
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct comparison of 3-centre and 4-centre HBr elimination pathways in methyl-substituted vinyl bromides†
Shubhrangshu
Pandit
,
Balázs
Hornung
and
Andrew J.
Orr-Ewing
 *
*
School of Chemistry, University of Bristol, Cantock's Close, Bristol BS8 1TS, UK. E-mail: a.orr-ewing@bristol.ac.uk
First published on 21st September 2016
Abstract
Elimination of HBr from UV-photoexcited vinyl bromides can occur through both 3-centre and 4-centre transition states (TSs). The competition between these pathways is examined using velocity map imaging of HBr (v = 0–2, J) photofragments. The three vinyl bromides chosen for study have methyl substituents that block either the 3-centre or the 4-centre TS, or leave both pathways open. The kinetic energy distributions extracted from velocity map images of HBr from 193 nm photolysis of the three vinyl bromide compounds are approximately described by a statistical model of energy disposal among the degrees of freedom of the photoproducts, and are attributed to dissociation on the lowest electronic state of the molecule after internal conversion. Dissociation via the 4-centre TS gives greater average kinetic energy release than for the 3-centre TS pathway. The resonance enhanced multi-photon ionization (REMPI) schemes used to detect HBr restrict measurements to J ≤ 7 for v = 2 and J ≤ 15 for v = 0. Within this spectroscopic range, the HBr rotational temperature is colder for the 4-centre than for the 3-centre elimination pathway. Calculations of the intrinsic reaction coordinates and RRKM calculations of HBr elimination rate coefficients provide mechanistic insights into the competition between the pathways.
1. Introduction
Halogen-substituted alkenes, RHC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHX (with R denoting an organic group and X a halogen atom) are examples of bi-chromophoric molecules in which the longest wavelength ultraviolet (UV) absorption bands excite electron density into the π* molecular orbital associated with the CC bond or a σ* orbital centred on the C–X bond. A variety of relaxation pathways can follow from absorption of UV radiation by the CC or C–Br chromophores. These pathways include cis/trans isomerization about the CC bond, and direct and indirect fragmentation channels. Photo-initiated cis–trans isomerization of alkenes is an important chemical process in biological systems.1 For example, the chemical activity of photoreceptor proteins2 and some light-driven molecular motors3 is a consequence of the twisting motion of two methylene groups connected by a CC bond during relaxation from the photoexcited state.
CHX (with R denoting an organic group and X a halogen atom) are examples of bi-chromophoric molecules in which the longest wavelength ultraviolet (UV) absorption bands excite electron density into the π* molecular orbital associated with the CC bond or a σ* orbital centred on the C–X bond. A variety of relaxation pathways can follow from absorption of UV radiation by the CC or C–Br chromophores. These pathways include cis/trans isomerization about the CC bond, and direct and indirect fragmentation channels. Photo-initiated cis–trans isomerization of alkenes is an important chemical process in biological systems.1 For example, the chemical activity of photoreceptor proteins2 and some light-driven molecular motors3 is a consequence of the twisting motion of two methylene groups connected by a CC bond during relaxation from the photoexcited state.
The photochemistry associated with the CC chromophores of simple alkenes has been extensively studied. For example, after internal conversion back to the electronic ground state, internally excited ethene eliminates molecular H2 from one of its methylene groups.4 Calculations by Barbatti et al. indicated that 42% of the H2 molecules are eliminated after cis/trans isomerization, and identified pathways for H-atom migration between the two methylene groups.5
The photochemistry of vinyl halides (VX) exhibits both atomic and molecular elimination via competing fragmentation channels. The direct loss of an X atom involves C–X bond breaking on an electronically excited state, whereas internal conversion of the photoexcited VX molecules produces ground electronic state molecules with large amounts of internal (vibrational) energy.6,7 Elimination of HX from these hot ground state molecules can then occur through two alternative pathways: (1) H and X originate from the same C atom, with HX elimination via a 3-centre (3-C) transition state (TS); and (2) H and X are lost from two adjacent C atoms via a 4-centre (4-C) TS.6 Gordon and coworkers considered the competition between migration of H atoms across the CC bond and 3-C or 4-C elimination, but argued against significant migration on the basis of their studies of HCl and DCl elimination from CH2CDCl.8
Here, we seek to distinguish the 3-C and 4-C pathways by characterizing the properties of HBr photofragments from methyl-substituted vinyl bromides. The positions of methyl substitution are chosen to block selectively either the 3-C or 4-C elimination pathway. Methyl migration along a CC bond is associated with a large energy barrier and computed to be much slower than HBr elimination, so does not influence the measurement outcomes.
The rich photochemistry of the vinyl halides and vinyl cyanides is a consequence of the presence of several closely spaced excited electronic states, some of which contribute to the broad UV absorption bands observed in the wavelength range from 160–220 nm. The precise wavelengths spanned by these bands depend on the RHCCHX substituents (X = F, Cl, Br, I, CN; R = H, alkyl). These excited states correspond to electronic transitions from the ground state to πCCπCC*, nXσCX*, nXπCC* and πCCσCX* valence states as well as Rydberg states of VX.6,9 Of these excitations, the πCC* ← πCC transition gives rise to the strongest UV absorption band, and is centred at a wavelength of 193 nm for vinyl bromide (VBr).9Fig. 1 contains a schematic diagram, based on previous calculations,10,11 of the lowest lying electronic states of VBr responsible for the UV absorption and atomic and molecular fragmentation processes. In the optically accessed state with πCCπCC* character, the ethylenic component relaxes from planarity in the Franck–Condon region along the torsional angle coordinate around the CC bond.12,13 The two CH2 groups rotate towards a twisted-orthogonal geometry corresponding to the minimum energy structure on the excited state PES. Within ∼30 fs the torsional angle approaches ∼80° where the πCCπCC* states crosses the πCCσCBr* state.14 This πCCσCBr* state is purely repulsive along the C–Br stretching coordinate, promoting C–Br bond cleavage.11 The twisted ethylenic motion in the πCCπCC* state can also avoid the crossing to the πCCσCBr* state, instead further relaxing towards a conical intersection with the ground state at a twisted-pyramidal geometry of one methylene group.12 The internal conversion probability at this conical intersection in VBr is estimated to lie in the range 0.44–0.64.11 The geometry near the conical intersection differs from the equilibrium structure in the ground state and the internal conversion produces highly internally excited vinyl bromide which can eliminate either H2 or HBr.15
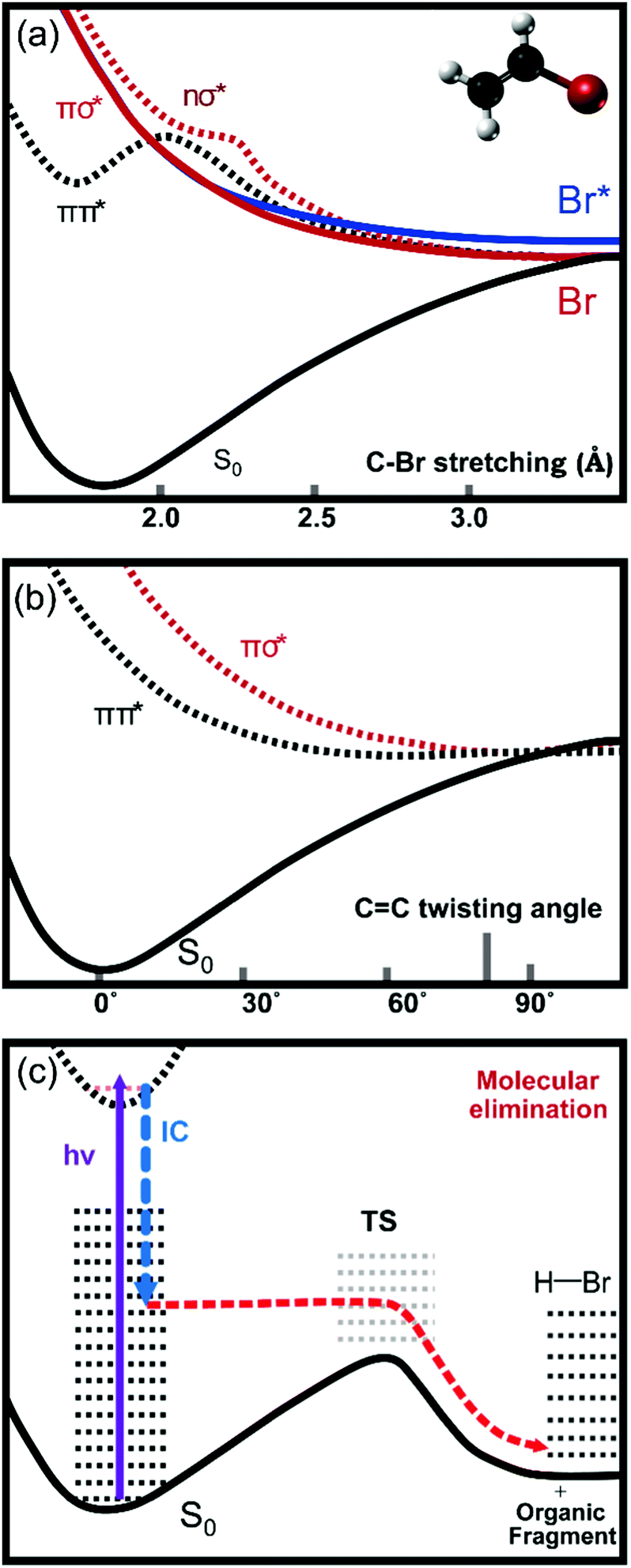 | ||
| Fig. 1 Schematic diagrams of cuts through the electronic potential energy surfaces responsible for different dissociation processes in vinyl bromide. The potential energy curves in panels (a) and (b) are adapted from ref. 10 and 11. The πCCπCC* state is a bound state along the C–Br stretching coordinate (a) but has a steep downward gradient along the CC torsional coordinate (b). During twisting motion of the methylene groups, the πCCπCC* state crosses the πCCσCBr* state at a torsional angle of ∼80°. This πCCσCBr* state is purely repulsive along the C–Br stretching coordinate. Further relaxation along the CC torsional coordinate leads the πCCπCC* state towards a conical intersection with the ground state. After internal conversion, internally excited parent molecules on the ground state potential energy surface can eliminate HBr (c). The internal conversion step conserves the energy of the molecule, but only the fraction along the molecular elimination coordinate is represented in the figure. | ||
