
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoinduced water splitting in pyridine water clusters†
Natalia
Esteves-López
,
Stephane
Coussan
,
Claude
Dedonder-Lardeux
and
Christophe
Jouvet
*
CNRS, Aix-Marseille Université, Physique des Interactions Ioniques et Moléculaires (PIIM) UMR-7345, Marseille, France. E-mail: Christophe.jouvet@univ-amu.fr
First published on 19th August 2016
Abstract
Ab initio calculations predict that pyridine (Py) can act as a photo-catalyst to split water by the absorption of a UV photon following the reaction Py–H2O + hν → PyH˙ + OH˙. To test this prediction, we performed two types of experiments: in the first, we characterize the electronic spectroscopy of the PyH˙ radical in the gas phase. In the second, we evidence the reaction through the UV excitation of molecular Py–(H2O)n clusters obtained in a supersonic expansion and monitoring the PyH˙ reaction product. The results show unambiguously that PyH˙ is produced, and thus that water is split using pyridine as a photo-catalyst.
In this paper we show that pyridine can act as a photo-catalytic molecular system, which can dissociate the water covalent bond with UV C light. A water molecule is a ubiquitous system to produce H2, however VUV light is needed to photo-dissociate the H–OH covalent bond (186 nm, 6.66 eV). Since most of the sunlight reaching the earth is in the visible spectral region, we cannot dissociate the H–OH bond under normal sunlight conditions and it is necessary to design a system that could break the H–OH bond with visible light.
Recent theoretical calculations1–3 show that, when the mixed pyridine water complex (Py–H2O) is excited to its first optically active state (1ππ*, ∼4.8 eV), water splitting occurs through the following reaction, Py–H2O + hν → PyH˙ + OH˙, generating pyridinyl and hydroxyl radicals. The energy for the UV excitation of Py–H2O is 1.8 eV lower than the energy necessary to excite and photo-dissociate water directly. Liu et al. further reported that similar water splitting occurs in the triplet state (T1) of Py–H2O.3 Thus, pyridine acts as a photo-catalyst but to be useful the photo-catalyst should be recovered by the absorption of a second photon. Indeed, the calculations also indicate that the pyridinyl radical can release the additional hydrogen atom when irradiated with visible light (∼600 nm, ∼2 eV). The pyridinyl radicals obtained by the irradiation of pyridine in water have been postulated in the homogeneous reduction of CO2 by pyridine.4 The ground state of the PyH˙–OH˙ diradical structure has been calculated to be the ion pair structure PyH+ OH−.1 Recently, water splitting has been observed in negatively charged [Py–(H2O)n]− clusters.5,6
Molecular clusters have been extensively used to evidence the primary processes involved in excited states of molecules in a well-defined environment. These studies have allowed to evidence very clearly some photochemical reactions such as proton transfer through molecular wires,7–10 hydrogen transfer to solvents in mixed molecule–solventn clusters,11–23 solvent isomerization,24–27 characterization of intermediate states of reactions28–31 and chiral discrimination.32 The case of pyridine–water clusters is peculiar since some attempts have been made to characterize their spectroscopy but without success.33 This is already good indication that some fast non-radiative processes occur, which prevent the detection of Py–H2O clusters through multiphoton ionization or laser induced fluorescence (LIF).
To demonstrate directly the photochemical reaction in Py–H2O clusters, and compare with ab initio calculations, the experiment has been conducted in the gas phase, using molecular clusters of pyridine with water produced in a supersonic expansion. After excitation to the 1ππ* state of pyridine, two reaction products are expected to be produced, the OH˙ radical and the hydrogenated pyridine (pyridinyl radical PyH˙). In the gas phase the radical pair can separate freely. As will be seen, the ionization potential (IP) of the PyH˙ radical is calculated to be rather low, which should make its detection quite easy. It should then be easier to detect the PyH˙ radical as a reaction product of excited Py–(H2O)n clusters than to detect OH˙ through laser induced fluorescence, which has a low detection efficiency (typically 10−3). The experimental scheme used involves the excitation of Py–(H2O)n clusters and the detection of the PyH˙ radical through ionization by one or two photons.
Since the PyH˙ radical spectroscopy has not yet been studied, the experiment will be split into two parts, the first one being the spectroscopic characterization of the cold PyH˙ radical, and the second one the detection of PyH˙ as a reaction product after the excitation of the Py–(H2O)n clusters. Additional calculations are performed to get information on the nature of the excited states of the PyH˙ radical and its ionization potential.
Experimental
The setup that we used is composed of a pulsed supersonic expansion combined with a time of flight mass spectrometer. The setup has been previously described34 and is quite standard. The discharge source is the same as used in previous experiments to produce either cold neutral van der Waals species35 or more recently protonated molecular ions.36 In a few words, a pulsed high voltage discharge located a few mm after the pulsed nozzle produces PyH˙ radicals from a gas mixture of He/H2/pyridine. The radicals are cooled down in the expansion and ionized between the extraction plates of the reflectron Time of Flight (TOF) mass spectrometer (Jordan TOF Products, Inc.). Additional experiments were conducted with fully deuterated pyridine (hereafter named dPy).In the second type of experiments, the discharge is removed and Py–(H2O)n (or dPy–(D2O)n) clusters are produced by expanding a mixture of water and pyridine (at their vapor pressure at room temperature) with He (between 1 and 5 atm). Two laser beams are temporally and spatially synchronized between the extracting plates of the mass spectrometer, the pump laser excites the clusters and the probe laser detects the PyH˙ (or dPyD˙) radicals.
