
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular origin of photoluminescence of carbon dots: aggregation-induced orange-red emission†
Venkatesh
Gude
,
Ananya
Das
,
Tanmay
Chatterjee
and
Prasun K.
Mandal
*
Department of Chemical Sciences, Indian Institute of Science Education and Research (IISER) Kolkata, Mohanpur, West Bengal 741246, India. E-mail: prasunchem@iiserkol.ac.in
First published on 16th September 2016
Abstract
The molecular origin of the photoluminescence of carbon dots (CDs) is not known. This restricts the design of CDs with desired optical properties. We have synthesized CDs starting from carbohydrates by employing a simple synthesis method. We were able to demonstrate that the CDs are composed of aggregated hydroxymethylfurfural (HMF) derivatives. The optical properties of these CDs are quite unique. These CDs exhibit an excitation-independent PL emission maximum in the orange-red region (λmaxem ∼ 590 nm). These CDs also exhibit excitation as well as monitoring wavelength-independent single exponential PL decay. These observations indicate that only one type of chromophore (HMF derivative) is present within the CDs. Several HMF derivatives are aggregated within the CDs; therefore, the aggregated structure cause a large Stokes shift (∼150 nm). By several control experiments, we showed that the same aggregated chromophore unit (HMF derivative), and not the individual fluorophores, is the fluorescing unit. The emission maximum and the single exponential PL lifetime are independent of the polarity of the medium. The existence of a low-lying trap state could be reduced quite significantly. A model has been proposed to explain the interesting steady state and dynamical photoluminescence behaviour of the CDs. As the molecular origin of their photoluminescence is known, CDs with desired optical properties can be designed.
Introduction
Carbon dots (CDs) are recognized as excellent, zero-dimensional bio-compatible and non-toxic nanomaterials in comparison to semi-conductor quantum dots for bio-imaging applications.1 However, most literature reports focus on CDs that emit blue or green light, limiting their scope for bioapplications.2–5 Hence, it is necessary to prepare CDs that emit orange or red light.6,7 Control over the emission wavelength can be achieved only when the molecular origin of CD photoluminescence is understood. Understanding the molecular origin of this phenomenon is possible when starting from small precursor molecules, using simple experimental conditions, so that the mechanism of formation of the CDs can be experimentally verified. A few reports have described polymeric/graphitic units as being responsible for the PL of CDs.4,5,8,9 However, no detailed description of the mechanism of formation of the species responsible for the PL of CDs has been reported. Hence, it is very pertinent to investigate the molecular origin of the PL of CDs.From an optical spectroscopic point of view, most literature reports describe excitation wavelength-dependent emission maxima.10 Incomplete solvation,11 the presence of trap states12 and the red edge effect13 have been shown to be responsible for the observed behaviour. In most literature reports, the PL decay has been shown to be multi-exponential in nature.2–5 The applicability of CDs with multi-exponential PL decay is highly restricted in experiments such as FRET and FLIM. As the molecular origin of the PL of CDs is not known, the multi-exponential nature of PL decay cannot be understood. In a recent report, the presence of fluorescent dyes in CDs was proposed as a reason why CDs exhibit PL.9,14 However, if the fluorescence of the dye is responsible for the PL of CDs, the purpose of using CDs as nanomaterials showing PL is lost. There are other reports showing that unsaturated ligands/functional groups are responsible for the PL of CDs.9,15 However, from the spectroscopic point of view, this claim does not have any standing.
In this manuscript, we report the synthesis of orange-red-emitting CDs from inexpensive non-toxic chemicals such as sucrose, fructose and glucose using inexpensive laboratory equipment. The size of the CDs is less than 5 nm. It was shown that aggregated hydroxymethylfurfural (HMF) derivatives are responsible for the PL of the CDs. The mechanism of formation of furfural derivatives from sucrose, fructose, etc. was provided. Support for the molecular origin was provided by NMR spectroscopy, MS, etc. The mechanism of formation of the aggregated structures was provided. Unlike most literature reports, the PL emission maximum is independent of the excitation wavelength, and the PL decay is single exponential in nature.6,12,14,16 Several control experiments have been performed to support the molecular origin of the PL as well as the optical behaviour of the CDs.
Synthesis of CDs
2 g of sucrose, 2 mL of water and 4 mL of H3PO4 were placed in a two-neck round bottom flask and heated at 70 °C for 10 minutes (Scheme 1). During this time, the colourless solution first became yellow, then became brown, and finally turned black. After cooling the reaction mixture to ambient temperature, it was neutralized with dilute NaOH solution (checked with pH paper). This neutralized solution was extracted with dichloromethane (DCM). The dark yellow organic layer was separated. Excess solvent was distilled using a rotary evaporator. A slurry was prepared using silica gel and subjected to purification by column chromatography, using DCM as the eluent. The colour of the column-purified CDs solution was faint brown under sunlight; when exposed to UV light of 365 nm, the solution exhibited orange-red emission, as shown in Scheme 1.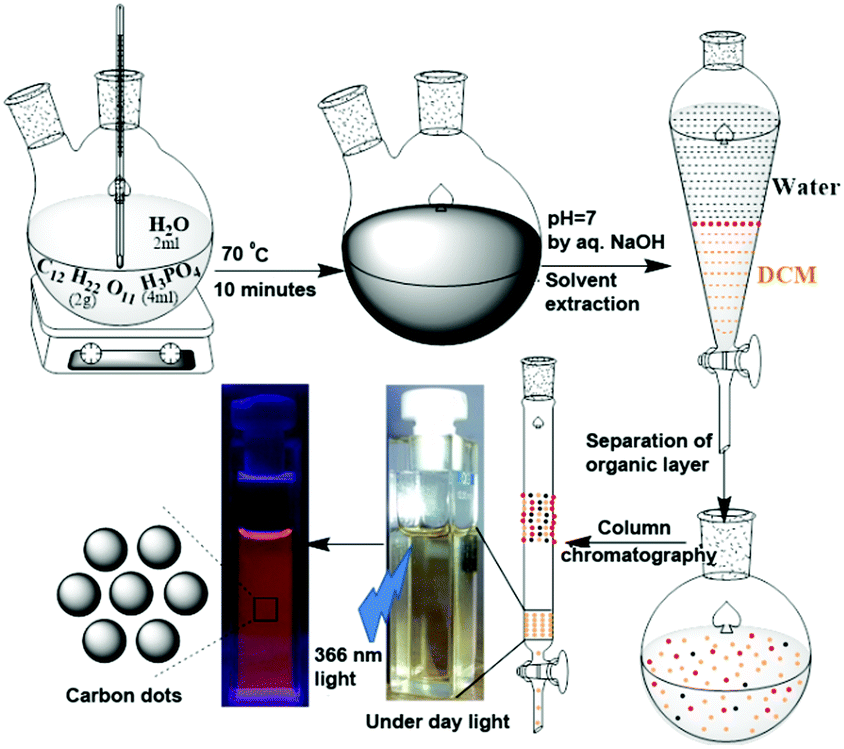 | ||
| Scheme 1 Preparation and purification of orange-red emitting carbon dots using sucrose and H3PO4. | ||
Results and discussion
The column-purified CDs (sucrose-derived CDs, abbreviated as S-CDs) were characterized by spectroscopic techniques. The FT-IR spectrum of the purified S-CDs is shown in Fig. 1a. The peak at 3125 cm−1 corresponds to sp2 (–C–H); the peaks at 2923 and 2852 cm−1 correspond to the asymmetric and symmetric stretching of sp3 (–C–H), respectively; the intense peak at 1668 cm−1 can be assigned to conjugated carbonyl functional groups; the peaks at 1588 and 1519 cm−1 are due to the –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C– functional group; and the peaks in the region of 1390 to 1000 cm−1 are due to various modes of the –C–O–C– functional group. Thus, the presence of unsaturated sp2 carbons, –C–H, CO and –C–O–C– ether linkages are demonstrated.