The branching between excited state and ground state photochemistry is governed by the energies and orbital characters of the excited states and the locations of conical intersections, and hence by the identities of the halogen atom substituents.16 In vinyl fluoride (VF), non-bonding electrons are mostly localized on the F atom and the πCC orbital is the highest occupied molecular orbital (HOMO).9 In contrast, the non-bonding iodine electrons and the πCC MO interact strongly in vinyl iodide (VI) and the HOMO is of I (5p⊥) character.9 Vinyl chloride (VCl) and vinyl bromide lie somewhere between these extremes.9
The photodissociation of VF at 193 nm17 and 157 nm18 predominantly gives HF, with almost no F atom photoproducts. Both atomic Cl and molecular HCl fragments are seen from the 193 nm photodissociation of VCl.8,19–25 Similarly, both atomic and molecular photofragments have been identified from thermal decomposition26 and photodissociation7,27,28 of VBr. Wodtke et al.27 reported HBr to be a minor photofragment, with a branching ratio of Br/HBr = 1.28 ± 0.05. However, Johnson and Price29 observed vinyl radical (the partner fragment to a Br atom) only as a minor product in their flash photolysis experiments with photon wavelengths in the range 150–200 nm. HBr and H2 have also been recognized as the main photofragments in matrix isolation experiments.30 A trajectory calculation by Abrash et al.15 on vinyl bromide photodissociation at 193 nm predicted the product distributions to be: H2 (48%), HBr (44%), Br (5%) and H (3%). In contrast to the other vinyl halides, photofragmentation of vinyl iodide leads only to the formation of halogen atoms.16,31
All the previous studies suggest that atomic fragmentation can occur either from repulsive πCCσCX*, nXσCX* states or the ground electronic state. However, the molecular fragmentation processes take place only from the electronic ground state. The hot vinyl bromide produced by internal conversion to the ground state was computed to have a lifetime in the range 470 fs to 2 ps, depending on its internal energy.15,32 HBr elimination can occur through a 3-C or 4-C transition state; the 3-C elimination produces HBr and a vinylidene diradical as initial photoproducts, whereas HBr and acetylene come from the 4-C path. The thermochemistries of these two process are distinctly different because ground-state acetylene lies ∼200 kJ mol−1 below the lowest energy of vinylidene,33 but the unstable vinylidene fragment isomerizes to internally excited acetylene within 40–200 fs.34,35 Both channels give vibrationally as well as rotationally hot HBr (v = 0–6, J = 0–40) fragments.28,36,37 Berry,6 from his flash photolysis experiments, and later Gordon and co-workers22 from their velocity aligned Doppler spectroscopy measurements, argued that the 4-C mechanism is the dominant elimination pathway. However, IR emission studies of HBr fragments from vinyl bromide dissociation indicated a preference for the 3-C pathway,28,37 and were supported in this deduction by classical trajectory calculations.25,35,36,38
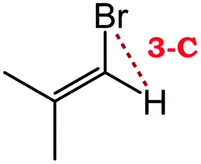
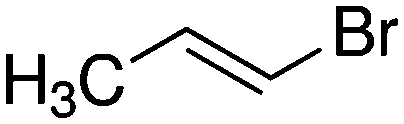
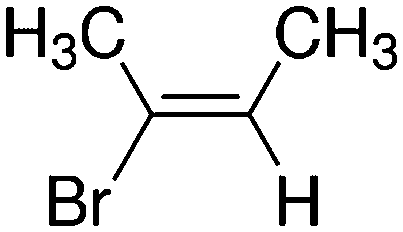
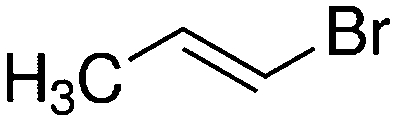
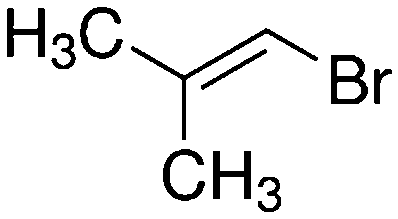
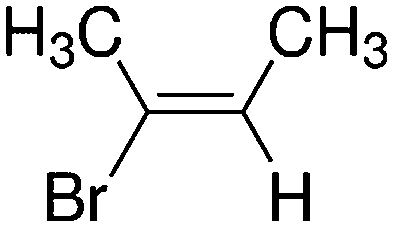
Despite extensive experimental and theoretical efforts over the last four decades, details of the photochemistry of vinyl bromide in the deep UV region remain ambiguous. Here, we report use of state selective velocity map imaging (VMI) of Br/Br* and HBr fragments from the photodissociation of three higher analogues of vinyl bromide, (E)-1-bromo-1-propene [EBP], 1-bromo-2-methyl-1-propene [BMP] and (E)-2-bromo-2-butene [EBB]. These molecules are illustrated in Table 1 and are chosen to distinguish 3-C and 4-C elimination pathways and their dynamical signatures; in the case of EBP, both pathways are open, but methyl groups in BMP and EBB block the 4-C and 3-C elimination routes respectively. In EBB, a second 4-C TS is also accessible, as shown in Table 1, and we distinguish it by the label 4′-C. We adopt an abbreviated notation throughout of BMP-3, EBB-44 and EBP-34 that identifies both the molecule and the types of TS available to it for HBr elimination.
| Parent molecule | Name | 3-Centre path | 4-Centre path | 4′-Centre path |
|---|---|---|---|---|
| a The 5-C channel is ignored because of a higher activation energy (see Section 4.1). b Both 3-C and 4-C channels are available to the E-isomer, whereas cis–trans isomerization to the Z-isomer restricts the HBr elimination to the 3-C channel. c Both 4-C and 4′-C channels are available to the Z-isomer, but cis–trans isomerization restricts HBr elimination to the 4′-C channel in the E-isomer. | ||||
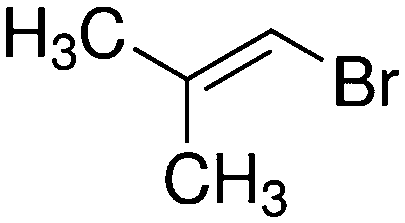
|
BMP-3a |
|
||
|
EBP-34b |
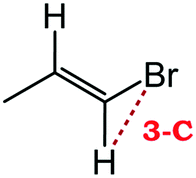
|
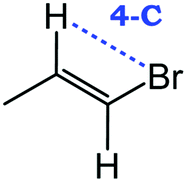
|
|
|
EBB-44c |
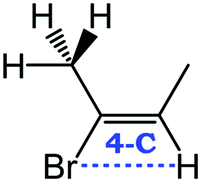
|
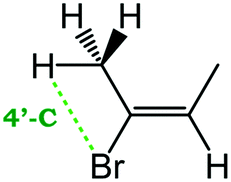
|
|
Our experimental measurements obtain total kinetic energy release (TKER) distributions for the two partner photofragments from velocity images of either atomic Br and Br* or molecular HBr, the latter with vibrational and rotational quantum state resolution. Unimolecular rate coefficients obtained from Rice–Ramsperger–Kassel–Marcus (RRKM) theory provide an estimate of the branching between 3-C and 4-C HBr elimination channels. We contrast the experimental TKER distributions for signatures of 3-C vs. 4-C elimination pathways, and compare with predictions from models that assume a statistical distribution of the total available internal energy among all degrees of freedom.
2. Experimental
The velocity map images of both atomic and molecular photofragments from (E)-1-bromo-1-propene, 1-bromo-2-methyl-1-propene and (E)-2-bromo-2-butene were recorded on a VMI machine incorporating a time-of-flight mass spectrometer (TOF-MS). The experimental setup has been reported in detail elsewhere.39 All three liquid precursors, EBP (containing copper as stabiliser, 99%), BMP (98%) and EBB (96%) were obtained from Sigma-Aldrich and further purified by the freeze–pump–thaw method. The vapour pressures were used to prepare ∼7–20% gas samples40 diluted in 1.5 bar of argon. A molecular beam was generated by expansion of the dilute gas samples from a Parker Hannifin (Series 9) pulsed nozzle at a repetition rate of 10 Hz. The resulting expansion was further collimated coaxially with the time-of-flight axis by a 1 mm diameter skimmer. The vacuum in the chamber was maintained below 4 × 10−7 Torr during Br/Br* photofragment measurements and below 9 × 10−7 Torr during experiments probing HBr photofragments.The parent molecules were photolysed at a wavelength of 193 nm using the output from a pulsed ArF excimer laser (Lambda Physik) with a pulse rate of 10 Hz (≤10 mJ per pulse). The unpolarized pump light was focused by a 25 cm focal length lens into the chamber approximately 10 mm in front of the repeller plate of the TOF MS. Br and Br* were detected using (2 + 1) resonance enhanced multiphoton ionization (REMPI) at 266.6 nm (4p 2P3/2 → 5p 4P3/2) and 266.67 nm (4p 2P1/2 → 5p 4S3/2) respectively. HBr (v = 0–2) was ionized using (2 + 1) REMPI, two-photon resonant with the F1Δ ← X1Σ+ transition. HBr (v = 0) was also probed by (2 + 1) REMPI via the g3Σ−(0+) state. The probe laser setup has been discussed in detail previously.40 A time delay of 10–20 ns was set between pump and probe pulses to minimize unwanted background signals. The typical probe laser energy was <1 mJ per pulse during Br/Br* imaging and ∼2.0 mJ per pulse during HBr imaging. The Doppler-broadened absorption lines of both Br/Br* and HBr necessitated a scan of the probe wavelength range to image all photofragments with the same efficiency. The two-colour signals were recorded on a shot-to-shot basis to subtract probe-laser-only signals. Some pump-laser-only signals in the Br/Br* image were incorporated in the analysis and showed different kinetic energy distributions from the two-colour signals.
