
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanoparticle electrochemistry
Peter H.
Robbs
and
Neil V.
Rees
*
School of Chemical Engineering, University of Birmingham, Edgbaston, Birmingham, B15 2TT, UK. E-mail: n.rees@bham.ac.uk
First published on 17th August 2016
Abstract
This perspective article provides a survey of recent advances in nanoscale electrochemistry, with a brief theoretical background and a detailed discussion of experimental results of nanoparticle based electrodes, including the rapidly expanding field of “impact electrochemistry”. Following this, recent advances in experimental study of exotic nanoparticle electrodes are reviewed.
Introduction
The plethora of physical,1,2 chemical3–6 and catalytic7–10 properties of nanoparticles have driven extensive research into nanomaterials. In the realms of electrochemistry the nanoscale has been subject to much interest in both fundamental11 and applied12–15 science. The benefits of decreasing scale in electrochemistry can, simplistically, be attributed to enhanced mass transport and greatly reduced capacitive charging. The improved signal![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :noise ratio allows for greater sensitivity towards trace species analysis and better resolution of competitive signals in electroanalysis.16,17
:noise ratio allows for greater sensitivity towards trace species analysis and better resolution of competitive signals in electroanalysis.16,17
This perspective review will focus primarily on advances in novel theoretical and experimental electrochemistry at nanoparticle electrodes, since 2014.18 Some of the underlying theory of electrochemistry at nanoparticles will be briefly summarised as a reminder for the reader.
Theoretical aspects of nanoelectrochemistry
As the critical dimension of an electrode decreases to the (sub)micron scale, the classical theories of electrochemistry can still adequately explain the observed phenomena. However, as the electrode radius shrinks to below ∼10 nm, the Butler–Volmer and (continuum) Marcus–Hush formalisms can fall short of a complete description. The detailed theory of electrochemistry at this scale has been reviewed extensively11,19,20 but merits an overview.The effective thickness of the diffusion layer (δeff) at the nanoelectrode|electrolyte interface can be estimated by:
| δeff = r0/A | (1) |
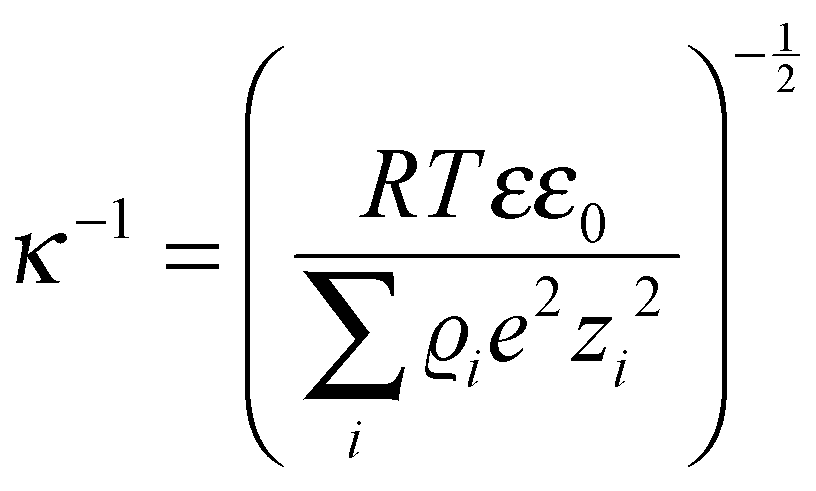 | (2) |
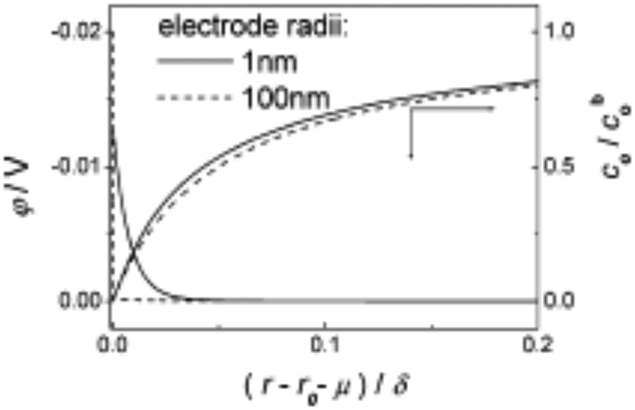 | ||
| Fig. 1 Theoretical model of the diffusion layer and EDL in the interfacial region of 1 and 100 nm radii electrodes. Parameters of model reduction reaction were: concentration of analyte and supporting electrolyte were 5 mM and 0.5 M respectively, analyte charge = −1, k0 = 1.0 cm s−1, D = 1 × 10−5 cm s−2, α = β = 0.5 and E0′ = 0. Full details of the computational work is available in ref. 23. Reproduced with permission of the American Chemical Society. | ||
This overlap can cause non-classical phenomena to complicate analysis at planar nanodisk electrodes, primarily for r0 ≤ 5 nm.11 The overlap of EDL and diffusion layer can violate the electroneutrality assumption, altering the mass transfer to the smaller nanoelectrodes. Chen et al.26 have described that conventional mass transport theories have been found to apply to nanoelectrodes to some extent. It was shown that the limiting current (Ilim) varied linearly with the concentration of [Fe(CN6)]3− for all electrode radii considered, however Ilim was shown to be depressed at electrodes where r0 < 10 nm. For r0 = 5 nm, Ilim was ∼20% lower than at larger electrodes (r0 = 9.7 μm and 16 nm).26 In this context, near wall hindered diffusion of analyte molecules was not considered.