The lasers used are YAG pumped nanosecond OPO lasers (EKSPLA model-NT342B), which have a 10 Hz repetition rate, a resolution (FWHM) of 8 cm−1 and a minimum scanning step of 0.02 nm. The lasers are focused in the middle of the extraction zone of the time of flight mass spectrometer. These lasers can be scanned from visible to UV in three regions: from 410 to 709 nm (OPO signal), from 296 to 409.9 nm (frequency mixing) and from 225 to 295.9 nm (frequency doubling).
Calculations are also performed using the Turbomole package,37 at the TD-DFT (B3LYP) and RI-CC2 levels with the aug-cc-pVDZ basis set, to get information on the PyH˙ ionization potential and on the excited states of the radical.
Results and discussion
Spectroscopic investigation of the pyridinyl radical (PyH˙)
Fig. 1 shows the mass spectra obtained when the probe laser is set at 225 nm with the discharge on or off. In the absence of discharge (red trace) the ion at m/z = 79 corresponds to the pyridine radical cation Py+ produced by the two-photon ionization of pyridine and the small peak at m/z = 80 corresponds to the pyridine isotope with a 13C atom. When the discharge is on, an intense peak at m/z = 80 with its isotopic companion at m/z = 81 is obtained indicating that protonated pyridine PyH+ is observed by the ionization of the PyH˙ radical. | ||
| Fig. 1 Mass spectra obtained with a mixture of He/H2/pyridine with the discharge source on (black) or off (red), the ionization laser being set at 225 nm. | ||
The spectroscopy of the PyH˙ radical is obtained by monitoring the intensity of the m/z = 80 mass peak (PyH+) while scanning the laser from visible to UV as displayed in Fig. 2. The onset of the very strong signal at 234 nm (5.31 eV) and the observation of a continuum at higher energy correspond clearly to the ionization potential of the PyH˙ radical. To comfort this assertion, the IP of PyH˙ has been calculated using the DFT method with the B3LYP functional and the aug-cc-pVDZ basis set (including the difference in zero point energy ΔZPE), and is found at 235 nm (5.28 eV).
 | ||
| Fig. 2 Spectroscopy of the PyH˙ radical produced in the discharge source and cooled down in the supersonic expansion. At high energy the molecule is ionized by one photon. At lower energies, i.e. for excitation wavelengths between 300 and 440 nm, the spectrum is obtained by resonance enhanced one-color two-photon ionization (R2PI). The main bands have been assigned by comparison with ab initio calculations (see the text). | ||
Some well resolved transitions are observed in the visible and UV A spectral regions. The first band is observed at 425.6 nm (23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 496 cm−1 or 2.91 eV) and it requires two photons to reach the IP of PyH˙. One can detect quite easily the states that can be ionized with two photons i.e. those which have their absorption below 468 nm (half the IP). Lower energy states would need the absorption of three photons to be detected, and nothing has been observed at energies lower than 425.6 nm. The dPyD˙ spectrum shows the same transitions slightly red-shifted and is given in Fig. 5c.
496 cm−1 or 2.91 eV) and it requires two photons to reach the IP of PyH˙. One can detect quite easily the states that can be ionized with two photons i.e. those which have their absorption below 468 nm (half the IP). Lower energy states would need the absorption of three photons to be detected, and nothing has been observed at energies lower than 425.6 nm. The dPyD˙ spectrum shows the same transitions slightly red-shifted and is given in Fig. 5c.
The ground state optimized structure has been obtained from calculations at the RI-CC2 (aug-cc-pVDZ) level and shows a C2v symmetry. Since the experimental spectrum shows weak Franck Condon activity and strong band origins, the radical should not be too much distorted from this symmetry in the excited states. The excited state optimizations have then been calculated under C2v geometry constraints for states which have a significant oscillator strength (>10−4). The calculated transition energies are systematically higher by 0.3 eV than the experimental transitions (see Table 1). This is the typical uncertainty of CC2 calculations for closed-shell molecules, while the uncertainty for open-shell species cannot be estimated at present due to very few experimental and theoretical data. Based on these calculations, we propose the assignment of Fig. 2. It should be mentioned that the first transition observed corresponds to the fourth excited state and the third state of B2 symmetry. We tried to calculate the excited state vibrations but in the absence of symmetry the open shell/CC2 calculation of such a high excited state failed.
| Experimental | State label | C 2v optimized transition (eV) | Oscillator strength | |
|---|---|---|---|---|
| nm | eV | |||
| 425.6 | 2.91 | 32B2 | 3.20 | 1 × 10−2 |
| 391.2 | 3.17 | 22A1 | 3.37 | 4 × 10−3 |
| 356.0 | 3.48 | 42B2 | 3.58 | 3 × 10−3 |
| 320.0 | 3.88 | 52B2 | 4.14 | 2 × 10−4 |
It should be mentioned that the PyH˙ signal is not very strong because the discharge method is not so efficient to produce the radical, so that the laser is rather powerful and some saturation of the vibronic transitions probably occurs, which may explain why some excited states with a low oscillator strength can be detected. As can be seen in the ESI† (Table S1), there is not so much choice to assign the states and one should keep in mind that open shell excited state calculations may not be so accurate, and that the Franck–Condon factors could not be calculated.