C– functional group; and the peaks in the region of 1390 to 1000 cm−1 are due to various modes of the –C–O–C– functional group. Thus, the presence of unsaturated sp2 carbons, –C–H, CO and –C–O–C– ether linkages are demonstrated.
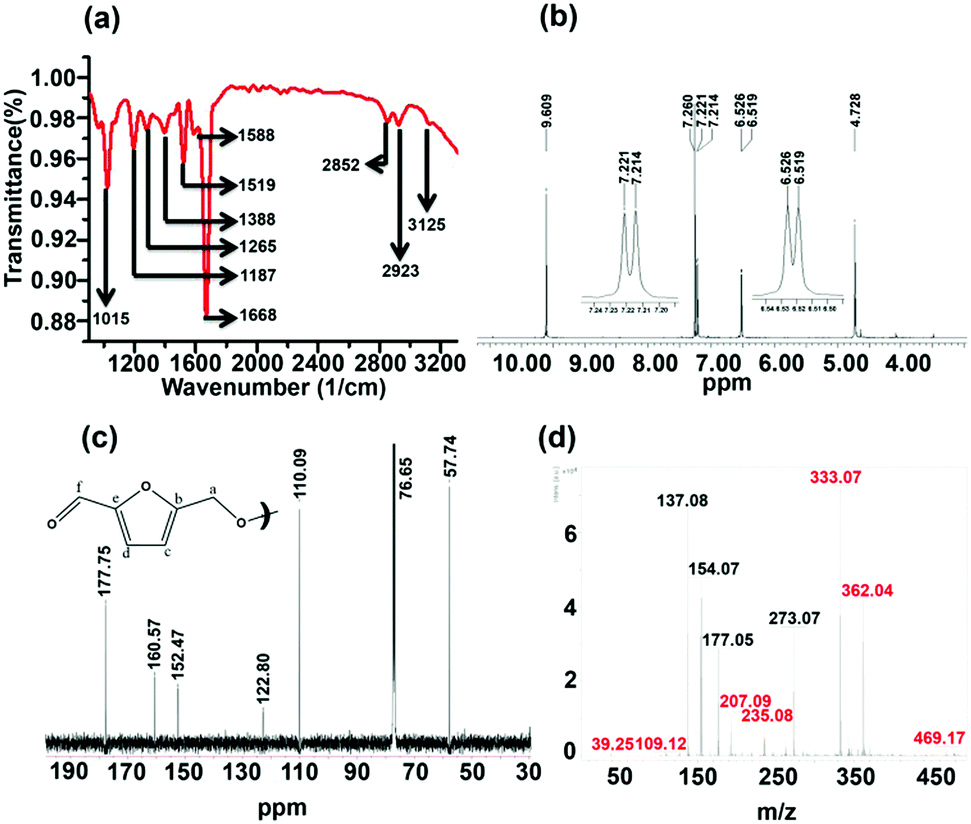 | ||
| Fig. 1 (a) FT-IR, (b) 1H NMR, (c) 13C NMR and (d) MALDI-mass spectra of sucrose-derived CDs (S-CDs). | ||
The 1H NMR spectrum of the S-CDs was recorded using a 500 MHz spectrometer in CDCl3 solvent and is shown in Fig. 1b. The chemical shift (δ) value at 4.728 ppm is due to the –OCH2 group protons; the signals at 6.519 and 6.526 ppm (d, J = 3.5 Hz) and 7.214 and 7.221 ppm (d, J = 3.5 Hz) correspond to the protons attached to sp2 (aromatic) carbons. The equal magnitudes of the coupling constants (J) indicate that these protons are influenced by the spin of each other. Further, the ‘J’ value of 3.5 Hz hints that those protons are not attached to the aromatic phenyl ring. However, the J value of 3.5 Hz could be due to protons attached to a furan-type system. The peak at 9.609 ppm indicates that the proton is attached to a carbonyl group. It is known that the dehydration of carbohydrate molecules such as sucrose, glucose and fructose in acidic medium produces 5-hydroxymethylfurfural (HMF) as a major component.17 The NMR spectrum of the S-CDs matches quite well with the reported NMR spectrum of HMF17 in the region from 4.5 to 10 ppm, except for the expected signal for the proton of the hydroxyl (–OH) group, which lies between 5 and 6 ppm. This indicates that the S-CDs are composed of an HMF derivative in which the –OH functional group is absent but the aldehyde functional group is present.
