
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Strong 1D localization and highly anisotropic electron–hole masses in heavy-halogen functionalized graphenes†
Lukas Eugen
Marsoner Steinkasserer
*,
Alessandra
Zarantonello
and
Beate
Paulus
Institut für Chemie und Biochemie, Freie Universität Berlin, Takustraße 3, D-14195 Berlin, Germany. E-mail: marsoner@zedat.fu-berlin.de
First published on 5th September 2016
Abstract
While halogenation of graphene presents a fascinating avenue to the construction of a chemically and physically diverse class of systems, their application in photovoltaics has been hindered by often prohibitively large optical gaps. Herein we study the effects of partial bromination and chlorination on the structure and optoelectronic properties of both graphane and fluorographene. We find brominated and chlorinated fluorographene derivatives to be as stable as graphane with a detailed investigation of the systems band structure revealing significant 1D localization of the charge carriers as well as strongly electron–hole asymmetric effective masses. Lastly using G0W0 and BSE, we investigate the optical adsorption spectra of the aforementioned materials whose first adsorption peak is shown to lie close to the optimal peak position for photovoltaic applications (≈1.5 eV).
I. Introduction
The need for clean and sustainable energy has become one of the main driving forces of scientific research in the 21st century. Amongst the large number of proposed solutions to the energy crisis, solar cell technology has received a great amount of attention promising to replace large parts of fossil-fuel based energy production in the future. For this to be realized though, the availability of cheap and efficient solar cell materials is of great importance. While classical solar cells are mainly based on silicon, 2D materials, notably graphene1–5 and MoS26–9 have recently emerged as possible alternatives, allowing for the construction of ultrathin photovoltaic devices.10Though graphene itself has sparked the interest of the photovoltaic community, its chemical modifications, most prominently its fully hydrogenated form (graphane11,12) and its fully fluorinated form (fluorographene13,14), have not yet found applications in solar cell technology due to their prohibitively large optical gaps.15–17 Another problem plaguing all possible applications of 2D materials in solar cells is the presence of strongly bound electron–hole pairs (excitons) created upon optical excitation.18,19 To achieve large photocurrents the electron–hole pairs should be easily separable which makes the application of 2D materials to solar cells challenging.
Herein we present a possible solution to both the aforementioned problems via the introduction of heavy halogen atoms into graphane and fluorographene. While, as we will show, these modifications help to significantly redshift the optical adsorption of the aforementioned materials, the strong asymmetry in their charge-carrier masses could be exploited to overcome the problem of strong exciton binding, allowing for an efficient separation of electron–hole pairs and in turn high quantum yields in future solar cells. We will consider in detail some of the main properties required for a viable solar cell i.e. its stability, optical adsorption spectrum and exciton binding energy using single particle methods (DFT) as well as many-body methods i.e. GW20 and the Bethe–Salpeter equation (BSE),21 to accurately account for both electron–electron as well as electron–hole interactions.
Our study is based on earlier work performed by Karlický, Zbořil and Otyepka22 though we extend on the structures proposed by them and take a more in-depth look at the systems structural, electronic as well as optical properties. The systems are based on 1 × 2 supercells of graphane and fluorographene in which every second row of H/F atoms along the zigzag direction has been substituted by Br/Cl and we considered both symmetric as well as asymmetric substitutions and functionalization on only one of the two faces of the graphane/fluorographene layer.
II. Computational details
Structure optimizations on all systems were performed using the CRYSTAL14 program23,24 together with the M06-2X25 functional using the POB-triple-ζ basis set proposed by Peintinger et al.26 In the case of Br and Cl, HSE0327–30 band gap calculations on the relaxed structures were done employing the Stuttgart triple-ζ basis set as modified for use in periodic calculations by Steenbergen et al.,31 together with the associated quasirelativistic pseudopotentials.32,33 For C, F and H, basis sets were constructed according to the procedure described by Usvyat.34 In all cases the description of the vacuum region was enhanced by adding ghost atoms containing a 1 s function with an exponent of 0.06 a0−1, 1 Å above the position of the halogen atoms.Using the obtained structures we performed electronic structure calculations using the GPAW35–37 code. It is well known that DFT alone is unable to accurately describe electron–electron interactions which results in a systematic underestimation of the band gap. To remedy this issue we performed G0W0 calculations based on our DFT results which provides a first-principles description of electron correlation resulting in a significant improvement in the accuracy of the band gap.20,38–41 Still, G0W0 band gaps only allow one to calculate a system's quasiparticle gap, i.e. the difference between the ionization potential and electron affinity. This leaves out interactions in electron–hole pairs created when a system is excited through the adsorption of light. These effects are of particular importance in low-dimensional systems where the screening of the electron–hole Coulomb interaction is significantly reduced as compared to the 3D case.18,19,42,43 To account for both electron–electron as well as electron–hole interactions we therefore employed the BSE method21,42,44 on top of our G0W0 results. BSE is able to account for excitonic effects missing in the G0W0 treatment and we use it to calculate the adsorption spectrum of our systems in the optical limit (q → 0) which is given by the imaginary part of the frequency dependent dielectric function. Both GW45–47 and BSE48,49 calculations have been widely and successfully applied to study the optical properties of materials.