The first two lower excited states have quite similar vibrational spectra. The active vibrations at 380/371 cm−1 and 1122/1005 cm−1 are typical of the active vibrations in pyridine and can be tentatively assigned to the 16a1 and 121 modes observed at 323 and 955 cm−1 in the excited state of pyridine and at 314 and 1105 cm−1 in the radical cation.38
Excitation of Py–(H2O)n or dPy–(D2O)n clusters: direct evidence of the water splitting reaction
The experiments have been conducted for hydrogenated and fully deuterated species dPy, and the experimental results are basically the same. Py–(H2O)n clusters produced in the supersonic jet are excited in the energy region of the pyridine 1ππ* state (around 255 nm) and PyH˙ is probed by ionization at 225 nm. The probe laser at 225 nm ionizes efficiently PyH˙ with one photon and excites very weakly the parent clusters (see Fig. S1 in the ESI†). The delay between lasers is 600 ns, to discriminate PyH˙ radicals from the 13C isotope of pyridine (both have the same mass) and to discriminate the reaction in the excited state from a reaction in the ionic state of the clusters due to a two-photon absorption at 255 nm (the PyH+ peak in the upper trace of Fig. 3). Besides, such a delay preludes the absorption of the probe laser from the excited states of the clusters, which have a much shorter lifetime. The ions issued from the ionization of PyH˙ by the probe laser are then observed 600 ns after the ones produced by the pump laser since time t = 0 for the time of flight is synchronized with the pump laser. The formation of pyridinyl is clearly shown in Fig. 3 in which the signal of PyH+ is absent if the pump laser is off and the probe laser is on (middle trace), while when both lasers are present (lower trace) the PyH+ ion signal coming from the direct ionization of PyH˙ radicals issued from the excitation of Py–(H2O)n clusters is the most intense peak. | ||
| Fig. 3 Mass spectra recorded after excitation of the Py–(H2O)n clusters. Upper trace: pump laser at 255 nm alone: the Py–(H2O)n clusters are excited and the PyH+ ion is obtained through one-color two-photon absorption. Middle trace: probe laser at 225 nm alone: nearly no ion signal since Py and Py–(H2O)n clusters absorb very weakly at this wavelength. Lower trace: pump (255 nm) and probe (225 nm) lasers with the probe laser delayed by 600 ns: the most intense peak is the PyH+ ion produced by excitation of the clusters and ionization of the PyH˙ radical via a one-photon process. | ||
In the upper trace of Fig. 3, the PyH+ ion is observed through a one-color two-photon scheme at 255 nm. This signal is not linked to the reaction in clusters with water but to the reaction of pyridine cluster ions. The same experiment has been performed in absence of water in the supersonic expansion. To ensure that there is no water in the tubing bringing the He/Py gas to the pulsed valve, some desiccant (CaCl2) has been introduced in the line, and absolutely no water complex could be obtained. Under these conditions, the signal corresponding to the ionization of the PyH˙ radical by the probe laser is negligible, whereas the PyH+ signal coming from the reaction in (Py)n+ clusters (n = 2 or more) in their ionic state can still be observed (Fig. S2, ESI†). The comparison between pump/probe mass spectra observed in the presence and absence of water clusters is shown in Fig. S3 (ESI†).