Finally, the electrode size has an effect on electron transfer (ET) kinetics. At nanoelectrodes much faster ET kinetics can be probed, but the classic Butler–Volmer (BV) approach is not wholly appropriate. The BV formalism is more accurate at smaller overpotentials (η), where η is close to E0f, which has found general application to study at macro and microelectrodes. This derives from the underlying linearization of the free energy curves of reactants and products, which is most applicable close to E0f (i.e. small η). In addition, the BV model only considers ET to/from a narrow range of electronic states near the Fermi level of the electrode from/to the electroactive species. These two factors render the BV approach less accurate for ET at nanoelectrodes, where the enhanced mass transport can shift the ET to significantly higher η.11
The quantum Marcus–Hush (MH) and Marcus–Hush–Chidsey (MHC) formalisms can predict more reliable k0 values at the larger η of nanoelectrodes. The MHC theory in particular, considers the probability of the ET involving the density of states in the electrode (suitably weighted via Fermi–Dirac statistics) for inclusion into the Marcus Theory. Both MH and MHC formalisms predict similar rate constants, for large reorganisation energies (λ), at potential below the inversion predicted by classical MH theory. Complete discussion of the applicability of the different kinetic analyses can be found in the reviews by Chen and Liu11 and Zhang et al.19,20 Feldberg has derived relations to quantify the conditions where BV and MHC results diverge or agree, based on γ and λ (where γ = k0r0/D).27 Where the inequality (3) is true, the results are indistinguishable
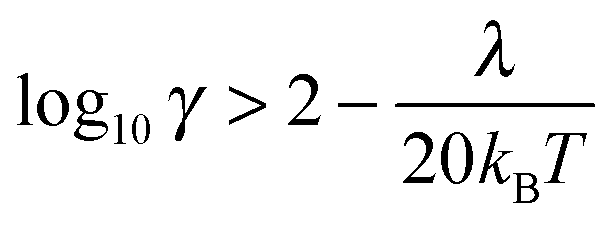 | (3) |
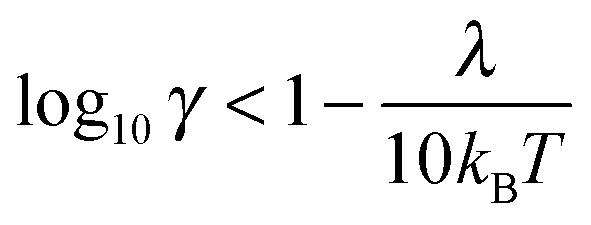 | (4) |
Nanoparticle arrays
Single disk nanoelectrodes can be challenging to reproduce by a variety of different methods.28 Modern micro and nanofabrication technologies are readily capable of fabricating nanoelectrode arrays (NEAs),29–31 however, the procedures are complex and can be very time consuming. A simple alternative approach is to use nanoparticles as a nanoparticle modified electrode.32 These can be manufactured by facile nanoparticle casting or electrodeposition methods to create irregular arrays. Arrays can provide an insight into electrochemistry at nanoparticles, at a fundamental level,32,33 as well as significant practical applicative use in ions in sensing.12The ideal nanoparticle modified working surface can be approximated as an array of spherical nanoelectrodes on an extended plane.13,32 The diffusion layer of each particle in a NEA can be estimated using conventional theories. It follows that the diffusion layer extends into the bulk over time and can interact and overlap, between particles depending on surface coverage, or spacing.13,34 As such, the interfacial structure at the NEA dictates the voltammetric response, which can be qualitatively divided into 4 ideal response categories (Fig. 2).
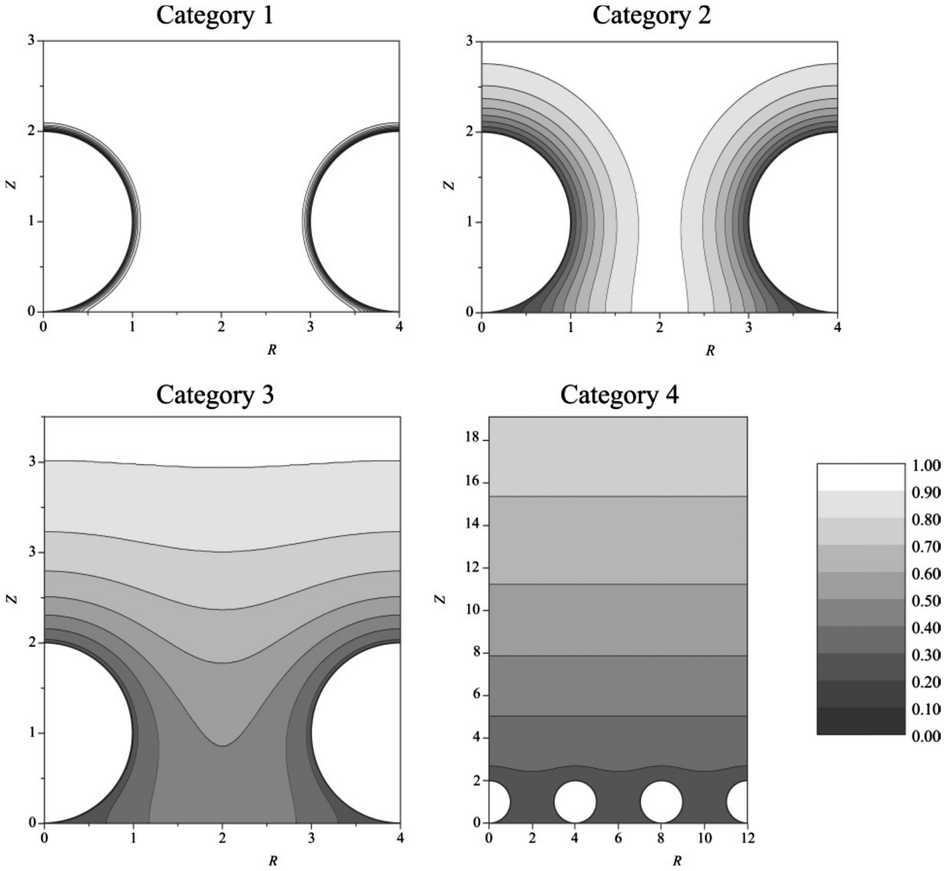 | ||
| Fig. 2 Simulated concentration profiles at nanoparticle electrode arrays at different dimensionless scan rates (σ). Category 1: σ = 1000. Category 2: σ = 10. Category 3: σ = 1. Category 4: σ = 0.01. For all categories, spherical diffusion domain boundary radius = 2. Reproduced from ref. 13, with permission of the American Chemical Society. | ||
Mass transport in category 1 is convergent, as with (sub)micro electrodes, while at category 4 planar diffusion contributes increasingly to the voltammetric response. As the planar contribution increases, the voltammogram changes from a steady state response to a peak;35 micro to macro-type response. There is a fifth conceptual case of the diffusion domain approximation, where a stochastic dispersion of nanoparticles could group into case 3 or 4 type regions, separated by case 1 or dead zones. This fifth case can make detailed analysis challenging because of the need for deconvolution of the different ensemble responses.
Electrodes modified with irregular NEAs have found extensive application in sensors, for detection of analytes including heavy metals,36–40 pollutants41 and biomolecules and pharmaceuticals.42–46 Nanoparticles have been applied to electroanalysis in this way, because of the high degree of selectivity toward analytes, such that trace species like As(III) and Hg(II) have been determined in the presence of interfering species like Cu(II),47 The lowest limit of detection (LOD) known to the authors for As(III) was 0.0096 ppb at an AuNP modified glassy carbon electrode, reported by Compton et al. in 2004.48 Other commonly studied heavy metals include Pb(II) (LOD: 0.008 ppb at dimercaptosuccinic acid capped Fe3O4 NP modified carbon paste electrode)49 and Cd(II) (LOD: 0.7 ppb at a single walled carbon nanotube film electrode).50
Common biomolecules studied include dopamine (DA), Ascorbic acid (AA) and uric adic (UA), all of which can cause interference with one another if analysed collectively.51,52 Using anodized titanium sheet electrodes modified with decorated TiO2 nanotubes, Cai et al.51 detected DA and UA in the presence of AA in several matrices of different acidities (pH 7, 5, 3) with a LOD of ca. 30 nM DA.