The VMI ion optics were configured for DC-slice velocity-map imaging of the Br+ or HBr+ ions. Images were accumulated for m/z = 79 (79Br+) and 82 (H81Br+) ions and converted to radial velocity distributions using pixel-to-speed conversion factors obtained by multi-photon excitation of O2 at 224.999 nm,41 Cl atoms from the 355 nm photolysis of Cl2, and Br atoms from the 532 nm photolysis of Br2. The degree of slice imaging depends on the recoil velocities of the photofragments, with only partial slicing of the Newton sphere for slow Br or HBr. A partial slicing analysis method was therefore used to extract the velocity distributions from the unprocessed radial distributions.42
3. Calculations
Calculations of the optimized structures of reactants, products and transition states used unrestricted Møller–Plesset second-order perturbation theory (MP2). Dunning type aug-cc-pVDZ basis sets were placed on the C and H atoms,43 but only the valence electrons of the Br atoms were treated explicitly. Electrons in the lower lying shells of Br were taken into account by using an effective core potential (ECP).44 Single point energies were calculated at the MP2-optimized reactant, product and TS geometries at the coupled cluster singles and doubles with perturbative triples, CCSD(T) level using the aug-cc-pVDZ basis set with an ECP on the Br atoms. The TS geometries were also optimized, with basis set superposition error (BSSE) corrected by the Boys–Bernardi counterpoise method.45 The Gaussian09 suite of codes46 was used to carry out all of these ab initio electronic structure calculations. The vibrational frequencies of the reactants, products and transition states were calculated at the MP2/aug-cc-pVDZ level with an ECP on the Br atoms. BSSE corrected vibrational frequencies differed by less than 1% from the uncorrected values. The intrinsic reaction paths (IRCs)47,48 were calculated at the same level of theory both with and without BSSE correction.Unimolecular decomposition rates were determined using the RRKM formalism.49 The vibrational densities of states were calculated using the standard Beyer–Swinehart algorithm with 1 cm−1 vibrational energy bin size.49 The product HBr and Br translational energy distributions were modelled assuming a statistical distribution of the total available energy, corresponding to the difference between the photon energy and reaction energy, among all of the degrees of freedom (see Section 4.2 for details). Each simulation considered only a single (3-C, 4-C or 4′-C) reaction pathway.
4. Results and discussion
Velocity map images were accumulated for both atomic (Br/Br*) and molecular (HBr) elimination channels from the 193 nm photolysis of the three vinylic bromide compounds EBP-34, BMP-3 and EBB-44. The analysis of the Br/Br* images reveals information about excited electronic state dissociation dynamics, whereas the HBr products arise from competing elimination pathways on the ground electronic state. We focus here on the HBr elimination mechanisms, with the results for atomic (Br/Br*) dissociation presented and discussed in the ESI† (Section S.2). The dynamics of the C–Br bond cleavage in alkyl-substituted vinyl bromides and the subsequent dissociation of internally hot radical co-fragments were the subject of prior investigations by Butler and coworkers.50,51The initially excited πCCπCC* state and the nearby πCCσCBr*, nXσCBr* states do not asymptotically correlate to HBr in its electronic ground state.22 However, HBr elimination can take place from the ground state of each of the parent molecules and is associated with a large reaction barrier. This elimination can occur through a number of transition states in the molecules of interest, as shown in Table 1, producing different organic fragments. For vinyl bromide, the 3-C TS to HBr elimination makes the vinylidene diradical whereas acetylene is the product from the 4-C TS pathway. Most previous studies argued for vinylidene as the major elimination cofragment of HX from 193 nm photolysis of vinyl chloride and vinyl bromide.25,28,35,37,38,52 Two of the three molecules examined here were selected because methyl substitution at specific sites of the CC–Br moiety blocks either the 3-C or 4-C HBr elimination pathways. If distinct dynamical signatures derive from the two different types of TS, they should be evident in comparisons of asymptotic measurements of HBr from these two precursors. The third molecule studied has both 3-C and 4-C HBr loss pathways available, and the balance of competition between the two pathways may be resolvable from velocity map images of the product HBr.
4.1 Transition state geometries and energies
The different thermochemistries of all possible elimination reactions were deduced from the optimized structures of reactants and products using the calculation methods described in Section 3. Table 2 summarizes the reaction enthalpies and the total available energies for different reaction pathways after 193 nm photoexcitation.| Molecule | TS | Photon energy (kJ mol−1) | Reaction energy (kJ mol−1) | Transition state energy (kJ mol−1) | Available energy (kJ mol−1) |
|---|---|---|---|---|---|
|
3-C | 620 | 265 | 264 | 355 |
| 4-C | 620 | 66 | 286 | 554 | |
|
3-C | 620 | 297 | 277 | 323 |
| 5-C | 620 | 205 | — | 415 | |
|
4-C | 620 | 116 | 285 | 504 |
| 4′-C | 620 | 147 | 285 | 473 | |
The relative energies of different reactants, products, and transition states obtained from our calculations are presented diagrammatically in Fig. 2. There is only modest exit-channel energy release from the TS for the 3-C elimination, and the TS geometry is close to the equilibrium geometry of the products. The C–Br, C–H and H–Br bond distances in the optimized TS geometries are presented in Table 3. In the 3-C TS, the H and Br atoms both move away from the C-atom, and the H moves toward the Br atom along a mode with imaginary frequency, whereas the structure of the remaining part is very close to the substituted vinylidene geometry of the product. The H–Br bond distance (1.48–1.51 Å) is ∼0.1 Å longer than the equilibrium HBr molecule bond distance of 1.41 Å.53
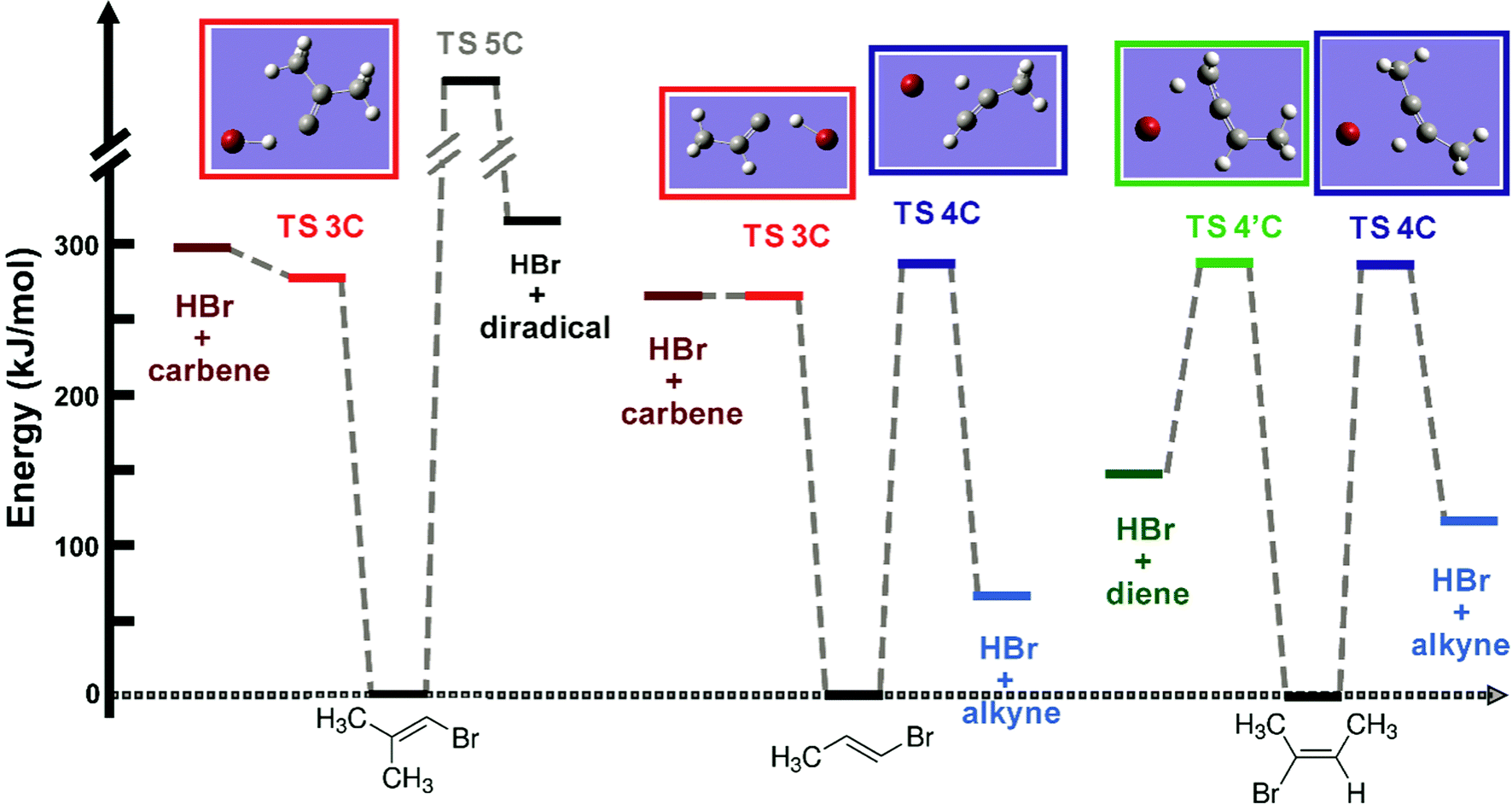 | ||
| Fig. 2 Zero-point-energy corrected CCSD(T)/aug-cc-VDZ//MP2/aug-cc-pVDZ relative energies of different reactants, transition states and products. The TS energies are corrected for BSSE. The optimized transition state structures are presented in the boxes at the top of the figure. The energy of the 5-C TS was not determined in our calculations, but is deduced to be >330 kJ mol−1. | ||
| Molecule | Mechanism | r(H–Br)/Å | r(H–C)/Å | r(C–Br)/Å |
|---|---|---|---|---|
| a The equilibrium bond lengths are r(H–Br) = 1.41 Å in HBr products, and r(H–C) = 1.09 Å and r(C–Br) = 1.90 Å for typical bonds to an sp2-hybridized C atom. | ||||
|
3-C | 1.51 | 1.56 | 2.95 |
| 4-C | 2.04 | 1.24 | 2.62 | |
|
3-C | 1.48 | 1.72 | 3.13 |
|
4-C | 1.98 | 1.23 | 2.83 |
| 4′-C | 2.07 | 1.21 | 2.76 | |
In contrast, there is a large reverse energy barrier of 150–200 kJ mol−1 for the 4-C process, which makes the 4-C elimination channel more exothermic than the 3-C pathway by ∼200 kJ mol−1. The 3-C and the 4-C transition state energies are very close to each other, but the 4-C TS structure is more tightly constrained: the ∠C–C–Br and ∠C–C–H bond angles contract from ∼120° to ∼90° and the CC bond length shortens. The 4′-C TS only features as a possible reaction pathway in EBB-44, and has energetics similar to the 4-C TS. In the 4′-C TS structure, the three carbon atoms are almost linear and the C–C–C bending motion is hindered. The ∠C–C–Br and ∠C–C–H bond angles also contract to ∼90°.
