Open-ended response theory with polarizable embedding: multiphoton absorption in biomolecular systems†
Received
30th July 2016
, Accepted 19th September 2016
First published on 19th September 2016
Abstract
We present the theory and implementation of an open-ended framework for electric response properties at the level of Hartree–Fock and Kohn–Sham density functional theory that includes effects from the molecular environment modeled by the polarizable embedding (PE) model. With this new state-of-the-art multiscale functionality, electric response properties to any order can be calculated for molecules embedded in polarizable atomistic molecular environments ranging from solvents to complex heterogeneous macromolecules such as proteins. In addition, environmental effects on multiphoton absorption (MPA) properties can be studied by evaluating single residues of the response functions. The PE approach includes mutual polarization effects between the quantum and classical parts of the system through induced dipoles that are determined self-consistently with respect to the electronic density. The applicability of our approach is demonstrated by calculating MPA strengths up to four-photon absorption for the green fluorescent protein. We show how the size of the quantum region, as well as the treatment of the border between the quantum and classical regions, is crucial in order to obtain reliable MPA predictions.
1 Introduction
Molecular properties are fundamental for understanding and interpreting experimental observations in chemistry and physics. Linear and nonlinear optical properties provide important information about the behavior of a molecule in the presence of electromagnetic fields, while properties involving nuclear motion can be used to determine molecular structure. Properties combining both of these categories enable prediction and interpretation of well-known spectroscopic phenomena such as infrared and Raman spectroscopy.1–6 One way of calculating molecular properties is by the use of quantum-chemical response theory.6–8 A recent formulation of response theory,9 and its subsequent implementation based on recursive routines,10 allows any response property that can be formulated as a derivative of the molecular quasienergy to be calculated at the Hartree–Fock (HF) and Kohn–Sham density-functional theory (KS-DFT) levels. The implementation has recently been extended to enable the calculation of single residues of response functions,11 which is needed for the evaluation of multiphoton absorption (MPA) strengths.
Before the developments of the present work, the open-ended response framework was formulated and implemented in a purely quantum-mechanical picture, which limited its applicability to relatively small or medium-sized molecular systems considered in vacuo. This treatment is often inadequate, since the majority of experiments are performed in an environment that can significantly perturb the properties of the molecule of interest. The absence of a proper description of the interaction between the system studied and its molecular environment can lead to discrepancies when comparing experimental observations to calculated results, ranging from minor deviations12–14 to qualitative disagreement.15–17 It is therefore important to develop models that can adequately describe the effects of the surrounding environment on the system being studied, e.g. embedding models, and a substantial amount of work has therefore been carried out in this area. We refer to a review by Gomes and Jacob for a recent account of embedding methods for electronic excitation processes.18 Classical embedding models can be roughly separated into continuum models,19 where the molecular environment is described by a structureless dielectric medium, and hybrid quantum mechanics/molecular mechanics (QM/MM) models,20,21 where the explicit molecular structure of the environment is retained. The effects from the environment on the quantum part are described by an embedding potential which is added to the electronic Hamiltonian.
In this work, we present an extension of the open-ended response theory approach to include environment effects based on the polarizable embedding (PE) model,22,23 which is a combined fragmentation and QM/MM-based embedding approach (see ref. 24 for a recent perspective). This model has been formulated with the particular aim of accurately modeling environment effects in calculations of local response properties. Importantly, polarization of the environment is included to give a more physically sound coupling between the quantum region, its molecular environment, and the external field(s).25,26 Several other polarizable QM/MM-based models have been formulated for optical property calculations (see e.g.ref. 27–32). The accuracy of the calculated results depends crucially on the ability of the embedding potential to reproduce the electrostatic potential. The embedding potential used in the PE model is therefore derived from ab initio calculations on the fragments defining the environment. This leads to high-quality electrostatic potentials, as has been demonstrated both for solvents22,33,34 and proteins.35 The open-ended formulation presented here is limited to electric response properties, i.e. polarizabilities, any order of hyperpolarizabilities, and MPA strengths through the single-residues functionality. We note, however, that although we will only discuss properties involving electric dipole perturbations, the formulation is also applicable to the calculation of properties that involve general electric multipole perturbations.
To demonstrate the applicability of this approach, we have calculated MPA properties of the green fluorescent protein (GFP). Multiphoton absorption is a nonlinear process, i.e. the signal depends nonlinearly on the intensity of the applied field, where the combined and simultaneous absorption of more than one photon leads to an excitation. Experimental observations have been reported up to five-photon absorption (5PA).36 Technological applications, at present mostly based on two-photon absorption (2PA), benefit from the long wavelength of the involved photons and the intrinsic three-dimensional resolution of the process. Applications include three-dimensional data storage,37,38 photodynamic therapy39 and photoactivated drug delivery.40 Another important application of MPA is multiphoton microscopy,41–44 which enables high-resolution in vivo imaging. Multiphoton microscopy takes advantage of the long wavelength of photons in two-41 and three-photon42,43 processes to penetrate deeper into biological tissue and to reduce photodamage. Fluorescent proteins such as GFP and its mutants are frequently used fluorophores in multiphoton microscopy43,44 owing to their high absorption cross sections,44 extensive palette of available absorption and fluorescence wavelengths,44,45 and ability to be genetically encoded to monitor in vivo gene expression and protein localization.45,46 Even though four-photon absorption (4PA) has been experimentally observed in GFP47 and red fluorescent proteins,48 two- and three-photon processes operate in the most advantageous spectral window for imaging in living tissue.43 Three-photon absorption (3PA) requires a high laser intensity, but is especially useful for fluorophores that absorb in the ultraviolet range rather than in the visible region.42,43 The calculation of 2PA in biological molecules was very recently reviewed by Salem, Gedik and Brown.49 It has been shown experimentally50,51 that the protein environment critically affects the 2PA signal of the chromophores of fluorescent proteins. Drobizhev et al. have shown that red fluorescent protein mutants with the same chromophore can have 2PA cross sections with differences as large as a factor of five.50 This has been explained by differences in the local effective field around the chromophore in the different mutants.51
The work presented here is an important step towards computational predictions of MPA in biological systems. Specifically, we have combined an open-ended density-matrix-based formulation of response theory with the PE model, which thus allows calculations of response and multiphoton absorption properties of large molecular systems. The theoretical details of the implementation are provided in Section 2. In addition, we have performed initial investigations of some important aspects related to multiscale modeling of multiphoton absorption. The results are presented and discussed in Section 4 with the computational details given in Section 3.
2 Theory
We begin this section by showing the foundations of the PE model22,23 and then move on to give a brief introduction to the open-ended response theory framework.9,10 Finally, we present the expressions that incorporate the contributions that arise from the introduction of a polarizable embedding potential in the open-ended formulation of response theory. The theory is formulated for hybrid KS-DFT and reduces trivially to HF theory. Furthermore, it is expressed in a density-matrix formulation in an atomic-orbital (AO) basis,9 the latter feature making the theory amenable to linear-scaling approaches for the quantum region.
2.1 Polarizable embedding
The PE model is an embedding method based on a combination of fragmentation and QM/MM methodologies that concentrates on an accurate inclusion of environment effects in the calculation of response properties. To achieve this, the total molecular system is partitioned into a quantum and a classical region, where the quantum region in this work is described by KS-DFT, and where the effects from the classical region are modeled through an embedding potential that consists of distributed multipole moments and polarizabilities, which describe the static and induced charge distributions, respectively. These embedding potential parameters are obtained by fragmenting the classical region into small computationally manageable fragments, from which distributed parameters are derived based on quantum-chemical calculations. This is a straightforward task for classical regions that consist of small molecules. However, for large molecules where the fragmentation makes it necessary to cut covalent bonds, we employ a fragmentation method that involves overlapping fragments.52,53
The combined PE-DFT energy, expressed in terms of the density matrix in the AO basis, can be partitioned as
| | | E(D) = EDFT(D) + EPE(D), | (1) |
where
EDFT(
D) is the KS energy of the polarized quantum region and
EPE(
D) is the PE energy. The KS energy is given by
| | 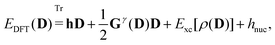 | (2) |
where
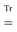
denotes tracing of each term on the right-hand side, the one-electron matrix
h describes the KS kinetic energy and the electron–nuclear attraction, the two-electron matrix
Gγ contains the electronic Coulomb and
γ-fractional exchange interactions,
Exc[
ρ(
D)] is the exchange–correlation energy written as a functional of the density which in turn depends on the density matrix, and
hnuc is the nuclear interaction energy. We will not go into further detail about these standard terms but will instead turn our attention to the PE energy.
The PE energy consists of the contributions due to interactions between the quantum region and both the permanent and induced charge distributions of the classical region, i.e.
| | | EPE(D) = EesPE(D) + EindPE(D), | (3) |
where
EesPE(
D) is the electrostatic energy from the interactions between the permanent multipole moments and the quantum-region electrons and nuclei, and where
EindPE(
D) is the induction energy associated with the polarization of the classical region.
The electrostatic interaction energy can be written compactly using a multi-index notation§
| | 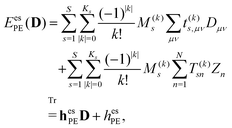 | (4) |
where the summation over
s runs over the
S sites in the classical region,
k is a multi-index,
Ks is the truncation level of the multipole expansion, and
M(k)s is a component of a |
k|th-order multipole moment located at site
s. The superscript (
k) notation is used to indicate order and Cartesian component. For example, in the case of multipoles,
k = (0, 0, 0) specifies a monopole (
i.e. charge),
k = (0, 1, 0) corresponds to the
y-component of a dipole, and so on for higher-order multipoles and different Cartesian components. Furthermore,
μ and
ν are indices of AO basis functions belonging to the quantum region,
Dμν is an element of the AO density matrix, and
t(k)s,μν is a one-electron interaction integral defined as
| | 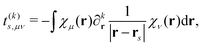 | (5) |
which involves partial derivatives of the potential operator. Finally,
eqn (4) also involves a sum over the
N nuclei in the quantum region and
Zn is thus the charge of the
nth nucleus. The interaction between multipoles and nuclei is described
via the so-called interaction tensors whose elements are defined by
| | 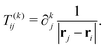 | (6) |
The first part of
eqn (4) is the contribution to the energy that is due to the interaction between the electrons in the quantum region and the multipoles in the classical region, and the second part of
eqn (4) is the energy contribution that is due to the interaction of the nuclei in the quantum region and the multipoles.
The polarization of the classical region gives rise to the induction energy contribution given by
| | 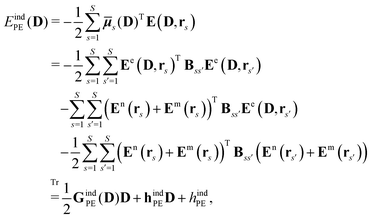 | (7) |
where
![[small mu, Greek, macron]](https://www.rsc.org/images/entities/b_i_char_e0cd.gif) s
s(
D) is an induced dipole located at site
s and
E(
D,
rs) is the electric field at this site generated by the quantum-region electrons and nuclei, and the permanent multipole moments in the classical region. The second equality is obtained by making use of the fact that the induced dipoles and electric fields can be separated into contributions stemming from the electrons, nuclei and permanent multipole moments (indicated by superscripts e, n, and m, respectively), and that an induced dipole can be expressed as
| | 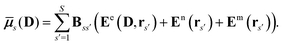 | (8) |
Eqn (8) introduces the
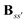
matrices that are 3 × 3 subblocks of the (symmetric) classical linear response matrix,
55| |  | (9) |
which contains the inverse of the electronic dipole–dipole polarizabilities in the diagonal blocks and second-order interaction tensors in the off-diagonal blocks (where the superscript (2) in this case denotes the tensor rank). The final equality in
eqn (7) is obtained by taking into consideration the fact that the electronic electric field is defined by
| | 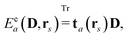 | (10) |
where the index
a indicates a Cartesian component of the field, and where
ta(
rs) is a one-electron matrix whose elements are defined as
| | 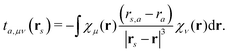 | (11) |
The PE contributions to the KS matrix are determined by minimizing the total energy in eqn (1) with respect to variations in the electronic density. Consequently, only terms that involve the electron density enter into the effective KS operator
| | | FPE = hesPE + GindPE(D) + hindPE. | (12) |
From this expression, it can be seen that the quadratic dependence of the induction energy in
eqn (7) on the electron density translates into a KS matrix contribution that takes a form similar to the Coulomb term in the vacuum KS matrix, but involving products of one-electron integrals rather than two-electron integrals.
2.2 Open-ended calculation of response functions
In this section, we give an introduction to the open-ended density-matrix-based formulation of response theory9 which has been implemented using recursive programming techniques.10 A more detailed presentation of this theory, without consideration of environment effects, can be found in previous papers.9,10
Our starting point is to express response functions as derivatives of a perturbation-strength-differentiated quasienergy derivative Lagrangian which is time-averaged and evaluated at zero perturbation strengths. A linear response function 〈〈A;B〉〉ωb can then be expressed as
| | 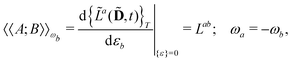 | (13) |
in an AO density-matrix parametrization. The superscripts (·)
abc… denote perturbation-strength differentiation with respect to perturbations
a,
b,
c… with associated frequencies
ωa,
ωb,
ωc…. A tilde above a symbol denotes evaluation at general perturbation strengths, while the same symbol without a tilde denotes evaluation at zero perturbation strength. The quasienergy derivative Lagrangian
![[L with combining tilde]](https://www.rsc.org/images/entities/i_char_004c_0303.gif) a
a(
![[D with combining tilde]](https://www.rsc.org/images/entities/b_char_0044_0303.gif)
,
t) can be expressed as
| | 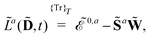 | (14) |
where
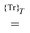
indicates that the trace and time-average of each term on the right-hand side are taken, and where we introduced the overlap matrix
![[S with combining tilde]](https://www.rsc.org/images/entities/b_char_0053_0303.gif)
and a generalization of the energy-weighted density matrix
![[W with combining tilde]](https://www.rsc.org/images/entities/b_char_0057_0303.gif)
defined as
| | 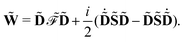 | (15) |
Eqn (14) and (15) introduce the generalized KS energy
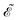
and KS matrix
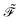
, given by
| | 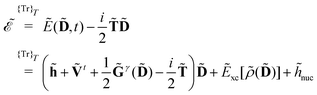 | (16) |
and
| | 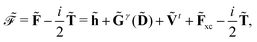 | (17) |
respectively. In
eqn (16) and (17), we introduced the anti-Hermitian time-differentiated overlap matrix
![[T with combining tilde]](https://www.rsc.org/images/entities/b_char_0054_0303.gif)
and the one-electron matrix describing the time-dependent perturbation
Ṽt. We remark that for the electric response properties, which are the topic of this work, contributions from
are all zero, and we have included these terms in the above expressions only for the sake of completeness.