Nanoparticle impacts
While the nanoparticle modified electrodes discussed above are valuable tools, particularly for electroanalysis, it can be computationally demanding to deconvolute signals arising from inhomogeneity in the array structure. The electrochemical study of single nanoparticles, first studied by Heyrovsky et al.53–56 in 1995 and developed by Bard et al. in 200757 has gained increasing attention as a means of probing the electrochemistry at single nanoparticles.18,58 The underlying theory has been established, based on simplifying the nanoparticle-electrode impact to a spherical nanoelectrode in contact with an infinite plane. Ilim can be approximated using classical electrochemical theory for a sphere on an extended plane,57,59 where the critical time is on the order of microseconds; the limiting current is reached in t ≈ rNP2/DO. The transient current response during an impact event can be analysed according to the Shoup–Szabo equation.60–66 The rapid attainment of Ilim means the peak current of a transient can be recorded at a series of different applied potentials to model a voltammetric response of a single nanoparticle64,67–69 of radius rNP. Depending on the experimental conditions, the impinging particle can contact and catalyse a reaction, or be directly subject to a redox process; indirect and direct particle voltammetry respectively. A more detailed discussion of the analytical approaches and value of both general modes is available elsewhere.18Computational results
The contact between the particle and electrode has been found to be more complicated than a simple electrical contact.The interface at the substrate electrode, as the active particle approaches, is quantized (Fig. 3), with probabilities associated to the particle passing into the different regions close to the surface.70,71 One of the implications of the quantized interface is the occurrence of electron tunnelling when the particle approaches to within the tunnelling region.71 Once in the tunnelling region of the substrate, nanoparticles have been shown to be subject to a common colloidal phenomenon;72,73 near-wall hindered diffusion.65,70 As a particle approaches close to an infinite plane (the substrate electrode), diffusion in the z direction, away from the wall is limited, resulting in an ‘hydrodynamic adsorption’,70 which can have noticeable experimental effects.74–76 Apart from computational intrigue, an electrocatalytic particle hydrodynamically adsorbed to the substrate will show qualitatively different current transients (Fig. 4)76 as well as modulate the contact between the particle and substrate.77,78
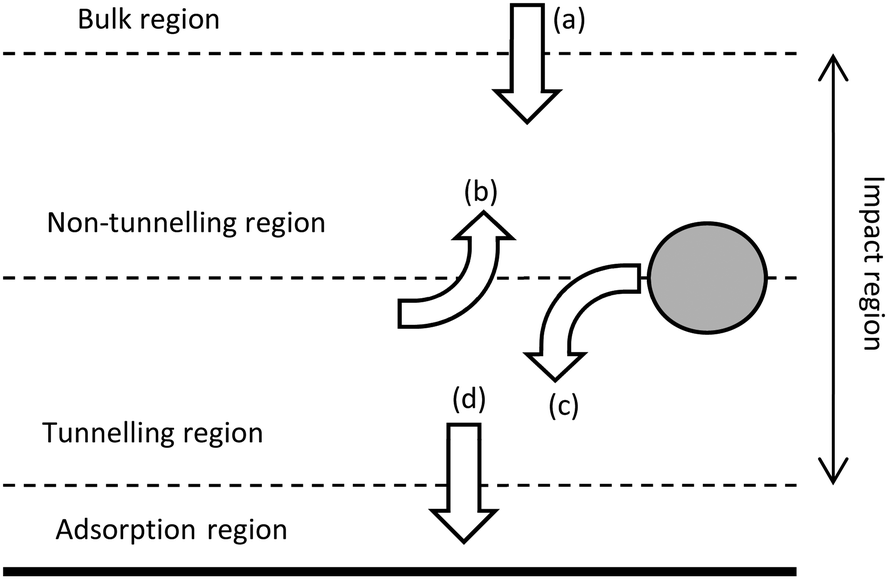 | ||
| Fig. 3 Schematic of the different regions at the electrode surface (solid line) and the associated probabilities that a particle will (a) diffuse into the non-tunnelling region from the bulk, (b) diffuse from the tunnelling to the non-tunnelling region, (c) move from non-tunnelling to tunnelling regions and (d) enter the adsorption region from the tunnelling region. Not to scale. Replicated from ref. 70 and 71 with permission from the Royal Society of Chemistry and Wiley VCH respectively. | ||
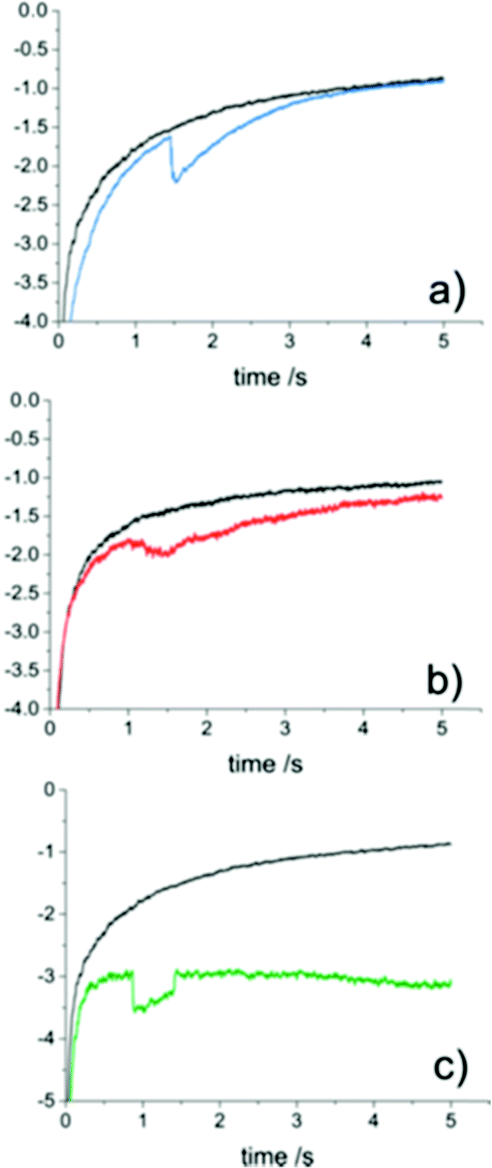 | ||
| Fig. 4 Different current transient waveforms for proton reduction (10 mM HClO4, 0.6 M NaClO4) at Au NPs on a carbon UME (r0 = 4.6 μm) in the absence (black) and presence (coloured) of au NPs (rNP = 9.8 ± 6.0 nm). (a) Step on-gradual off. (b) Gradual on-gradual off. (c) Step on-step off. Reproduced from ref. 76 with permission of the American Chemical Society. | ||
The state of agglomeration of nanoparticles has been investigated both computationally and experimentally, based on the assumption that agglomerates can be approximated as nanoparticles; ragg ≈ Nrmono where N is the number of monomers in the agglomerate. Initial experiments suggest that destructive anodic particle coulometry (APC) quantitatively gave the state of agglomeration.79 Later work80 investigated the reversibility of the process, suggesting Ag NPs undergo reversible agglomeration in KCl, however the decreased flux of larger particles limits the likelihood of observing agglomerate impacts.81