The spectral signatures of both processes are totally different (see Fig. 4). The PyH+ signal issued from the ionic reaction obtained in a water free expansion exhibits a smooth increase going from 225 nm up to 245 nm (Fig. 4b). When water is added in the expansion, the excitation of Py–(H2O)n clusters leads to the formation of a pyridinyl radical, which displays a step function type spectrum, no signal between 245 and 234 nm and a strong signal between 234 and 225 nm, as observed when pyridinyl radicals produced in the discharge source are ionized (Fig. 4a). Thus, there is a clear and unambiguous signature of the processes involved in the experiment. In pure pyridine clusters, there is a ionic reaction leading to PyH+, while in expansion containing water, excited pyridine–water clusters react to produce the pyridinyl radical, clearly identified by its ionization threshold. The water splitting reaction is then clearly demonstrated.
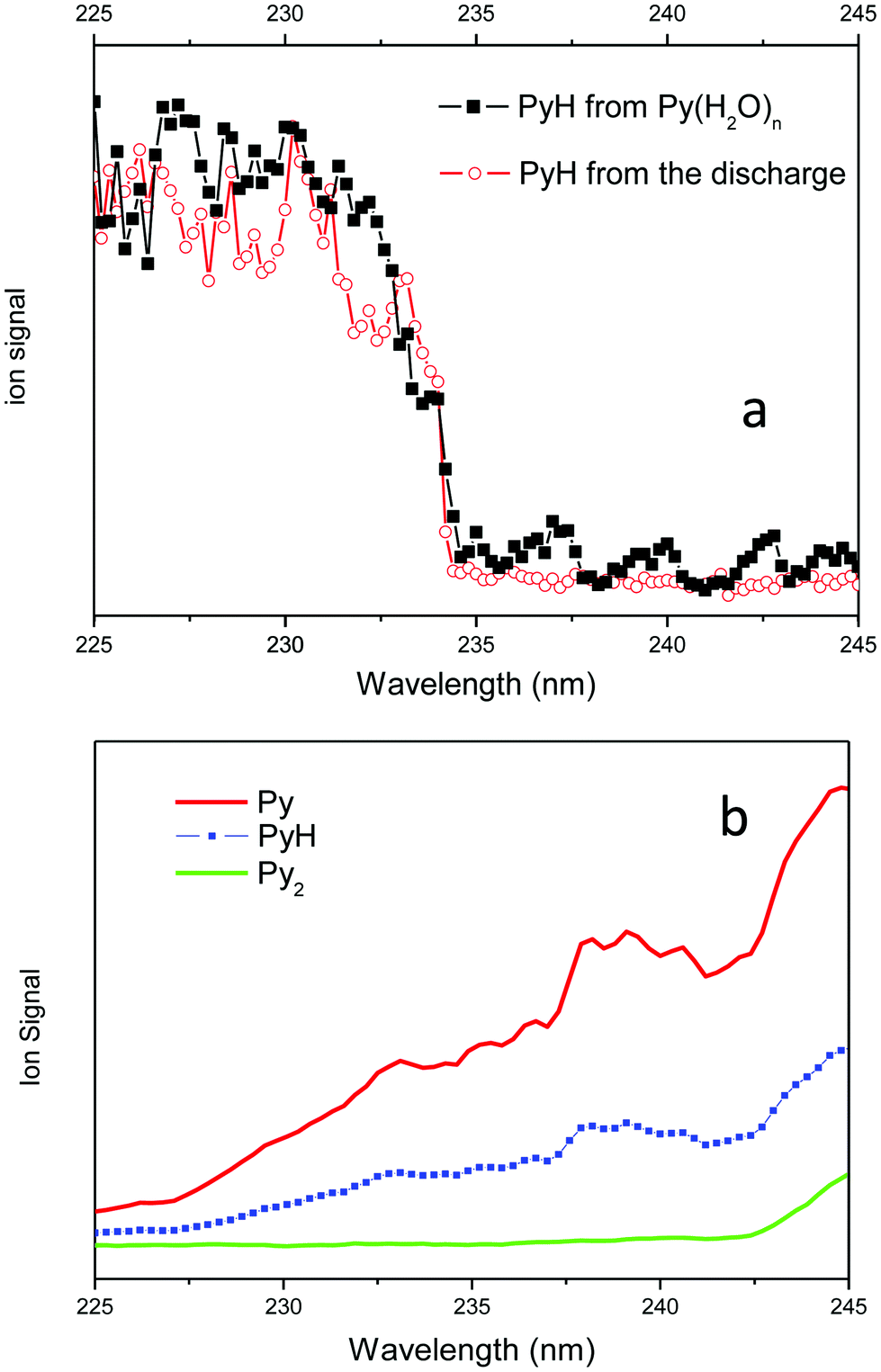 | ||
| Fig. 4 (a) PyH+ signal coming from the ionization of the pyridinyl radical: in red pyridinyl radicals produced by the discharge source; in black pyridinyl radicals obtained from the reaction of pyridine–water clusters excited at 255 nm and probed by a second laser. (b) Py+, PyH+ and Py2+ signals obtained from multiphoton ionization of Py, PyH˙ and Py2 obtained from an expansion of pyridine in He without water. In this case the PyH+ spectrum is similar to the Py+ spectrum. | ||
Another method to detect the PyH˙ radical is through the two-photon ionization scheme in the visible spectral region. In Fig. 5 are presented the pump probe PyH˙ and dPyD˙ (fully deuterated pyridinyl) action spectra in comparison with the spectra obtained for the cold species issued from the discharge source. This unambiguously ascertains that the PyH+ signal detected with the probe laser is due to the ionization of the PyH˙ radical. It can be noticed that the action spectra resulting from the reaction in clusters are broadened as compared to the spectra of the cold species produced in the discharge source, which implies that the PyH˙ or dPyD˙ reaction products are produced with some internal energy. Note that the dPyD˙ vibronic bands are red-shifted as compared to the PyH˙ bands because the heavier deuterium mass induces an isotopic shift reducing the vibrational frequencies.
 | ||
| Fig. 5 Two-photon ionization spectra, comparison between: (a) PyH˙ issued from the discharge source, (b) PyH˙ from the reaction of Py–(H2O)n excited at 255 nm, (c) dPyD˙ from the discharge source, (d) dPyD˙ from the reaction of the dPy–(D2O)n clusters excited at 255 nm. | ||
For which cluster size does the reaction occur?
We could expect to identify the parent ion from the action spectrum recorded in probing PyH˙ while scanning the pump laser. The PyH˙ action spectrum is compared to the R2PI spectrum of the Py–(H2O) or (Py)2 complex in Fig. S4 (ESI†). As for the free molecule the spectra are broad and not well resolved making the identification of the parent molecule difficult, even though the PyH˙ action spectrum and the Py–(H2O) R2PI spectra are similar.Since we can record the one-color two-photon absorption/ionization spectrum at the mass of the Py–H2O complex, the complex does not react very quickly, otherwise it could not absorb the second photon necessary to ionize. In fact, there is a kind of contradiction to use spectroscopy to determine the reacting cluster size since, if the cluster reacts quickly, it will not be ionized thus not observed. Along these lines, it can be noted that the Py–(H2O)2 complex stays very weak and the Py–(H2O)3 cluster is barely observed. However, the similarity between the PyH˙ and Py–H2O spectra, shown in Fig. S4 (ESI†), indicates that pyridine–water clusters are responsible for the reaction.
The only way to get some idea of the clusters responsible for the reaction is by changing expansion conditions.
The pressure effect measurements shown in Fig. 6 have been recorded to see if we could correlate the PyH˙ signal with some Py–(H2O)n cluster sizes since the mean cluster size increases with the backing pressure.