Introducing the notation
| | 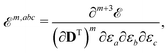 | (18) |
response functions in the
n + 1 rule formulation can now be obtained by straightforward differentiation of
eqn (14), so that, for example, a quadratic response function can be expressed as
| | 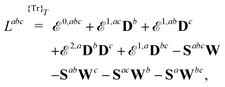 | (19) |
where the tracing of matrix products is for instance given by
| | 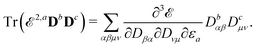 | (20) |
Formulations based on other choices than the
n + 1 rule, which can be any between and including the
n + 1 and 2
n + 1 rules,
56 are also possible by the introduction of Lagrangian multipliers. For the sake of compactness, we introduce the notation
and
where adjungation is done before any field strength differentiation. The idempotency condition for the density matrix can be expressed with the matrix
Y, so that
and we express the time-dependent SCF condition as the matrix
Z, defined as
| | 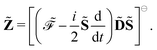 | (24) |
Introducing the Lagrangian multipliers
and
| | 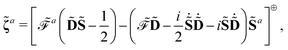 | (26) |
the general expression for an arbitrary response property can be written as
| | 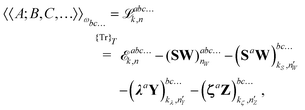 | (27) |
where the subscripts
k and
n are related to truncations of terms that involve the matrices
D,
F, and
S perturbed with respect to perturbation tuples including and not including perturbation
a, respectively. In particular, for the energy-type contribution
![[scr E, script letter E]](https://www.rsc.org/images/entities/char_e140.gif) abc…k,n
abc…k,n, all terms involving
D perturbed to higher order than
k for perturbation tuples involving perturbation
a, and to higher order than
n for perturbation tuples not involving perturbation
a can be removed from the expression. We refer to
ref. 9 for additional details about such truncations, but note that for a property that is formulated as an
N′th order derivative of the energy, it generally holds that
k +
n =
N − 1, where
k can be chosen as an integer in the interval
k ∈ [0, (
N − 1)/2] where integer division of (
N − 1)/2 is applied for even values of
N. We denote the choice between the various possible pairwise values of
k and
n at a given order
N as the (
k,
n) rule choice, constituting a generalization of the choice of truncation rule. In this generalization, the previously mentioned
n + 1 and 2
n + 1 rules are two of several possible choices, the number of such choices depending on the number of available choices of
k.
The evaluation of eqn (27) at a given (k, n) rule choice and the identification and calculation of the necessary perturbed D, F, and S matrices can be done by employing a series of algorithms that to a varying extent involve recursion. A comprehensive overview of these algorithms is given in ref. 10 and 11, and we restrict ourselves here to briefly introducing the elements that are relevant for the inclusion of the environment effects detailed in the next section.
We denote the density and KS matrices that are perturbed with respect to a perturbation tuple bN as 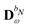 and
and 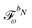 , respectively. The algorithmic approach rests on the separation of
, respectively. The algorithmic approach rests on the separation of 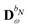 into particular and homogeneous parts
into particular and homogeneous parts 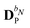 and
and 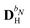 , respectively, as
, respectively, as
| | 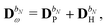 | (28) |
where
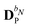
and
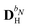
are determined at different stages during the execution of the algorithm. The perturbed KS matrix
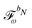
can be partitioned as
| | 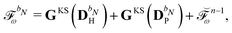 | (29) |
where
GKS contains the sum of two-electron and exchange–correlation contributions as
| | | GKS(M) = Gγ(M) + Gxc(M), | (30) |
and where
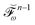
contains all contributions to
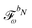
that do not depend on
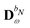
. Through an approach that involves solving a linear equation system involving
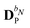
to obtain parameters that can be used to determine
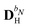
, the
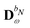
and
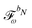
matrices of interest can be obtained.
Multiphoton transition moments can be calculated from the single residues of the response functions.7 A thorough presentation of this procedure in the context of open-ended response theory has been given in ref. 11, and since the addition of environment effects through the PE model enters in the same way for single residues of response functions as for the response functions themselves, we consider it sufficient to only include a presentation of the latter in this work.
2.3 Polarizable embedding in the open-ended response framework
In the following, we will show that environment effects described by the PE model can be included in calculations of electric response properties by rather modest modifications of the approach presented in Section 2.2.
In the presence of an external electric field, the local field experienced by the quantum region is modified by the interactions between the classical region and the external field. In addition to the static reaction field in the unperturbed energy and KS matrix in eqn (3) and (12), respectively, additional contributions arise as a consequence of the dipole polarization in the classical region induced by the perturbed quantum region and the external electric field. The former gives rise to a dynamic reaction field, and the latter to an effective external field (EEF), which describes the effective field in the classical region devoid of the quantum part.26 This EEF effect is similar in origin to the cavity-field effects introduced in the context of polarizable continuum models.57–60 The EEF effect enters in the definitions of the response and transition properties of the quantum region and increases with increasing number of photons.26 It should therefore be taken into account in calculations of properties that involve the external field. This is achieved by replacing the electric field vector in eqn (8), which generates the induced dipoles, with its time-dependent analogue augmented with the time-dependent external field Ẽt, so that
| | | Ẽ(,rs) = Ẽe(,rs) + Ẽn(rs) + Em(rs) + Ẽt(rs), | (31) |
where the electric dipole approximation was assumed in the last term. The external field can be considered to be uniform in the long-wavelength limit and we can therefore omit its dependency on the position,
i.e.Ẽt(
rs) =
Ẽt.
When including the environment effects in the response theory framework, it is convenient to separate the energy and KS matrix contributions according to their degree of dependence on the density matrix. For the PE energy, this dependence can be either zeroth-, first- or second-order, and the corresponding contributions are denoted by ẼPE,0, ẼPE,1, and ẼPE,2, which can be written as
| | ẼPE,0 = ![[h with combining tilde]](https://www.rsc.org/images/entities/i_char_0068_0303.gif) esPE + indPE + (1)EEF + (2)EEF, esPE + indPE + (1)EEF + (2)EEF, | (32) |
| | 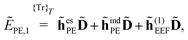 | (33) |
and
| | 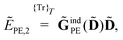 | (34) |
respectively. The EEF contributions are the additional terms, involving the external electric field, which enter into the time-dependent analogue of
eqn (7). The
(1)EEF term couples the external field to the fields from the nuclei and permanent multipole moments so that
| | 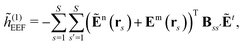 | (35) |
and it thus depends linearly on the external field, whereas
(2)EEF depends quadratically on the external field, so that
| | 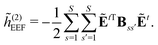 | (36) |
The EEF contribution that couples the external field to the electronic field, which therefore has a first-order dependence on the density matrix and depends linearly on the external field, is defined by
| | 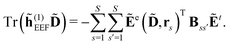 | (37) |
For the KS matrix contributions, the dependence on the density matrix can be either zeroth- or first-order, and the respective contributions are called ![[F with combining tilde]](https://www.rsc.org/images/entities/b_char_0046_0303.gif) PE,0 and PE,1, and are given by
PE,0 and PE,1, and are given by
| | PE,0 = ![[h with combining tilde]](https://www.rsc.org/images/entities/b_char_0068_0303.gif) esPE + indPE + (1)EEF esPE + indPE + (1)EEF | (38) |
and
respectively. The PE contributions to the generalized KS energy and KS matrix can then be included by adding the right-hand sides of
eqn (32)–(34),
(38) and (39) to
eqn (16) and (17), respectively, so that
| | 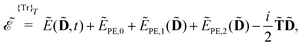 | (40) |
and
| | 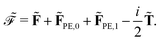 | (41) |
When calculating the properties considered in this work, the above contributions are differentiated with respect to one or more perturbation strengths. Upon differentiation of
eqn (32)–(34) with respect to a tuple of
N perturbations, it follows from the (
k,
n) rule that at most
N − 1 perturbations can be assigned to differentiation of density matrices. Therefore, at least one perturbation strength in the tuple must differentiate a non-density-matrix term. Taking this into consideration and furthermore considering the density-matrix dependence of each term, the parts of
eqn (32)–(39) that do not vanish upon differentiation with respect to a tuple
bN of perturbations
a,
b,
c,…, are
| | 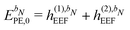 | (42) |
| | 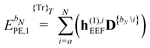 | (43) |
and
| | 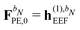 | (44) |
| | 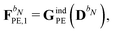 | (45) |
where
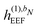
and
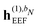
are zero if
N > 1,
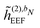
is zero if
N > 2, and where the summation in
eqn (43) runs over all perturbations in
bN. In
eqn (43), the collection {
bN\
i} denotes the
bN perturbation tuple except perturbation
i, and the terms in this equation may be subject to further truncation depending on the choice of (
k,
n) rule. Consequently, for electric response properties, the inclusion of a polarizable environment in the response property scheme of Section 2.2 is accomplished relatively straightforwardly by calculating the terms in
eqn (42)–(45) and adding them in the appropriate places.
3 Computational details
3.1 Molecular structures
Structures for GFP with a neutral and anionic chromophore were taken from ref. 61. The structures were obtained from a classical molecular dynamics simulation using the empirical force field of Reuter et al.62 starting from the 1GFL crystal structure.63 In total 50 snapshots were obtained at intervals of 300 ps from a 15 ns production run. The geometry of the chromophore was optimized within the frozen protein environment as described in ref. 61, to which we refer for further details on the preparation of the molecular structures. The protein model is solvated in water and includes all water molecules within 8 Å from any Cα atom.61
Three different sizes of the quantum region were investigated. For the smallest, the quantum region was chosen as in previous work61,64–66 and consisted of residues 65 to 67 as well as the backbone C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O from residue 64 and the backbone NH from residue 68 (Fig. 1a). The medium quantum region contained, in addition to this, the CαH from residues 64 and 68 (Fig. 1b) and is the same quantum region as used in the cluster model in ref. 66 and 67. The large quantum region contained the complete residues 64 to 68 as well as the backbone COCαH from residue 63 and backbone NHCαH from residue 69 (Fig. 1c). Hydrogen caps were added in all cases.
O from residue 64 and the backbone NH from residue 68 (Fig. 1a). The medium quantum region contained, in addition to this, the CαH from residues 64 and 68 (Fig. 1b) and is the same quantum region as used in the cluster model in ref. 66 and 67. The large quantum region contained the complete residues 64 to 68 as well as the backbone COCαH from residue 63 and backbone NHCαH from residue 69 (Fig. 1c). Hydrogen caps were added in all cases.
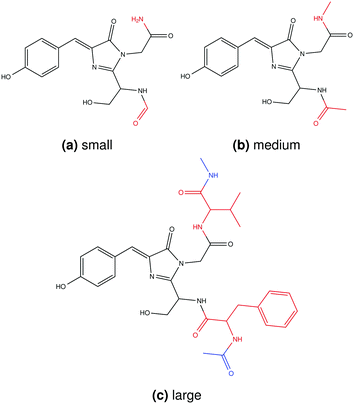 |
| | Fig. 1 Chemical structures of the quantum regions investigated in this work. Atoms from residues 65–67 (chromophore) are shown in black, from residues 64 and 68 in red, and from residues 63 and 69 in blue. | |
3.2 Generation of embedding potentials
The embedding potential parameters for GFP were calculated as described in ref. 61, based on the extensive tests reported in ref. 64. For the small quantum region (Fig. 1a), this means that the parameters were the same as in ref. 61. The potentials consisted of LoProp68 atom-centered multipoles (up to quadrupoles) and anisotropic dipole–dipole polarizabilities, calculated in Molcas.69,70 These embedding potential parameters were calculated with KS-DFT (B3LYP71–74/6-31+G*75–77) using the molecular fractionation with conjugate caps approach by Zhang and Zhang52 that was extended to multipoles and polarizabilities by Söderhjelm and Ryde.53 The basis set was recontracted to an atomic natural orbital type basis as required by the LoProp approach.68
3.3 Multiphoton absorption calculations
All MPA property calculations were performed with a modified version of the Dalton quantum chemistry program,78,79 using a local version of the recursive and open-ended response code10 introduced in Section 2.2. These programs are linked to the PE library (PElib),80 which makes use of the Gen1Int library81,82 to calculate perturbed one-electron integrals. The exchange–correlation contributions were obtained using the XCint83 and XCFun84 libraries.
Vertical excitation energies and MPA transition moments for the π → π* excitation in GFP were calculated with the range-separated CAM-B3LYP exchange–correlation functional.85 This functional is better suited for the description of charge-transfer excitations than common hybrid functionals86 despite a general overestimation of the excitation energies.87 The basis set used in the calculations was chosen consistently with the KS-DFT-derived embedding potential parameters, namely 6-31+G*.75–77 This basis set was previously found to represent a good compromise between computational cost and accuracy for one- and two-photon absorption calculations on the same molecular systems.64 We therefore expect that the trends found in this work are representative, but recommend the use of a larger basis set for quantitative work. The MPA moments include the EEF effect26 as described in Section 2.3, unless otherwise specified.
Since the GFP chromophore has two covalent bonds to the rest of the protein, two quantum–classical cuts are necessary for each of the quantum regions in Fig. 1. There are several schemes proposed for handling these quantum–classical boundaries.21 In this work we have used the link-atom approach where hydrogen caps were inserted on both sides of the cut, and sites in the classical region that are closer than 1.4 Å to any quantum-region atom were removed. Their charges were distributed to preserve the total charge of the protein whereas their higher-order multipoles and polarizabilities were removed. The redistribution scheme was tested by redistributing the charges to either the nearest one, two, or three sites, or to all sites in the classical region (see results in Section 4.1 and Tables S3 and S4 in the ESI†). The redistribution to all classical sites essentially corresponds to removal of the affected charges at the quantum–classical border because of the number of sites in the system considered here (>8000), except that it preserves the total charge. For all other calculations presented in this work, we used the latter scheme where charges were redistributed to all remaining sites in the classical region.
The tests on the redistribution scheme (Section 4.1) and EEF effect (Section 4.2 and Table S7†) were performed on a single snapshot. The tests on the size of the quantum region (Section 4.1) were performed on five different snapshots. Averaging over more structures and comparing to the large quantum region (Fig. 1c) is not feasible due to the size of the large quantum region in combination with the high computational cost of especially 3PA and 4PA calculations. Excitation energies and 1PA–4PA strengths of the π → π* transition were calculated for the medium quantum region as averages over 50 snapshots (Section 4.3).