Hydrodynamic systems
Under conventional experimental conditions, the mass transport of nanoparticles is by Brownian motion, as described by Stokes–Einstein equation. The flux of the particles is largely assumed to be independent of the electrical field in the cell, because of the supporting electrolyte, such that the only external field acting on the particles is gravity. A few research groups have made some initial advancements into increasing the flux of particles to the substrate electrode by using convection, specifically microfluidic channels,82,83 magnetically stirred and microjet84 electrochemical cells. A key advantage of hydrodynamic (micro)electrodes is the flow rate and regime are readily controllable experimentally, so the mass transport to an electrode is easily managed. At first consideration, it could be assumed that as the flow rate increases, the particle impact frequency would increase, up to the limit of the equipment deployed. It was in fact found that the collision frequency increased with flow rate to a maximum,82,84 in both configurations (Fig. 5). Under magnetic stirring, Jian et al.84 recorded a near binary change in impact frequency; when the stirring was off, there were negligible impacts recorded (Fig. 6), which was ascribed to the lower mass transfer.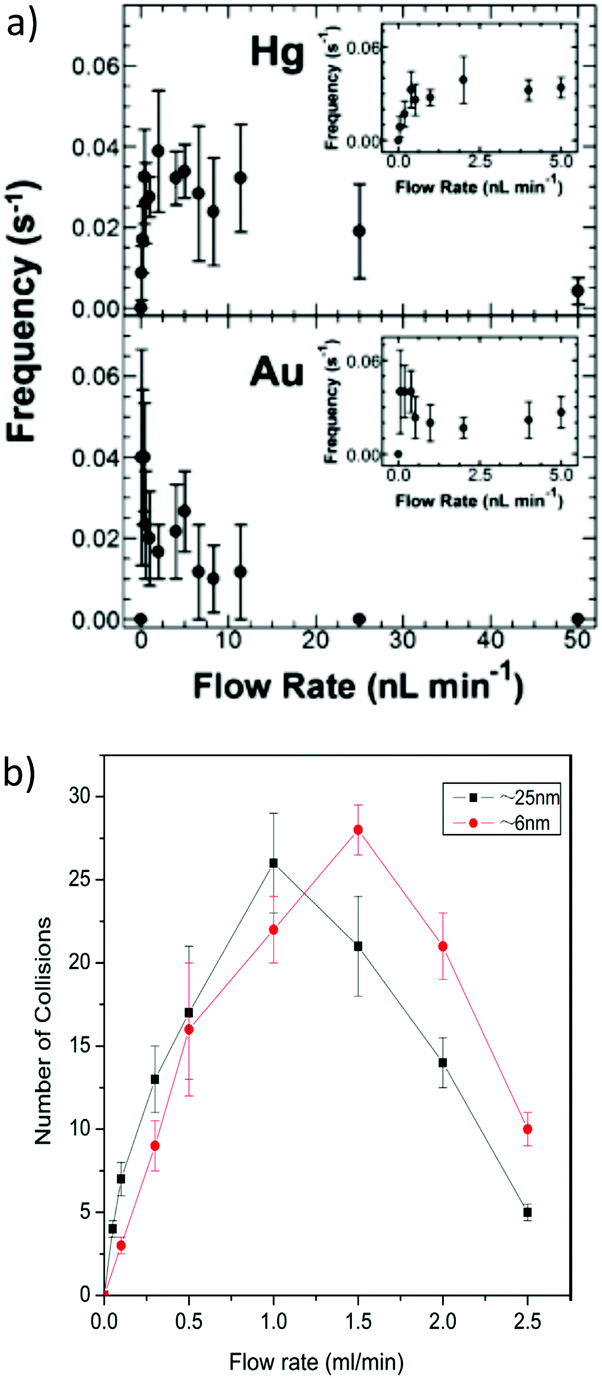 | ||
| Fig. 5 Change of impact frequency with flow rate at (a) Hg/Pt (top) and Au (bottom) microband UMEs and (b) face on microjet at an Hg/Pt UME. For full interpretation of figures, see the text details in the references. Reproduced from ref. 82 and 84 with permission of the American Chemical Society and Elsevier respectively. | ||
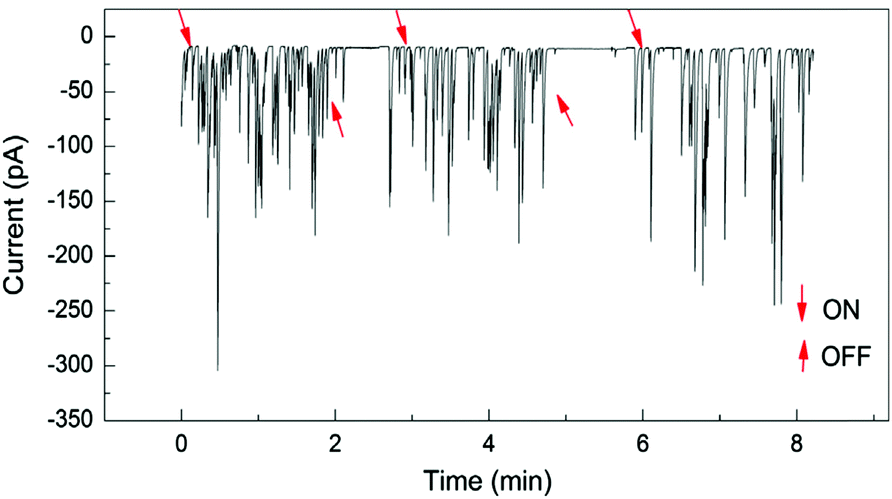 | ||
| Fig. 6 Pt NP (25 nm) impacts at a Hg/Pt UME at −0.04 V (vs. SCE) under pulsed magnetic stirring, in phosphate buffered (50 mM, pH 7.5) N2H4 (15 mM) solution. The arrows designated ON and OFF denote approximately where the stirring was switched on or off respectively. Reproduced from ref. 84 with permission of Elsevier. | ||
Exotic nanoparticle electrodes
Analysis of hetero-nanostructures, such as core@shell and decorated nanoparticles by impact methods is still in its infancy, compared to the field of nanoparticle electrochemistry. At the time of writing, there have only been 3 hetero-nanostructures probed, to the best of the authors' knowledge, covering supported particles77,78,83 and core@shell88 structures.
Investigation of N-doped carbon nanotube supported palladium particles (Pd/N-CNT) demonstrated the motion of the nanotube at the interface.48,49 The physical movement at the substrate, due to the hydrodynamic adsorption, could interfere with the electrical contact between the substrate and impinging particle, whereas the particle modified electrode showed a more stable current response. The other of the supported heteroparticles investigated were decorated magnetite, where the superparamagnetic magnetite particle supports were exposed to an external magnetic field.83,89 The magnetite support particles investigated were decorated with both platinum83 and Prussian Blue89 and were studied in a microfluidic and conventional configurations, respectively. In both cases the flux and consequently frequency of impacts was increased by application of an external magnetic field. In the microfluidic experiment, it was noted that the convective forces also increased the collision frequency,82 but to a lesser extent than the applied magnetic field. To add to the extensive range of nanoparticles that can be sized using nanoimpact methods, Au@Ag core@shell particles have been analysed by Holt et al.88 by APC; the charge transferred by exhaustive oxidation was used to derive the size of the particles. In the same experiment, the chemistry of core, shell and alloy materials was investigated, by modelling a CV for the desired reaction, within reason, or analysing the transient decays. This type of analysis is yet to be investigated in depth, but could prove to be an invaluable advancement in the field, particularly with supported electrocatalysts for use in better understood systems like proton reduction, or methanol oxidation.