Given the cost of the G0W0 calculations at the dense k-grids needed for well-converged BSE results, BSE calculations following G0W0 were performed employing the scissor approximation i.e. shifting the unoccupied DFT bands by the energy difference between the DFT and G0W0 gap. To avoid confusion, BSE calculations based on DFT orbitals where the unoccupied states have been shifted to reproduce the G0W0 gap will be labeled with the superscript G0W0e.g. BSE@GLLB-SCG0W0. We tested the validity of this approach for two smaller (1 × 1) test systems and found results of full BSE@G0W0 calculations to agree to within 0.05 eV with those obtained by applying the scissor approximation.
Lastly, as will be seen later, the PBE50 functional severely underestimates band gaps for the systems considered herein and, as we suspect, for systems containing strongly localized electrons in general. It therefore provides a poor starting-point for G0W0 calculations which assume the DFT one-particle wavefunctions to be close to the true quasiparticle wavefunctions. A possible solution consists in the use of screened hybrid functionals (e.g. HSE03/HSE06) as a starting point for G0W0 and this approach has already been successfully applied to a number of systems in the literature.16,51 Such calculations are though quite costly computationally as compared to GGA calculations and quickly become prohibitively expensive for larger systems. To circumvent these problems we investigated a low-cost alternative to hybrid functional calculations using the GLLB-SC functional52 which provides a computationally efficient approximation to the EXX-OEP, resulting in a better description of the electronic ground state for the case of highly localized systems.
The GLLB-SC functional can further be used to calculate the quasiparticle band gap of an N-electron system i.e. the difference of the ionization potential and electron affinity as the sum of the Kohn–Sham gap and the derivative discontinuity.52,53 This approach has been shown to give band gaps in excellent agreement with experimental results54 at a computational cost close to that of GGA. GLLB-SC + Δxc gaps would therefore seem the ideal starting point for subsequent G0W0 calculations. As pointed out by Yan et al.44 though inclusion of the derivative discontinuity in the calculation of the dielectric constant at the RPA level, i.e. excluding electron–hole interactions, leads to a systematic underestimation of static screening. Yan et al. focused on BSE calculations where they showed BSE based on GLLB-SC + Δxc orbitals and eigenvalues performed better when excluding the derivative discontinuity in the calculation of the dielectric constant. Still the same is not necessarily true for BSE@G0W0 using GLLB-SC as a starting point. While an underestimation of the dielectric constant at the G0W0 level does lead to an increase in the quasiparticle gap, this in turn will result in decreased screening at the BSE level, increasing the exciton binding energy and thereby redshifting the position of the first excitation peak.18,42
Given this uncertainty with regards to the best computational method, we have opted to provide results both including and excluding the derivative discontinuity (i.e. G0W0@GLLBSC and G0W0@GLLB-SC + Δxc). As we will see later, while results are obviously different, the particular choice of computational method does not influence our overall conclusions.
III. Results and discussion
A. Structural properties and stability
We mentioned in the introduction that, while some of the structures considered in this work were originally proposed by Karlický et al.,22 others have, to the best of our knowledge, never been studied before. We will therefore begin our discussion by briefly laying out the systems as well as discuss their predicted stability compared to better-known graphene halides.Fig. 1 shows a schematic representation of all the systems considered in this work as structurally optimized using the M06-2X functional. Comparative structure-relaxations using the PBE functional resulted in only slight differences (see ESI†). While the initial study by Karlický et al. considered only single unit cells for the pure Br systems (BrFBr and BrHBr) our investigation on 1 × 2 supercells showed the systems to undergo significant buckling of the bromine atoms. This deformation is indicated for the case of BrFF, BrFBr and BrFCl in Fig. 1. It results in a notable increase in the band gap, e.g. while the BrFBr gap is equal to ≈0.85 eV at the GLLB-SC + Δxc level if no buckling of the bromines is allowed, the gap increases to 1.52 eV after relaxation in the supercell. This increase is likely caused by the dealignment of the bromine atoms which dominate the systems valence band maximum (VBM). It is consequently stronger in BrFBr as compared to BrFCl where buckling causes an increase in the band gap of ≈0.5 eV (going from 0.75 to 1.24 eV) as compared to the BrFBr increase of ≈0.7 eV. This difference is in line with the increase in the Br–C–C–Br dihedral angle from 9.6° to 10.7° as shown in Fig. 1 leading us to believe that it is in fact a dealignment between the Br atoms causing the increase in the band gap.