3.4 Analysis of the data
The rotational averaging of the MPA transition moments S to obtain the MPA strengths 〈δMPA〉 was performed by the method described by Friese, Beerepoot and Ruud,88 so that| | 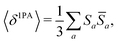 | (46) |
| | 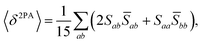 | (47) |
| | 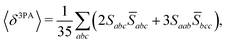 | (48) |
| | 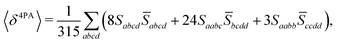 | (49) |
where the overbar indicates complex conjugation. For the SCF-based theories considered in this work, the transition moments are real-valued and thus equal to their complex conjugates. Rotationally averaged MPA strengths are given in the ESI† in atomic units, which for M-photon absorption are11| | | [〈δMPA〉] = a2M0·E2(M−1)h, | (50) |
where a0 and Eh are the atomic units for length and energy, respectively. In addition, the dimensionless oscillator strength f was calculated from the rotationally averaged 1PA strength 〈δ1PA〉 in eqn (46) and the photon frequency ω, as
The 2PA cross section σ2PA in GM units (1 GM = 10−50 cm4 s photon−1) was obtained from the rotationally averaged 2PA strength 〈δ2PA〉 in eqn (47) as
| | 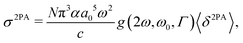 | (52) |
where
α is the fine structure constant and
c is the speed of light. The integer
N was set to 4 to compare with single-beam experiments as discussed in
ref. 89. The lineshape function
g(2
ω,
ω0,
Γ) is here a Lorentzian broadening function with a half-width at half-maximum of
Γ = 0.1 eV (0.00367
Eh) describing homogeneous broadening processes. When evaluated at the maximum of the peak (2
ω =
ω0), the Lorentzian broadening function reduces to a constant as
| | 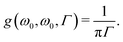 | (53) |
We note that even though the value of 0.1 eV has been used in many works (discussed in
ref. 89), there is no rigorous theoretical foundation for this value.
In relation to the test calculations regarding the size of the quantum region, the mean absolute percentage deviation (MAPD) is calculated for different sizes of the quantum region as an average over five snapshots as
| | 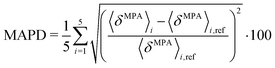 | (54) |
where
i is the number of the snapshot and the large quantum region is used as a reference (
Fig. 1c).
4 Results and discussion
4.1 Required size of the quantum region and redistribution scheme
We have tested different sizes of the quantum region in order to investigate the sensitivity of the results to the location of the quantum–classical border and to find the necessary minimum size of the quantum region needed to obtain reliable results. All numerical results are tabulated in the ESI.† The small and medium quantum regions (Fig. 1) differ only by the addition of two methyl groups in the latter. The computational time for the MPA calculations only increases very little when going from the small to medium region. The large quantum region contains, in addition to the medium one, the complete Phe64 and Val68 residues (shown in red in Fig. 1). Since this leads to a large increase in the computational time (approximately a factor of two when going from medium to large), this quantum region is primarily meant as a reference to test against and not necessarily as a feasible choice for further work. The tests of the different sizes of the quantum region in the polarizable environment are therefore limited to five snapshots of the neutral and anionic GFP.
Tabulated results for excitation energies and MPA strengths are shown in Tables S1 and S2.† The differences in excitation energies are minimal between the different quantum regions, especially for the neutral chromophore. The differences do not exceed 0.03 eV (neutral) and 0.06 eV (anionic) for the small and 0.01 eV (neutral and anionic) for the medium quantum region when using the results on the large quantum region as the reference. The calculations on the neutral and anionic GFP are thus affected slightly differently by the size of the quantum region. We have previously shown that the PE model can accurately describe the influence of the immediate surroundings of the chromophore on the excitation energies by comparing CC2 calculations on cluster models to the corresponding PE-CC2 calculations (using the small quantum region in Fig. 1a).66 Here, we have also tested different schemes for the redistribution of the embedding potential parameters from classical sites that are within 1.4 Å of one of the quantum-region atoms, i.e. those at the quantum–classical boundary (Tables S3 and S4†). The excitation energies are not much affected, with differences not exceeding 0.04 eV (neutral) and 0.05 eV (anionic) between different redistribution schemes. Comparison with ref. 61 suggests that these small differences in excitation energies cancel when averaging over a large number of snapshots. Indeed, the average excitation energy of the neutral GFP chromophore (based on 50 snapshots) is equal (3.51 eV) in this work (medium quantum region, redistribution of charges to all sites) and in ref. 61 (small quantum region, redistribution of all multipoles and polarizabilities to one site) with very similar standard deviations (0.037 and 0.038 eV, respectively). The average excitation energy of the anionic chromophore (based on 50 snapshots) is also similar in this work (3.01 eV) and in ref. 61 (3.00 eV) with similar standard deviations (0.035 and 0.034 eV, respectively). The insensitivity of the excitation energy to the size of the quantum region and the treatment of the quantum–classical boundary can be explained by the molecular orbitals (MOs) that dominate the transition. The π → π* excitation investigated here is dominated by a transition from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO), where both MOs are located on the conjugated part of the chromophore. Enlarging the quantum region beyond a certain minimum size only modestly affects those MOs, making the excitation energy rather insensitive to the size of the quantum region. We conclude that it is unproblematic to use the small quantum region for the calculation of excitation energies, as we have done in previous work.61,66
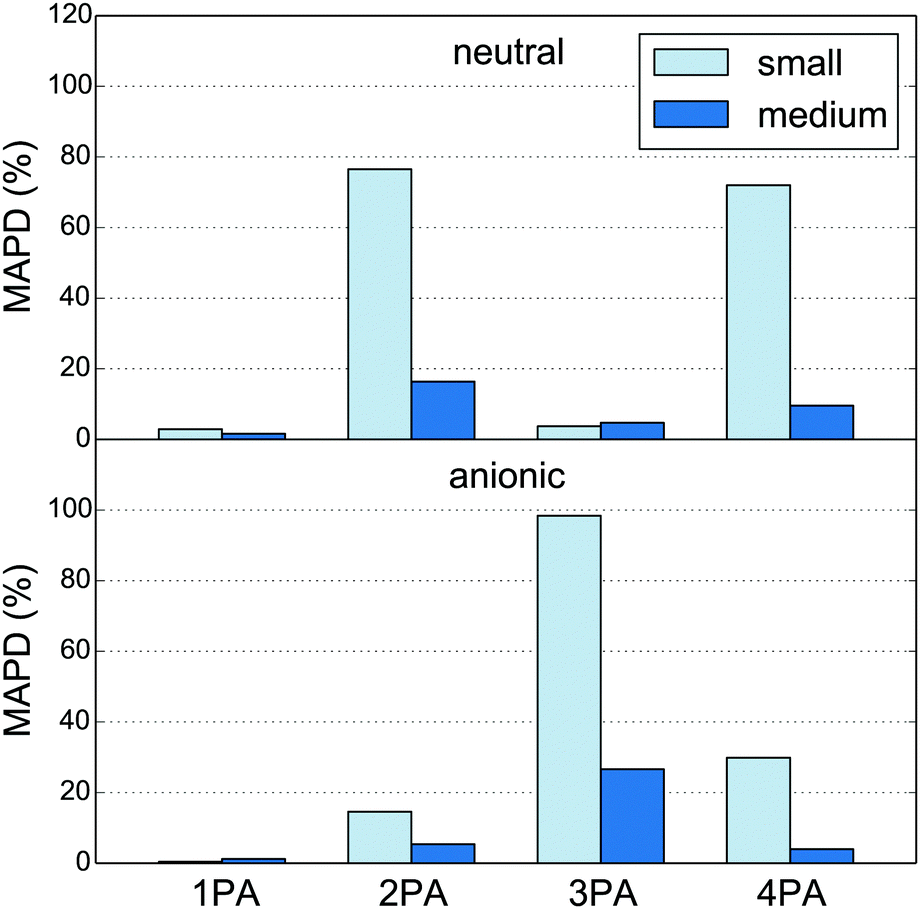
It is interesting to consider the performance of the PE-DFT approach on less localized properties such as 2PA, 3PA and 4PA strengths. The absolute percentage deviations of the MPA strengths, relative to the large quantum region, are calculated as averages over five snapshots (MAPD; eqn (54)) and shown in Fig. 2. It is clear from this figure that 2PA, 3PA and 4PA strengths can be much more affected by the size of the quantum region, and thus are less localized, than the 1PA strength. Indeed, as was the case for the excitation energies, the 1PA strength is barely affected by the size of the quantum region, indicating that the small quantum region can also be used safely for the calculation of 1PA strengths in fluorescent proteins as done in earlier work.90 For MPA strengths, the MAPD decreases significantly when the quantum region is enlarged from small to medium. The decrease is from 76% to 16% for 2PA and from 72% to 10% for 4PA in the neutral chromophore and from 98% to 27% for 3PA in the anionic chromophore. The sensitivity of the results to the size of the quantum region can be explained by the number and location of the MOs involved in the MPA processes. A large number of MOs on and around the conjugated system of the chromophore can be involved in a two-, three- or four-step process from the HOMO to the LUMO. The new MOs that appear as a result of extending the quantum region can thus affect the calculated MPA strengths. The results in Fig. 2 indicate that the additional MOs play a significant role in the MPA process with an even number of photons for the neutral chromophore and with an odd number of photons for the anionic chromophore. The 1PA process, however, is dominated by a one-step HOMO to LUMO transition where both MOs are located on the conjugated system. The addition of MOs on and around the conjugated system will thus not significantly affect the 1PA strength. The MOs that involve the peptide bonds close to the quantum–classical boundary (see Fig. 1) are also modeled more realistically by the medium quantum region, where the Cα atom is substituted by a methyl carbon rather than a hydrogen atom.
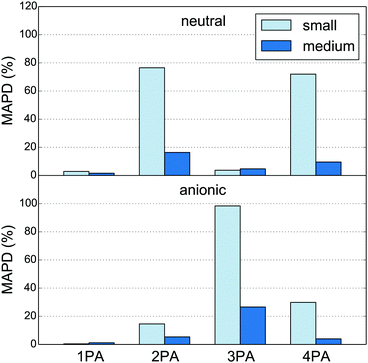 |
| | Fig. 2 Mean absolute percentage deviation (MAPD, eqn (54)) of MPA strengths calculated for the small and medium quantum regions (Fig. 1) relative to those for the large quantum region (see Tables S1 and S2† for the numbers). The charges of the classical sites within 1.4 Å of any atom in the quantum region have been redistributed to all other classical sites. The numbers are based on five snapshots of the neutral (top) and anionic (bottom) GFP chromophores in the polarizable environment of the protein. | |
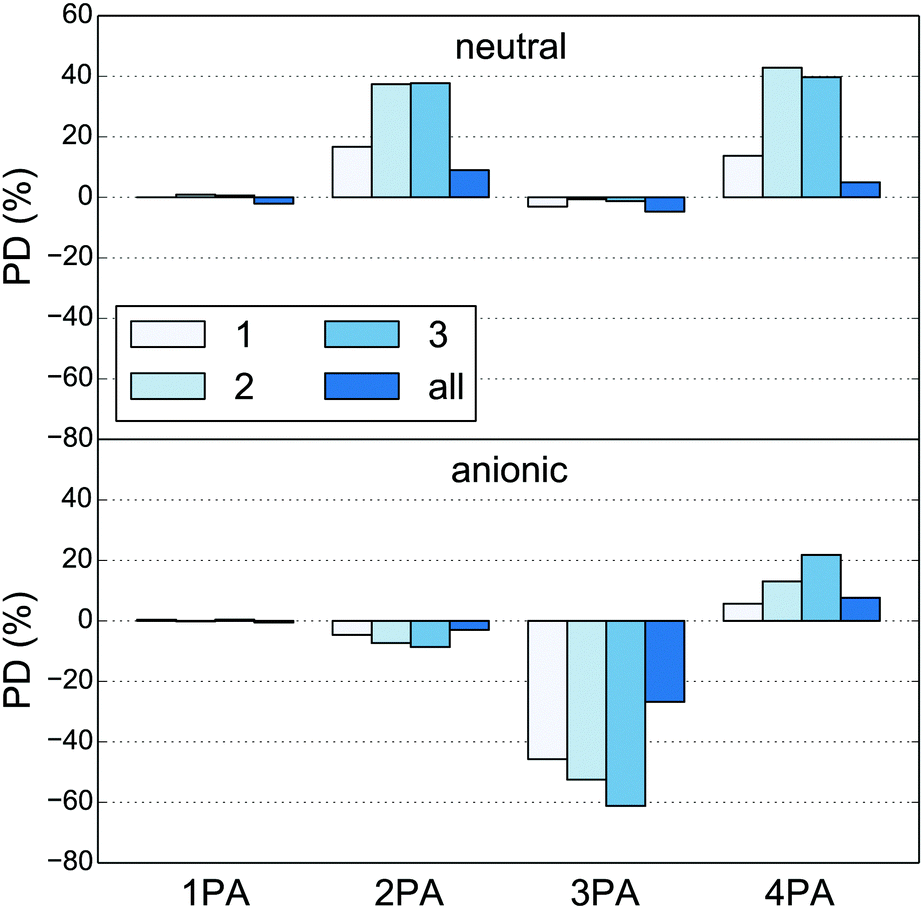
In this work we use the hydrogen link-atom approach in the separation of the quantum and classical regions. In this approach, where heavier atoms are replaced by hydrogens, it is necessary to consider how to treat overlapping atoms, i.e. classical atoms that have been replaced by hydrogens in the quantum region, in order to avoid double-counting. Furthermore, due to the close proximity of classical sites to the quantum link atom, there is a risk of overpolarization. The specific treatment of the quantum–classical border affects the polarization of both the quantum and the classical regions, mainly in the area around the border. The aim is to reproduce the electron density as close as possible to the quantum–classical border. While this is in principle possible, it is not necessarily feasible without some form of parametrization. Optimally, one would therefore increase the size of the quantum region until no effect from the specific choice of redistribution scheme is observed. However, since this option is limited by current algorithms and technology, we instead investigate the sensitivity of the MPA strengths with respect to simple redistribution schemes. Percentage deviations (PD) for the MPA strengths of the medium quantum region relative to the large quantum region are shown in Fig. 3 for redistribution of charges to the nearest one, two, three, or all other classical sites, while all other parameters are deleted. All numbers for the small, medium and large quantum regions are tabulated in Tables S3 and S4.† Errors for 1PA strengths do not exceed 3%, indicating that this property is rather insensitive to the quantum–classical border treatment, or, equivalently, that the size of the quantum regions are appropriate for this property. The MPA strengths, on the other hand, can be dramatically affected by the number of sites to which the charges are redistributed. Comparing the MPA strengths of the large and medium quantum regions (Tables S3 and S4†), we observe that the spread between the different redistribution schemes decreases, but even for the large quantum region there are substantial differences. This indicates that a larger quantum region is needed for quantitative calculations when using such simple redistribution schemes. Fig. 3 shows that the difference between the MPA strengths calculated using the medium and large quantum region is smallest on average when charges are redistributed to all other sites. The PDs are less than 10% for both the neutral and anionic chromophore except for the 3PA strength of the anionic chromophore where the PD is 27%, which is still significantly less than for the other redistribution schemes. We have therefore chosen this scheme for the remaining calculations presented in this work. We stress that redistribution and deletion of embedding potential parameters can potentially be avoided by using other schemes to treat the quantum–classical border, such as the frozen molecular orbital approach of Friesner and coworkers,91,92 or the optimized effective atom-centered potentials by von Lilienfeld et al.,93,94 both of which, however, require parametrization for different chemical species.