The 13C NMR spectrum of the CDs was recorded in CDCl3 solvent and is shown in Fig. 1c. From this spectrum, it can be found that there are 6 different types of carbons present in the CDs. The peak at 57.74 ppm indicates a carbon which is connected to an electronegative element such as O; this is assigned as ‘a’ (Fig. 1c). The peak centred at 76.65 ppm corresponds to the carbon present in CDCl3. The peaks present in the region of 110 to 160 ppm correspond to sp2 (–CC–) carbons. The peaks at 110.09 and 122.80 ppm are assigned as ‘c’ and ‘d’, and the peaks at 152.47 and 160.57 ppm are assigned as ‘b’ and ‘e’ (Fig. 1c). The peak at 177.75 ppm corresponds to the carbonyl carbon and is assigned as ‘f’. This 13C NMR spectrum completely matches that of the reported 13C NMR spectrum of HMF.18 Thus, from the 1H and 13C NMR analyses, it can be understood that the CDs are composed of an HMF derivative, and the absence of the hydroxyl proton (in 1H NMR) hints that the HMF derivative must be symmetric in nature; it is perhaps formed by the removal of one water molecule from two HMF molecules.
The validity of the above claim can be understood by mass spectral analysis. In order to investigate the molecular weight of the S-CDs, the Matrix Assisted Laser Desorption Ionization (MALDI) technique was used. Here, we preferred 2,5-dihydroxy benzoic acid (DHB) as the matrix. The DHB mass spectra are shown in Fig. S2 (ESI†). The m/z values corresponding to the matrix are shown in Table S1 (Fig. S4, ESI†). The mass spectra of the S-CDs with the DHB matrix are shown in Fig. S3 (ESI†). No prominent signal is observed above 500 m/z. This proves that the CDs are not composed of very large molecular structures; this indicates that the S-CDs are not polymeric in nature, nor do they contain a graphitic core. A pertinent magnified portion of the mass spectrum is shown in Fig. 1d. Signals related to the CDs have been extracted (after comparison with the DHB signals) and are marked in red (Fig. 1d and Table S1, ESI†).
Considering the synthetic procedure, NMR and mass spectral analyses, we propose a mechanism for the formation of the HMF derivative (Scheme 2). It is known from the literature that hydrolysis of sucrose produces glucose and fructose. Glucose undergoes isomerization to form fructose, which further produces a greater percentage of HMF under acidic conditions.19–21 Although the precursor molecule is sucrose, fructose is the component that is responsible for the formation of the HMF derivative under the experimental conditions. Here, H3PO4 acts as a dehydrating agent and produces H+ ions which act as a catalyst; therefore, this reaction is considered to be acid-catalyzed dehydration. HMF consists of two reactive functional groups, a primary alcoholic and an aldehyde group. The NMR analysis suggests that the CD structure contains the aldehyde group. In acidic medium, the hydroxyl group of HMF interacts with fructose, leading to the formation of the HMF derivative. This HMF derivative can also be produced from two HMF molecules, as shown in Scheme 2. This HMF derivative is symmetrical in structure. Because of the formation of this ether linkage between two HMF molecules, no –OH proton signal is observed in the 1H NMR spectrum between 5 to 6 ppm; rather, the presence of a C–O–C bond was indicated in the IR spectrum. The molecular weight of this HMF derivative is 234, which exactly matches the observed mass spectroscopic signal obtained at 235 [M − H+] (Fig. 1d). The fragmented chemical species responsible for the lower m/z values of the mass spectra shown in Fig. 1d and Fig. S3 (ESI†) have been depicted in Scheme 3. The m/z values of 333 and 362 could be assigned to S-CDs associated with the matrix. Similar association/adduct formation has been reported in the literature.22,23 The m/z value of 469 [2M − H+] corresponds to the dimer of the HMF derivative [m/z = 234]. Thus, from the IR, NMR and mass spectral analyses, it can be concluded that an HMF derivative is present in the S-CDs.