 | ||
| Fig. 1 Summary of all systems considered with the corresponding designation which will be used throughout this work. A top-view of the 2 × 1 supercell construction as well as a side view are also shown. In the latter the Br-buckling as well as the resulting Br–C–C–Br dihedral angle have also been indicated. | ||
In order to verify the stability of the systems shown in Fig. 1, we calculated reaction energies starting from graphane (GrH), fluorographene (GrF) as well as chlorographene (GrCl) and bromographene (GrBr) and the hydrogen/halogen molecules i.e. H2, F2, Cl2 and Br2 as Estab. = (Eprod. − Ereac.)/NC, with NC being the number of carbon atoms in the system, in analogy to the method used by Karlický et al.22
While only M06-2X results are shown in Table 1, PBE provides very similar numbers and results are given in the ESI.† We see that all systems are more stable than GrCl with fluorographene-based systems (BrFF, BrFBr and BrFCl) being more stable than their graphane-based counterparts (BrHH, BrHBr and BrHCl). BrFCl and BrFF in particular are predicted to be more stable than even GrH and BrHBr's stability, though lower, lies only slightly below that of GrH.
| GrH | GrF | GrCl | GrBr | |
|---|---|---|---|---|
| BrFF | −67 | 105 | −203 | −311 |
| BrFBr | 18 | 189 | −119 | −227 |
| BrFCl | −4 | 167 | −141 | −249 |
| BrHH | 43 | 214 | −93 | −202 |
| BrHBr | 69 | 240 | −68 | −176 |
| BrHCl | 49 | 220 | −88 | −196 |
B. Electronic properties
Having briefly considered the thermodynamic stabilities of the heavy-halogen substituted graphene derivatives we will now turn to their electronic properties. We will focus our detailed analysis on the more stable fluorinated derivatives, beginning by looking at both the PBE as well as the GLLB-SC band structures. The corresponding plots are shown in Fig. 2 and allow us to make some very interesting observations: | ||
| Fig. 2 PBE and GLLB-SC band structures for BrFF, BrFBr and BrFCl. The inset on the lower-right further shows a schematic representation of the first Brillouin zone together with the labels for the high-symmetry points used in the band structure plots. Note that we have not included the derivative discontinuity in the figures so as to allow for a direct comparison between the KS-band structures. | ||
(1) PBE predicts significantly lower band gaps than GLLB-SC in all cases with BrFF being predicted to be conducting at the PBE level while a gap is present in the GLLB-SC results.
(2) All structures show strong band-dispersion along the Γ → M′ and Γ → K directions while bands close to the Fermi energy (EF) are nearly flat along the Γ → M direction.
(3) Substitution of Br by Cl leads to an increased splitting of the bands close to the Fermi energy.
Let us start by considering the first of these observations: electrons occupying conduction and valence bands close to the Fermi energy are highly localized along one spatial direction in all systems as evidenced by the low dispersion of these bands along the Γ → M direction while the dispersive nature of the bands along Γ → M′ indicates a large degree of delocalization. This characteristic feature of the band structure results in a strong 1D localization of the occupied bands close to the Fermi energy (see also Fig. 3). This means the overlap between charge densities in different unit cells is large along one spatial direction whereas it is small to negligible along the other. The failure of PBE to correctly describe the systems band gap is therefore likely attributable to the known failures of GGA-functionals in describing localized systems of electrons.55 GLLB-SC on the other hand, through approximating the OEP-EXX functional, better describes the important on site interactions and yields finite band gaps for all three systems shown in Fig. 2.