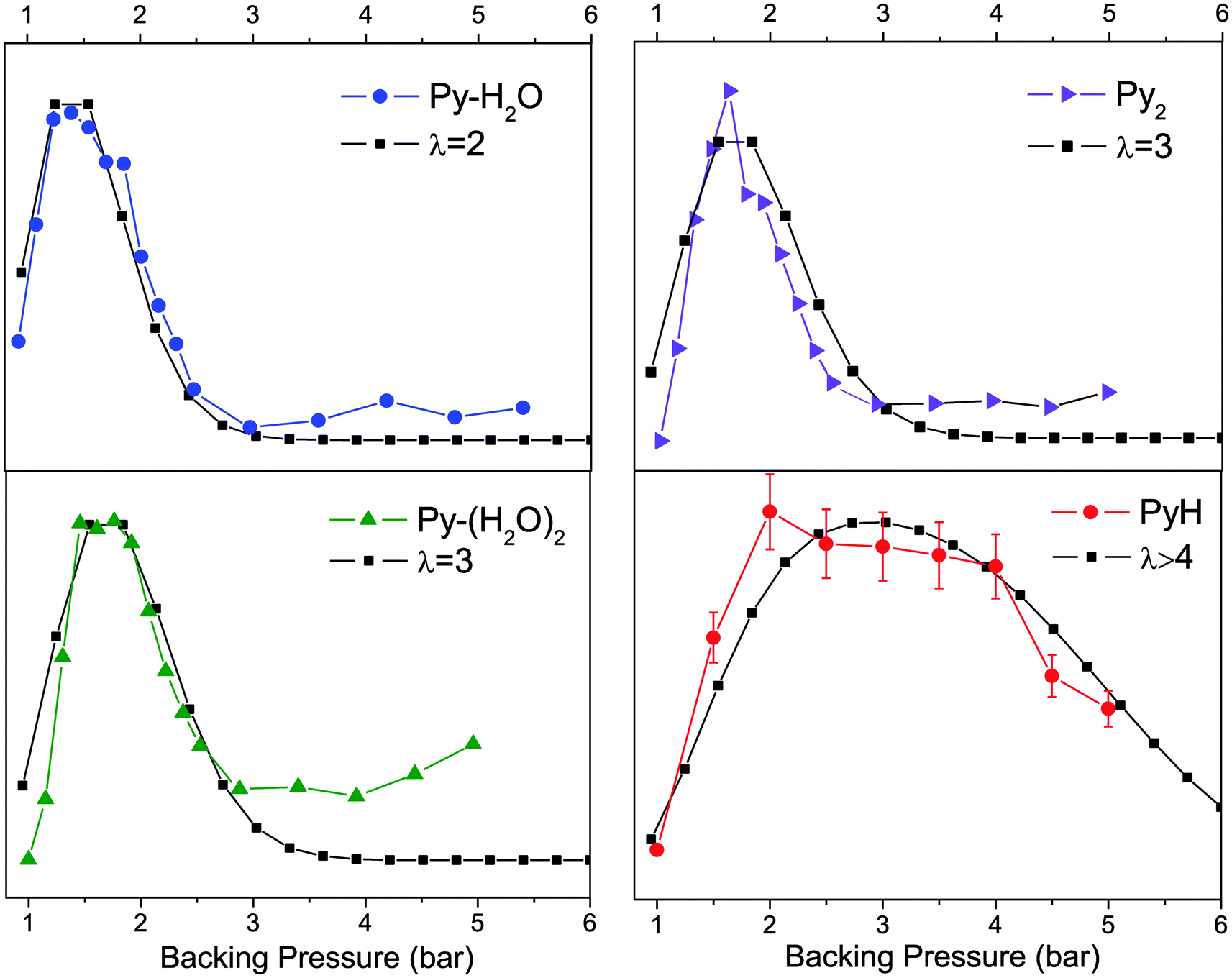 | ||
| Fig. 6 Variation of the ion intensities as a function of the backing pressure. When the pressure increases the cluster size increases. In red the PyH˙ signal seems to indicate that large clusters are involved in the reaction. | ||
The variation of the population of a given cluster size with the backing pressure should follow a Poisson distribution. To calibrate the signal versus the Poisson distribution, we assume that the Py–H2O complex follows a second order Poisson distribution P(λ = 2), and fitted the observed signal with this distribution (Fig. 6 left upper panel). As can be seen, the Py–(H2O)2 complex follows a P(λ = 3) distribution, which provides some confidence in the approach. The PyH˙ signal (lower right panel) clearly does not follow the same variation of the signal as the 1–1 or 1–2 complexes but can be fitted with a sum of distributions with P(λ ≥ 4) to be reproduced. At the same time we did not observe Py–(H2O)n clusters with n > 3, which probably means that these clusters react faster. This situation is in line with the experiments concerning pyridine–water anions, where it is suggested that with n > 3, only structures exhibiting proton transfer [PyH˙OH]−(H2O)n−1 are present, best described as (PyH˙OH−) (H2O)n−1.5,6
Why are large (n ≥ 4) clusters necessary to induce the reaction? The 1–1 complex is observed though a one-color two-photon ionization scheme, which implies a relatively long excited state lifetime i.e. a barrier to the reaction: in the absence of a barrier the reaction rate would be very fast, the excited state lifetime very short and the complex would not be ionized. The simplest explanation for the reactivity of larger clusters would be the lowering of the barrier as the cluster size increases. This effect is well known in the case of ionic reactions in which the transition state is stabilized by solvent molecules. But we are dealing with a radical reaction, which is a lot less sensitive to the solvent effect. As a matter of fact Reimers et al. calculated that the barrier is not strongly influenced by the solvent,1 so that this effect should be marginal.