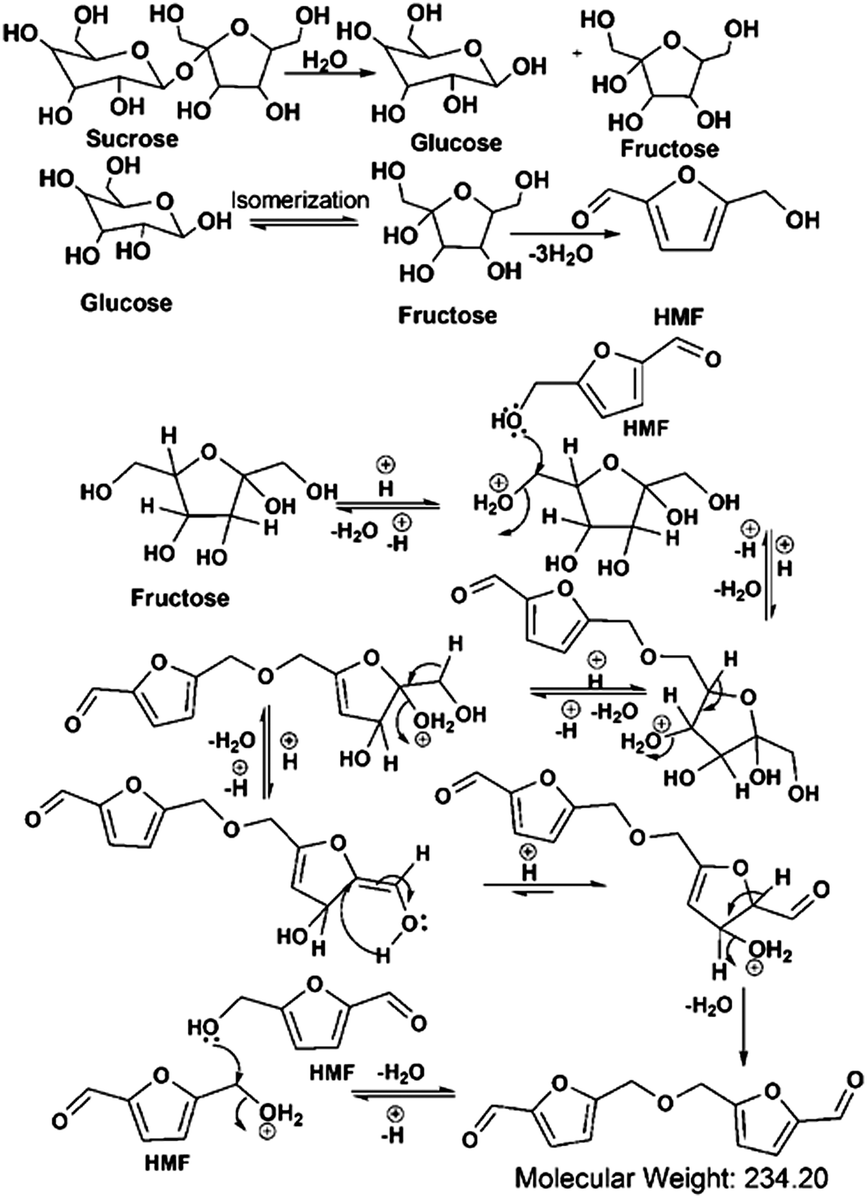 | ||
| Scheme 2 Mechanism of formation of the HMF derivative in carbon dots using sucrose and H3PO4. | ||
 | ||
| Scheme 3 Fragmentation pattern of the chromophore of the carbon dots and its adduct formation with the matrix. | ||
A transmission electron microscopy (TEM) image of the S-CDs is shown in Fig. 2a; it indicates that the S-CD particles are spherical in shape. An HRTEM image is shown in the inset of this figure. A distinct lattice spacing of 0.37 nm is shown in the inset. The size distribution histogram obtained from the TEM images is shown in Fig. 2b. The average size of the particles is around 4.5 nm. The lattice spacing (∼0.37 nm) closely matches the (002) interlayer spacing value of graphite.2–5
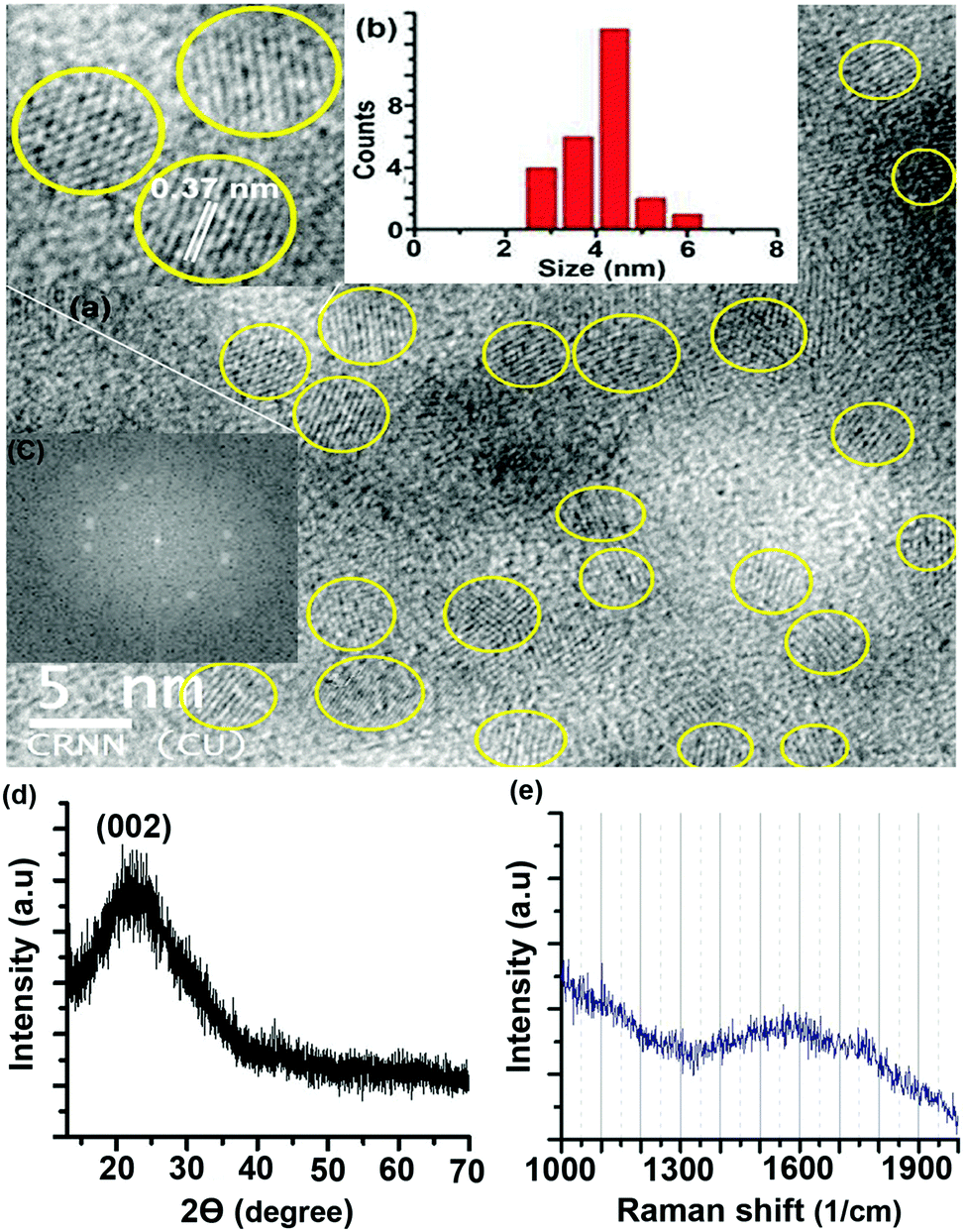 | ||
| Fig. 2 (a) TEM image (HRTEM image in inset), (b) size distribution histogram, (c) fast Fourier transform pattern of the HRTEM image, (d) XRD pattern and (e) Raman spectrum of the S-CDs. | ||
The fast Fourier transform (FFT) pattern of the HRTEM image is shown in Fig. 2c. The bright spot pattern clearly indicates crystallinity or regular order among the constituents of the CD. The X-ray diffraction (XRD) pattern of the S-CDs (Fig. 2d) shows a broad peak centred at 23°, which corresponds to a d-spacing value of around 0.37 nm. This XRD pattern matches well with literature reports of CDs obtained from different precursors and synthesis methods with different PL emission colours.5,7,24,25 The d-spacing value obtained from XRD exactly matches the lattice spacing value calculated from the TEM image. This d-spacing value (0.37 nm) is very close to 0.33 nm, which corresponds to the (002) plane of bulk graphite. The Raman spectrum of the S-CDs (Fig. 2e) shows a broad peak in the region of 1300 to 1900 cm−1 with a maximum at 1550 cm−1, which corresponds to the –CC– stretching vibration of the HMF derivative. The broadness of the band indicates the presence of other functional groups attached to sp2-C atoms. Similar broad Raman bands for CDs have been reported in the literature.24,25 It is also reported that the –CC– stretching mode frequency of furfural derivatives is observed in the region of 1500 to 1550 cm−1 in the Raman spectrum.26 In the present case, the maximum at 1550 cm−1 of the broad Raman spectrum of the CDs corresponds to the C(sp2)–C(sp2) stretching motion of the HMF derivative. Thus, the presence of the HMF derivative in the S-CDs is confirmed from IR, NMR, MS and Raman spectroscopic techniques. HMF derivatives can form aggregated structures through non-covalent interactions such as dipole–dipole, π–π stacking, and van der Waals interactions (Fig. 3); similar aggregation has been hypothesized in the literature.26–28
 | ||
| Fig. 3 Plausible aggregation pattern of HMF derivatives in the S-CDs. | ||
The geometry-optimized structure of the HMF derivative is shown in Fig. S5 (ESI†). As can be seen from this figure, two furfural moieties are anti-parallel to each other. It is known that the presence of a polar group at the end an organic molecule results in an anti-parallel short range order among the constituents, even in the solid state or in the condensed phase.29,30 The HMF derivative contains a terminal polar aldehyde functional group; hence, a short range anti-parallel orientation can be assumed to form between the molecules (Fig. 3). Thus, from the synthesis mechanism and characterisation, it can be seen that the CDs are composed of aggregated HMF derivatives. Earlier researchers speculated that when carbohydrates are used, furfural derivatives may polymerize to form CDs; however, no proof was given.31 We have shown that the S-CDs do not have a polymeric furfural structure; rather, they are composed of aggregated HMF derivatives.