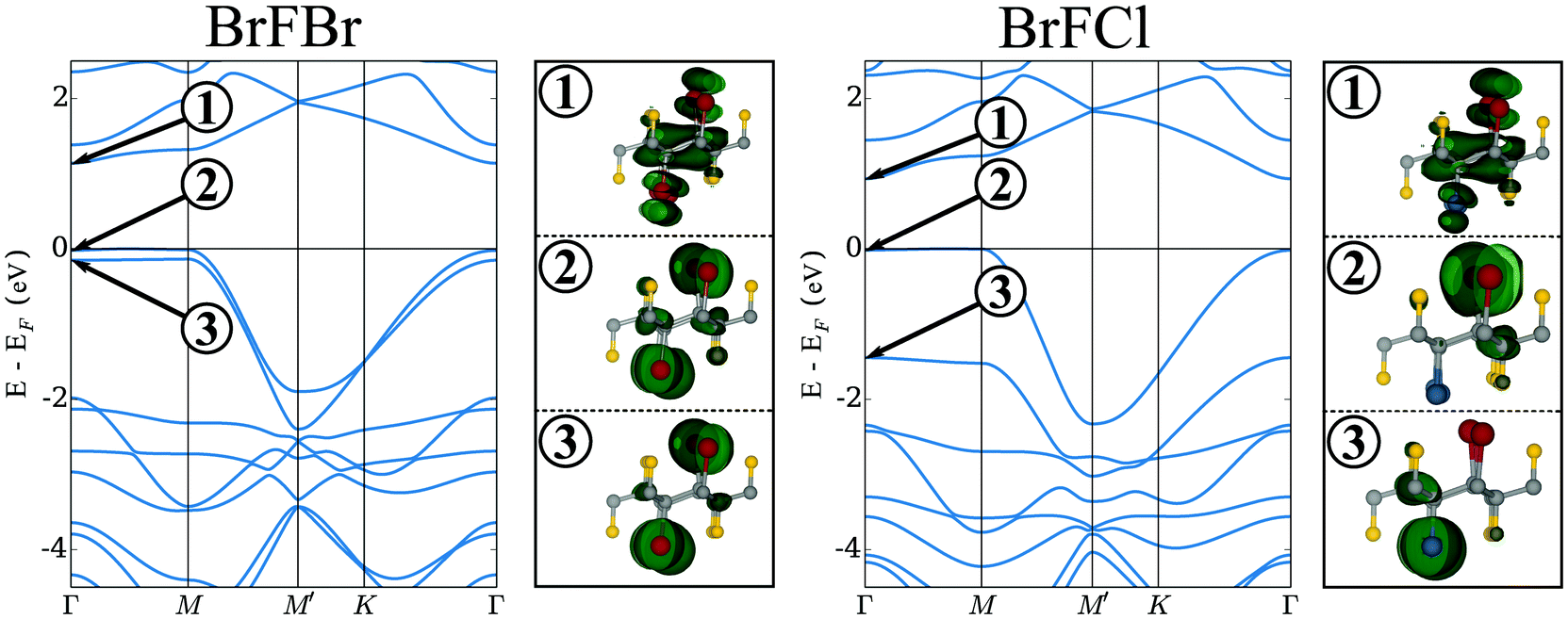 | ||
| Fig. 3 DFT band structures at the GLLB-SC level for the BrFBr and BrFCl system are shown together with charge-density isosurfaces at a level of 8 × 10−2 Å−3 emphasizing the strong 1D localization in the occupied bands close to EF. | ||
We now compare band gap values at the GLLB-SC and GLLB-SC + Δxc level to band gaps calculated using the HSE03 screened hybrid functional within the CRYSTAL14 code. Results are summarized in Table 2. As expected, GLLB-SC is able to correctly reproduce the trends seen in the HSE03 results, predicting all three systems to be semi-conducting. Upon including the derivative discontinuity, the agreement is further improved with band gaps for BrFBr and BrFCl being close to identical in the two methods. The difference is somewhat larger in the case of BrFF, though we are unsure as to what causes this discrepancy. This good agreement between results at the HSE03 and GLLB-SC + Δxc level combined with the demonstrated success of HSE03 as a starting point for G0W0 calculations16,51 makes GLLB-SC + Δxc seem to be an excellent starting point for G0W0 calculations given the low computational cost of GLLB-SC as compared to hybrid-functional calculations.
| BrFF | BrFBr | BrFCl | |
|---|---|---|---|
| HSE03 | 0.73 | 1.46 | 1.26 |
| GLLB-SC | 0.40 | 1.16 | 0.95 |
| GLLB-SC + Δxc | 0.52 | 1.54 | 1.25 |
| G 0 W 0@GLLB-SC | 3.14 | 3.52 | 3.47 |
| G 0 W 0@GLLB-SC + Δxc | 3.29 | 3.96 | 3.82 |
We stress though that, lacking experimental validation, it is unclear whether or not GLLB-SC + Δxc underestimates the macroscopic dielectric constants in our systems as has been shown for both HSE0351 as well as GLLB-SC + Δxc44 in a series of other materials and if so, how this affects the quality of G0W0 results. For this reasons we have performed both G0W0@GLLB-SC as well as G0W0@GLLB-SC + Δxc calculations which might serve as two limiting cases for the true quasiparticle gap. The results of these are also given in Table 2 and, unsurprisingly, show an opening of the G0W0 gap upon inclusion of the derivative discontinuity.
Returning now to the band structures shown in Fig. 2 it is worth considering the nature of the highly-localized states close to the Fermi energy (EF). To do so in Fig. 3 we have shown the band structures of BrFBr and BrFCl together with charge-density plots representing the three bands close to EF at the Γ-point. In BrFBr both of the closely-spaced occupied bands show strong localization on the chains of bromine atoms along the 2D structure while the unoccupied band forming the conduction band minimum (CBM) is largely delocalized over the entirety of the layer. Upon Br → Cl substitution, while the band forming the CBM is not visibly altered, the two closely-spaced, occupied bands lying close to EF in BrFBr are split significantly. The origin of this split can again be understood from looking at the charge density plots. While the two bands in question are delocalized over the Br atoms lying on both sides of the 2D layer in BrFBr, they become localized on only one side in the case of BrFCl with the band localized on the Cl-side being pushed down in energy with respect to the Br-localized one.