HBr elimination from the BMP-3 molecule can in principle occur via a 5-C transition state. A structure for this TS was not found, but it is deduced to lie higher in energy than the 3-C and two types of 4-C TSs because it leads to a diradical product located 330 kJ mol−1 above the ground state. The 5-C elimination pathway can therefore be ruled out as a significant HBr loss channel. We also considered, and ruled out, the possibility of migration of a methyl group across the CC bond in the internally hot vinyl bromide compounds. At the same level of theory as the calculations discussed above, the TS barrier heights for these migrations are estimated to be 344 kJ mol−1 and 312 kJ mol−1 for EBP-34 and BMP-3 respectively. The corresponding RRKM rate coefficient for methyl migration within internally hot EBP-34 is two orders of magnitude smaller than the 4-C and 3-C HBr elimination rate coefficients. Similarly, isomeric scrambling of BMP-3 through methyl migration is 7–70 times slower than HBr elimination. Further details are provided in the ESI† (Section S.1).
4.2 Translational energy distributions
Velocity map images of HBr fragments from all three vinyl bromide derivatives were recorded state-selectively in different ro-vibrational levels (v = 0–2, J = 0–6). Representative raw images of HBr (v = 2, J = 1) fragments from all three parent molecules are shown in Fig. 3. The photolysis laser in our experiments was unpolarized, hence the angular distributions of HBr from all three molecules are isotropic. In previous studies of vinyl halides, the molecular fragments showed almost no anisotropy when photolysed by a polarized pump laser, with the exception of the work by Wodtke et al.27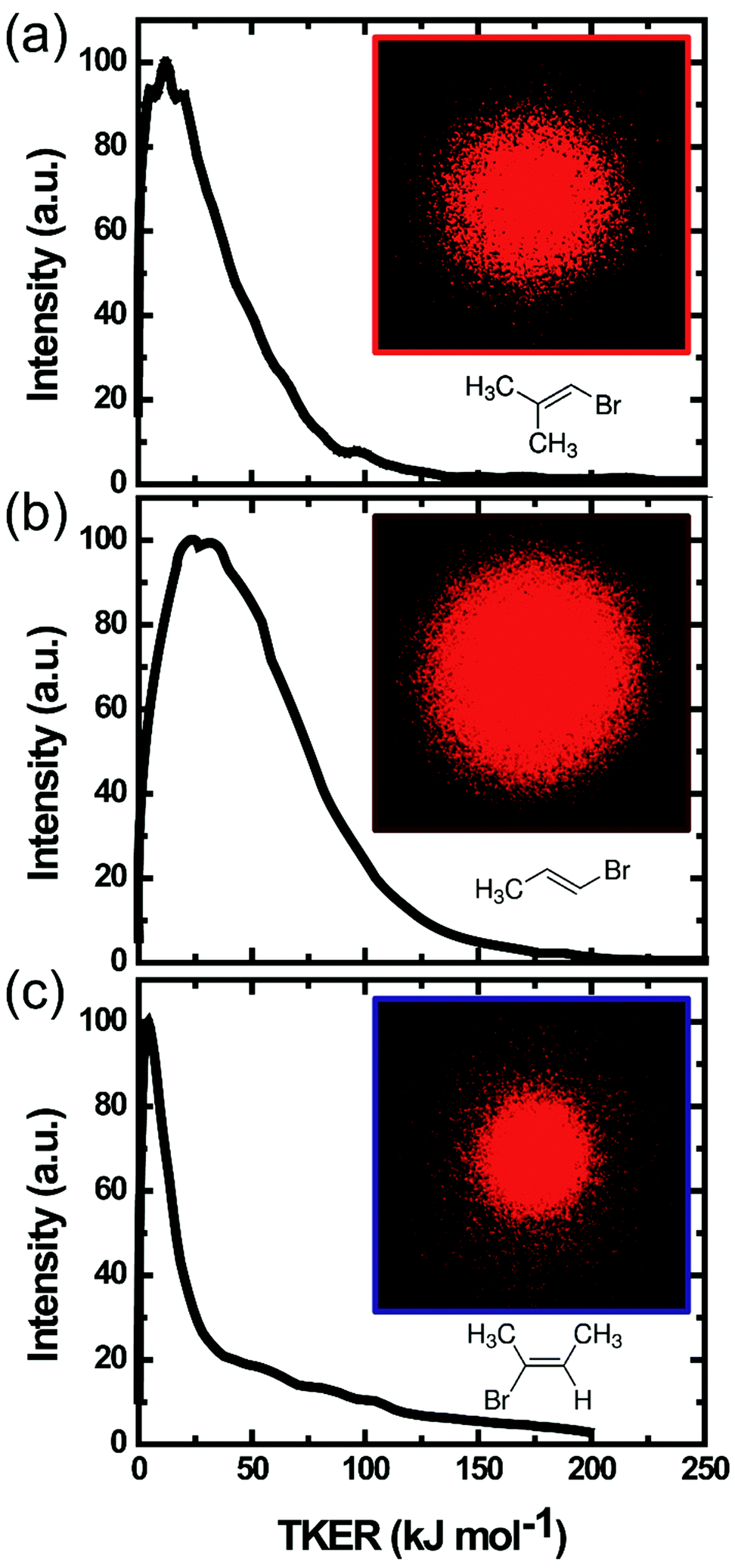 | ||
| Fig. 3 Velocity map images (insets) of HBr (v = 2, J = 1) fragments from 193 nm photolysis of three methyl-substituted vinyl bromides: (a) BMP-3, (b) EBP-34 and (c) EBB-44 and the derived TKER distributions, averaged over all recoil angles. | ||
Slow fragments appearing near the centres of the images were only partially sliced by our VMI instrument. There is effectively no slicing of the ion packet for fragments registering in the images at radii up to 6 pixels (or 0.1 kJ mol−1 TKER), and partial slicing out to TKERs of ∼10 kJ mol−1. Hence, a partial-slicing analysis algorithm was used to extract kinetic energy distributions from the raw images,42 but the parts of the distributions below 10 kJ mol−1 should be interpreted with some caution. The total kinetic energy distributions of HBr and the partner organic fragment, averaged over all recoil angles, are plotted in Fig. 3. All three molecules give products with low TKER compared to the total available energies listed in Table 2.
For a given parent molecule, the TKER distributions measured for HBr (v = 2) are identical irrespective of the probed rotational level (see ESI,† Section S.3.1). TKER distributions were also compared for HBr formed in vibrational levels v = 0–2, for which the maximum energy available to product translation differs by up to 80 kJ mol−1. Despite the changes in available energy, the TKER distributions are almost invariant with HBr vibrational excitation (see ESI,† Section S.3.2). As only a small fraction of the total available energy enters product translation, signatures of a small change in the maximum available energy will be hard to discern in the total kinetic energy distributions.
Previous studies suggested that the photoexcited parent molecules require only a few vibrational periods (e.g. 40 ± 10 fs for vinyl chloride)14 for internal conversion to the ground state, but may spend sufficient time (<2 ps) on the ground-state PES before HBr elimination for redistribution of internal energy among all degrees of freedom. The experimental TKER distributions are compared in Fig. 4 to a model prediction in which the difference between the photon and reaction energy is assumed to be distributed statistically among the translational, vibrational and rotational degrees of freedom of the product. Since each experiment probes a specific rovibrational state of the HBr, the available energy (Eavail) of the products was reduced by the corresponding vibrational and rotational energies of HBr in the simulations. The statistical product translation energy distribution was calculated using eqn (9.11) of ref. 49. This method approximates the TKER distribution as the product of the vibrational and rotational densities of states of the organic fragment at an internal energy of Eavail – TKER and the translational density of states of the products, which is proportional to TKER1/2.49,54 The vibrational density of states was computed using the Beyer–Swinehart algorithm, and the rotational term considered only the degrees of freedom of the polyatomic product because HBr was probed quantum-state specifically.