Carbon nanoparticles and organic nanoparticles
Carbon based NPs have not been overlooked in these investigations insofar as the first direct use of an organic amplifier species was the non-destructive tag-redox coulometry90 (TRC), where an inert particle is modified with an electrochemically active species. Graphene nanoflakes have been observed91 in a similar manner,92 where the flakes were grafted with a ferrocene derivative (Fc′) as a redox tag. The unmodified graphene nanoflakes showed only capacitive current from particle charge/discharge.91 The Fc′ modified graphene showed both faradaic and capacitive currents, where the latter was approximately 10 fold lower.92 Other carbon nanomaterials acting as nanoparticle electrodes include fullerene clusters (nano-C60), reported by Stuart et al.93 and carbon nanotubes grafted onto an AuNP reported by Park et al.94 The nano-C60 experiments were conducted using direct electrochemistry of the fullerene clusters, while the carbon nanotubes made use of the area increase, when the particle contacted the electrode, with ferrocene as the indicator species.The first fully organic nanoparticles analyse by this technique were re-precipitated indigo dye nanoparticles.95 The particle size determined was quantitative, as confirmed by dynamic light scattering (DLS). Indigo can undergo a 2 electron reduction to leuco-indigo (Fig. 7a)96 but aromatic hydrocarbons can also be stable to irreversible oxidation reactions and nanoimpact analysis has been shown to be robust toward the analysis of bifunctional magnetite nanoparticles. Following from that, oil blue dye particles, which can undergo 2 separate 1-electron oxidation and reduction reactions (Fig. 7b) was studied.97 The voltammetric response of oil blue dye, both in dry acetonitrile and deionised water (pH 6.8) was shown to be comparable to a substituted anthraquinone, and as such undergoes irreversible redox reactions wherein the charge transferred again allowed a quantitative size distribution to be derived, by both APC and CPC.
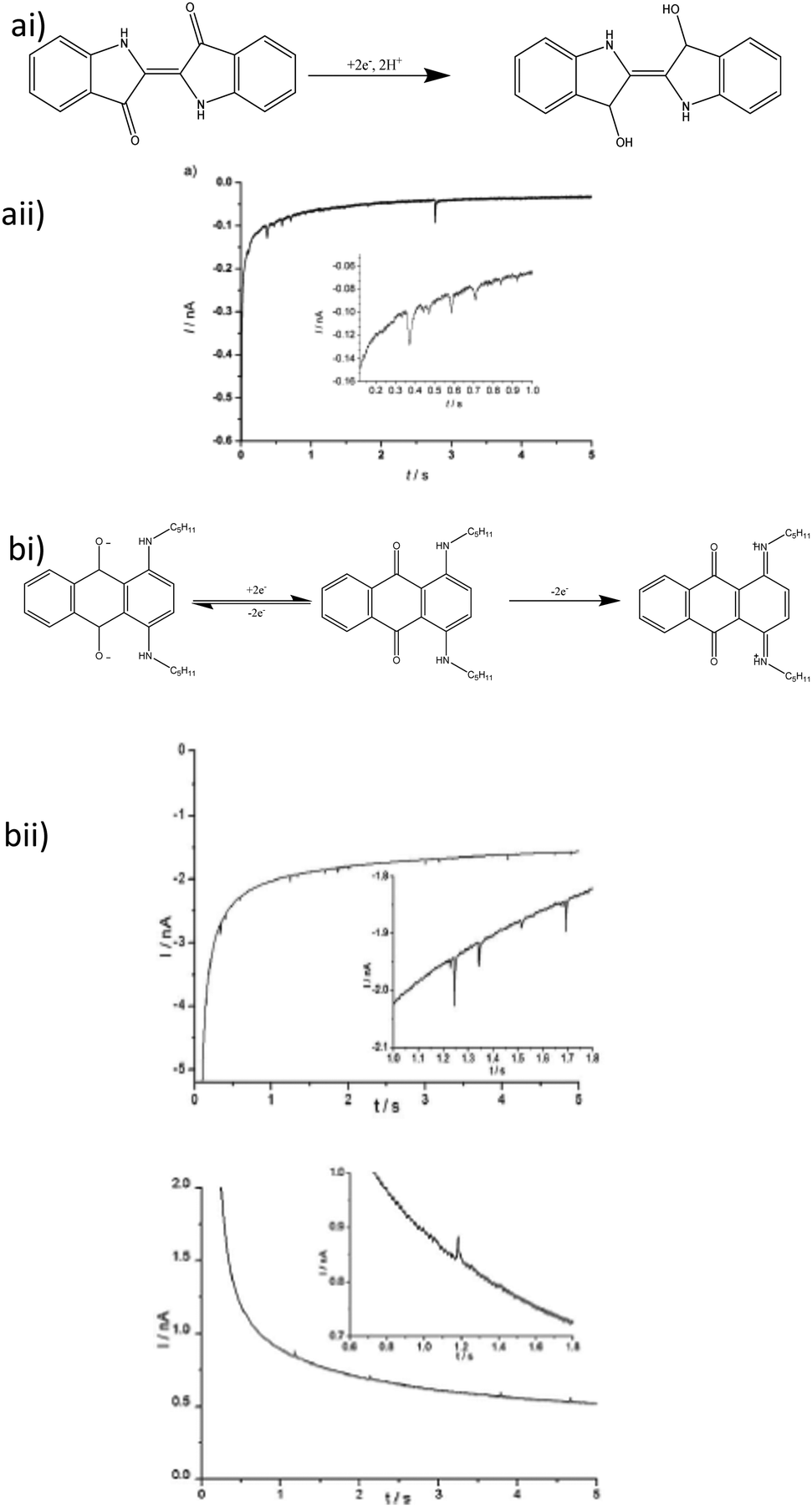 | ||
| Fig. 7 (a) Indigo dye impact electrochemistry (i) reduction mechanism and (ii) impact current transient. (b) oil blue dye nanoimpacts (i) redox chemistry of oil blue dye and (ii) both cathodic (bottom left) and anodic (bottom right) impact transients for oil blue dye particles. All insets are magnified sections to show impact current spikes in more detail. Reproduced from ref. 95 and 97 with permission of Wiley VCH and the Royal Society of Chemistry respectively. | ||
Micelles and soft particles
Above the critical micelle concentration (CMC), surfactants can encapsulate electroactive species. A relatively simple example is cetyltrimethyl ammonium bromide (CTAB), wherein the encapsulated Br− counterions are oxidised.98 It was shown that the current spikes only appear at [CTAB] > CMC98,99 in electrolyte solution. The encapsulation of reactive species has been well studied recently,100–103 where the micelles formed act as nanoreactors containing as little as attolitres103 of analyte. Typical inorganic redox active species other than bromide have been uncommon in soft particle impacts, but Lebègue et al.100 measured the response of [Fe(CN6)]4− encapsulated in a phospholipid vesicle. Electrochemically active biomolecules have been studied as vesicle reactors as well; the kinetic constant and turnover frequency of vitamin B12 mediated dehalogenation of trichloroethylene determined to be 29 ± 4 mM and 225 ± 32 s−1 respectively.101 Interestingly, ‘blank’ vesicles impinging on an electrode give rise to a step on-step off type signal (vide supra), where the absolute limiting current decreases.100 The current steps observed by Lebègue et al.100 were qualitatively the same as those observed by impacts of insulating latex104 polystyrene105,106 and silica106 particles.Practical considerations for experimentalists
It is challenging to develop a general set of rules for a successful nanoimpact experiment due to the multiple factors to account for. However, consideration of these factors, although complex, can aid in the design and troubleshooting of an experiment. Key factors affecting both direct and indirect type collision processes include:Particle number concentration
In order to achieve a convenient, practical impact rate, whilst maintaining dispersion and minimising the probability of simultaneous impacts, a low number concentration is needed, with numerous studies using the nM to pM range. In concert with the ionic strength of the solution, high number concentrations can contribute to agglomeration and aggregation of the colloid.107,108Supporting electrolyte concentration
Experiments require a sufficiently high supporting electrolyte concentration to suppress migration, however the ionic strength of the solution has a clear effect on the state of particle agglomeration or aggregation.79,80,107–110Signal![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :noise ratio
:noise ratio
Notwithstanding minimisation of electrical and instrumental noise levels, the magnitude of the current signals must be considered. In terms of direct experiments, the signal:noise ratio places a lower limit on the size of the particle,111 and for indirect experiments this limits the analyte concentration.110 For both direct and indirect experiments, increasing the number of electrons transferred during a reaction can improve the signal:noise ratio.