Having analyzed the local nature of the bands close to EF qualitatively we now move to a more quantitative assessment by considering the associated charge carrier effective masses. Here we have obtained electron (m−) and hole (m+) effective masses using the GLLB-SC functional by fitting splines to bands at the CBM/VBM along the high-symmetry directions. The effective masses are then obtained by computing the corresponding second derivatives. The results of this analysis are shown in Table 3. It is worth mentioning at this point that tests on the 1 × 1 systems show that the inclusion of electron–electron interactions within G0W0 does not significantly alter the results obtained using GLLB-SC.
| GLLB-SC | |||
|---|---|---|---|
| m −/me | m +/me | ||
| BrFF | Γ → M | 0.9 | 51.2 |
| Γ → M′ | 0.6 | 0.3 | |
| BrFBr | Γ → M | 1.6 | 14.0 |
| Γ → M′ | 0.8 | 0.3 | |
| BrFCl | Γ → M | 1.1 | 44.1 |
| Γ → M′ | 0.7 | 0.4 | |
The effective masses now allow us to draw some interesting conclusions regarding the behavior of electrons/holes created upon photoexcitation: electrons are largely unconstrained along both Γ → M as well as Γ → M′ and so their density will rapidly delocalize over the 2D layer. Holes on the other hand, while having low effective masses along the Γ → M′ direction, are heavily constrained along Γ → M with effective masses being around two orders of magnitude higher than those along Γ → M′. This strong anisotropy in effective masses will result in rapid delocalization of the hole-density along Γ → M′ (i.e. the direction parallel to the rows of Br atoms) combined with strong localization along Γ → M (i.e. the direction orthogonal to the rows of Br atoms). This behavior is not only interesting in and of itself but might conceivably be exploited in splitting excitons created within the systems by appropriately varying the external potential in the two spatial directions. For completeness we mention that the band gap in all structures is technically indirect with the VBM lying between M and Γ though this does not significantly influence our conclusions given the flatness of the bands along that same direction.
IV. Optical properties
Since we are interested in the optical properties of our systems in particular as they relate to possible applications in photovoltaics, effects due to exciton binding cannot be neglected, especially as they are expected to be of even greater importance in low-dimensional systems such as those considered here18,19 as compared to 3D ones. We therefore performed BSE calculations employing both G0W0@GLLB-SC as well as G0W0@GLLB-SC + Δxc as a starting point. Fig. 4 shows the resulting spectra in the optical limit (q → 0) at different levels of theory and for different directions of the incoming photons. As expected from the systems band structure shown in Fig. 2, the lowest-lying exciton is localized along the![[Y with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0059_20d1.gif) -direction while the first peak along
-direction while the first peak along ![[X with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0058_20d1.gif) lies at ≈2.49 eV showing very low intensity as compared to the peak along . Comparing the curves shown on the right-hand side of Fig. 4 we can further see that, as expected for a 2D material, electron–hole interactions play a significant role in determining the position of the first optical adsorption peaks with spectra at the RPA level being strongly blue-shifted compared to their BSE counterparts.
lies at ≈2.49 eV showing very low intensity as compared to the peak along . Comparing the curves shown on the right-hand side of Fig. 4 we can further see that, as expected for a 2D material, electron–hole interactions play a significant role in determining the position of the first optical adsorption peaks with spectra at the RPA level being strongly blue-shifted compared to their BSE counterparts.