A second hypothesis can be suggested. Ab initio calculations show that the crossing between the ground and the reacting excited state occurs at an OH distance of 1.9/1.8 Å, i.e. for NH around 1.1/1.2 Å if the NO distance is fixed, i.e. if the OH fragment did not move during the H transfer. As seen in Fig. 7, the excited state correlates with the PyH˙ + OH˙ ground state. Due to the H/OH mass ratio, the oxygen atom does not move very quickly as compared to the H atom. In order to be trapped in the weak PyH˙ potential well, the excess energy has to be removed quite efficiently and the reaction path represented by the dotted blue arrow will cross only once the conical intersection leading back to the initial Py–H2O system. This is probably the role of the other water molecules in the cluster. In the absence of such a dissipating bath the H atom will bounce on the N atom and recombine in the most stable Py–H2O form through the conical intersection (C), and further dissociate into Py + H2O.
 | ||
| Fig. 7 Schematic representation of the reaction mechanism. There is a competition between the stabilization of PyH˙ and the recombination in Py–H2O, which depends on how fast the excess energy is released and how fast the OH˙ radical is leaving the cluster. The dotted blue arrow represents the schematic reaction pathway showing the efficient energy relaxation induced by the solvent molecules, which prevents multiple passages over the conical intersection C. The potential curves are adapted from ref. 2. | ||
In summary of this part, the PyH˙ photo-product does not seem to come from the 1–1 complex and the fact that the 1–1 complex can be ionized means that its excited state lifetime is relatively long (longer than a few ps,…). It seems that larger clusters are necessary, probably because they react more efficiently and because the surrounding water molecules can dissipate the excess energy necessary to trap the PyH˙ product.
Photodissociation of PyH˙
In order to use pyridine as a photo-catalyst, one should ensure that the absorption of a photon by PyH˙ can lead to H loss and recovery of the catalyst. Calculations2 indicate that there is a πσ* state quasi dissociative along the NH coordinate, which crosses the ππ* excited state and leads to H atom loss. The barrier seems to be quite low, the excitation of the first observed transition at 425.6 nm (2.91 eV) is above the dissociation limit in Py + H (1.5 eV), thus PyH˙ photodissociation is energetically allowed.In the first step, we tried to evidence this process in exciting cold PyH˙ issued from the discharge source. The detection scheme requires the discharge to produce PyH˙, one photon in the 420 nm range to excite PyH˙ and a probe photon at 255 nm to ionize Py by resonant one-color two-photon absorption. The result was disappointing and no Py could be evidenced in this experiment.
In the second step, we studied the dissociation process from hot PyH˙ issued from the reaction in the clusters. In this case, the first laser at 255 nm excites the clusters, and the second powerful laser in the visible region is used to excite and ionize PyH˙ (via R2PI) and to ionize Py through a four-photon absorption process. The results are presented in Fig. 8.
 | ||
| Fig. 8 (a) Hydrogenated pyridine: lower panel, the two-photon excitation/ionization spectrum of cold PyH˙ produced in the discharge; middle panel, the action spectrum of PyH˙ produced after the excitation of Py–(H2O)n clusters at 255 nm and detecting PyH+ by R2PI; upper panel, the action spectrum of PyH˙ produced after excitation of Py–(H2O)n clusters and detecting Py+. (b) Deuterated pyridine: lower panel, two-photon excitation/ionization spectrum of cold dPyD˙ produced in the discharge; middle panel, the action spectrum of dPyD˙ produced after the excitation of dPy–(D2O)n clusters at 255 nm and detecting dPyD+ by R2PI; upper panel, the action spectrum of dPyD˙ produced after the excitation of dPy–(D2O)n clusters and detecting dPy+. | ||
The first set of data concerning the hydrogenated system is the richest (Fig. 8a). When PyH˙ is produced by reaction in Py–(H2O)n clusters, the first band at 23500 cm−1 is observed only in the PyH˙ channel and not in the Py channel. At 23600 cm−1 a threshold is observed in the Py channel (arrow in the upper panel of Fig. 8a) and the second band at 23880 cm−1 appears in both Py and PyH˙ channels. A third vibrational band is observed very weakly in the cold PyH˙ spectrum (from the discharge) and very strongly in the Py channel. In the case of the deuterated species, no structure is observed in the Py channel, only a broad band, which is also observed in the dPyD˙ channel underneath the vibronic bands.
These data can be rationalized in the following way: the dissociative πσ* state has a different symmetry than the optically active state ππ*. Thus, out of plane vibrations or high rotational excitations are necessary to induce a coupling between these states. In the cold PyH˙ from the discharge, the rotational excitation is low and the out of plane vibrations may not be very active, so that no dissociation is observed. The out of plane vibrations could be populated by the vibrational redistribution but in such a small cyclic molecule with a relatively small vibrational energy excess,39,40 this process should not be competitive with the radiative process.
For the hydrogenated species, the fragmentation threshold is clearly observed at 23600 cm−1 and the observation of a common vibrational band at 23880 cm−1 in all the spectra is good evidence that the Py observed comes from the excitation of PyH˙. An alternative explanation could have been the excitation of the clusters at 255 nm, relaxation toward the Py triplet state and evaporation of water molecules, followed by the ionization of the triplet by the probe laser. Since the probe laser is delayed in time by 600 ns and since it seems that the Py triplet is relatively short lived (72 ns),1,41 this mechanism will not match our measurements.