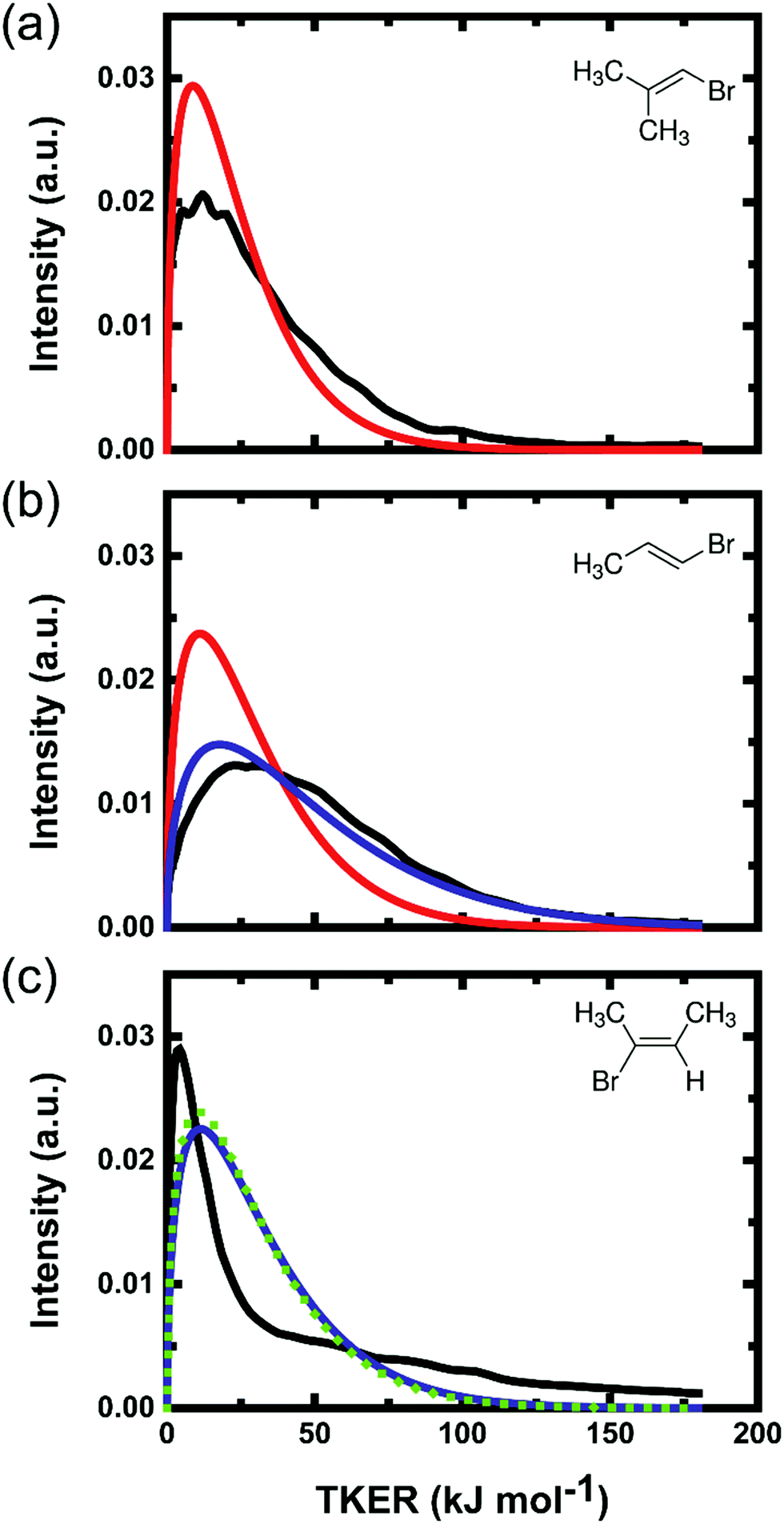 | ||
| Fig. 4 Total kinetic energy release distributions corresponding to formation of HBr (v = 2, J = 1) fragments from photolysis of (a) BMP-3, (b) EBP-34, and (c) EBB-44 at 193 nm. The black lines are experimental data after partial-slicing analysis of velocity map images. The coloured lines are statistical model predictions for the 3-C TS (solid red), 4-C TS (solid blue) and 4′-C TS (dotted green). The area under each curve is normalized to unity. | ||
Both the experimental and the calculated energy distributions show very low product total translational energy for all three systems. Low TKERs of the products of the 3-C elimination pathway can be understood from the modest energy difference between the 3-C TS and the separated products. The total available energy is much greater for products of the 4-C elimination, but TKERs are still low for this channel. Our calculations suggest a large geometrical change is required to go from the 4-C TS to the HBr and alkyne co-product ground states. Hence, we might expect that most of the available energy will go to the vibrational modes, and to some extent to the rotational modes of the products. TKER distributions of HBr (v = 0–2) fragments from the three different parent molecules are compared further in Section 4.4.
4.3 Internal energy distributions
Dai and coworkers reported IR emission experiments that observed HBr fragments from vinyl bromide photolysis at 193 nm with vibrational excitation up to vmax = 6.28 In the current study, our (2 + 1) REMPI detection scheme observes HBr (v = 0–2) fragments, and we cannot reliably deduce vibrational branching ratios or compare HBr (v) yields from different parent molecules. However, a qualitative comparison of uncorrected image intensities suggests that more HBr fragments populate v = 1 and v = 2 vibrational levels than v = 0 for all three vinyl bromide derivatives. In a second IR emission study of vinyl bromide photodissociation, Lin et al. reported a branching ratio of HBr (v = 1)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HBr (v = 2) of 0.30: 0.30 that is consistent with our qualitative deduction.37
:HBr (v = 2) of 0.30: 0.30 that is consistent with our qualitative deduction.37
REMPI spectra of HBr (v = 0–2) from all three precursors were acquired to estimate the rotational temperatures of these molecular fragments. HBr rotational levels up to J = 7 for BMP-3 and EBP-34 and J = 5 for EBB-44 were seen in the F1Δ–X1Σ+ REMPI spectra, but rotational levels up to J = 15 were observed for HBr (v = 0) using the alternative g3Σ+(0+)–X1Σ+(2 + 1) REMPI scheme. Fig. 5 shows a representative g–X REMPI spectrum of HBr (v = 0) from BMP-3. We focus here on the analysis of REMPI spectra (from the F–X transition) and velocity map images for HBr (v = 2) products, and further analysis of the additional REMPI spectra from the g–X transition is included in the ESI† (see Section S.4).
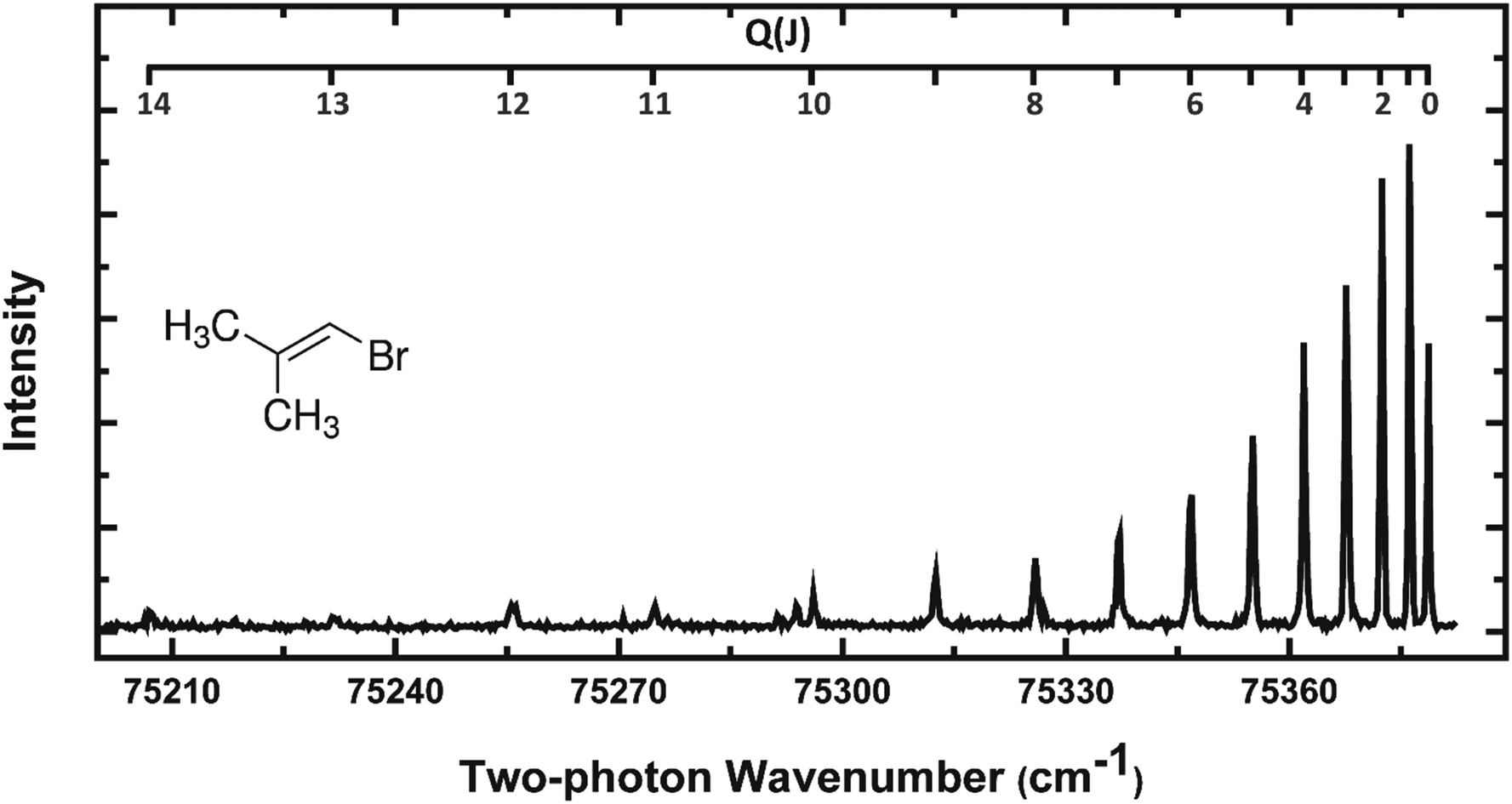 | ||
| Fig. 5 The Q branch region of the g3Σ−(0+) ← X1Σ+(2 + 1) REMPI spectrum of HBr (v = 0) from photodissociation of BMP-3 at 193 nm. | ||
Perturbations or predissociation of the Rydberg states reached by the two-photon excitation step in (2 + 1) REMPI spectroscopy of hydrogen halides restrict the rotational levels that can be probed in this way.55 Within our restricted range of observation, the rotational temperatures of the HBr (v = 0–2) products from all three parent molecules are ∼100 K or lower, and decrease from v = 0 to v = 2. We are unable to determine experimental correction factors linking REMPI line intensities to relative populations of ro-vibrational levels for vibrationally excited HBr, hence our analysis of the experimental line intensities gives only an estimate of rotational temperatures. Previous studies of vinyl bromide photolysis by Lin et al. revealed a significant portion of the HBr products with considerably higher rotational excitation than is evident in Fig. 5.37 The portions of the rotational distributions that we observe appear to be only a part of the whole picture of the rotational excitation of HX from vinyl halide photolysis. For example, He et al. measured rotational temperatures of 2000 K and 1400 K for HCl (v > 0) from vinyl chloride and dichloroethylene using HCl REMPI spectroscopy.56 Furthermore, Lin et al. obtained average rotational temperatures of 3400 K and 5300 K for v = 1–5 of HBr from vinyl bromide and HCl from vinyl chloride respectively from IR emission spectra.37,52 Our measurements are better suited to interrogation of the low-J products which are difficult to observe under nascent conditions in the IR emission experiments because of contamination by collisional cooling. Rotational temperatures of HBr fragments from the three different vinylic bromides are compared in more detail in Section 4.4.