 | ||
Fig. 4 The left-hand figure shows the imaginary part of the frequency dependent dielectric function (Im![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ε(ω)) as calculated at the BSE@GLLB-SCG0W0 level for BrFF, BrFBr and BrFCl. For the case of BrFF spectra obtained for two orthogonal directions of the incident photons are shown. The inset in the left-hand figure shows the atomic structure of BrFF together with the definition of the two directions of incident photons used in the plot. The right-hand figure on the other hand shows the calculated optical spectrum of BrFF at four different levels of theory. In all cases spectra are obtained applying a Lorentzian broadening of 0.1 eV. ε(ω)) as calculated at the BSE@GLLB-SCG0W0 level for BrFF, BrFBr and BrFCl. For the case of BrFF spectra obtained for two orthogonal directions of the incident photons are shown. The inset in the left-hand figure shows the atomic structure of BrFF together with the definition of the two directions of incident photons used in the plot. The right-hand figure on the other hand shows the calculated optical spectrum of BrFF at four different levels of theory. In all cases spectra are obtained applying a Lorentzian broadening of 0.1 eV. | ||
Another interesting observation that can be made from Table 4 is the fact that substituting one of the bromine atoms in the unit cell by chlorine and finally fluorine i.e. moving from BrFBr to BrFCl and BrFF, significantly affects the position of the first excitation peak. While Cl → F substitution leaves the exciton binding energy (i.e. the difference between the fundamental and the optical gap) nearly unaltered at ≈1.8 eV, interchanging Cl/F with Br (i.e. going from BrFCl/BrFF to BrFBr) lowers it by ≈0.2 eV. This change is made all the more interesting by the fact that the change in the G0W0 gap is 0.34 eV and 0.53 eV upon Cl → F substitution using G0W0@GLLB-SC and G0W0@GLLB-SC + Δxc respectively, while the G0W0@GLLB-SC/G0W0@GLLB-SC + Δxc gaps of BrFBr and BrFCl only differ by 0.05/0.14 eV. To understand the origin of the effect we return briefly to the discussion of the band structures shown in Fig. 3. While pure bromine systems show two almost degenerate bands at the Fermi energy, one of these bands is significantly lowered in energy upon substituting bromine by chlorine which leads to the exciton becoming localized in only one of the two bands. As this band is localized on only one side of the 2D layer in BrFCl and BrFF as compared to both sides of the layer in BrFBr, the spatial localization of the exciton increases which in turn leads to an increase in the exciton binding energy. Finally, we note that the first adsorption peak for all three systems lies close to the optimal peak position for photovoltaic applications at ≈1.5 eV.56,57 In particular BrFF, which shows a first adsorption peak at ≈1.3–1.4 eV (depending on the level of theory) and displays predicted stability higher than that of graphane constitutes a very promising candidate for application in future solar technology.
| BrFF | BrFBr | BrFCl | |
|---|---|---|---|
| G 0 W 0@GLLB-SC | 3.14 | 3.52 | 3.47 |
| G 0 W 0@GLLB-SC + Δxc | 3.29 | 3.96 | 3.82 |
| BSE@GLLB-SCG0W0 | 1.27 | 1.86 | 1.65 |

|
1.41 | 2.26 | 1.96 |
V. Conclusions
Herein we have studied the thermodynamic stability and optoelectronic properties of a series of heavy-halogen substituted graphane and fluorographene derivatives, based on the work originally done by Karlický, Zbořil and Otyepka.22 Graphane-based systems show predicted stabilities lower than graphane, whilst still exceeding that of chlorographane. Fluorographene-based systems on the other hand were shown to display stabilities on par with and even exceeding that of graphane.We studied in detail the electronic structure of the aforementioned systems, showing them to display strong 1D-localization of charge carriers with a marked electron–hole asymmetry which we hypothesize might be exploited in separating electron–hole pairs created upon photoexcitation.
Employing the GLLB-SC functional we were able to obtain band gaps which closely reproduce HSE03 results while keeping computational costs to approximately those of GGA calculations. Given the success of the G0W0@HSE03 approach in predicting the band gaps of a number of materials51 we believe it to provide an excellent starting point for G0W0 calculations, combining low computational requirements with good accuracy.
Lastly we investigated the optical spectra of the systems including electron–hole interactions on top of G0W0 by solving the Bethe–Salpeter equation. The first optical adsorption peak for all the systems considered lies close to the optimal peak position given by the Shockley–Queisser limit i.e. ≈1.5 eV.56,57 Especially BrFF, who's first adsorption peak lies at ≈1.3–1.4 eV and which shows a predicted stability higher than that of graphane constitutes a very promising candidate for application in future solar technology.
Acknowledgements
LEMS acknowledges financial support by the Studienstiftung des deutschen Volkes e.V., the Deutsche Forschungsgemeinschaft within the Priority Program (SPP) 1459 (Graphene) as well as the International Max Planck Research School “Complex Surfaces in Material Sciences”. The High Performance Computing Network of Northern Germany (HLRN) and computer facilities of the Freie Universität Berlin (ZEDAT) are acknowledged for computer time. The authors are indebted to Kirsten Trøstrup Winther and Filip Anselm Rasmussen (both Copenhagen) for providing the latest development version of the GPAW-GW code and help with its use. We also like to express our gratitude towards Thomas Olsen (Copenhagen) for many fruitful discussions regarding the BSE-calculations. Lastly we thank Johannes Voss (SUNCAT), Jean Christophe Tremblay, Johannes Budau, Gunter Hermann, Vincent Pohl and Marcel Quennet (all Berlin) for enlightening discussions and suggestions regarding this manuscript. ASE35 and XCrySDen58–60 were used to create images of atomic structures throughout this work, VESTA61 was used to visualize charge densities, while plots were created using Matplotlib.62References
- X. Wang, L. Zhi and K. Müllen, Nano Lett., 2008, 8, 323–327 CrossRef CAS PubMed.
- X. Miao, S. Tongay, M. K. Petterson, K. Berke, A. G. Rinzler, B. R. Appleton and A. F. Hebard, Nano Lett., 2012, 12, 2745–2750 CrossRef CAS PubMed.