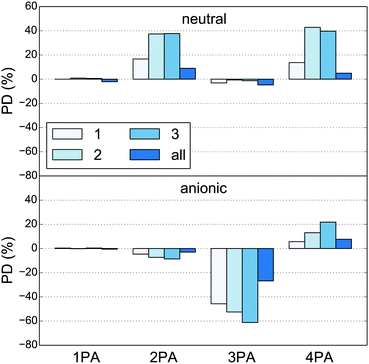 |
| | Fig. 3 Percentage deviation (PD) of MPA strengths calculated for the medium quantum region (Fig. 1b) relative to those calculated for the large quantum region (see Tables S3 and S4† for the numbers). The charges of the classical sites within 1.4 Å of any quantum-region atom have been redistributed to either one, two, three or all other classical sites. The numbers are based on one snapshot (#1 in the ESI†) of the neutral (top) and anionic (bottom) GFP chromophores. | |
4.2 Influence of the polarizable environment
Calculated excitation energies and 1PA to 4PA strengths are shown for five different snapshots of the neutral and anionic GFP chromophores in Tables S5 and S6† respectively, both in the absence and the presence of the protein environment. For the neutral chromophore, inclusion of the protein environment leads to a red shift (of 0.09 to 0.16 eV) in the π → π* excitation energy, as has also been observed in earlier work.61,64,66,95 The excitation energy of the anionic chromophore is less affected, with changes ranging from a red shift of 0.01 eV to a blue shift of 0.06 eV between the different snapshots. The small shift in the excitation energy of the anionic chromophore can be explained by a cancellation of the blue shift due to electrostatic and ground-state polarization effects (static reaction field) and the red shift due to dynamic polarization effects (dynamic reaction field), as previously described95 and apparent from the data in Table S7.†
Tables S5 and S6† also reveal that the MPA strengths generally decrease when adding the polarizable environment. In previous work that did not include the EEF effect, we have found an increase in the 1PA oscillator strengths in the different protonation states of GFP,64,90 in DsRed96 and in various other fluorescent proteins.90 In addition, 2PA cross sections were found to increase when adding the polarizable environment to the neutral and anionic GFP64 and DsRed96 chromophores also when corrected for the dependence on the photon energy (see eqn (52)). It has been shown for DsRed that inclusion of the EEF effect, i.e. defining the properties with respect to the external field, leads to a decrease in the 1PA and 2PA strengths compared to the chromophore in isolation, and to a considerably improved agreement with supermolecular calculations.26 The percentage change of the MPA strengths relative to the isolated quantum region are shown in Fig. 4 (see also Table S7†). Indeed, we see here that neglecting the EEF effect leads to an increase of MPA strengths in all cases and thus a qualitatively wrong change with respect to the isolated chromophore. In conclusion, we find that the EEF effect is important to obtain both qualitative and quantitative correct results, and in particular for higher-order MPA strengths.
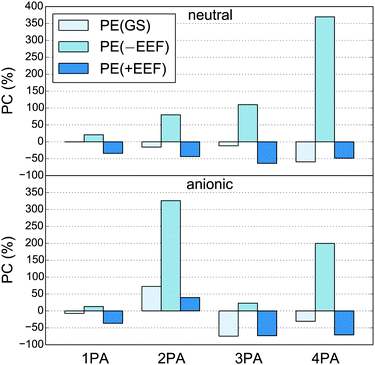 |
| | Fig. 4 Percentage change (PC) of MPA strengths for the medium quantum region calculated in the polarizable environment relative to the MPA strengths of the medium quantum region in isolation (see Table S7† for the numbers). The MPA strengths are calculated with only ground-state polarization [PE(GS)], with full polarization and without the EEF effect [PE(−EEF)], and with full polarization and with the EEF effect [PE(+EEF)]. The numbers are based on one snapshot (#1 in the ESI†) of the neutral (top) and anionic (bottom) GFP chromophores. | |
4.3 Configurational sampling
Excitation energies and 1PA–4PA strengths are shown in Table 1 as averages over 50 snapshots from a classical molecular dynamics simulation (using medium quantum region and redistribution of charges to all sites). Standard deviations (shown in the same table) are roughly of the same order of magnitude as the strengths themselves for 2PA–4PA, emphasizing the importance of configurational sampling for this molecular system. The 1PA strengths, on the other hand, are much less affected by the sampling, with standard deviations that are more than an order of magnitude lower than the strength itself. We note that the spectral broadening of the excitation energies is decreased by approximately a factor three as a result of the geometry optimization of the chromophore,61 which will also have a similar effect on the MPA strengths.
Table 1 Excitation energies (in eV), one-, two-, three and four-photon absorption strengths (in atomic units), averaged over 50 snapshots. Standard deviations are shown in parentheses
|
|
Neutral |
Anionic |
|
E
exc
|
3.51 (0.04) |
3.01 (0.04) |
| 1PA |
1.84 (0.13) |
3.23 (0.12) |
| 2PA |
405 (238) |
1160 (398) |
| 3PA (104) |
838 (111) |
85 (94) |
| 4PA (109) |
18 (15) |
64 (18) |
4.4 Limitations
The computational protocol followed here does not allow for a quantitative comparison with available experimental data for several reasons. First and foremost, current exchange–correlation kernels used in time-dependent DFT do not appear to be able to predict quantitatively accurate MPA strengths. Indeed, it has been shown that CAM-B3LYP underestimates the 2PA strength of the GFP chromophore by about a factor of two compared to EOM-CCSD.89 Part of the problem can be related to the unreliable prediction of the excited-state dipole moment of π → π* excitations by commonly employed exchange–correlation functionals,97 which affects the MPA strength for noncentrosymmetric molecules through difference dipole moments. In addition to difference dipole moments, transition dipole moments between excited states also play a role in MPA, adding to the uncertainty of whether current DFT functionals are accurate enough for the calculation of absolute MPA strengths. There is a clear need for benchmarking DFT MPA strengths, which, however, is limited by the lack of accurate and efficient correlated methods for calculating MPA strengths. Secondly, reliable MPA experimental data are needed for a reliable comparison with calculated data. Experimental 2PA cross sections of fluorescent proteins show a large variation between different experimental studies, as discussed in ref. 98. Comparison between calculated and measured values also places demands on the presentation of the experimental data. Ideally, an MPA spectrum should be available, or the reported cross sections should at least correspond to the absorption maximum. This is not the case for available experimental 3PA and 4PA cross sections on GFP.47 Moreover, enough details about the experimental setup should be presented to allow for a correct comparison with calculations. In fact, comparison with different experimental setups requires the use of different factors to convert MPA strengths (in atomic units) to cross sections (in macroscopic units), as discussed for 2PA in ref. 89. Therefore, thorough knowledge of both the experimental and computational details is needed for a correct comparison of measured and calculated results. Thirdly, we have here calculated energies and absorption strengths associated with vertical excitations, assuming that the one-photon and multiphoton absorption strengths are resulting from exactly the same (vertical) electronic excitation. Non-Condon effects, however, have been shown to be significant in 2PA and in particular in shifts between 1PA and 2PA spectra.99,100 Deviation from the Franck–Condon behavior is thus likely to play a role in both the location and the amplitude of the peaks in the calculated MPA spectra and is therefore important to consider if comparison with experimental spectra is attempted.
The problematic comparison is illustrated by the large discrepancy between the calculated and experimental 2PA cross section for the neutral chromophore. The former is 1.8 GM, using eqn (52) and the average excitation energy and 2PA strength from Table 1 (see Section 3.4 for details on the conversion). This is an order of magnitude smaller than the experimentally measured cross sections reported in ref. 47 and 98. For comparison, we have previously compared experimental and calculated excitation energies using the same multiscale approach. Relative trends between different fluorescent proteins were reproduced even without conformational averaging. However, the calculations overestimated the experimental excitation energies by almost 0.4 eV for neutral and anionic GFP, which we ascribed to imperfections in the used quantum method rather than in the PE model.61 We also note that the π → π* excitation, which is a HOMO to LUMO transition with both MOs localized on the conjugated system of the chromophore, is not necessarily the one with the highest MPA strength. This has been shown before for 2PA by experiments98 and calculations both in isolation101 and in a protein environment.64 The same has been observed in the present work.
5 Conclusions and outlook
We have extended an open-ended density-matrix-based response theory formulation and its corresponding implementation to include a multiscale description of molecular environment effects modeled by the polarizable embedding model, which enables calculations of electric response and multiphoton absorption properties to any order for molecules embedded in realistic molecular environments. The present implementation is applicable to response properties involving electric dipole and higher-order multipole perturbations. To demonstrate the implementation, we have calculated one-, two-, three-, and four-photon absorption strengths of GFP, making this work the first to explore three- and four-photon absorption cross sections of a chromophore in an explicit (bio)molecular environment. The developments presented here are therefore particularly useful in the development of chromophores for multiphoton microscopy.
We have examined several practical aspects that are important to consider in multiscale modeling applications: convergence with respect to the size of the quantum region, treatment of the quantum–classical border, and the importance of configurational sampling. Concerning the size of the quantum region, we have found that the small quantum region shown in Fig. 1a is appropriate for calculating the excitation energy and 1PA strength of the (local) π → π* transition in GFP. The MPA strengths, however, are much more sensitive to the size of the quantum region, as well as to different ways of treating the quantum–classical border. We have used the hydrogen link-atom approach at the quantum–classical border, which means there are parameters on classical sites that are in close proximity to the quantum link atom that need to be taken care of to avoid overpolarization. Here we have investigated simple redistribution schemes where charges that are closer than a specified threshold are redistributed to other classical sites. We found that for MPA strengths it is necessary to either use large quantum regions or, alternatively, to improve the treatment of the quantum–classical border, although the latter was not investigated here. Furthermore, configurational sampling was found to be very important for the investigated MPA strengths, where the standard deviations are on the same order of magnitude as the averages.
Finally, we have investigated how different approximations of the polarizable component of the embedding potential affect the MPA strengths. The most important observation is that including dynamic polarization effects without adding the effective external field effect can lead to a significant overestimation of the MPA strengths defined with respect to the external field strength.
In future developments of this methodology, we will seek to include environment effects for other categories of properties such as those involving geometric perturbation of the nuclei of the quantum system. These properties will enable the inclusion of environment effects in spectroscopic phenomena that involve molecular vibrations such as infrared and Raman spectroscopy and variants thereof. Further extensions to cover properties that involve magnetic perturbations, which will make it possible to predict and interpret NMR spectra, are also planned. In addition, work is underway to improve the embedding methodology. Specifically, we are extending the coupling to include the polarizable density embedding model which features improved short-range electrostatics and also models nonelectrostatic repulsion effects, i.e. exchange repulsion/Pauli repulsion.102
Acknowledgements
The authors thank Radovan Bast and Daniel H. Friese (University of Tromsø—The Arctic University of Norway) for technical support. A. H. S., M. T. P. B., M. R. and K. R. acknowledge support from the Research Council of Norway through a Centre of Excellence Grant (Grant No. 179568/V30) and from the European Research Council through a Starting Grant (Grant No. 279619). A. H. S. acknowledges financial support from Tromsø Forskningsstiftelse. N. H. L. acknowledges financial support from the Carlsberg Foundation (Grant No. CF15-0792). J. K. and J. M. H. O. acknowledge financial support from the Danish Council for Independent Research (DFF) through the Sapere Aude research career program (Grant no. DFF – 0602-02122B, DFF – 1323-00744, and DFF – 1325-00091). J. K. acknowledges the VILLUM Foundation for support. Computation/simulation for the work described in this paper was supported by the DeiC National HPC Center (SDU) and by the Norwegian Supercomputer Program (Grant No. NN4654K).
References
-
E. B. Wilson, Jr., J. C. Decius and P. C. Cross, Molecular Vibrations: The Theory of Infrared and Raman Vibrational Spectra, Dover publications Inc., New York, 1980 Search PubMed.
-
L. D. Barron, Molecular Light Scattering and Optical Activity, Cambridge University Press, Cambridge, United Kingdom, 2nd edn, 2004 Search PubMed.
- V. Barone, J. Chem. Phys., 2005, 122, 014108 CrossRef PubMed.
- C. Puzzarini, J. F. Stanton and J. Gauss, Int. Rev. Phys. Chem., 2010, 29, 273–367 CrossRef CAS.
- R. Bast, U. Ekström, B. Gao, T. Helgaker, K. Ruud and A. J. Thorvaldsen, Phys. Chem. Chem. Phys., 2011, 13, 2627–2651 RSC.
- T. Helgaker, S. Coriani, P. Jørgensen, K. Kristensen, J. Olsen and K. Ruud, Chem. Rev., 2012, 112, 543–631 CrossRef CAS PubMed.
- J. Olsen and P. Jørgensen, J. Chem. Phys., 1985, 82, 3235–3264 CrossRef CAS.
- K. Sasagane, F. Aiga and R. Itoh, J. Chem. Phys., 1993, 99, 3738–3778 CrossRef CAS.
- A. J. Thorvaldsen, K. Ruud, K. Kristensen, P. Jørgensen and S. Coriani, J. Chem. Phys., 2008, 129, 214108 CrossRef PubMed.
- M. Ringholm, D. Jonsson and K. Ruud, J. Comput. Chem., 2014, 35, 622–633 CrossRef CAS PubMed.
- D. H. Friese, M. T. P. Beerepoot, M. Ringholm and K. Ruud, J. Chem. Theory Comput., 2015, 11, 1129–1144 CrossRef CAS PubMed.
- E. G. McRae, J. Phys. Chem., 1957, 61, 562–572 CrossRef CAS.
- J. Kongsted and B. Mennucci, J. Phys. Chem. A, 2007, 111, 9890–9900 CrossRef CAS PubMed.
-
Solvation Effects on Molecules and Biomolecules, ed. S. Canuto, Springer, Netherlands, 2008 Search PubMed.
- K. O. Sylvester-Hvid, K. V. Mikkelsen, P. Norman, D. Jonsson and H. Ågren, J. Phys. Chem. A, 2004, 108, 8961–8965 CrossRef CAS.
- J. Neugebauer, Angew. Chem., Int. Ed., 2007, 46, 7738–7740 CrossRef CAS PubMed.
- C. Cappelli and B. Mennucci, J. Phys. Chem. B, 2008, 112, 3441–3450 CrossRef CAS PubMed.
- A. S. P. Gomes and C. R. Jacob, Annu. Rep. Prog. Chem., Sect. C: Phys. Chem., 2012, 108, 222–277 RSC.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
- A. Warshel and M. Levitt, J. Mol. Biol., 1976, 103, 227–249 CrossRef CAS PubMed.