Instrumentation
The potentiostat employed must have low noise levels and ultralow (<1 nA) detection. In addition, it must have fast rise times and sampling rates, compared to the timescale of the impact current transients. The sensitivity and rise times must be appropriate to the experiment, but can often be adjusted according to the objectives. Careful electrical grounding of equipment and the use of a Faraday cage is imperative.Conclusions and outlook
Nanoelectrodes have been widely used to study fast electrode kinetics through a wide range of experimental techniques. Nanoparticle modified electrodes offer a facile starting point for analysis, but nanoimpact methods can give more detailed insight into electrochemistry at single particles. Nanoimpact methods have been extensively used to analyse a wide range of nanomaterials, but the field has scope to develop further. Fast reaction kinetics and synthetic methods have received less attention than computational methods and physical analysis under nanoimpact regimes and offer further means of probing surface chemistry at nanocolloids.List of abbreviations
| AA | Ascorbic acid |
| APC | Anodic particle coulometry |
| BV | Butler–Volmer |
| C 0 | Bulk concentration |
| CMC | Critical micelle concentration |
| CPC | Cathodic particle coulometry |
| CTAB | Cetyltrimethylammonium bromide |
| CV | Cyclic voltammetry |
| DA | Dopamine |
| DLS | Dynamic light scattering |
| D x | Diffusion coefficient of species x |
| EDL | Electrical double layer |
| E 0f | Formal potential |
| ET | Electron transfer |
| Fc′ | Ferrocene derivative |
| I lim | Limiting current |
| LOD | Limit of detection |
| LTD | Layered transition metal dichalcogenide |
| MH | Marcus–Hush |
| MHC | Marcus–Hush–Chidsey |
| NEA | Nanoelectrode array |
| NP | Nanoparticle |
| r 0 | Nominal electrode radius |
| r agg | Radius of aggregate particle |
| r mono | Radius of monomer component of aggregate(s) |
| r NP | Nanoparticle radius |
| TRC | Tag-redox coulometry |
| UA | Uric acid |
| δ eff | Effective diffusion layer thickness |
| λ | Reorganisation energy |
| η | Overpotential |
Acknowledgements
The Authors acknowledge financial support from the EPSRC, through the Centre for Doctoral Training in Fuel Cells and their Fuels.Notes and References
- E. Roduner, Chem. Soc. Rev., 2006, 35, 583–592 RSC.
- G. Schmid, Chem. Rev., 1992, 92, 1709–1727 CrossRef CAS.
- C. Rao, G. Kulkarni, P. Thomas and P. Edwards, Chem. – Eur. J., 2002, 8, 28–35 CrossRef CAS.
- B. K. Teo, J. Cluster Sci., 2013, 25, 5–28 CrossRef.
- M.-C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS PubMed.
- B. Gates, Chem. Rev., 1995, 95, 511–522 CrossRef CAS.
- Z. Xu, F. Xiao, S. Purnell, O. Alexeev, S. Kwai, S. Deutsch and B. Gates, Nature, 1994, 372, 346–348 CrossRef CAS.
- J. Greeley, J. K. Nørskov and M. Mavrikakis, Annu. Rev. Phys. Chem., 2002, 53, 319–348 CrossRef CAS PubMed.
- B. F. G. Johnson, Coord. Chem. Rev., 1999, 190–192, 1269–1285 CrossRef CAS.
- A. Choplin and F. Quignard, Coord. Chem. Rev., 1998, 178–180, 1679–1702 CrossRef CAS.
- S. Chen and Y. Liu, Phys. Chem. Chem. Phys., 2014, 16, 635–652 RSC.
- D. W. M. Arrigan, Analyst, 2004, 129, 1157–1165 RSC.
- I. Streeter, R. Baron and R. G. Compton, J. Phys. Chem. C, 2007, 111, 17008–17014 CAS.
- M. V. Mirkin, W. Nogala, J. Velmurugan and Y. Wang, Phys. Chem. Chem. Phys., 2011, 13, 21196–21212 RSC.
- H. G. Park and J. K. Holt, Energy Environ. Sci., 2010, 3, 1028–1036 CAS.
- X. Xie, D. Stueben and Z. Berner, Anal. Lett., 2005, 38, 2281–2300 CrossRef CAS.
- C. M. Li and W. Hu, J. Electroanal. Chem., 2013, 688, 20–31 CrossRef CAS.
- N. V. Rees, Electrochem. Commun., 2014, 43, 83–86 CrossRef CAS.
- S. M. Oja, M. Wood and B. Zhang, Anal. Chem., 2013, 85, 473–486 CrossRef CAS PubMed.
- S. M. Oja, Y. Fan, C. M. Armstrong, P. Defnet and B. Zhang, Anal. Chem., 2016, 88, 414–430 CrossRef PubMed.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, John Wiley & Sons, Inc., New York, 2nd edn, 2001, pp. 137–155 Search PubMed.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, John Wiley & Sons, Inc., New York, 2nd edn, 2001, pp. 534–579 Search PubMed.
- R. He, S. Chen, F. Yang and B. Wu, J. Phys. Chem. B, 2006, 110, 3262–3270 CrossRef CAS PubMed.
- E. J. F. Dickinson and R. G. Compton, J. Phys. Chem. C, 2009, 113, 17585–17589 CAS.
- X. Yang and G. Zhang, Nanotechnology, 2007, 18, 335201 CrossRef.
- S. Yu, L. Yuwen, L. Zhixiu, X. Lu, W. Aili and C. Shengli, J. Phys. Chem. C, 2009, 113, 9878–9883 Search PubMed.