- J. Wu, H. A. Becerril, Z. Bao, Z. Liu, Y. Chen and P. Peumans, Appl. Phys. Lett., 2008, 92, 263302 CrossRef.
- Z. Liu, Q. Liu, Y. Huang, Y. Ma, S. Yin, X. Zhang, W. Sun and Y. Chen, Adv. Mater., 2008, 20, 3924–3930 CrossRef CAS.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- H.-P. Komsa, J. Kotakoski, S. Kurasch, O. Lehtinen, U. Kaiser and A. V. Krasheninnikov, Phys. Rev. Lett., 2012, 109, 035503 CrossRef PubMed.
- R. Ganatra and Q. Zhang, ACS Nano, 2014, 8, 4074–4099 CrossRef CAS PubMed.
- M.-L. Tsai, S.-H. Su, J.-K. Chang, D.-S. Tsai, C.-H. Chen, C.-I. Wu, L.-J. Li, L.-J. Chen and J.-H. He, ACS Nano, 2014, 8, 8317–8322 CrossRef CAS PubMed.
- X. Gu, W. Cui, H. Li, Z. Wu, Z. Zeng, S.-T. Lee, H. Zhang and B. Sun, Adv. Energy Mater., 2013, 3, 1262–1268 CrossRef CAS.
- M. Bernardi, M. Palummo and J. C. Grossman, Nano Lett., 2013, 13, 3664–3670 CrossRef CAS PubMed.
- S. Ryu, M. Y. Han, J. Maultzsch, T. F. Heinz, P. Kim, M. L. Steigerwald and L. E. Brus, Nano Lett., 2008, 8, 4597–4602 CrossRef CAS PubMed.
- D. C. Elias, R. R. Nair, T. M. G. Mohiuddin, S. V. Morozov, P. Blake, M. P. Halsall, A. C. Ferrari, D. W. Boukhvalov, M. I. Katsnelson, A. K. Geim and K. S. Novoselov, Science, 2009, 323, 610–613 CrossRef CAS PubMed.
- R. Zbořil, F. Karlický, A. B. Bourlinos, T. A. Steriotis, A. K. Stubos, V. Georgakilas, K. Šafářová, D. Jančík, C. Trapalis and M. Otyepka, Small, 2010, 6, 2885–2891 CrossRef PubMed.
- R. R. Nair, W. Ren, R. Jalil, I. Riaz, V. G. Kravets, L. Britnell, P. Blake, F. Schedin, A. S. Mayorov, S. Yuan, M. I. Katsnelson, H.-M. Cheng, W. Strupinski, L. G. Bulusheva, A. V. Okotrub, I. V. Grigorieva, A. N. Grigorenko, K. S. Novoselov and A. K. Geim, Small, 2010, 6, 2877–2884 CrossRef CAS PubMed.
- D. K. Samarakoon, Z. Chen, C. Nicolas and X.-Q. Wang, Small, 2011, 7, 965–969 CrossRef CAS PubMed.
- F. Karlický and M. Otyepka, J. Chem. Theory Comput., 2013, 9, 4155–4164 CrossRef PubMed.
- F. Karlický and M. Otyepka, Ann. Phys. (Berlin, Ger.), 2014, 526, 408–414 CrossRef.
- J.-H. Choi, P. Cui, H. Lan and Z. Zhang, Phys. Rev. Lett., 2015, 115, 066403 CrossRef PubMed.
- S. Latini, T. Olsen and K. S. Thygesen, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 245123 CrossRef.
- L. Hedin, Phys. Rev., 1965, 139, A796 CrossRef.
- E. E. Salpeter and H. A. Bethe, Phys. Rev., 1951, 84, 1232 CrossRef.
- F. Karlický, R. Zbořil and M. Otyepka, J. Chem. Phys., 2012, 137, 034709 CrossRef PubMed.
- R. Dovesi, R. Orlando, A. Erba, C. M. Zicovich-Wilson, B. Civalleri, S. Casassa, L. Maschio, M. Ferrabone, M. De La Pierre, P. D'Arco, Y. Noël, M. Causà, M. Rerat and B. Kirtman, Int. J. Quantum Chem., 2014, 114, 1287–1317 CrossRef CAS.
- R. Dovesi, V. R. Saunders, C. Roetti, R. Orlando, C. M. Zicovich-Wilson, F. Pascale, B. Civalleri, K. Doll, N. M. Harrison, I. J. Bush, P. D'Arco, M. Llunell, M. Causà and Y. Noël, CRYSTAL14 User's Manual, University of Torino, Torino, 2014 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- M. F. Peintinger, D. V. Oliveira and T. Bredow, J. Comput. Chem., 2013, 34, 451–459 CrossRef CAS PubMed.
- A. V. Krukau, O. A. Vydrov, A. F. Izmaylov and G. E. Scuseria, J. Chem. Phys., 2006, 125, 224106 CrossRef PubMed.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2006, 124, 219906 CrossRef.