- H. M. Senn and W. Thiel, Angew. Chem., Int. Ed., 2009, 48, 1198–1229 CrossRef CAS PubMed.
- J. M. Olsen, K. Aidas and J. Kongsted, J. Chem. Theory Comput., 2010, 6, 3721–3734 CrossRef CAS.
- J. M. H. Olsen and J. Kongsted, Adv. Quantum Chem., 2011, 61, 107–143 CrossRef CAS.
- N. H. List, J. M. H. Olsen and J. Kongsted, Phys. Chem. Chem. Phys., 2016, 18, 20234–20250 RSC.
- K. Sneskov, T. Schwabe, O. Christiansen and J. Kongsted, Phys. Chem. Chem. Phys., 2011, 13, 18551–18560 RSC.
- N. H. List, H. J. Aa. Jensen and J. Kongsted, Phys. Chem. Chem. Phys., 2016, 18, 10070–10080 RSC.
- M. A. Thompson, J. Phys. Chem., 1996, 100, 14492–14507 CrossRef CAS.
- L. Jensen, P. Th. van Duijnen and J. G. Snijders, J. Chem. Phys., 2003, 118, 514–521 CrossRef CAS.
- S. Yoo, F. Zahariev, S. Sok and M. S. Gordon, J. Chem. Phys., 2008, 129, 144112 CrossRef PubMed.
- P. Söderhjelm, C. Husberg, A. Strambi, M. Olivucci and U. Ryde, J. Chem. Theory Comput., 2009, 5, 649–658 CrossRef PubMed.
- C. Curutchet, A. Muñoz-Losa, S. Monti, J. Kongsted, G. D. Scholes and B. Mennucci, J. Chem. Theory Comput., 2009, 5, 1838–1848 CrossRef CAS PubMed.
- F. Lipparini, C. Cappelli and V. Barone, J. Chem. Theory Comput., 2012, 8, 4153–4165 CrossRef CAS PubMed.
- T. Schwabe, J. M. H. Olsen, K. Sneskov, J. Kongsted and O. Christiansen, J. Chem. Theory Comput., 2011, 7, 2209–2217 CrossRef CAS PubMed.
- M. T. P. Beerepoot, A. H. Steindal, N. H. List, J. Kongsted and J. M. H. Olsen, J. Chem. Theory Comput., 2016, 12, 1684–1695 CrossRef CAS PubMed.
- J. M. H. Olsen, N. H. List, K. Kristensen and J. Kongsted, J. Chem. Theory Comput., 2015, 11, 1832–1842 CrossRef CAS PubMed.
- Q. Zheng, H. Zhu, S.-C. Chen, C. Tang, E. Ma and X. Chen, Nat. Photonics, 2013, 7, 234–239 CrossRef CAS.
- D. A. Parthenopoulos and P. M. Rentzepis, Science, 1989, 245, 843–845 CAS.
- A. S. Dvornikov, E. P. Walker and P. M. Rentzepis, J. Phys. Chem. A, 2009, 113, 13633–13644 CrossRef CAS PubMed.
- A. V. Kachynski, A. Pliss, A. N. Kuzmin, T. Y. Ohulchanskyy, A. Baev, J. Qu and P. N. Prasad, Nat. Photonics, 2014, 8, 455–461 CrossRef CAS.
- J. Croissant, M. Maynadier, A. Gallud, H. P. N'Dongo, J. L. Nyalosaso, G. Derrien, C. Charnay, J.-O. Durand, L. Raehm, F. Serein-Spirau, N. Cheminet, T. Jarrosson, O. Mongin, M. Blanchard-Desce, M. Gary-Bobo, M. Garcia, J. Lu, F. Tamanoi, D. Tarn, T. M. Guardado-Alvarez and J. I. Zink, Angew. Chem., Int. Ed., 2013, 52, 13813–13817 CrossRef CAS PubMed.
- W. Denk, J. H. Strickler and W. W. Webb, Science, 1990, 248, 73–76 CAS.
- S. Maiti, J. B. Shear, R. M. Williams, W. R. Zipfel and W. W. Webb, Science, 1997, 275, 530–532 CrossRef CAS PubMed.
- C. Xu, W. Zipfel, J. B. Shear, R. M. Williams and W. W. Webb, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 10763–10768 CrossRef CAS.
- W. R. Zipfel, R. M. Williams and W. W. Webb, Nat. Biotechnol., 2003, 21, 1369–1377 CrossRef CAS PubMed.
- R. Y. Tsien, Annu. Rev. Biochem., 1998, 67, 509–544 CrossRef CAS PubMed.
- M. Chalfie, Y. Tu, G. Euskirchen, W. W. Ward and D. C. Prasher, Science, 1994, 263, 802–805 CAS.
- L.-C. Cheng, N. G. Horton, K. Wang, S.-J. Chen and C. Xu, Biomed. Opt. Express, 2014, 5, 3427–3433 CrossRef PubMed.
- M. Drobizhev, C. Stoltzfus, I. Topol, J. Collins, G. Wicks, A. Mikhaylov, L. Barnett, T. E. Hughes and A. Rebane, J. Phys. Chem. B, 2014, 118, 9167–9179 CrossRef CAS PubMed.
-
M. A. Salem, M. Gedik and A. Brown, Two Photon Absorption in Biological Molecules, in Handbook of Computational Chemistry, ed. J. Leszcynskiet al., Springer, Dordrecht, 2015 DOI:10.1007/978-94-007-6169-8_47-1.
- M. Drobizhev, S. Tillo, N. S. Makarov, T. E. Hughes and A. Rebane, J. Phys. Chem. B, 2009, 113, 855–859 CrossRef CAS PubMed.
- M. Drobizhev, S. Tillo, N. S. Makarov, T. E. Hughes and A. Rebane, J. Phys. Chem. B, 2009, 113, 12860–12864 CrossRef CAS PubMed.
- D. W. Zhang and J. Z. H. Zhang, J. Chem. Phys., 2003, 119, 3599–3605 CrossRef CAS.
- P. Söderhjelm and U. Ryde, J. Phys. Chem. A, 2009, 113, 617–627 CrossRef PubMed.
-
X. Saint Raymond, Elementary Introduction to the Theory of Pseudodifferential Operators, CRC Press, 1991, vol. 3 Search PubMed.
- J. Applequist, J. R. Carl and K.-K. Fung, J. Am. Chem. Soc., 1972, 94, 2952–2960 CrossRef CAS.
- K. Kristensen, P. Jørgensen, A. J. Thorvaldsen and T. Helgaker, J. Chem. Phys., 2008, 129, 214103 CrossRef PubMed.
- R. Wortmann and D. M. Bishop, J. Chem. Phys., 1998, 108, 1001–1007 CrossRef CAS.
- R. Cammi, B. Mennucci and J. Tomasi, J. Phys. Chem. A, 1998, 102, 870–875 CrossRef CAS.
- L. Ferrighi, L. Frediani, C. Cappelli, P. Sałek, H. Ågren, T. Helgaker and K. Ruud, Chem. Phys. Lett., 2006, 425, 267–272 CrossRef CAS.
- S. Pipolo, S. Corni and R. Cammi, J. Chem. Phys., 2014, 140, 164114 CrossRef PubMed.
- M. T. P. Beerepoot, A. H. Steindal, J. Kongsted, B. O. Brandsdal, L. Frediani, K. Ruud and J. M. H. Olsen, Phys. Chem. Chem. Phys., 2013, 15, 4735–4743 RSC.
- N. Reuter, H. Lin and W. Thiel, J. Phys. Chem. B, 2002, 106, 6310–6321 CrossRef CAS.
- F. Yang, L. G. Moss and G. N. Phillips Jr., Nat. Biotechnol., 1996, 14, 1246–1251 CrossRef CAS PubMed.
- A. H. Steindal, J. M. H. Olsen, K. Ruud, L. Frediani and J. Kongsted, Phys. Chem. Chem. Phys., 2012, 14, 5440–5451 RSC.
- M. T. P. Beerepoot, A. H. Steindal, K. Ruud, J. M. H. Olsen and J. Kongsted, Comput. Theor. Chem., 2014, 1040–1041, 304–311 CrossRef CAS.
- T. Schwabe, M. T. P. Beerepoot, J. M. H. Olsen and J. Kongsted, Phys. Chem. Chem. Phys., 2015, 17, 2582–2588 RSC.
- V. R. I. Kaila, R. Send and D. Sundholm, Phys. Chem. Chem. Phys., 2013, 15, 4491–4495 RSC.
- L. Gagliardi, R. Lindh and G. Karlström, J. Chem. Phys., 2004, 121, 4494–4500 CrossRef CAS PubMed.
- G. Karlström, R. Lindh, P.-Å. Malmqvist, B. O. Roos, U. Ryde, V. Veryazov, P.-O. Widmark, M. Cossi, B. Schimmelpfennig, P. Neogrady and L. Seijo, Comput. Mater. Sci., 2003, 28, 222–239 CrossRef.
- F. Aquilante, L. De Vico, N. Ferré, G. Ghigo, P.-Å. Malmqvist, P. Neogrády, T. B. Pedersen, M. Pitoňák, M. Reiher, B. O. Roos, L. Serrano-Andrés, M. Urban, V. Veryazov and R. Lindh, J. Comput. Chem., 2010, 31, 224–247 CrossRef CAS PubMed.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. Von Ragué Schleyer, J. Comput. Chem., 1983, 4, 294–301 CrossRef CAS.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS.
- Dalton, a molecular electronic structure program, Release Dalton2015, see http://daltonprogram.org/.
- K. Aidas, C. Angeli, K. L. Bak, V. Bakken, R. Bast, L. Boman, O. Christiansen, R. Cimiraglia, S. Coriani, P. Dahle, E. K. Dalskov, U. Ekström, T. Enevoldsen, J. J. Eriksen, P. Ettenhuber, B. Fernández, L. Ferrighi, H. Fliegl, L. Frediani, K. Hald, A. Halkier, C. Hättig, H. Heiberg, T. Helgaker, A. C. Hennum, H. Hettema, E. Hjertenæs, S. Høst, I.-M. Høyvik, M. F. Iozzi, B. Jansík, H. J. Aa. Jensen, D. Jonsson, P. Jørgensen, J. Kauczor, S. Kirpekar, T. Kjærgaard, W. Klopper, S. Knecht, R. Kobayashi, H. Koch, J. Kongsted, A. Krapp, K. Kristensen, A. Ligabue, O. B. Lutnæs, J. I. Melo, K. V. Mikkelsen, R. H. Myhre, C. Neiss, C. B. Nielsen, P. Norman, J. Olsen, J. M. H. Olsen, A. Osted, M. J. Packer, F. Pawlowski, T. B. Pedersen, P. F. Provasi, S. Reine, Z. Rinkevicius, T. A. Ruden, K. Ruud, V. V. Rybkin, P. Sałek, C. C. M. Samson, A. S. de Merás, T. Saue, S. P. A. Sauer, B. Schimmelpfennig, K. Sneskov, A. H. Steindal, K. O. Sylvester-Hvid, P. R. Taylor, A. M. Teale, E. I. Tellgren, D. P. Tew, A. J. Thorvaldsen, L. Thøgersen, O. Vahtras, M. A. Watson, D. J. D. Wilson, M. Ziolkowski and H. Ågren, WIREs Comput. Mol. Sci., 2014, 4, 269–284 CrossRef CAS PubMed.
- The PE library developers, PElib: The Polarizable Embedding library (development version), 2015, http://gitlab.com/pe-software/pelib-public.
-
B. Gao, Gen1Int (version 0.2.1), 2012, http://gitlab.com/bingao/gen1int Search PubMed.
- B. Gao, A. J. Thorvaldsen and K. Ruud, Int. J. Quantum Chem., 2011, 111, 858–872 CrossRef CAS.
-
R. Bast, XCint library, 2016, http://dftlibs.org/xcint/ Search PubMed.
- U. Ekström, L. Visscher, R. Bast, A. J. Thorvaldsen and K. Ruud, J. Chem. Theory Comput., 2010, 6, 1971–1980 CrossRef PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- M. J. G. Peach, P. Benfield, T. Helgaker and D. J. Tozer, J. Chem. Phys., 2008, 128, 044118 CrossRef PubMed.
- N. H. List, J. M. Olsen, T. Rocha-Rinza, O. Christiansen and J. Kongsted, Int. J. Quantum Chem., 2012, 112, 789–800 CrossRef CAS.
- D. H. Friese, M. T. P. Beerepoot and K. Ruud, J. Chem. Phys., 2014, 141, 204103 CrossRef PubMed.
- M. T. P. Beerepoot, D. H. Friese, N. H. List, J. Kongsted and K. Ruud, Phys. Chem. Chem. Phys., 2015, 17, 19306–19314 RSC.
- A. Pikulska, A. H. Steindal, M. T. P. Beerepoot and M. Pecul, J. Phys. Chem. B, 2015, 119, 3377–3386 CrossRef CAS PubMed.
- D. M. Philipp and R. A. Friesner, J. Comput. Chem., 1999, 20, 1468–1494 CrossRef CAS.
- R. B. Murphy, D. M. Philipp and R. A. Friesner, J. Comput. Chem., 2000, 21, 1442–1457 CrossRef CAS.
- O. A. von Lilienfeld, I. Tavernelli, U. Rothlisberger and D. Sebastiani, Phys. Rev. Lett., 2004, 93, 153004 CrossRef PubMed.
- O. A. von Lilienfeld, I. Tavernelli, U. Rothlisberger and D. Sebastiani, J. Chem. Phys., 2005, 122, 014113 CrossRef PubMed.
- C. Daday, C. Curutchet, A. Sinicropi, B. Mennucci and C. Filippi, J. Chem. Theory Comput., 2015, 11, 4825–4839 CrossRef CAS PubMed.
- N. H. List, J. M. H. Olsen, H. J. Aa. Jensen, A. H. Steindal and J. Kongsted, J. Phys. Chem. Lett., 2012, 3, 3513–3521 CrossRef CAS PubMed.
- J. Bednarska, A. Roztoczyńska, W. Bartkowiak and R. Zaleśny, Chem. Phys. Lett., 2013, 584, 58–62 CrossRef CAS.
- M. Drobizhev, N. S. Makarov, S. E. Tillo, T. E. Hughes and A. Rebane, Nat. Methods, 2011, 8, 393–399 CrossRef CAS PubMed.
- E. Kamarchik and A. I. Krylov, J. Phys. Chem. Lett., 2011, 2, 488–492 CrossRef CAS.
- N. Lin, Y. Luo, K. Ruud, X. Zhao, F. Santoro and A. Rizzo, ChemPhysChem, 2011, 12, 3392–3403 CrossRef CAS PubMed.
- R. Nifosì and Y. Luo, J. Phys. Chem. B, 2007, 111, 14043–14050 CrossRef PubMed.