It has been reported in the literature that when H3PO4 is used, P-doping can occur;32 however, in our case, according to a 31P NMR experiment (Fig. S6, ESI†) and to the mass spectra, no indication of P-doping could be observed. The energy dispersive analysis by X-rays (EDAX) spectrum of the S-CDs (recorded on an Au-coated silicon wafer) (Fig. S7, ESI†) indicates the presence of carbon and oxygen atoms but not P in the CDs.
Absorption spectra of the S-CDs in two different solvents are shown in Fig. 4a. The absorption spectra have two bands, one from 250 to 300 nm and another from 350 to 450 nm. We performed a series of control experiments in order to understand the origin of the two absorption bands. As can be seen from the ESI† (Fig. S8a), the dilute solution of furfural has an absorption band from 250 to 300 nm. As the concentration of furfural increases, a new absorption band at a higher wavelength, extending up to 500 nm, appears.
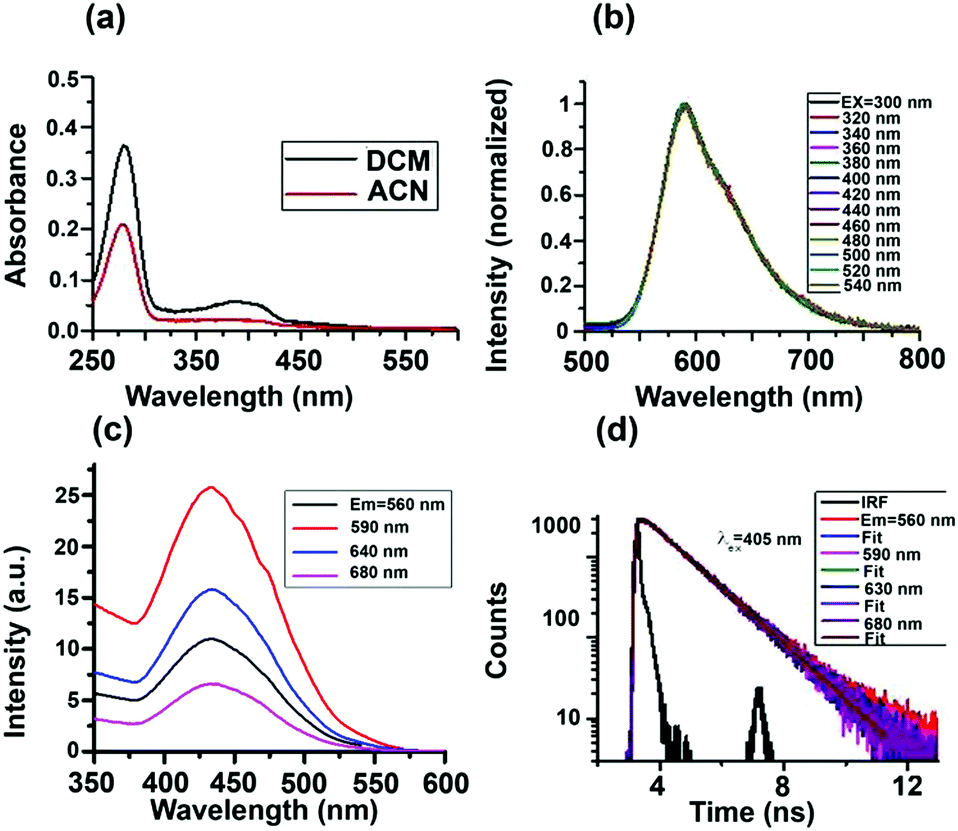 | ||
| Fig. 4 (a) Absorption, (b) emission, (c) excitation and (d) PL decay curves of the S-CDs in DCM. | ||
Comparing the absorption spectra of the S-CDs with the control experiment of different concentrations of furfural, we can conclude that absorption band of S-CDs in the region from 250 to 300 nm corresponds to the furfural moiety (π–π*), and the higher wavelength absorption band corresponds to aggregated forms of furfural or the HMF derivative.
Intensity normalised emission spectra of the S-CDs in DCM at different excitation wavelengths are shown in Fig. 4b. Unlike the majority of literature reports,2–5 the S-CDs exhibited excitation wavelength-independent emission. When the excitation wavelength is below 300 nm, although the absorbance is high, the emission intensity of the S-CDs (as well as that of the control experiment with furfural) is very low. However, when excited above 350 nm, the emission intensity of the S-CDs (as well as that of the control experiment with furfural, Fig. S8b, ESI†) increases; the maximum intensity is obtained when the excitation wavelength is ∼420 to 440 nm. The emission maximum remains fixed at 590 nm for the S-CDs irrespective of the excitation wavelength (Fig. 4b). Thus, our observation is that at lower concentrations of furfural, feeble emission is observed; meanwhile, for the aggregated furfural/HMF derivative, strong emission is observed. We recorded the excitation spectra for both the S-CDs and the control experiment. The excitation spectra are independent of the monitoring wavelength, with an excitation maximum at ∼440 nm (Fig. 4c). The excitation wavelength-independent emission spectra and the monitoring wavelength-independent excitation spectra indicate a single type of chromophoric structure. Moreover, emission is obtained when the concentration is quite high for both the control experiments and the S-CDs.