4.4 Comparison of the HBr elimination pathways in BMP-3, EBB-44 and EBP-34
The elimination of HBr from BMP-3 through a 3-C transition state produces a vinylidene-like isobutylidene diradical which is expected to be short lived because the lifetime of vinylidene is 40–200 fs with respect to isomerization to highly excited vibrational levels of acetylene. This isomerization timescale is shorter than the timescale for the ground-state vinyl bromide fragmentation.34 Hence, Huang et al. argued for a concerted dissociation and isomerization process in which a fraction of the energy released by the vinylidene to acetylene conversion can be transferred into translation of the two separating fragments, giving a component of non-statistical behaviour.22 Our measured TKER distribution does not show any clear signatures of this type of concerted dissociation dynamics.
Mechanistic insights for the 3-C HBr elimination can be drawn from the computed transition state frequencies and the types of motions with which they are associated. The disappearing vibrational mode corresponds to the H atom moving towards the Br atom, and the bond length of 1.48 Å between the departing H and Br atoms at the 3-C TS is slightly longer than the equilibrium HBr bond length (1.414 Å).53 The optimized structure of the carbon frame is close to the equilibrium structure of the isobutylidene product. These geometric parameters, coupled with the dissociation of energized molecules on the ground electronic state point towards the isobutylidene product from the 3-C elimination, and perhaps also the HBr, showing a statistical internal energy distribution. In support of this reasoning, QCT calculations predict a Boltzmann-like vibrational population distribution of HCl from vinyl chloride via the 3-C process,25 but our qualitative interpretation of HBr vibrational level populations (Section 4.3) argues against a Boltzmann distribution over vibrational levels in HBr. Energy conservation arguments demonstrate that the organic fragments partnering HBr (v = 2) must be vibrationally and rotationally very hot, with ∼250 kJ mol−1 of internal energy relative to the isobutylidene ground state, and Miller et al. showed that the organic fragments from 2-bromo-1-butene photolysis are prone to secondary dissociation.51
The observable HBr (v = 0–2) fragments from BMP-3 photodissociation are rotationally cold, and over the range of J levels accessible to our REMPI measurements (Jmax = 7 for v = 1 and v = 2 and Jmax = 15 for v = 0), are well described by a single temperature for a given vibrational level. The estimated rotational temperatures (obtained from the F–X REMPI spectra) from linear fits to Boltzmann plots are 155 ± 7 K and 121 ± 13 K for v = 0 and v = 2 respectively.
Our electronic structure calculations show that the H–Br distances in the 4-C and 4′-C TSs are 1.98 and 2.07 Å respectively, which are ∼0.6 Å longer than the equilibrium HBr bond length. Decomposition of the TS will therefore promote vibrational excitation of the HBr product. In addition, deformational motions of the carbon framework are predicted during both 4-C eliminations. Hence, we expected vibrational excitation of both products of the 4-C and 4′-C elimination, and dynamical calculations predict an inverted vibrational population of HBr from 4-C elimination in vinyl bromide.38
The rotational temperatures of HBr fragments from EBB-44 obtained from linear fits to Boltzmann plots of line intensities from F–X REMPI spectra are 124 ± 6 K and 62 ± 1 K for v = 0 and v = 2 vibrational states respectively. In contrast, Lin et al. reported bimodal rotational distributions of HBr (v < 6) from vinyl bromide photolysis. These authors argued for rotationally hotter HBr fragments from the 3-C TS,37 and comparison of the rotational temperatures we measure from EBB-44 and BMP-3 supports this suggestion, at least for the low J region of the distribution we can observe. QCT calculations have not been able to capture the bimodal behavior in the overall rotational population distributions from VBr photolysis, and instead predict higher rotational excitation of HBr from the 4-C channel.38 Indeed, all HBr fragments which are generated via the 4-C channel were predicted to have rotational quantum numbers J ≥ 10.38 In contrast, HBr with rotational quantum numbers only up to J = 4 were detected in our study of EBB-44.
4.4.2.1 Comparison of 4-C and 4′-C pathways. The question remains of the relative importance of 4-C and 4′-C TS pathways in the elimination of HBr from EBB-44. Decomposition of the bimodal TKER distribution of Fig. 4(c) indicates that the branching ratio of higher to lower TKER components from EBB-44 is 0.63
:0.37. We have carried out RRKM calculations of microcanonical rate coefficients for the two pathways, based on the parent molecule and TS structure calculations described in Section 3. The results are presented in Table 4 and the ratio of calculated rate coefficients suggest a branching ratio of 0.55:0.45 for HBr elimination from EBB-44 via 4-C and 4′-C pathways. Parsons et al. attributed a similar preference in HCl elimination from 2-chloropropene to the difference in the ease of CCC bending motion within the 4-C and 4′-C TSs.57 QCT calculations suggested HCl from 2-chloropropene should be translationally hotter by ∼10 kJ mol−1 when formed via the 4-C than the 4′-C TS,36 and that 4-C elimination of HCl from vinyl chloride and 2-chloropropene gives average TKERs of ∼83 kJ mol−1 and a distribution extending up to 230 kJ mol−1.35,36 We therefore propose that the higher TKER fragments from EBB-44 evident in Fig. 4(c) come from the 4-C pathway whereas the lower TKER fragments are mostly from the 4′-C elimination, and that the latter pathway is better described by the statistical model of energy disposal.
| Molecule | Mechanism | RRKM rate coefficient/109 s−1 |
|---|---|---|
|
3-C | 264 |
| 4-C | 46.8 | |
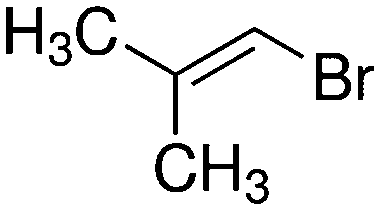
|
3-C | 3.91 |
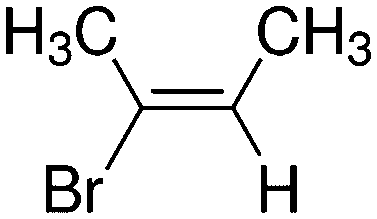
|
4-C | 0.24 |
| 4'-C | 0.20 | |
Intrinsic reaction coordinate scans establish which internal coordinates change during the course of the elimination reaction and how the internal energy varies along the path to products. Calculated IRC energies are shown in Fig. 6 for the 4-C and 4′-C elimination mechanisms available to EBB-44. The post-TS IRC paths for both elimination mechanisms can be divided into two distinct segments. Firstly, the H atom moves towards the Br atom, and then the HBr moiety departs from the organic fragment. The 4-C and 4′-C TSs are close in energy and both are bottle-necks along the reaction path. However, the 4′-C TS imposes somewhat tighter constraints on the carbon framework than the 4-C TS, hence the calculated unimolecular decomposition rate is slightly higher for the 4-C channel.
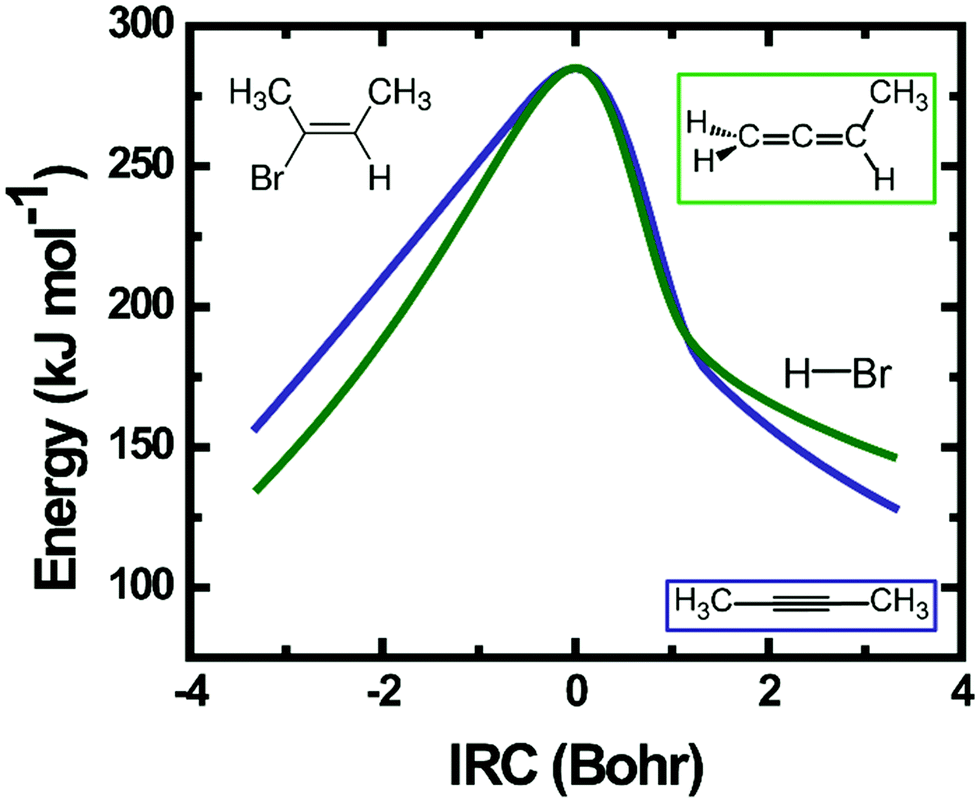 | ||
| Fig. 6 Computed intrinsic reaction coordinate energies for elimination of HBr from EBB-44. The blue curve represents the reaction path via the 4-C TS, and the green curve is for the 4′-C TS pathway. The procedure for the calculation is described in the main text. | ||
Fig. 7 compares a rotational Boltzmann plot for HBr (v = 2, J) photoproducts from EBP-34 with those for BMP-3 and EBB-44. The rotational excitation of HBr (v = 2) from EBP-34 is similar to that of BMP-3 but HBr (v = 2) from EBB-44 is rotationally colder. HBr (v = 2) is observed up to rotational level J = 7 from BMP-3 and EBP-34, whereas the highest level detected from EBB-44 is J = 4. Linear fits to the rotational Boltzmann plots for HBr (v = 0, 2) from EBP-34 give rotational temperatures of 214 ± 16 K and 140 ± 33 K for v = 0 and v = 2 vibrational states. The corresponding estimated rotational temperatures are 121 ± 13 K and 62 ± 1 K for HBr (v = 2) from BMP-3 and EBB-44 respectively, which suggest that the 3-C TSs give slightly hotter rotational population distributions compared to 4-C TSs in this low J region. The greater similarity between the EBP-34 and BMP-3 than EBB-44 rotational distributions for HBr (v = 2) products suggests that the 3-C elimination pathway in EBP-34 is the more important for production of HBr in this vibrational level.