- J. M. H. Olsen, C. Steinmann, K. Ruud and J. Kongsted, J. Phys. Chem. A, 2015, 119, 5344–5355 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Numerical data (excitation energies and MPA strengths) for the results presented in the figures and in the text. See DOI: 10.1039/c6cp05297e |
| ‡ Present address: Division of Theoretical Chemistry and Biology, School of Biotechnology, KTH Royal Institute of Technology, SE-106 91, Stockholm, Sweden. |
§ A multi-index k is an ordered 3-tuple of nonnegative integers that are associated with a component of a Cartesian coordinate as indicated by the subscript, i.e. k = (kx, ky, kz). The norm and factorial of a multi-index are defined as |k| = kx + ky + kz and k! = kx!·ky!·kz!, respectively. The multi-index partial derivative operator is defined as 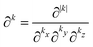 . Summing over a multi-index norm, e.g. |k|, implies a summation over all multi-indices whose norm equals |k|.54 . Summing over a multi-index norm, e.g. |k|, implies a summation over all multi-indices whose norm equals |k|.54 |
|
| This journal is © the Owner Societies 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Magnus
Ringholm
a,
Nanna Holmgaard
List‡
b,
Kenneth
Ruud
a,
Jacob
Kongsted
b and
Jógvan Magnus Haugaard
Olsen
a,
Magnus
Ringholm
a,
Nanna Holmgaard
List‡
b,
Kenneth
Ruud
a,
Jacob
Kongsted
b and
Jógvan Magnus Haugaard
Olsen

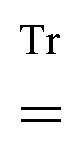 denotes tracing of each term on the right-hand side, the one-electron matrix h describes the KS kinetic energy and the electron–nuclear attraction, the two-electron matrix Gγ contains the electronic Coulomb and γ-fractional exchange interactions, Exc[ρ(D)] is the exchange–correlation energy written as a functional of the density which in turn depends on the density matrix, and hnuc is the nuclear interaction energy. We will not go into further detail about these standard terms but will instead turn our attention to the PE energy.
denotes tracing of each term on the right-hand side, the one-electron matrix h describes the KS kinetic energy and the electron–nuclear attraction, the two-electron matrix Gγ contains the electronic Coulomb and γ-fractional exchange interactions, Exc[ρ(D)] is the exchange–correlation energy written as a functional of the density which in turn depends on the density matrix, and hnuc is the nuclear interaction energy. We will not go into further detail about these standard terms but will instead turn our attention to the PE energy.
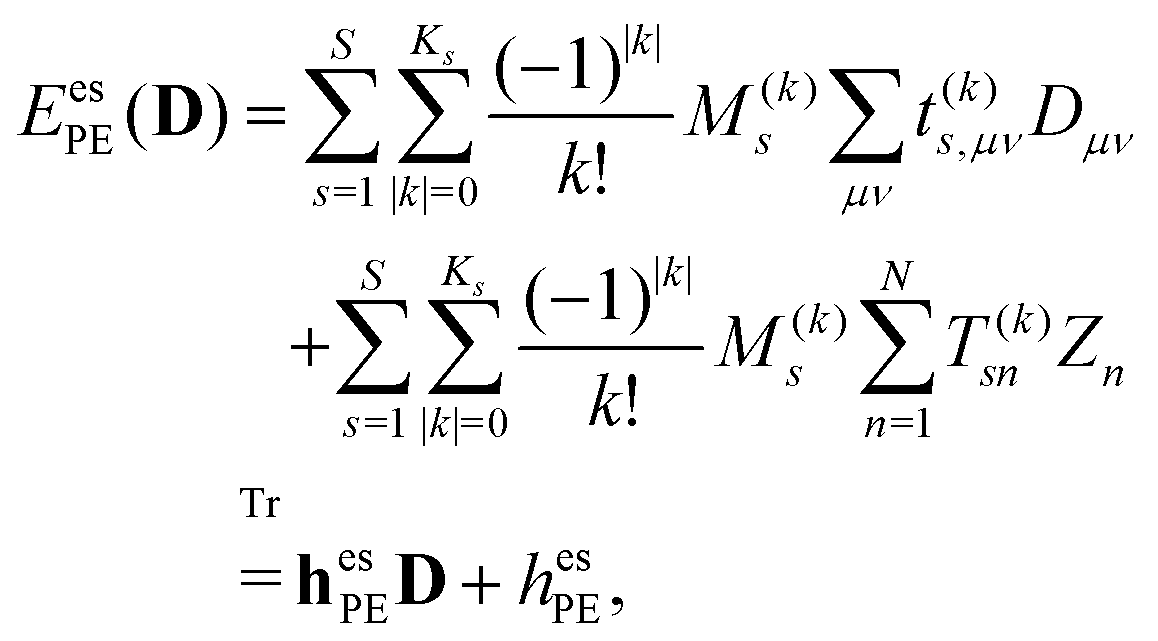
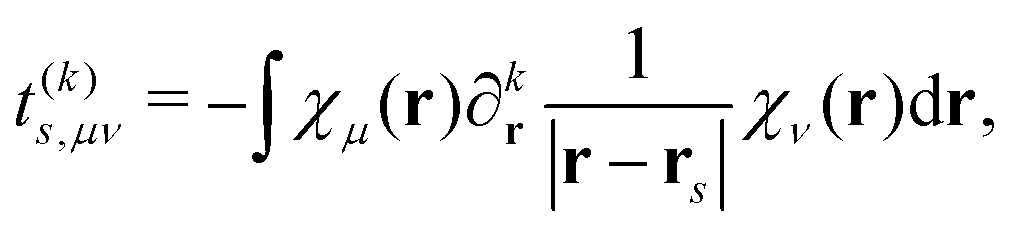
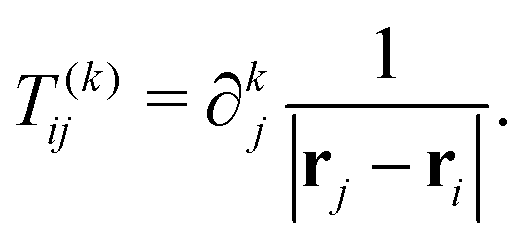
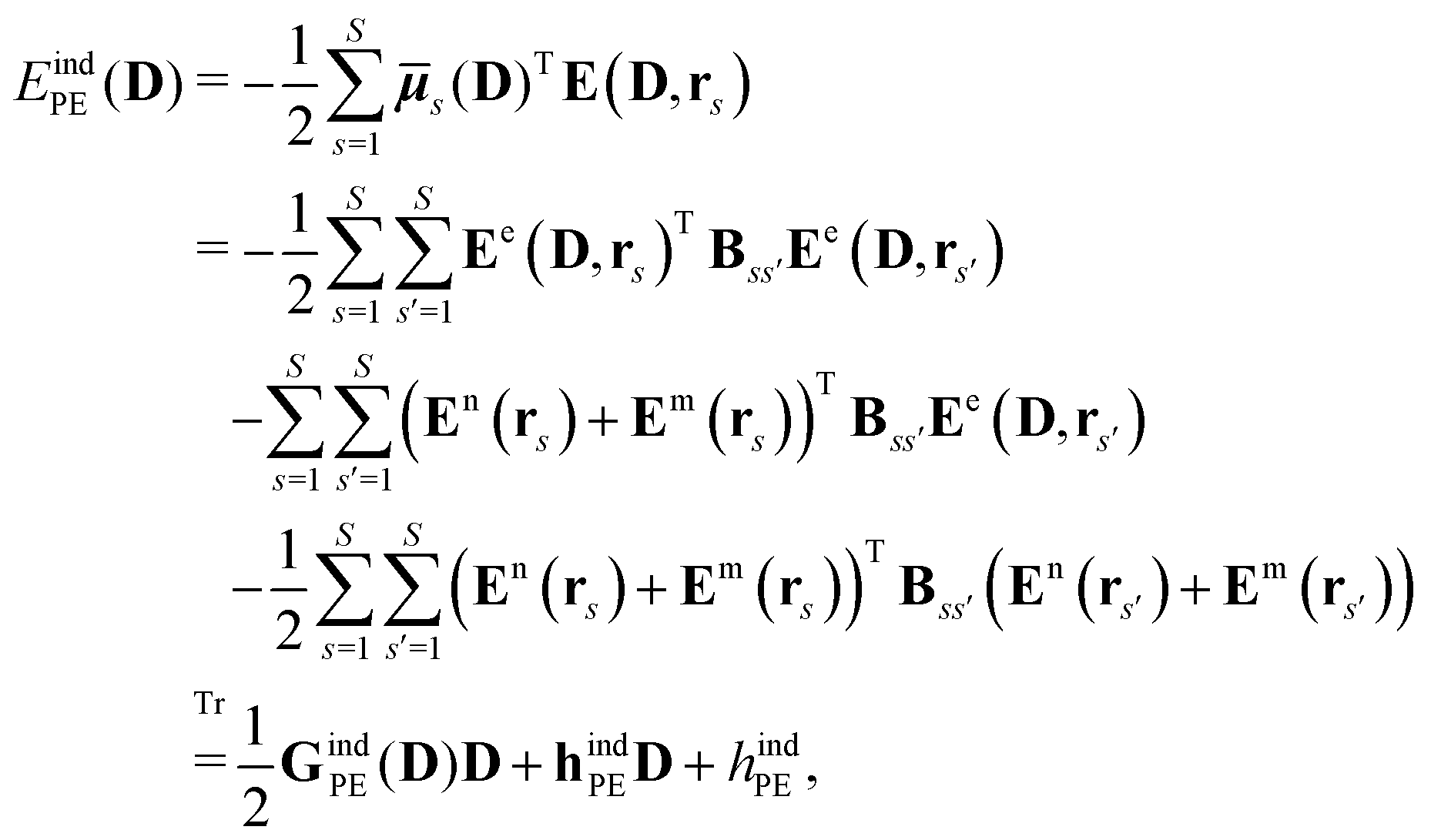
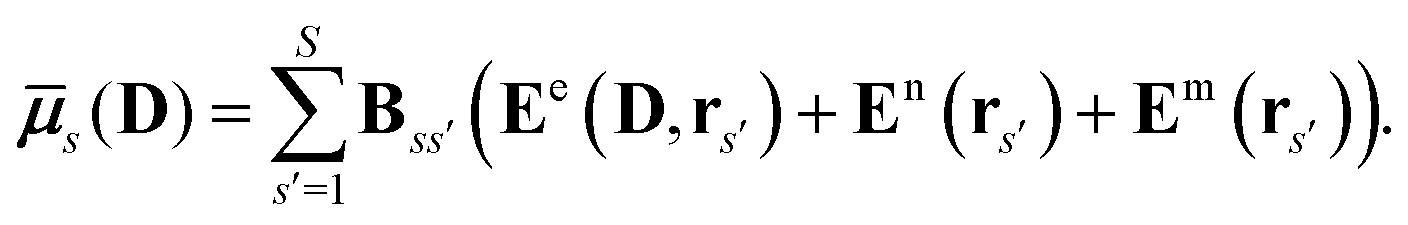