Also, unlike semiconductor quantum dots, the S-CDs demonstrate a considerable Stokes shift of 150 nm (excitation maximum = 440 nm and emission maximum = 590 nm). Because the HMF derivative has no charge transfer donor–acceptor moiety and no extended conjugation is present, it is expected that as the polarity of the medium is changed from low polarity DCM to high polarity ACN, the emission maximum will not change. We have observed that the excitation-independent emission maxima in both DCM and ACN are ∼590 nm (Fig. S10, ESI†). Thus, no polarity-dependent shift of the emission maximum is noted. In the absence of a charge transfer moiety or extended conjugation, the only possible reason for this large Stokes shift could be aggregation-induced emission.33,34 From these observations, we can conclude that the S-CDs are composed of a single type of aggregated chromophoric (HMF derivative) structure. Thus, the optical spectroscopic results support our model depicted in Fig. 3.
The fluorescence quantum yield (ϕFl) of the S-CDs was measured to be 15% (using coumarin 153 in ethanol as a standard). The reported ϕFl value of furfural is only ∼10−5.35 Thus, the steady state optical spectroscopic results support our aggregation-induced fluorescence model quite well. If our model of one type of dye (HMF derivative) aggregation-induced emission is correct, then in addition to excitation-independent emission, we should observe single exponential PL decay. This is exactly what was observed. The PL decay of the S-CDs in DCM is depicted in Fig. 4d and Fig. S9, ESI† (for ACN, see Fig. S10, ESI†). As can be seen from these figures, the PL decay is both excitation and monitoring wavelength independent (see also Table 1). These observations further strengthen the facts that the emission is induced by aggregation of the same HMF derivative and that there is no significant low-lying trap state emission.
| Solvent | λ em (nm) | Φ (%) | λ ex (nm) | Monitoring λem (nm) | τ (ns) |
|---|---|---|---|---|---|
| DCM | 590 | 15 | 405 | 680 | 1.26 |
| 630 | 1.25 | ||||
| 590 | 1.24 | ||||
| 560 | 1.29 | ||||
| 375 | 680 | 1.26 | |||
| 630 | 1.26 | ||||
| 590 | 1.26 | ||||
| 560 | 1.31 | ||||
| 340 | 590 | 1.23 | |||
| 281 | 590 | 1.24 | |||
| 405 | 680 | 1.16 | |||
| 630 | 1.15 | ||||
| 590 | 1.15 | ||||
| 560 | 1.16 | ||||
| ACN | 586 | 10 | 375 | 680 | 1.19 |
| 630 | 1.20 | ||||
| 590 | 1.19 | ||||
| 560 | 1.20 | ||||
| 340 | 590 | 1.16 | |||
| 281 | 590 | 1.30 |
Literature reports show that glucose, fructose and sucrose all produce HMF as their dehydration products. From the synthesis point of view, if the HMF derivative is the molecular aggregating unit for the S-CDs, then when either fructose or glucose is used as the starting material, we should obtain the same orange-red emitting CDs. Indeed, when we started from fructose and glucose, we obtained the same orange-red emitting CDs with exactly the same NMR, MS, TEM, and optical spectroscopic characterisation results (Fig. S12–S16, ESI†).
It is important to discuss why in most literature reports, CDs exhibit excitation wavelength-dependent emission maxima and multi-exponential PL decay. As can be seen from Scheme 4, if there are multiple species and they interact with trap states then as the absorption and emission from all these species are different, hence for different excitation wavelengths contribution of these species will be different towards emission and hence PL emission maximum will be dependent on excitation wavelength. Moreover, as the species are different, they will choose different pathways of PL decay. Furthermore, interaction of the excited states of these species with trap states will open different excited state decay pathways. Thus, the PL decay is expected to be multi-exponential in nature (Scheme 4a and b).
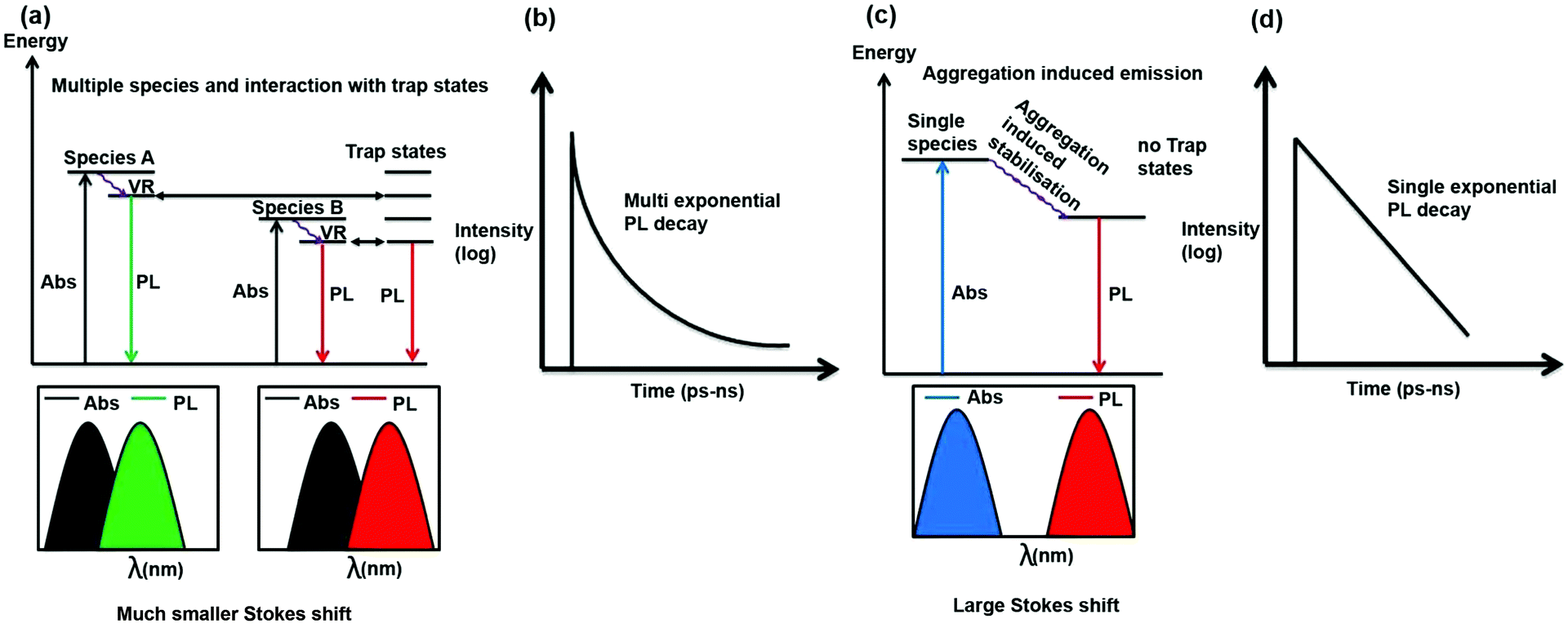 | ||
| Scheme 4 Schematic showing the cause of the excitation-dependent PL emission maximum and multi-exponential PL decay (a and b). It is also shown that when the PL emission is independent of the excitation wavelength, the PL decay will be single exponential and a large Stokes shift will be observed (c and d). | ||