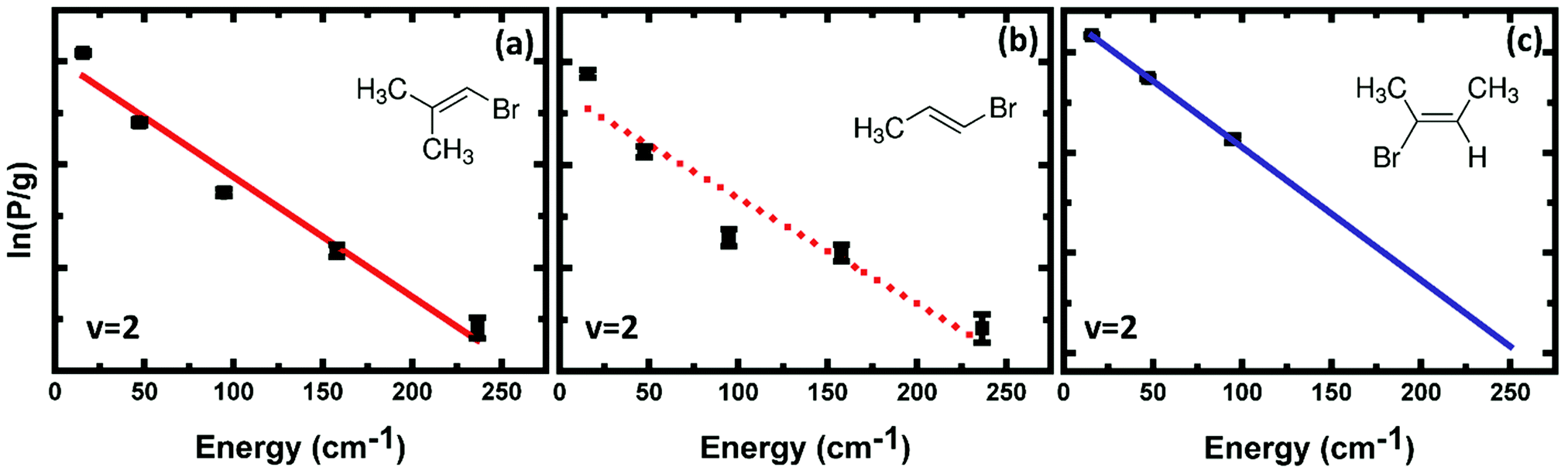 | ||
| Fig. 7 Boltzmann plots of the rotational population of HBr (v = 2) from the photodissociation of (a) BMP-3, (b) EBP-34, and (c) EBB-44 against energy of the HBr (v = 2) rotational level probed. The population intensities (P) are the integrated areas of individual peaks in the REMPI spectrum without any correction for the REMPI line strengths. The degeneracies of the rotational levels are denoted by g. The straight lines (solid, red), (dotted, red) and (solid, blue) are linear fits. | ||
4.4.3.1 Comparison of 3-C and 4-C pathways. BMP-3 dissociation is restricted to a 3-C TS and ∼10% of the total available energy is observed in translational degrees of freedom of the fragments. Elimination of HBr from EBB-44 occurs through competing 4-C and 4′-C TSs and is ∼180 kJ mol−1 more exothermic than the 3-C elimination from BMP-3. The average TKER of the faster component we earlier tentatively assigned to the 4-C TS corresponds to ∼18% of the total available energy. EBP-34 has both 3-C and 4-C pathways open to HBr elimination, and the average TKER of the products corresponds to ∼14% of the total available energy for the 3-C pathway, or ∼9% of that for the 4-C pathway. Only a small fraction of the HBr products have TKERs approaching the average value of 92 kJ mol−1 assigned to the 4-C elimination pathway in EBB-44. The RRKM rate coefficients reported in Table 4 suggest that HBr elimination from EBP-34 via the 3-C TS is ∼6 times faster than via the 4-C TS. On the basis of this evidence from both the TKER measurements and the RRKM calculations, we therefore propose that the 3-C pathway is the more significant HBr elimination channel from EBP-34. This proposition is supported by the evidence from REMPI spectra which show close correspondence of the rotational temperatures of HBr (v = 2) products from EBP-34 and BMP-3, and discrepancies in the case of EBB-44.
Calculated IRC energies for the 3-C and 4-C elimination reactions from EBP-34 are shown in Fig. 8. In the case of the 3-C elimination, the HBr molecule moves away from the propylidene fragment on a flat potential energy surface, the carbon frame structure is propylidene-like and the H atom moves toward the Br atom with only weak geometric constraints. In contrast, the 4-C TS geometry is more tightly constrained and the elimination reaction coordinate has similar topography to that computed for EBB-44 (Fig. 6). The loose transition state for the 3-C pathway favours this route to HBr elimination, as is reflected in RRKM calculations of unimolecular dissociation rate coefficients. The predicted lifetime of ground-electronic state EBP-34 with an internal energy corresponding to a 193 nm photon is only a few ps, whereas the internally excited EBB-44 is calculated to have a lifetime to HBr elimination of a few ns.
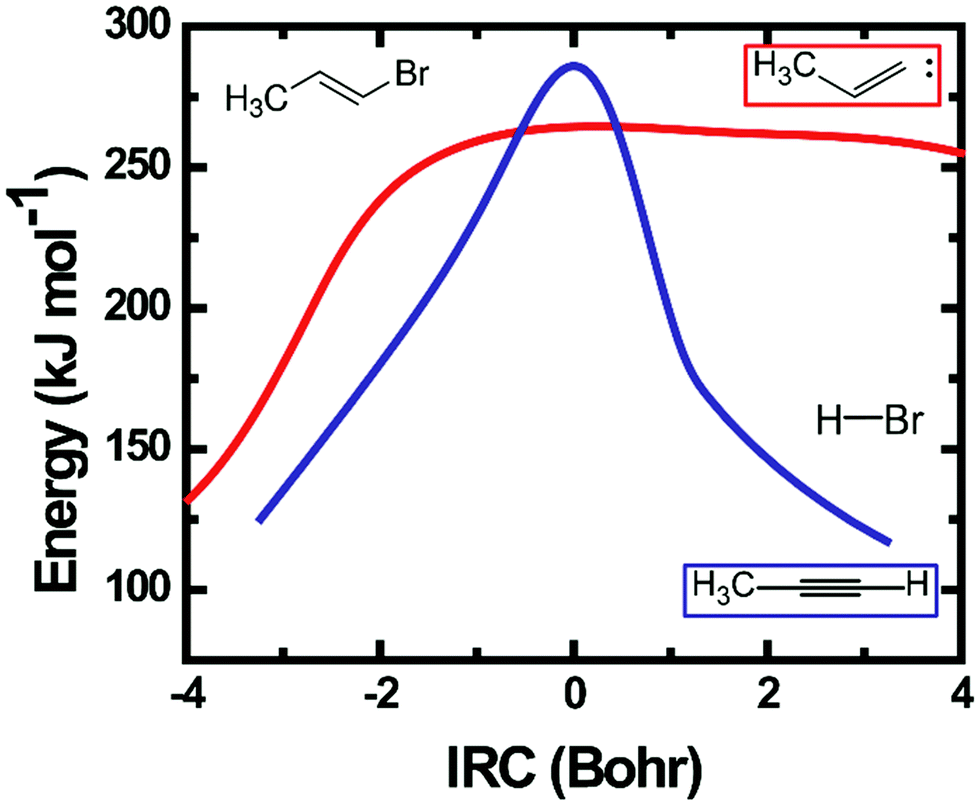 | ||
| Fig. 8 Computed intrinsic reaction coordinate energies for HBr elimination from EBP-34. The red curve represents the reaction path via a 3-centre transition state and the blue curve is for the 4-centre TS pathway. The calculations were performed using methods described in the main text. | ||
5. Conclusion
Velocity map images of HBr (v = 0 = 2, J = 0–7) from the 193 nm photolysis of three methyl-substituted vinyl bromides reveal total kinetic energy releases of this and its partner photofragment that are only a modest fraction of the total available energy. The elimination of HBr occurs from internally excited parent molecules after internal conversion to their ground electronic states, and some of the TKER distributions are well-described by statistical models of energy partitioning among the fragment degrees of freedom. The methyl substituents on two of the compounds studied block either a 3-centre or a 4-centre transition state for HBr elimination from contributing to the observed velocity images. Comparisons suggest that the product TKER is lower for the 3-C pathway than for 4-C elimination. REMPI spectra reveal the rotational excitation of HBr photoproducts in different vibrational levels, and point to a higher rotational energy when the HBr forms through a 3-C transition state. A third methyl substituted vinyl bromide compound studied has both 3-C and 4-C transition state pathways open to the elimination of HBr, and evidence from TKER distributions, rotational distributions and RRKM calculations indicates that the 3-C pathway to HBr and a propylidene diradical is the more important of the two.Acknowledgements
This work was supported by EPSRC Programme Grant EP/L005913/1. S. P. gratefully acknowledges the University of Bristol for award of a postgraduate scholarship. We thank Prof. Jeremy Harvey (KU Leuven) and Mitchell Quinn (University of Bristol) for valuable discussions.References
- N. J. Turro, Modern Molecular Photochemistry, University Science Books, Sausalito, CA, 1991 Search PubMed.