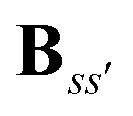 matrices that are 3 × 3 subblocks of the (symmetric) classical linear response matrix,55
matrices that are 3 × 3 subblocks of the (symmetric) classical linear response matrix,55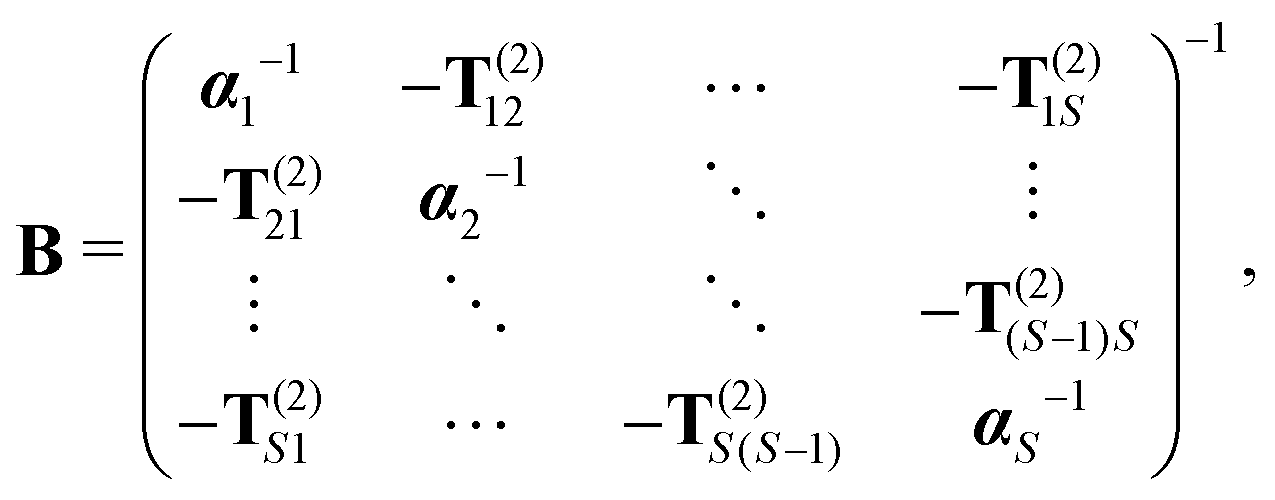
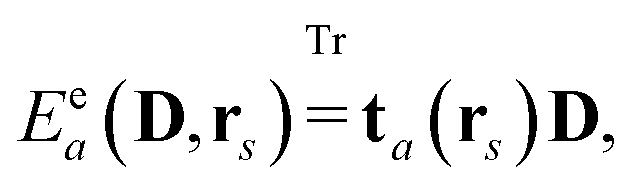
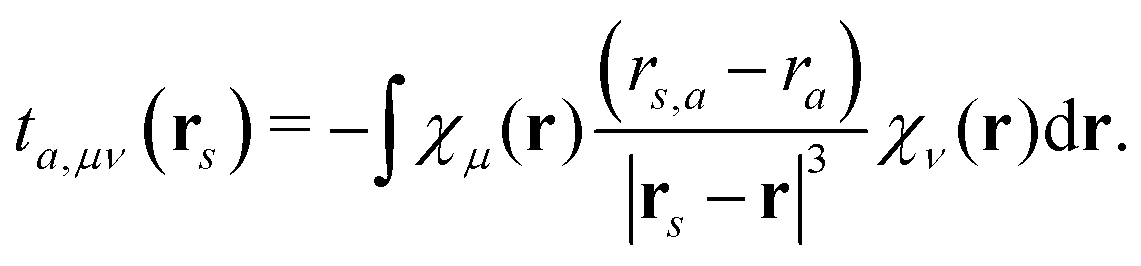
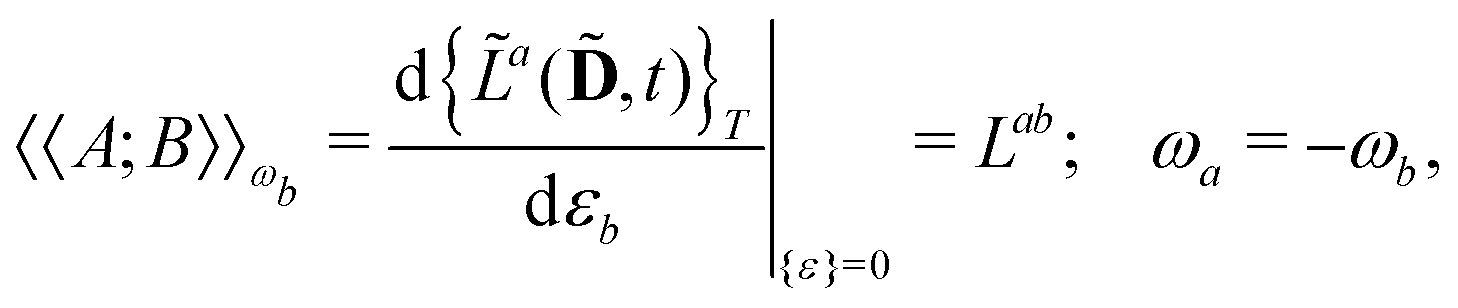
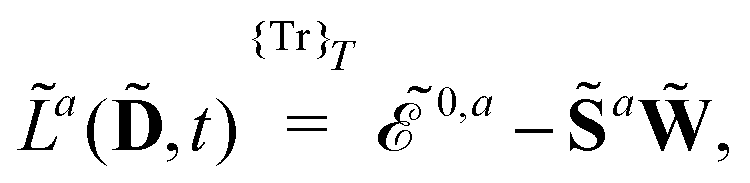
 indicates that the trace and time-average of each term on the right-hand side are taken, and where we introduced the overlap matrix
indicates that the trace and time-average of each term on the right-hand side are taken, and where we introduced the overlap matrix 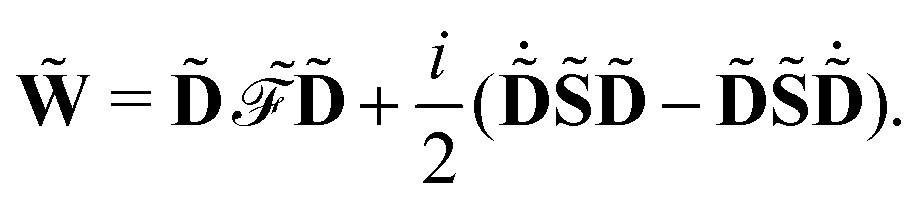
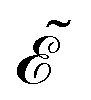 and KS matrix
and KS matrix 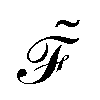 , given by
, given by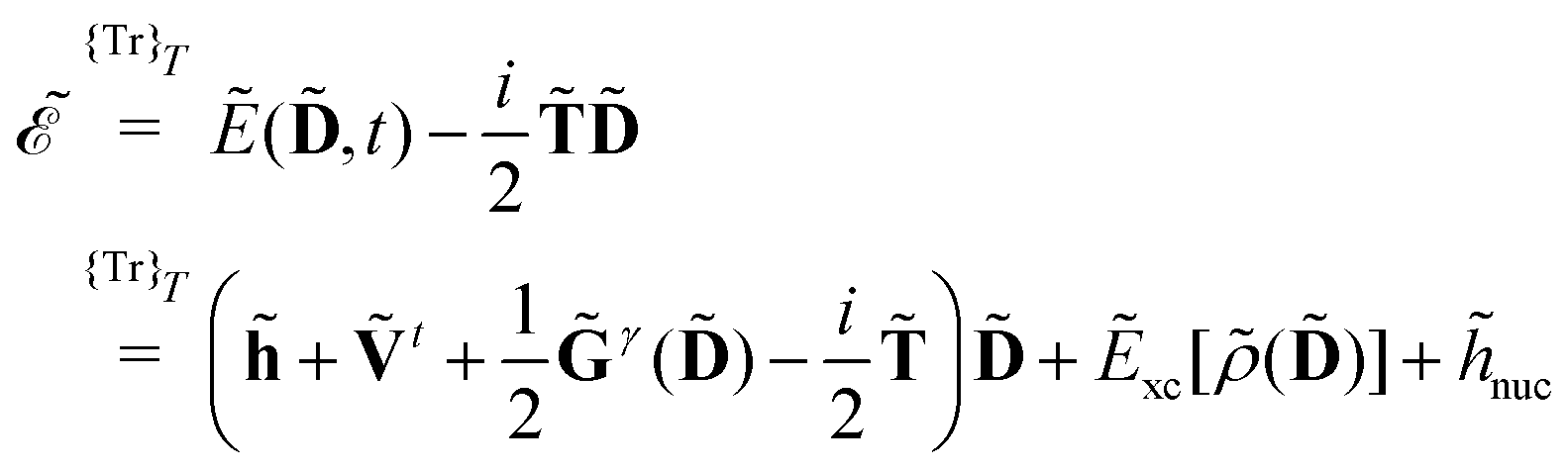
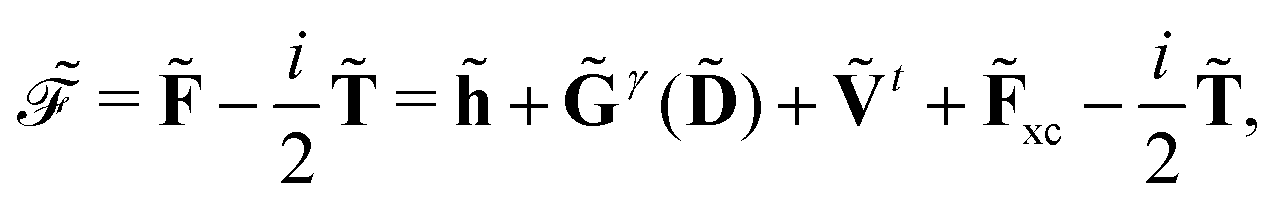

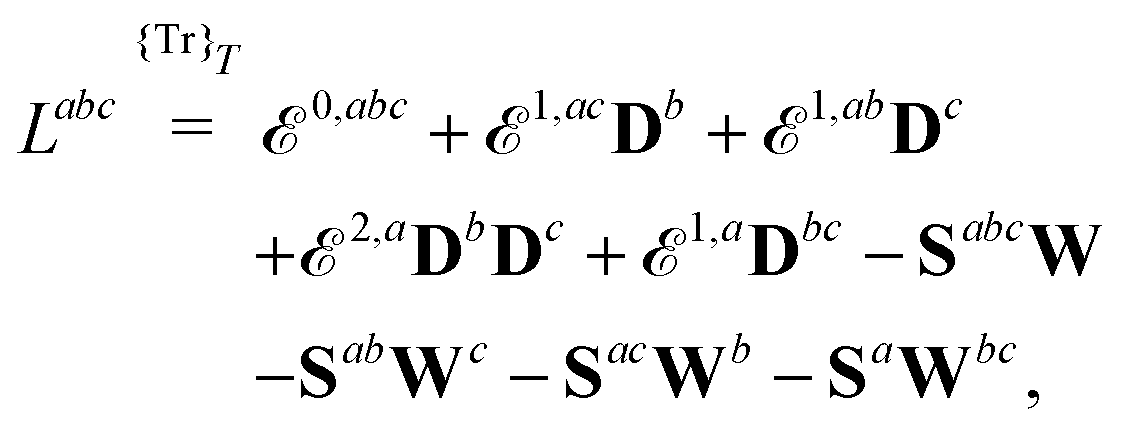

![[M with combining tilde]](https://www.rsc.org/images/entities/b_char_004d_0303.gif) ]⊖ =
]⊖ = 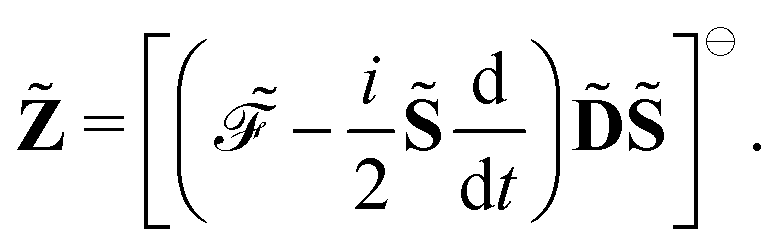
![[small lambda, Greek, tilde]](https://www.rsc.org/images/entities/b_i_char_e0df.gif) a = [
a = [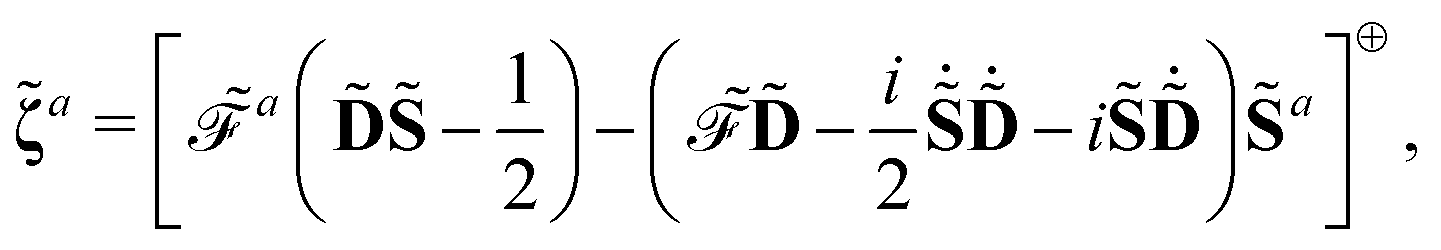
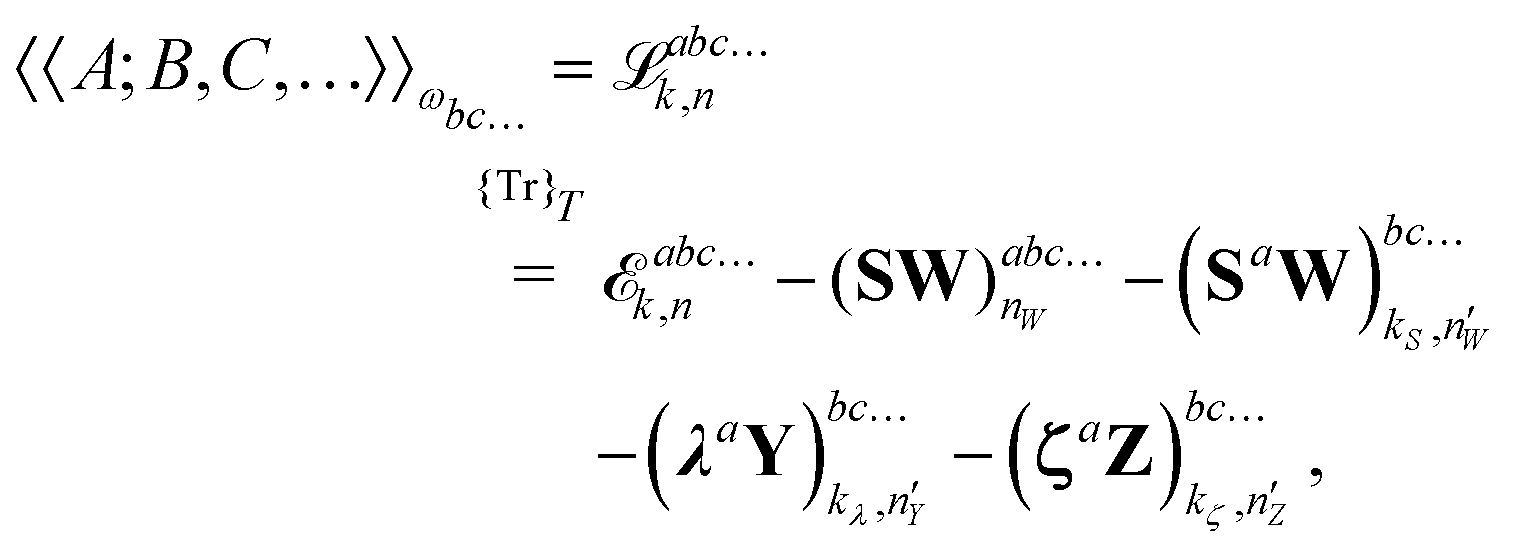
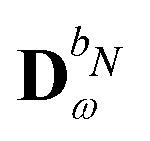 and
and  , respectively. The algorithmic approach rests on the separation of
, respectively. The algorithmic approach rests on the separation of 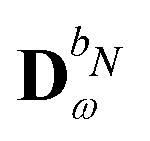 into particular and homogeneous parts
into particular and homogeneous parts 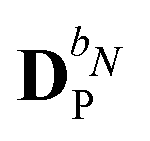 and
and 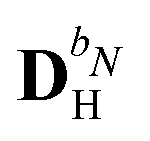 , respectively, as
, respectively, as
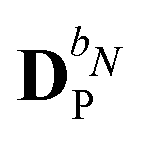 and
and 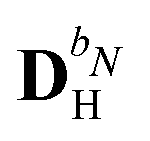 are determined at different stages during the execution of the algorithm. The perturbed KS matrix
are determined at different stages during the execution of the algorithm. The perturbed KS matrix 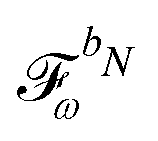 can be partitioned as
can be partitioned as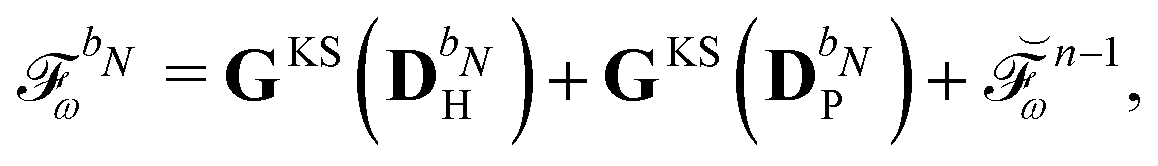
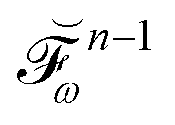 contains all contributions to
contains all contributions to  that do not depend on
that do not depend on  . Through an approach that involves solving a linear equation system involving
. Through an approach that involves solving a linear equation system involving 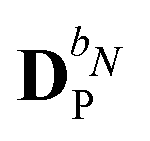 to obtain parameters that can be used to determine
to obtain parameters that can be used to determine 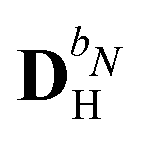 , the
, the 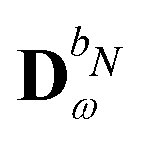 and
and 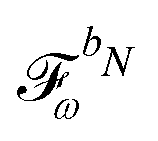 matrices of interest can be obtained.
matrices of interest can be obtained.
