PubMed.
- J. H. Seinfeld and S. N. Pandis, Atmospheric chemistry and physics: from air pollution to climate change, Wiley, Hoboken, N.J., 2nd edn, 2006 Search PubMed.
- B. J. Finlayson-Pitts and J. N. Pitts, Chemistry of the upper and lower atmosphere, Academic Press, San Diego, CA, 2000 Search PubMed.
- I. J. George and J. P. D. Abbatt, Nat. Chem., 2010, 2, 713–722 CrossRef CAS PubMed.
- J. H. Park, A. V. Ivanov and M. J. Molina, J. Phys. Chem. A, 2008, 112, 6968–6977 CrossRef CAS PubMed.
- U. Poschl and M. Shiraiwa, Chem. Rev., 2015, 115, 4440–4475 CrossRef PubMed.
- B. Minofar, P. Jungwirth, M. R. Das, W. Kunz and S. Mahiuddin, J. Phys. Chem. C, 2007, 111, 8242–8247 CAS.
- S. Enami, M. R. Hoffmann and A. J. Colussi, J. Phys. Chem. Lett., 2015, 6, 527–534 CrossRef CAS PubMed.
- S. Enami, M. R. Hoffmann and A. J. Colussi, J. Phys. Chem. A, 2014, 118, 4130–4137 CrossRef CAS PubMed.
- S. Enami and Y. Sakamoto, J. Phys. Chem. A, 2016, 120, 3578–3587 CrossRef CAS PubMed.
- C. T. Cheng, M. N. Chan and K. R. Wilson, J. Phys. Chem. A, 2016, 120, 5887–5896 CrossRef CAS PubMed.
- R. C. Chapleski, Y. Zhang, D. Troya and J. R. Morris, Chem. Soc. Rev., 2016, 45, 3731–3746 RSC.
- M. Shiraiwa, Y. Sosedova, A. Rouviere, H. Yang, Y. Y. Zhang, J. P. D. Abbatt, M. Ammann and U. Poschl, Nat. Chem., 2011, 3, 291–295 CrossRef CAS PubMed.
- P. A. J. Bagot, C. Waring, M. L. Costen and K. G. McKendrick, J. Phys. Chem. C, 2008, 112, 10868–10877 CAS.
- M. Roeselova, J. Vieceli, L. X. Dang, B. C. Garrett and D. J. Tobias, J. Am. Chem. Soc., 2004, 126, 16308–16309 CrossRef CAS PubMed.
- R. Vacha, P. Slavicek, M. Mucha, B. J. Finlayson-Pitts and P. Jungwirth, J. Phys. Chem. A, 2004, 108, 11573 CrossRef CAS.
- A. J. Ingram, C. L. Boeser and R. N. Zare, Chem. Sci., 2016, 7, 39–55 RSC.
- S. Enami and A. J. Colussi, J. Chem. Phys., 2013, 138, 184706 CrossRef PubMed.
- S. Enami, Y. Sakamoto and A. J. Colussi, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 623–628 CrossRef CAS PubMed.
- S. Enami and A. J. Colussi, J. Phys. Chem. B, 2013, 117, 6276–6281 CrossRef CAS PubMed.
- S. Enami, H. Mishra, M. R. Hoffmann and A. J. Colussi, J. Chem. Phys., 2012, 136, 154707 CrossRef PubMed.
- S. Enami, L. A. Stewart, M. R. Hoffmann and A. J. Colussi, J. Phys. Chem. Lett., 2010, 1, 3488–3493 CrossRef CAS.
- S. Enami, M. R. Hoffmann and A. J. Colussi, J. Phys. Chem. A, 2010, 114, 5817–5822 CrossRef CAS PubMed.
- S. Enami, M. R. Hoffmann and A. J. Colussi, J. Phys. Chem. Lett., 2010, 1, 1599–1604 CrossRef CAS.
- J. Cheng, M. R. Hoffmann and A. J. Colussi, J. Phys. Chem. B, 2008, 112, 7157–7161 CrossRef CAS PubMed.
- J. Cheng, C. Vecitis, M. R. Hoffmann and A. J. Colussi, J. Phys. Chem. B, 2006, 110, 25598–25602 CrossRef CAS PubMed.
- R. Volkamer, B. Klotz, I. Barnes, T. Imamura, K. Wirtz, N. Washida, K. H. Becker and U. Platt, Phys. Chem. Chem. Phys., 2002, 4, 1598–1610 RSC.
- L. M. Wang, R. R. Wu and C. Xu, J. Phys. Chem. A, 2013, 117, 14163–14168 CrossRef CAS PubMed.
- X. M. Pan, M. N. Schuchmann and C. von Sonntag, J. Chem. Soc., Perkin Trans. 2, 1993, 289–297, 10.1039/p29930000289.
- N. Tanaka and S. Itoh, Open J. Phys. Chem., 2013, 3, 7–13 CrossRef.