- M. A. van der Horst and K. J. Hellingwerf, Acc. Chem. Res., 2004, 37, 13–20 CrossRef CAS PubMed.
- R. A. van Delden, M. K. J. ter Wiel, M. M. Pollard, J. Vicario, N. Koumura and B. L. Feringa, Nature, 2005, 437, 1337–1340 CrossRef CAS PubMed.
- J. J. Lin, C. C. Wang, Y. T. Lee and X. Yang, J. Chem. Phys., 2000, 113, 9668–9677 CrossRef CAS.
- M. Barbatti, J. Paier and H. Lischka, J. Chem. Phys., 2004, 121, 11614–11624 CrossRef CAS PubMed.
- M. J. Berry, J. Chem. Phys., 1974, 61, 3114–3143 CrossRef CAS.
- H. Katayanagi, N. Yonekuira and T. Suzuki, Chem. Phys., 1998, 231, 345–353 CrossRef CAS.
- Y. Huang, Y. Yang, G. He and R. J. Gordon, J. Chem. Phys., 1993, 99, 2752–2759 CrossRef CAS.
- K. H. Sze, C. E. Brion, A. Katrib and B. El-Issa, Chem. Phys., 1989, 137, 369–390 CrossRef CAS.
- J. Michl and V. Bonacic-Koutecky, Electronic Aspects of Organic Photochemistry, Wiley, New York, Chichester, Brisbane, Toronto, Singapore, 1990 Search PubMed.
- G. J. Mains, L. M. Raff and S. A. Abrash, J. Phys. Chem., 1995, 99, 3532–3539 CrossRef CAS.
- L. Freund and M. Klessinger, Int. J. Quantum Chem., 1998, 70, 1023–1028 CrossRef CAS.
- M. Ben-Nun, J. Quenneville and T. J. Martínez, J. Phys. Chem. A, 2000, 104, 5161–5175 CrossRef CAS.
- P. Farmanara, V. Stert and W. Radloff, Chem. Phys. Lett., 1998, 288, 518–522 CrossRef CAS.
- S. A. Abrash, R. W. Zehner, G. J. Mains and L. M. Raff, J. Phys. Chem., 1995, 99, 2959–2977 CrossRef CAS.
- P. Zou, K. E. Strecker, J. Ramirez-Serrano, L. E. Jusinski, C. A. Taatjes and D. L. Osborn, Phys. Chem. Chem. Phys., 2008, 10, 713–728 RSC.
- K. Sato, S. Tsunashima, T. Takayanagi, G. Fijisawa and A. Yokoyama, Chem. Phys. Lett., 1995, 242, 401–406 CrossRef CAS.
- J. Tu, J. J. Lin, Y. T. Lee and X. Yang, J. Chem. Phys., 2002, 116, 6982–6989 CrossRef CAS.
- C. Wittig, I. Nadler, H. Reisler, M. Noble, J. Catanzarite and G. Radhakrishnan, J. Chem. Phys., 1985, 83, 5581–5588 CrossRef CAS.
- P. T. A. Reilly, Y. Xie and R. J. Gordon, Chem. Phys. Lett., 1991, 178, 511–516 CrossRef CAS.
- Y. Huang, G. He, Y. Yang, S. Hashimoto and R. J. Gordon, Chem. Phys. Lett., 1994, 229, 621–627 CrossRef CAS.
- Y. Huang, Y.-A. Yang, G. He, S. Hashimoto and R. J. Gordon, J. Chem. Phys., 1995, 103, 5476–5487 CrossRef CAS.
- K. Tonokura, L. B. Daniels, T. Suzuki and K. Yamashita, J. Phys. Chem. A, 1997, 101, 7754–7764 CrossRef CAS.
- D. A. Blank, W. Sun, A. G. Suits, Y. T. Lee, S. W. North and G. E. Hall, J. Chem. Phys., 1998, 108, 5414–5425 CrossRef CAS.
- E. Martínez-Núñez, A. Fernández-Ramos, S. A. Vázquez, F. J. Aoiz and L. Bañares, J. Phys. Chem. A, 2003, 107, 7611–7618 CrossRef.
- K. Saito, T. Yokubo, T. Fuse, H. Tahara, O. Kondo, T. Higashihara and I. Murakami, Bull. Chem. Soc. Jpn., 1979, 52, 3507–3510 CrossRef CAS.
- A. M. Wodtke, E. J. Hintsa, J. Somorjai and Y. T. Lee, Isr. J. Chem., 1989, 29, 383–391 CrossRef CAS.
- D.-K. Liu, L. T. Letendre and H.-L. Dai, J. Chem. Phys., 2001, 115, 1734–1741 CrossRef CAS.
- G. R. Johnston and D. Price, Dyn. Mass Spectrom., 1973, 3, 183 CAS.
- S. A. Abrash, C. M. Carr, M. T. McMahon and R. W. Zehner, J. Phys. Chem., 1994, 98, 11909–11917 CrossRef CAS.
- J. R. Cao, J. M. Zhang, X. Zhong, Y. H. Huang, W. Q. Fang, X. J. Wu and Q. H. Zhu, Chem. Phys., 1989, 138, 377–382 CrossRef CAS.
- H. E. Hunziker, H. Kneppe, A. D. McLean, P. Siegbahn and H. R. Wendt, Can. J. Chem., 1983, 61, 993–995 CrossRef CAS.
- S. Carter, I. M. Mills and J. N. Murrell, Mol. Phys., 2006, 41, 191–203 CrossRef.
- K. M. Ervin, J. Ho and W. C. Lineberger, J. Chem. Phys., 1989, 91, 5974–5992 CrossRef CAS.
- E. Martínez-Núñez, S. A. Vázquez, F. J. Aoiz, L. Bañares and J. F. Castillo, Chem. Phys. Lett., 2004, 386, 225–232 CrossRef.
- C.-M. Chang, Y.-H. Huang, S.-Y. Liu, Y.-P. Lee, M. Pombar-Pérez, E. Martínez-Núñez and S. A. Vázquez, J. Chem. Phys., 2008, 129, 224301 CrossRef PubMed.
- S.-R. Lin, S.-C. Lin, Y.-C. Lee, Y.-C. Chou, I.-C. Chen and Y.-P. Lee, J. Chem. Phys., 2001, 114, 7396–7406 CrossRef CAS.
- E. Martínez-Núñez and S. Vázquez, Chem. Phys. Lett., 2006, 425, 22–27 CrossRef.
- R. A. Rose, S. J. Greaves and A. J. Orr-Ewing, J. Chem. Phys., 2010, 132, 244312 CrossRef PubMed.
- S. Pandit, T. J. Preston, S. J. King, C. Vallance and A. J. Orr-Ewing, J. Chem. Phys., 2016, 144, 244312 CrossRef PubMed.
- D. H. Parker and A. T. J. B. Eppink, J. Chem. Phys., 1997, 107, 2357–2362 CrossRef CAS.
- A. V. Komissarov, M. P. Minitti, A. G. Suits and G. E. Hall, J. Chem. Phys., 2006, 124, 014303 CrossRef PubMed.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- K. A. Peterson, D. Figgen, E. Goll, H. Stoll and M. Dolg, J. Chem. Phys., 2003, 119, 11113–11123 CrossRef CAS.
- S. F. Boys and F. Bernardi, Mol. Phys., 2006, 19, 553–566 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, GAUSSIAN 09 (Revision E.01), Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- K. Fukui, J. Phys. Chem., 1970, 74, 4161–4163 CrossRef CAS.
- K. Fukui, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS.
- T. Baer and W. L. Hase, Unimolecular Reaction Dynamics, Oxford University Press, 1996 Search PubMed.
- M. L. Morton, J. L. Miller, L. J. Butler and F. Qi, J. Phys. Chem. A, 2002, 106, 10831–10842 CrossRef CAS.
- J. L. Miller, M. J. Krisch, L. J. Butler and J. Shu, J. Phys. Chem. A, 2005, 109, 4038–4048 CrossRef CAS PubMed.
- S.-R. Lin, S.-C. Lin, Y.-C. Lee, Y.-C. Chou, I.-C. Chen and Y.-P. Lee, J. Chem. Phys., 2001, 114, 160–168 CrossRef CAS.
- D. H. Rank, U. Fink and T. A. Wiggins, J. Mol. Spectrosc., 1965, 18, 170–183 CrossRef CAS.
- E. J. Feltham, R. H. Qadiri, E. E. H. Cottrill, P. A. Cook, J. P. Cole, G. G. Balint-Kurti and M. N. R. Ashfold, J. Chem. Phys., 2003, 119, 6017–6031 CrossRef CAS.
- S. Rudić, C. Murray, J. N. Harvey and A. J. Orr-Ewing, J. Chem. Phys., 2004, 120, 186–198 CrossRef PubMed.
- G. X. He, Y. A. Yang, Y. Huang and R. J. Gordon, J. Phys. Chem., 1993, 97, 2186–2193 CrossRef CAS.
- B. F. Parsons, L. J. Butler and B. Ruscic, Mol. Phys., 2002, 100, 865–874 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Calculations of rates of methyl migration in the energized, ground state molecules; VMI results and analysis of Br and Br* channels from 193 nm photolysis of the vinyl bromide derivatives; HBr v and J dependence of TKER distributions; results from HBr (v = 0) detection using the g–X REMPI transition. All experimental and computational data are archived in the University of Bristol's Research Data Storage Facility (DOI http://10.5523/bris.pmjkef9fnf6b194vbaeh3twq6). See DOI: 10.1039/c6cp05393a |
| This journal is © the Owner Societies 2016 |