The third band observed at 24300 cm−1 in the Py channel might be assigned to some out of plane vibrations, which is weakly seen on the spectrum of the cold radical but will enhance strongly the coupling between the ππ* and the πσ* states and thus increase the fragmentation efficiency.
The contrast with the deuterated system is quite impressive, no structure is observed in the Py fragmentation pathway (Fig. 8b). The H/D loss is probably controlled by some tunneling mechanism, and the deuterated species will tunnel less efficiently, or need a higher internal energy to cross efficiently the barrier as compared to the hydrogenated species. In this case, only very hot dPyD molecules can dissociate. As already stated, the Py channel detection occurs via a four-photon ionization process, which may induce artifacts in the detection scheme (accidental resonances). Detection using a third laser to ionize Py with two-photon absorption would be worthwhile.
Conclusions
The spectroscopy of the pyridinyl radical has been characterized at low temperature in a supersonic expansion and its ionization potential has been determined. Since PyH˙ can be probed through one photon ionization in the UV, pump probe experiments have demonstrated that the UV excitation of Py–(H2O)n clusters leads to the formation of a pyridinyl radical, thus we have evidenced the water splitting reaction using pyridine as a photo-sensitizer. We have also evidenced the role of the surrounding molecules, some extra water molecules in the clusters being necessary to stabilize the PyH˙ product. It would probably be quite instructive to detect the other product of the reaction, the OH˙ radical.More work has to be done to fully characterize this reaction, in particular the cluster sizes that lead to the reaction may be studied through infrared hole burning experiments, and the dynamics of the excited states may be investigated in the picosecond regime.
The last step of the process, i.e. the regeneration of the catalyst through the photo-detachment of the hydrogen atom (PyH˙ + hν → Py + H) has been studied. It seems that only hot PyH˙ radicals can lose hydrogen atoms. Other aromatic molecules42,43 are expected to lead to similar water splitting reactions and will be tested in the near future.
Acknowledgements
We are highly indebted to Pr A. L. Sobolewski and W. Domcke for triggering our interest to this reaction mechanism and for very helpful discussions. We acknowledge the use of the computing facility cluster GMPCS of the LUMAT federation (FR LUMAT 2764).References
- J. R. Reimers and Z.-L. Cai, Phys. Chem. Chem. Phys., 2012, 14, 8791–8802 RSC.
- X. Liu, A. L. Sobolewski, R. Borrelli and W. Domcke, Phys. Chem. Chem. Phys., 2013, 15, 5957 RSC.
- X. Liu, A. L. Sobolewski and W. Domcke, J. Phys. Chem. A, 2014, 118, 7788–7795 CrossRef CAS PubMed.
- F. Riboni, E. Selli and A. J. Colussi, J. Phys. Chem. A, 2015, 119, 4433–4438 CrossRef CAS PubMed.
- A. F. DeBlase, C. T. Wolke, G. H. Weddle, K. A. Archer, K. D. Jordan, J. T. Kelly, G. S. Tschumper, N. I. Hammer and M. A. Johnson, J. Chem. Phys., 2015, 143, 144305 CrossRef PubMed.
- Y. Wang, X. Zhang, S. Lyapustina, M. M. Nilles, S. Xu, J. D. Graham, K. H. Bowen, J. T. Kelly, G. S. Tschumper and N. I. Hammer, Phys. Chem. Chem. Phys., 2016, 18, 704–712 RSC.
- A. Müller, F. Talbot and S. Leutwyler, J. Am. Chem. Soc., 2002, 124, 14486–14494 CrossRef.
- T. Droz, R. Knochenmuss and S. Leutwyler, J. Chem. Phys., 1990, 93, 4520 CrossRef CAS.
- K. Sakota, C. Okabe, N. Nishi and H. Sekiya, J. Phys. Chem. A, 2005, 109, 5245–5247 CrossRef CAS PubMed.
- G. A. Pino, I. Alata, C. Dedonder, C. Jouvet, K. Sakota and H. Sekiya, Phys. Chem. Chem. Phys., 2011, 13, 6325–6331 RSC.
- C. Dedonder-Lardeux, D. Grosswasser, C. Jouvet and S. Martrenchard, PhysChemComm, 2001, 4, 21–23 RSC.
- G. A. Pino, C. Dedonder-Lardeux, G. Grégoire, C. Jouvet, S. Martrenchard and D. Solgadi, J. Chem. Phys., 1999, 111, 10747 CrossRef CAS.
- G. Grégoire, C. Dedonder-Lardeux, C. Jouvet, S. Martrenchard, A. Peremans and D. Solgadi, J. Phys. Chem. A, 2000, 104, 9087–9090 CrossRef.
- O. David, C. Dedonder-Lardeux, C. Jouvet and A. L. Sobolewski, J. Phys. Chem. A, 2006, 110, 9383–9387 CrossRef CAS PubMed.
- M. Miyazaki, R. Ohara, K. Daigoku, K. Hashimoto, J. R. Woodward, C. Dedonder, C. Jouvet and M. Fujii, Angew. Chem., Int. Ed., 2015, 54, 15089–15093 CrossRef CAS PubMed.
- N. Tsuji, S.-I. Ishiuchi, C. Jouvet, C. Dedonder-Lardeux, M. Miyazaki, M. Sakai and M. Fujii, ChemPhysChem, 2011, 12, 1928–1934 CrossRef CAS PubMed.
- G. A. Pino, A. N. Oldani, E. Marceca, M. Fujii, S. Ishiuchi, M. Miyazaki, M. Broquier, C. Dedonder and C. Jouvet, J. Chem. Phys., 2010, 133, 124313 CrossRef CAS PubMed.