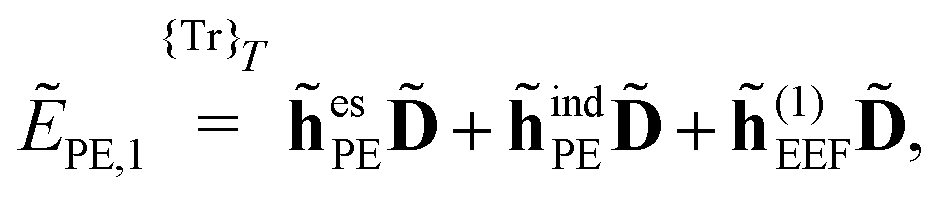
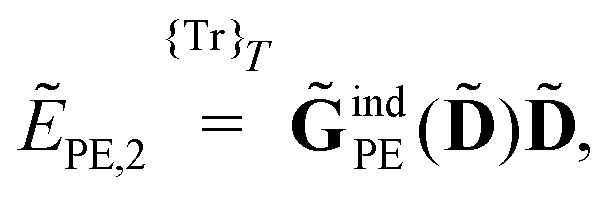
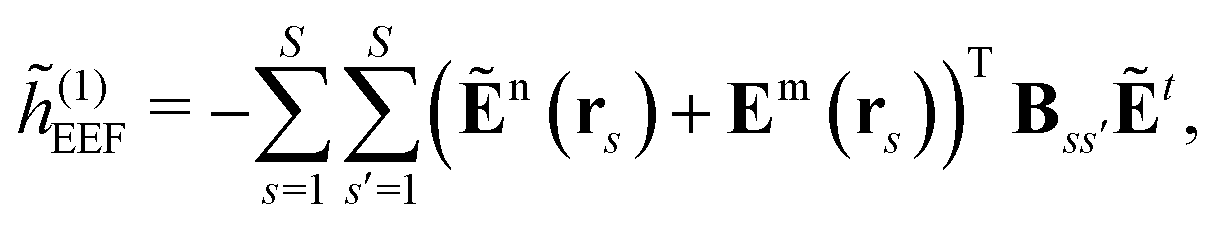
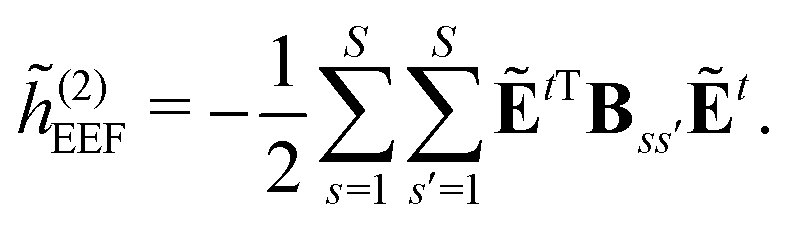
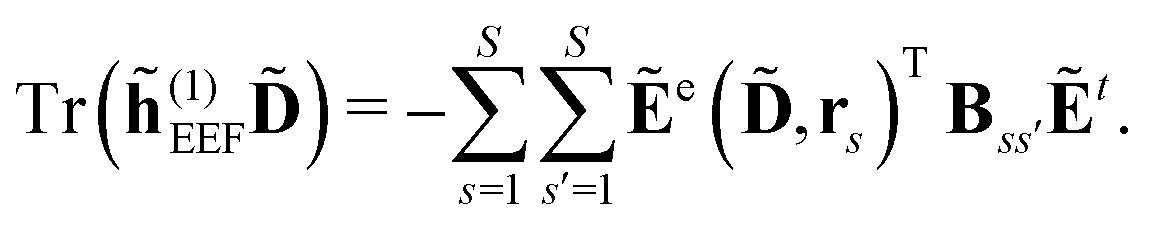
![[G with combining tilde]](https://www.rsc.org/images/entities/b_char_0047_0303.gif) indPE(
indPE(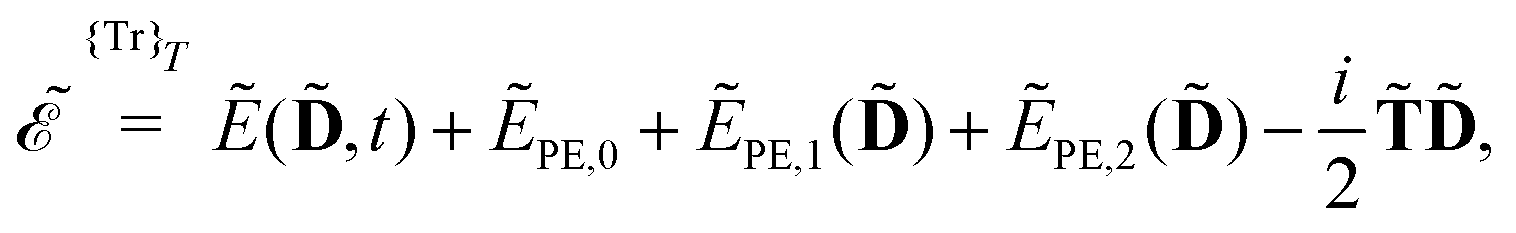
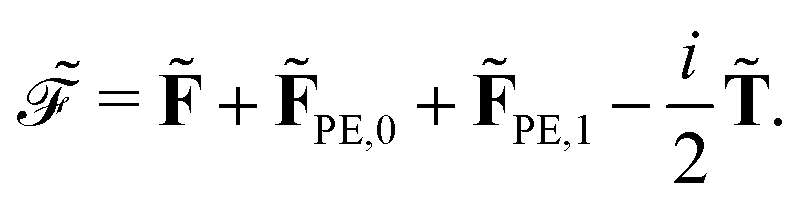
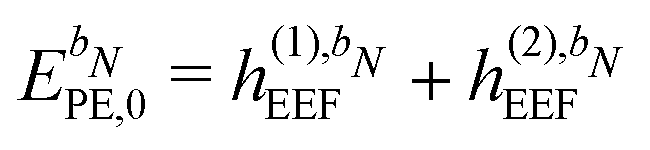
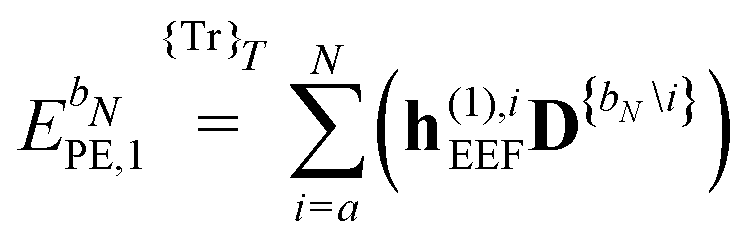
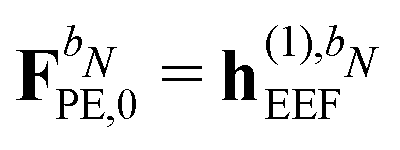
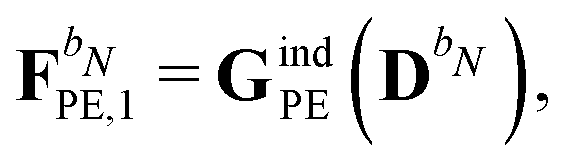
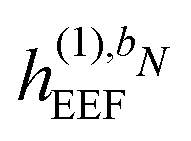 and
and 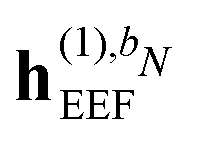 are zero if N > 1,
are zero if N > 1, 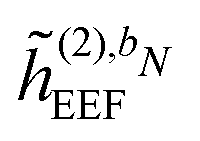 is zero if N > 2, and where the summation in eqn (43) runs over all perturbations in bN. In eqn (43), the collection {bN\i} denotes the bN perturbation tuple except perturbation i, and the terms in this equation may be subject to further truncation depending on the choice of (k, n) rule. Consequently, for electric response properties, the inclusion of a polarizable environment in the response property scheme of Section 2.2 is accomplished relatively straightforwardly by calculating the terms in eqn (42)–(45) and adding them in the appropriate places.
is zero if N > 2, and where the summation in eqn (43) runs over all perturbations in bN. In eqn (43), the collection {bN\i} denotes the bN perturbation tuple except perturbation i, and the terms in this equation may be subject to further truncation depending on the choice of (k, n) rule. Consequently, for electric response properties, the inclusion of a polarizable environment in the response property scheme of Section 2.2 is accomplished relatively straightforwardly by calculating the terms in eqn (42)–(45) and adding them in the appropriate places.

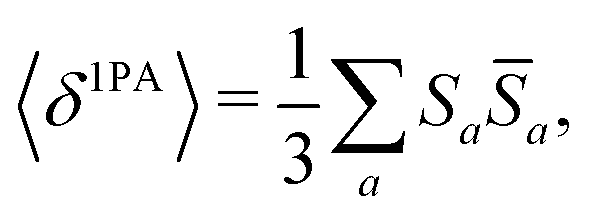
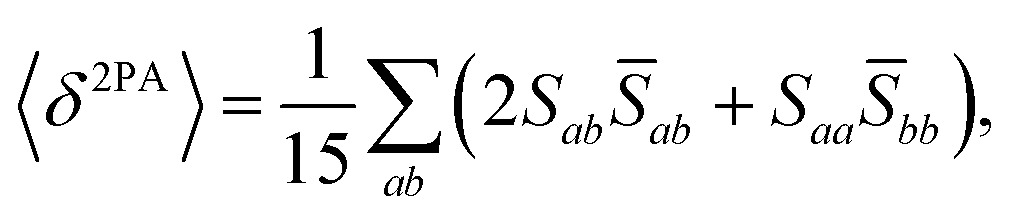
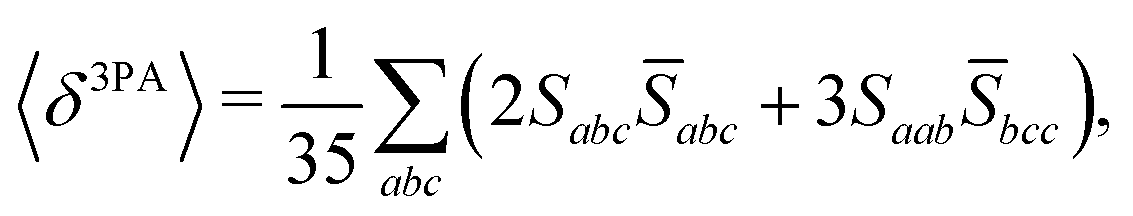
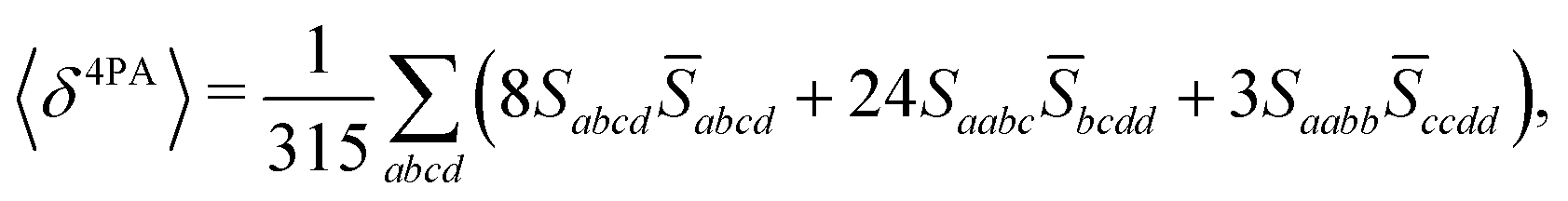
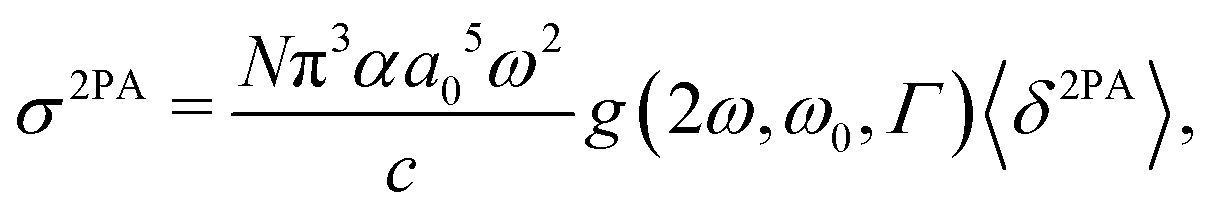
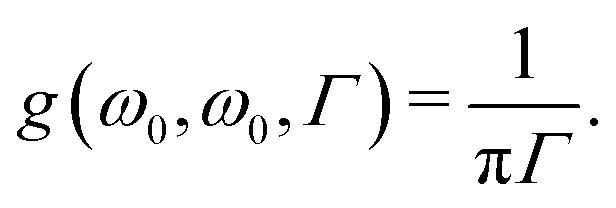
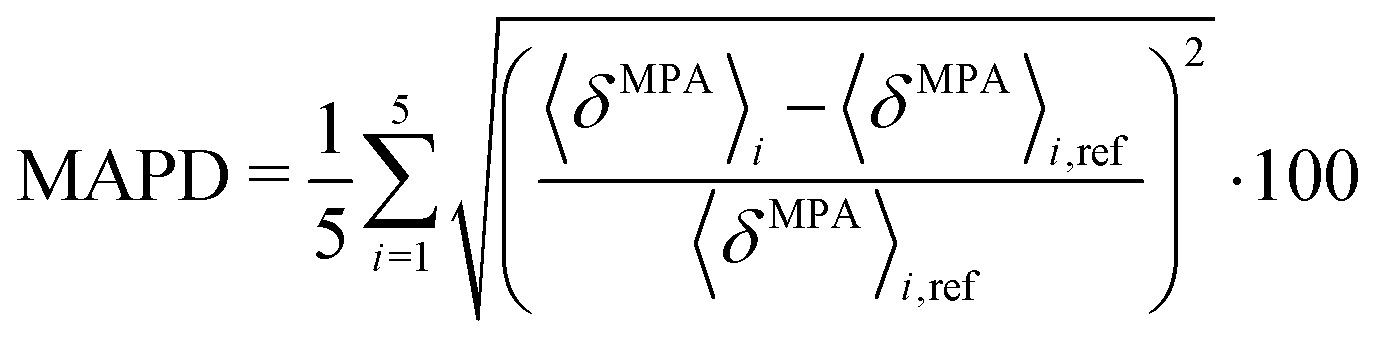

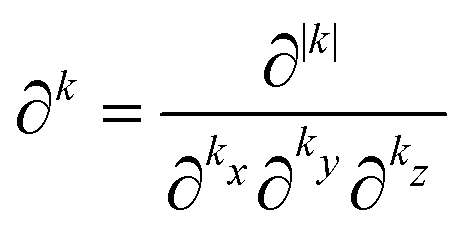 . Summing over a multi-index norm, e.g. |k|, implies a summation over all multi-indices whose norm equals |k|.54
. Summing over a multi-index norm, e.g. |k|, implies a summation over all multi-indices whose norm equals |k|.54