- S. W. Feldberg, Anal. Chem., 2010, 82, 5176–5183 CrossRef CAS PubMed.
- J. T. Cox and B. Zhang, Annu. Rev. Anal. Chem., 2012, 5, 253–272 CrossRef CAS PubMed.
- X. J. Huang, A. M. O'Mahony and R. G. Compton, Small, 2009, 5, 776–788 CrossRef CAS PubMed.
- Y. Chen and A. Pepin, Electrophoresis, 2001, 22, 187–207 CrossRef CAS PubMed.
- Y. Chen, Microelectron. Eng., 2015, 135, 57–72 CrossRef CAS.
- S. R. Belding, F. W. Campbell, E. J. F. Dickinson and R. G. Compton, Phys. Chem. Chem. Phys., 2010, 12, 11208–11221 RSC.
- F. W. Campbell, S. R. Belding, R. Baron, L. Xiao and R. G. Compton, J. Phys. Chem. C, 2009, 113, 14852–14857 CAS.
- N. Godino, X. Borrise, F. X. Munoz, F. J. del Campo and R. G. Compton, J. Phys. Chem. C, 2009, 113, 11119–11125 CAS.
- T. J. Davies, S. Ward-Jones, C. E. Banks, J. del Campo, R. Mas, F. X. Muñoz and R. G. Compton, J. Electroanal. Chem., 2005, 585, 51–62 CrossRef CAS.
- G. Aragay and A. Merkoci, Electrochim. Acta, 2012, 84, 49–61 CrossRef CAS.
- M. Li, H. Gou, I. Al-Ogaidi and N. Wu, ACS Sustainable Chem. Eng., 2013, 1, 713–723 CrossRef CAS.
- M. B. Gumpu, S. Sethuraman, U. M. Krishnan and J. B. B. Rayappan, Sens. Actuators, B, 2015, 213, 515–533 CrossRef CAS.
- G. March, T. Nguyen and B. Piro, Biosensors, 2015, 5, 241–275 CrossRef CAS PubMed.
- S. Thatai, P. Khurana, J. Boken, S. Prasad and D. Kumar, Microchem. J., 2014, 116, 62–76 CrossRef CAS.
- L. Rassaei, F. Marken, M. Sillanpää, M. Amiri, C. M. Cirtiu and M. Sillanpää, TrAC, Trends Anal. Chem., 2011, 30, 1704–1715 CrossRef CAS.
- B. J. Sanghavi, O. S. Wolfbeis, T. Hirsch and N. S. Swami, Microchim. Acta, 2015, 182, 1–41 CrossRef CAS PubMed.
- M. Holzinger, A. Le Goff and S. Cosnier, Front. Chem., 2014, 2, 63–73 Search PubMed.
- A. Chen and S. Chatterjee, Chem. Soc. Rev., 2013, 42, 5425–5438 RSC.
- W. Siangproh, W. Dungchai, P. Rattanarat and O. Chailapakul, Anal. Chim. Acta, 2011, 690, 10–25 CrossRef CAS PubMed.
- B. Uslu and S. a. Ozkan, Anal. Lett., 2011, 44, 2644–2702 CrossRef CAS.
- B. K. Jena and C. R. Raj, Anal. Chem., 2008, 80, 4836–4844 CrossRef CAS PubMed.
- X. Dai, O. Nekrassova, M. E. Hyde and R. G. Compton, Anal. Chem., 2004, 76, 5924–5929 CrossRef CAS PubMed.
- W. Yantasee, K. Hongsirikarn, C. L. Warner, D. Choi, T. Sangvanich, M. B. Toloczko, M. G. Warner, G. E. Fryxell, R. S. Addleman and C. Timchalk, Analyst, 2008, 133, 348–355 RSC.
- M.-P. N. Bui, C. A. Li, K. N. Han, X.-H. Pham and G. H. Seong, Anal. Sci., 2012, 28, 699–704 CrossRef CAS PubMed.
- S. Mahshid, C. Li, S. S. Mahshid, M. Askari, A. Dolati, L. Yang, S. Luo and Q. Cai, Analyst, 2011, 136, 2322–2329 RSC.
- T.-Q. Xu, Q.-L. Zhang, J.-N. Zheng, Z.-Y. Lv, J. Wei, A.-J. Wang and J.-J. Feng, Electrochim. Acta, 2014, 115, 109–115 CrossRef CAS.
- M. Heyrovsky and J. Jirkovsky, Langmuir, 1995, 11, 4288–4292 CrossRef CAS.
- M. Heyrovsky, J. Jirkovsky and M. Struplova-Bartackova, Langmuir, 1995, 11, 4309–4312 CrossRef CAS.
- M. Heyrovsky, J. Jirovsky and B. R. Muller, Langmuir, 1995, 11, 4293–4299 CrossRef CAS.
- M. Heyrovsky, J. Jirovsky and M. Struplova-Bartackova, Langmuir, 1995, 11, 4300–4308 CrossRef CAS.
- X. Xiao and A. J. Bard, J. Am. Chem. Soc., 2007, 129, 9610–9612 CrossRef CAS PubMed.
- N. V. Rees, Y.-G. Zhou and R. G. Compton, RSC Adv., 2012, 2, 379–384 RSC.
- P. A. Bobbert, M. M. Wind and J. Vlieger, Physica A, 1987, 141, 58–72 CrossRef.
- C. Batchelor-McAuley, E. J. F. Dickinson, N. V Rees, K. E. Toghill and R. G. Compton, Anal. Chem., 2012, 84, 669–684 CrossRef CAS PubMed.
- E. Kätelhön, E. E. L. Tanner, C. Batchelor-McAuley and R. G. Compton, Electrochim. Acta, 2016, 199, 297–304 CrossRef.
- W. Cheng and R. G. Compton, Trends Anal. Chem., 2014, 58, 79–89 CrossRef CAS.
- S. Eloul and R. G. Compton, J. Phys. Chem. C, 2015, 119, 27540–27549 CAS.
- E. J. E. Stuart, N. V. Rees and R. G. Compton, Chem. Phys. Lett., 2012, 531, 94–97 CrossRef CAS.
- E. O. Barnes, Y.-G. Zhou, N. V. Rees and R. G. Compton, J. Electroanal. Chem., 2013, 691, 28–34 CrossRef CAS.
- E. J. E. Stuart, Y.-G. Zhou, N. V. Rees and R. G. Compton, RSC Adv., 2012, 2, 6879–6884 RSC.
- J. M. Kahk, N. V. Rees, J. Pillay, R. Tshikhudo, S. Vilakazi and R. G. Compton, Nano Today, 2012, 7, 174–179 CrossRef CAS.
- B. Haddou, N. V Rees and R. G. Compton, Phys. Chem. Chem. Phys., 2012, 14, 13612–13617 RSC.
- Y.-G. Zhou, N. V. Rees and R. G. Compton, Angew. Chem., Int. Ed., 2011, 50, 4219–4221 CrossRef CAS PubMed.
- E. Kätelhön and R. G. Compton, Chem. Sci., 2014, 5, 4592–4598 RSC.
- E. Kätelhön and R. G. Compton, ChemElectroChem, 2015, 2, 64–67 CrossRef.