In the present case, there is only one type of fluorophore; hence, the emission maximum of the S-CDs is fixed. Thus, the PL emission maximum is excitation wavelength-independent and the PL decay is single exponential in nature (Scheme 4c and d). From a spectroscopic point of view, it is interesting to note that there is a significant Stokes shift (150 nm) in the present case. It is quite well known that for aggregation-induced emission,33,36 the Stokes shift is significantly large because of aggregation-induced energy stabilisation of the excited state. A similar magnitude of large Stokes shift (160 to 180 nm) has not only been observed in dyes but also in dyes that aggregate to form nanoparticles.37,38 If we look carefully at the excitation spectrum (Fig. 4c) instead of the absorption spectrum (the former being more sensitive than the latter), we will notice that the tail of the excitation spectrum is extended to 550 nm. However, in the excitation spectrum, the excitation maximum also remains the same (i.e. ∼440 nm) as that of the absorption spectrum. No new band is observed in the excitation spectrum. Thus, a large Stokes shift (∼150 nm) confirms that the emission is aggregation induced. PL decay is dependent on excitation wavelength only when more than one species or molecule is present in the ground state or there is incomplete solvation.11 In our case, we can show beyond a doubt that a single aggregated species is responsible for the PL of the CDs. A single species is responsible for the PL with a single absorption spectrum, and there is no new excited state phenomenon (similarity of absorption and excitation spectra); therefore, the PL decay is independent of the wavelength. Please also recall that unlike most literature reports, the PL decay is single exponential in nature. This further confirms that a single species is present in the excited state.
Excitation wavelength-dependent PL emission maxima and multi-exponential PL decay are possible for a few reasons: for example, if more than one species is present in the ground/excited state, or if there is incomplete solvation. In our case, there is clearly a single aggregated species in both the ground and excited states. The PL lifetime of the CDs is 1.2 ns. It is well documented in literature that in normal solvents with very low viscosity, such as ACN, the solvation is complete within 500 ps. Therefore, incomplete solvation is not a possibility. In our case, we monitored the entire range of the PL emission spectrum with a pulsed laser of ∼60 ps IRF (instrument response function), and we did not observe any signature of slow solvation. This confirms that there is no solvation with a time constant above 100 ps. Please recall that the PL lifetime of our CDs is 1.2 ns. Hence, incomplete solvation is not a possibility. Therefore, we have observed an excitation-independent emission maximum (following Kasha's rule), excitation-independent PL decay and single exponential PL decay.
We would like to point out here the S-CDs not only exhibit an excitation wavelength-independent PL emission maximum and excitation and monitoring wavelength-independent single exponential PL decay, but also exhibit an excitation power-independent PL emission maximum as well as an excitation power-independent single exponential PL decay and lifetime. This has been depicted in Fig. S11 (ESI†).
Conclusions
In conclusion, we have prepared orange-red emitting CDs from readily available sources such as sucrose, fructose and glucose at a low temperature of 70 °C. These CDs are around 5 nm in diameter. Based on several rigorous experimental analyses, we have proved that these CDs are composed of aggregated HMF derivatives. The presence of phosphorous in the S-CDs was not observed. The optical properties of these CDs are quite unique. These CDs exhibit an excitation wavelength-independent PL emission maximum (∼590 nm) and excitation as well as monitoring wavelength-independent single exponential PL decay. These observations point to the fact that only one type of chromophore (HMF derivative) is present within the CDs. Because several HMF derivatives are aggregated within the CDs, the aggregated structure causes a large Stokes shift (∼150 nm). By several control experiments, we showed that the same aggregated chromophore unit, not the individual fluorophores, is the fluorescing unit. The emission maximum and the single exponential PL lifetime are independent of the polarity of the medium. The existence of a low-lying trap state could be reduced quite significantly. A model has been proposed to explain the interesting steady state and dynamical photoluminescence behaviour of the CDs. As the molecular origin of their PL is known, CDs with desired optical properties can be designed.Acknowledgements
PKM thanks IISER-Kolkata for financial help and instrumental facilities. Support from the Fast-Track project (SR/FT/CS-52/2011) of DST-India is gratefully acknowledged. VG thanks IISER-Kolkata, AD and TC thank CSIR-India for their respective fellowships.Notes and references
- V. Georgakilas, J. A. Perman, J. Tucek and R. Zboril, Chem. Rev., 2015, 115, 4744–4822 CrossRef CAS PubMed.
- Y. Song, S. Zhu and B. Yang, RSC Adv., 2014, 4, 27184–27200 RSC.
- P. G. Luo, S. Sahu, S. T. Yang, S. K. Sonkar, J. Wang, H. Wang, G. E. LeCroy, L. Cao and Y. P. Sun, J. Mater. Chem. B, 2013, 1, 2116–2127 RSC.
- S. Y. Lim, W. Shen and Z. Gao, Chem. Soc. Rev., 2015, 44, 362–381 RSC.
- H. Li, Z. Kang, Y. Liu and S. T. Lee, J. Mater. Chem., 2012, 22, 24230–24253 RSC.