- J. Heyd and G. E. Scuseria, J. Chem. Phys., 2004, 121, 1187–1192 CrossRef CAS PubMed.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef CAS.
- K. G. Steenbergen, N. Gaston, C. Müller and B. Paulus, J. Chem. Phys., 2014, 141, 124707 CrossRef PubMed.
- M. Dolg, U. Wedig, H. Stoll and H. Preuss, J. Chem. Phys., 1987, 86, 866–872 CrossRef CAS.
- J. M. Martin and A. Sundermann, J. Chem. Phys., 2001, 114, 3408–3420 CrossRef CAS.
- D. Usvyat, J. Chem. Phys., 2015, 143, 104704 CrossRef PubMed.
- S. R. Bahn and K. W. Jacobsen, Comput. Sci. Eng., 2002, 4, 56–66 CrossRef CAS.
- J. J. Mortensen, L. B. Hansen and K. W. Jacobsen, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 035109 CrossRef.
- J. Enkovaara, C. Rostgaard, J. J. Mortensen, J. Chen, M. Dułak, L. Ferrighi, J. Gavnholt, C. Glinsvad, V. Haikola, H. A. Hansen, H. H. Kristoffersen, M. Kuisma, A. H. Larsen, L. Lehtovaara, M. Ljungberg, O. Lopez-Acevedo, P. G. Moses, J. Ojanen, T. Olsen, V. Petzold, N. A. Romero, J. Stausholm-Møller, M. Strange, G. A. Tritsaris, M. Vanin, M. Walter, B. Hammer, H. Häkkinen, G. K. H. Madsen, R. M. Nieminen, J. K. Nørskov, M. Puska, T. T. Rantala, J. Schiøtz, K. S. Thygesen and K. W. Jacobsen, J. Phys.: Condens. Matter, 2010, 22, 253202 CrossRef CAS PubMed.
- J. Yan, J. J. Mortensen, K. W. Jacobsen and K. S. Thygesen, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 245122 CrossRef.
- F. Hüser, T. Olsen and K. S. Thygesen, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 235132 CrossRef.
- F. A. Rasmussen, P. S. Schmidt, K. T. Winther and K. S. Thygesen, 2016, arXiv preprint arXiv:1511.00129.
- M. Pykal, P. Jurečka, F. Karlický and M. Otyepka, Phys. Chem. Chem. Phys., 2016, 18, 6351–6372 RSC.
- T. Olsen, S. Latini, F. Rasmussen and K. S. Thygesen, Phys. Rev. Lett., 2016, 116, 056401 CrossRef PubMed.
- Z. Chen and X.-Q. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 081405 CrossRef.
- J. Yan, K. W. Jacobsen and K. S. Thygesen, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 045208 CrossRef.
- M. Shishkin and G. Kresse, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 035101 CrossRef.
- W. G. Aulbur, L. Jönsson and J. W. Wilkins, Solid State Phys., 1999, 54, 1–218 Search PubMed.
- F. Aryasetiawan and O. Gunnarsson, Rep. Prog. Phys., 1998, 61, 237 CrossRef CAS.
- G. Onida, L. Reining and A. Rubio, Rev. Mod. Phys., 2002, 74, 601 CrossRef CAS.
- S. G. Louie, Contemp. Concepts of Condens. Matter Sci., 2006, 2, 9–53 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- F. Fuchs, J. Furthmüller, F. Bechstedt, M. Shishkin and G. Kresse, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 115109 CrossRef.
- M. Kuisma, J. Ojanen, J. Enkovaara and T. T. Rantala, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 115106 CrossRef.
- J. P. Perdew, R. G. Parr, M. Levy and J. L. Balduz Jr., Phys. Rev. Lett., 1982, 49, 1691 CrossRef CAS.
- I. E. Castelli, T. Olsen, S. Datta, D. D. Landis, S. Dahl, K. S. Thygesen and K. W. Jacobsen, Energy Environ. Sci., 2012, 5, 5814–5819 CAS.
- P. Mori-Sánchez, A. J. Cohen and W. Yang, Phys. Rev. Lett., 2008, 100, 146401 CrossRef PubMed.
- W. Shockley and H. J. Queisser, J. Appl. Phys., 1961, 32, 510–519 CrossRef CAS.
- O. D. Miller, E. Yablonovitch and S. R. Kurtz, IEEE J. Photovolt., 2012, 2, 303–311 CrossRef.
- A. Kokalj, Comput. Mater. Sci., 2003, 28, 155–168 CrossRef CAS.
- A. Kokalj, J. Mol. Graphics, 1999, 17, 176–179 CrossRef CAS.
- A. Kokalj and M. Causà, Scientific visualization in computational quantum chemistry, 2000 Search PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- J. D. Hunter, Comput. Sci. Eng., 2007, 9, 90–95 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp05188j |
| This journal is © the Owner Societies 2016 |