- Y. Kazoe and M. Yoda, Appl. Phys. Lett., 2011, 99, 124104 CrossRef.
- M. a. Bevan and D. C. Prieve, J. Chem. Phys., 2000, 113, 1228–1236 CrossRef CAS.
- E. Kätelhön, S. V. Sokolov and R. G. Compton, Sens. Actuators, B, 2016, 234, 420–425 CrossRef.
- S. Eloul, E. Kätelhön, C. Batchelor-McAuley, K. Tschulik and R. G. Compton, J. Electroanal. Chem., 2015, 755, 136–142 CrossRef CAS.
- L. S. Y. Ly, C. Batchelor-McAuley, K. Tschulik, E. Kätelhön and R. G. Compton, J. Phys. Chem. C, 2014, 118, 17756–17763 CAS.
- X. Li, C. Batchelor-Mcauley, S. A. I. Whitby, K. Tschulik, L. Shao and R. G. Compton, Angew. Chem., Int. Ed., 2016, 55, 4296–4299 CrossRef CAS PubMed.
- H. Hodson, X. Li, C. Batchelor-McAuley, L. Shao and R. G. Compton, J. Phys. Chem. C, 2016, 120, 6281–6286 CAS.
- J. Ellison, K. Tschulik, E. J. E. Stuart, K. Jurkschat, D. Omanović, M. Uhlemann, A. Crossley and R. G. Compton, ChemistryOpen, 2013, 2, 69–75 CrossRef CAS PubMed.
- S. V. Sokolov, K. Tschulik, C. Batchelor-McAuley, K. Jurkschat and R. G. Compton, Anal. Chem., 2015, 87, 10033–10039 CrossRef CAS PubMed.
- S. V. Sokolov, E. Kätelhön and R. G. Compton, J. Electroanal. Chem., 2016 DOI:10.1016/j.jelechem.2016.01.023.
- T. M. Alligrant, M. J. Anderson, R. Dasari, K. J. Stevenson and R. M. Crooks, Langmuir, 2014, 30, 13462–13469 CrossRef CAS PubMed.
- D. A. Robinson, J. J. Yoo, A. D. Casraneda, B. Gu, R. Dasari, R. M. Crooks and K. J. Stevenson, ACS Nano, 2015, 9, 7583–7595 CrossRef CAS PubMed.
- J. Jiang, X. Huang and L. Wang, J. Colloid Interface Sci., 2016, 467, 158–164 CrossRef CAS PubMed.
- K. Tschulik, B. Haddou, D. Omanović, N. V. Rees and R. G. Compton, Nano Res., 2013, 6, 836–841 CrossRef CAS.
- K. Shimizu, K. Tschulik and R. G. Compton, Chem. Sci., 2016, 7, 1–7 RSC.
- C. S. Lim, S. M. Tan, Z. Sofer and M. Pumera, ACS Nano, 2015, 9, 8474–8483 CrossRef CAS PubMed.
- L. R. Holt, B. J. Plowman, N. P. Young, K. Tschulik and R. G. Compton, Angew. Chem., Int. Ed., 2016, 55, 397–400 CrossRef CAS PubMed.
- G. P. Santos, A. F. Melo and F. N. Crespilho, Phys. Chem. Chem. Phys., 2014, 16, 8012–8018 RSC.
- N. V. Rees, Y.-G. Zhou and R. G. Compton, Chem. Phys. Lett., 2012, 525–526, 69–71 CrossRef.
- J. Poon, C. Batchelor-McAuley, K. Tschulik and R. G. Compton, Chem. Sci., 2015, 6, 2869–2876 RSC.
- H. Wu, Q. Lin, C. Batchelor-McAUley, L. M. Gonçalves, C. F. R. A. C. Lima and R. G. Compton, Analyst, 2016, 141, 2696–2703 RSC.
- E. J. E. Stuart, K. Tschulik, C. Batchelor-McAuley and R. G. Compton, ACS Nano, 2014, 8, 7648–7654 CrossRef CAS PubMed.
- J. H. Park, S. N. Thorgaard, B. Zhang and A. J. Bard, J. Am. Chem. Soc., 2013, 135, 5258–5261 CrossRef CAS PubMed.
- W. Cheng, X. F. Zhou and R. G. Compton, Angew. Chem., Int. Ed., 2013, 52, 12980–12982 CrossRef CAS PubMed.
- A. M. Bond, F. Marken, E. Hill, R. G. Compton and H. Hügel, J. Chem. Soc., Perkin Trans. 2, 1997, 1735–1742 RSC.
- X.-F. Zhou, W. Cheng and R. G. Compton, Nanoscale, 2014, 6, 6873–6878 RSC.
- H. S. Toh and R. G. Compton, Chem. Sci., 2015, 6, 5053–5058 RSC.
- K. Kuperkar, L. Abezgauz, K. Prasad and P. Bahadur, J. Surfactants Deterg., 2010, 13, 293–303 CrossRef CAS.
- E. Lebègue, C. M. Anderson, J. E. Dick, L. J. Webb and A. J. Bard, Langmuir, 2015, 31, 11734–11739 CrossRef PubMed.
- W. Cheng and R. G. Compton, Angew. Chem., Int. Ed., 2016, 55, 2545–2549 CrossRef CAS PubMed.
- W. Cheng and R. G. Compton, Angew. Chem., Int. Ed., 2015, 54, 7082–7085 CrossRef CAS PubMed.
- B. K. Kim, J. Kim and A. J. Bard, J. Am. Chem. Soc., 2015, 137, 2343–2349 CrossRef CAS PubMed.
- B. M. Quinn, P. G. Van't Hof and S. G. Lemay, J. Am. Chem. Soc., 2004, 126, 8360–8361 CrossRef CAS PubMed.
- A. Boika and A. J. Bard, Anal. Chem., 2015, 87, 4341–4346 CrossRef CAS PubMed.
- A. Boika, S. N. Thorgaard and A. J. Bard, J. Phys. Chem. B, 2013, 117, 4371–4380 CrossRef CAS PubMed.
- J. Eastman, in Colloid Science: Principles, Methods and Applications, ed. T. Cosgrove, Wiley-Blackwell, Hoboken N. J., 2nd edn, 2010, pp. 45–59 Search PubMed.
- A. R. Petosa, D. P. Jaisi, I. R. Quevedo, M. Elimelech and N. Tufenkji, Environ. Sci. Technol., 2010, 44, 6532–6549 CrossRef CAS PubMed.
- J. C. Lees, J. Ellison, C. Batchelor-Mcauley, K. Tschulik, C. Damm, D. Omanović and R. G. Compton, ChemPhysChem, 2013, 14, 3895–3897 CrossRef CAS PubMed.
- S. E. F. Kleijn, B. Serrano-Bou, A. I. Yanson and M. T. M. Koper, Langmuir, 2013, 29, 2054–2064 CrossRef CAS PubMed.
- T. R. Bartlett, S. V. Sokolov and R. G. Compton, ChemistryOpen, 2015, 4, 600–605 CrossRef CAS PubMed.
| This journal is © the Owner Societies 2016 |