- K. Jiang, S. Sun, L. Zhang, Y. Lu, A. Wu, C. Cai and H. Lin, Angew. Chem., Int. Ed., 2015, 54, 5360–5363 CrossRef CAS PubMed.
- S. K. Bhunia, A. Saha, A. Maity, S. C. Ray and N. R. Jana, Sci. Rep., 2013, 3, 1473 Search PubMed.
- M. O. Dekaliuk, O. Viagin, Y. V. Malyukin and A. P. Demchenko, Phys. Chem. Chem. Phys., 2014, 16, 16075–16084 RSC.
- S. Zhu, Y. Song, X. Zhao, J. Shao, J. Zhang and B. Yang, Nano Res., 2015, 8(2), 355–381 CrossRef CAS.
- M. Fu, F. Ehrat, Y. Wang, K. Z. Milowska, C. Reckmeir, A. L. Rogach, J. K. Stolarczyk, A. S. Urban and J. Feldmann, Nano Lett., 2015, 15, 6030–6035 CrossRef CAS PubMed.
- S. K. Cushing, M. Li, F. Huang and N. Wu, ACS Nano, 2014, 8, 1002–1013 CrossRef CAS PubMed.
- H. Ding, S. B. Yu, J. S. Wei and H. M. Xiong, ACS Nano, 2016, 10, 484–491 CrossRef CAS PubMed.
- S. Khan, A. Gupta, N. C. Verma and C. K. Nandi, Nano Lett., 2015, 15, 8300–8305 CrossRef CAS PubMed.
- Y. Song, S. Zhu, S. Zhang, Y. Fu, L. Wang, X. Zhao and B. Yang, J. Mater. Chem. C, 2015, 3, 5976–5984 RSC.
- L. Wang, S. J. Zhu, H. Y. Wang, S. N. Qu, Y. L. Zhang, J. H. Zhang, Q. D. Chen, H. L. Xu, W. Han, B. Yang and H. B. Sun, ACS Nano, 2014, 8(3), 2541–2547 CrossRef CAS PubMed.
- S. Zhuo, M. Shao and S. T. Lee, ACS Nano, 2012, 6, 1059–1064 CrossRef CAS PubMed.
- K. D. O. Vigier, A. Benguerba, J. Barrault and F. Jerome, Green Chem., 2012, 14, 285–289 RSC.
- G. Novodarszki, N. Retfalvi, G. Dibo, P. Mizsey, E. Csefalvay and L. T. Mika, RSC Adv., 2014, 4, 2081–2088 RSC.
- V. Choudhary, S. H. Mushrif, C. Ho, A. Andrzej, V. Niko-lakis, N. S. Marinkovic, A. I. Frenkel, S. I. Sandler and D. G. Vla-chos, J. Am. Chem. Soc., 2013, 135, 3997–4006 CrossRef CAS PubMed.
- S. P. Teong, G. Yi and Y. Zhang, Green Chem., 2014, 16, 2015–2026 RSC.
- N. Baccile, C. Falco and M. M. Titirici, Green Chem., 2014, 16, 4839–4869 RSC.
- X. Lou, M. Fransen, P. J. M. Stals, T. Mes., R. Bovee, J. J. L. Dongen and E. W. Meijer, J. Am. Soc. Mass Spectrom., 2013, 24, 1405–1412 CrossRef CAS PubMed.
- R. Zenobi and R. Knochenmuss, Mass Spectrom. Rev., 1998, 17, 337–366 CrossRef CAS.
- C. L. Li, C. C. Huang, A. P. Periasamy, P. Roy, W. C. Wu, C. L. Hsu and H. T. Chang, RSC Adv., 2015, 5, 2285–2291 RSC.
- S. Zhu, Q. Meng, L. Wang, J. Zhang, Y. Song, H. Jin, K. Zhang, H. Sun, H. Wang and B. Yang, Angew. Chem., Int. Ed., 2013, 52, 3953–3957 CrossRef CAS PubMed.
- C. Yao, Y. Shin, L. Q. Wang, C. F. Windisch, W. D. Samules, B. W. Arey, C. Wang, W. M. Risen and G. J. Exarhos, J. Phys. Chem. C, 2007, 111, 15141–15145 CAS.
- Y. Shin, L. Q. Wang, I. T. Bae, B. W. Arey and G. J. Exarhos, J. Phys. Chem. C, 2008, 112, 14236–14240 CAS.
- D. Jagadeesan and M. Eswaramoorthy, Chem. – Asian J., 2010, 5, 232–243 CrossRef CAS PubMed.
- R. A. Reddy and B. K. Sadashiva, J. Mater. Chem., 2004, 14, 310–319 RSC.
- B. K. Sadashiva, R. A. Reddy, R. Pratibha and N. V. Madhusudana, J. Mater. Chem., 2002, 12, 943–950 RSC.
- S. Sahu, B. Behera, T. K. Maiti and S. Mohapatra, Chem. Commun., 2012, 48, 8835–8837 RSC.
- Z. Q. Xu, L. Y. Yang, X. Y. Fan, J. C. Jin, J. Mei, W. Peng, F. L. Jiang, Q. Xiao and Y. Liu, Carbon, 2014, 66, 351–360 CrossRef CAS.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC.
- A. Qin and B. Z. Tang, Aggregation-induced emission: fundamentals, John Wiley & Sons Ltd, West Sussex, UK, 2014 Search PubMed.
- A. Gandini, P. A. Hackett and R. A. Back, Can. J. Chem., 1976, 54, 3089–3094 CrossRef CAS.
- L. Yan, Y. Zhang, B. Xu and W. Tian, Nanoscale, 2016, 8, 2471–2487 RSC.
- J. Zhang, R. Chen, Z. Zhu, C. Adachi, X. Zhang and C. S. Lee, ACS Appl. Mater. Interfaces, 2015, 7, 26266–26274 CAS.
- M. Gao, J. Chen, G. Lin, S. Li, L. Wang, A. Qin, Z. Zhao, L. Ren, Y. Wang and B. Z. Tang, ACS Appl. Mater. Interfaces, 2016, 8, 17878–17884 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of experimental and characterization techniques; MALDI-mass spectra, NMR spectra, EDAX spectrum, TEM image, steady state and time resolved PL parameters of carbon dots derived from sucrose, fructose and glucose. See DOI: 10.1039/c6cp05321a |
| This journal is © the Owner Societies 2016 |