- A. Carrera, I. B. Nielsen, P. Carcabal, C. Dedonder, M. Broquier, C. Jouvet, W. Domcke and A. L. Sobolewski, J. Phys. Chem. A, 2009, 130, 024302 CAS.
- V. Stert, L. Hesse, H. Lippert, C. P. Schulz and W. Radloff, J. Phys. Chem. A, 2002, 106, 5051–5053 CrossRef CAS.
- S. Ishiuchi, K. Daigoku, M. Saeki, M. Sakai, K. Hashimoto and M. Fujii, J. Chem. Phys., 2002, 117, 7077 CrossRef CAS.
- S. Ishiuchi, M. Sakai, K. Daigoku and T. Ueda, Chem. Phys. Lett., 2001, 347, 87–92 CrossRef CAS.
- S.-I. Ishiuchi, K. Daigoku, K. Hashimoto and M. Fujii, J. Chem. Phys., 2004, 120, 3215–3220 CrossRef CAS PubMed.
- O. David, C. Dedonder-Lardeux, C. Jouvet, H. Kang, S. Martrenchard, T. Ebata and A. L. Sobolewski, J. Chem. Phys., 2004, 120, 10101 CrossRef CAS PubMed.
- A. Courty, M. Mons and I. Dimicoli, J. Phys. Chem. A, 1998, 102, 6590–6600 CrossRef CAS.
- M. Miyazaki, A. Takeda, S. Ishiuchi, M. Sakai, O. Dopfer and M. Fujii, Phys. Chem. Chem. Phys., 2011, 13, 2744–2747 RSC.
- K. Tanabe, M. Miyazaki, M. Schmies, A. Patzer, M. Schütz, H. Sekiya, M. Sakai, O. Dopfer and M. Fujii, Angew. Chem., Int. Ed., 2012, 51, 6604–6607 CrossRef PubMed.
- H. M. Kim, K. Y. Han, J. Park, G.-S. Kim and S. K. Kim, J. Chem. Phys., 2008, 128, 041104 CrossRef PubMed.
- C. Jouvet and B. Soep, Chem. Phys. Lett., 1983, 96, 426–428 CrossRef CAS.
- W. H. Breckenridge, C. Jouvet and B. Soep, J. Chem. Phys., 1986, 84, 1443 CrossRef CAS.
- M. Boivineau, J. Le Calve, M. C. Castex and C. Jouvet, Chem. Phys. Lett., 1986, 130, 208 CrossRef CAS.
- M. Dantus, M. J. Rosker and A. H. Zewail, J. Chem. Phys., 1987, 87, 2395–2397 CrossRef CAS.
- A. Zehnacker-Rentien and M. A. Suhm, Angew. Chem., Int. Ed., 2008, 47, 6970–6992 CrossRef PubMed.
- J. Wanna, J. A. Menapace and E. R. Bernstein, J. Chem. Phys., 1986, 85, 1795–1805 CrossRef CAS.
- C. Jouvet, C. Lardeux-Dedonder, M. Richard-Viard, D. Solgadi and A. Tramer, J. Phys. Chem. A, 1990, 94, 5041–5048 CrossRef CAS.
- C. Dedonder-Lardeux, C. Jouvet, M. Richard-Viard and D. Solgadi, J. Chem. Phys., 1990, 91, 2828 CrossRef.
- I. Alata, R. Omidyan, M. Broquier, C. Dedonder and C. Jouvet, Chem. Phys., 2012, 399, 224–231 CrossRef CAS.
- TURBOMOLE V6.3 2011, a Dev. Univ. Karlsruhe Forschungszentrum Karlsruhe GmbH, 1989-2007, TURBOMOLE GmbH, since 2007; available from http://http//www.turbomole.com.
- M. Riese, Z. Altug and J. Grotemeyer, Phys. Chem. Chem. Phys., 2006, 8, 4441–4448 RSC.
- J. B. Hopkins, D. E. Powers and R. E. Smalley, J. Chem. Phys., 1980, 73, 683–687 CrossRef CAS.
- R. E. Smalley, Annu. Rev. Phys. Chem., 1983, 34, 129 CrossRef CAS.
- K. Enomoto, J. A. LaVerne, S. Seki and S. Tagawa, J. Phys. Chem. A, 2006, 110, 9874–9879 CrossRef CAS PubMed.
- X. Liu, T. N. V. Karsili, A. L. Sobolewski and W. Domcke, J. Phys. Chem. B, 2015, 119, 10664–10672 CrossRef CAS PubMed.
- T. N. V. Karsili, D. Tuna, J. Ehrmaier and W. Domcke, Phys. Chem. Chem. Phys., 2015, 17, 32183–32193 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Table S1: calculated vertical transition energies for PyH˙ in C2v symmetry. Fig. S1: action spectra of Py–H2O, the pump laser is scanned and the detection is set on the Py–H2O+ mass (lower trace), or on the PyH+ mass with the probe laser at 225 nm (upper trace). Fig. S2: mass spectra obtained for pyridine without water through a one-color two-photon excitation/ionization scheme. Fig. S3: comparison between mass spectra obtained with the pump/probe scheme in an expansion with and without water. Fig. S4: excitation/action spectra of the species in the jet: homodimer, Py–H2O and Py one-color two-photon ionization spectra and pump/probe (225 nm) PyH˙ and dPyD˙ action spectra. See DOI: 10.1039/c6cp04398d |
| This journal is © the Owner Societies 2016 |