- G. W. Klein, K. Bhatia, V. Madhavan and R. H. Schuler, J. Phys. Chem., 1975, 79, 1767–1774 CrossRef CAS.
- M. A. Oturan and J. Pinson, J. Phys. Chem., 1995, 99, 13948–13954 CrossRef CAS.
- C. Yan, S. Kocevska and L. N. Krasnoperov, J. Phys. Chem. A, 2016, 120, 6111–6121 CrossRef CAS PubMed.
- C. von Sonntag and H. P. Schuchmann, Angew. Chem., Int. Ed., 1991, 30, 1229–1253 CrossRef.
- P. Davidovits, C. E. Kolb, L. R. Williams, J. T. Jayne and D. R. Worsnop, Chem. Rev., 2006, 106, 1323–1354 CrossRef CAS PubMed.
- P. Davidovits, C. E. Kolb, L. R. Williams, J. T. Jayne and D. R. Worsnop, Chem. Rev., 2011, 111, PR76–PR109 CrossRef CAS PubMed.
- C. E. Kolb, R. A. Cox, J. P. D. Abbatt, M. Ammann, E. J. Davis, D. J. Donaldson, B. C. Garrett, C. George, P. T. Griffiths, D. R. Hanson, M. Kulmala, G. McFiggans, U. Poschl, I. Riipinen, M. J. Rossi, Y. Rudich, P. E. Wagner, P. M. Winkler, D. R. Worsnop and C. D. O'Dowd, Atmos. Chem. Phys., 2010, 10, 10561–10605 CrossRef CAS.
- S. Enami, M. R. Hoffmann and A. J. Colussi, J. Phys. Chem. Lett., 2015, 6, 3935–3943 CrossRef CAS PubMed.
- G. V. Buxton, C. L. Greenstock, W. P. Helman and A. B. Ross, J. Phys. Chem. Ref. Data, 1988, 17, 513–886 CrossRef CAS.
- H. Herrmann, Chem. Rev., 2003, 103, 4691–4716 CrossRef CAS PubMed.
- J. A. Howard and K. U. Ingold, Can. J. Chem., 1967, 45, 785–792 CrossRef CAS.
- N. Narita and T. Tezuka, J. Am. Chem. Soc., 1982, 104, 7316–7318 CrossRef CAS.
- M. P. DeMatteo, J. S. Poole, X. F. Shi, R. Sachdeva, P. G. Hatcher, C. M. Hadad and M. S. Platz, J. Am. Chem. Soc., 2005, 127, 7094–7109 CrossRef CAS PubMed.
- D. S. Hollman, A. C. Simmonett and H. F. Schaefer, Phys. Chem. Chem. Phys., 2011, 13, 2214–2221 RSC.
- J. M. Anglada, M. Martins-Costa, J. S. Francisco and M. F. Ruiz-Lopez, Acc. Chem. Res., 2015, 48, 575–583 CrossRef CAS PubMed.
- R. J. Buszek, J. S. Francisco and J. M. Anglada, Int. Rev. Phys. Chem., 2011, 30, 335–369 CrossRef CAS.
- E. Voehringer-Martinez, B. Hansmann, H. Hernandez, J. S. Francisco, J. Troe and B. Abel, Science, 2007, 315, 497–501 CrossRef CAS PubMed.
- Y. J. Feng, T. Huang, C. Wang, Y. R. Liu, S. Jiang, S. K. Miao, J. Chen and W. Huang, J. Phys. Chem. B, 2016, 120, 6667–6673 CrossRef CAS PubMed.
- G. A. Cisneros, K. T. Wikfeldt, L. Ojamae, J. B. Lu, Y. Xu, H. Torabifard, A. P. Bartok, G. Csanyi, V. Molinero and F. Paesani, Chem. Rev., 2016, 116, 7501–7528 CrossRef CAS PubMed.
- G. R. Medders and F. Paesani, J. Am. Chem. Soc., 2016, 138, 3912–3919 CrossRef CAS PubMed.
- S. A. Epstein, D. Shemesh, V. T. Tran, S. A. Nizkorodov and R. B. Gerber, J. Phys. Chem. A, 2012, 116, 6068–6077 CrossRef CAS PubMed.
- K. Badali, S. Zhou, D. Aljawhary, M. Antiñolo, W. Chen, A. Lok, E. Mungall, J. Wong, R. Zhao and J. Abbatt, Atmos. Chem. Phys., 2015, 15, 7831–7840 CAS.
- E. Vidrio, C. H. Phuah, A. M. Dillner and C. Anastasio, Environ. Sci. Technol., 2009, 43, 922–927 CrossRef CAS PubMed.
- H. Tong, A. M. Arangio, P. S. Lakey, T. Berkemeier, F. Liu, C. J. Kampf, W. H. Brune, U. Pöschl and M. Shiraiwa, Atmos. Chem. Phys., 2016, 16, 1761–1771 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional data and experimental details. See DOI: 10.1039/c6cp06652f |
| This journal is © the Owner Societies 2016 |