
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCalculations of the light absorption spectra of porphyrinoid chromophores for dye-sensitized solar cells†
Raúl
Mera-Adasme‡
ab,
Wen-Hua
Xu‡
cd,
Dage
Sundholm
*c and
Fernando
Mendizabal
*ae
aDepartamento de Química, Facultad de Ciencias, Universidad de Chile, P. O. Box 653, Las Palmeras 3425, Ñuñoa, Santiago, Chile. E-mail: hagua@uchile.cl
bDepartamento de Ciencias del Ambiente, Facultad de Qúimica y Biología, Universidad de Santiago de Chile, Av. Libertador B. O'Higgins 3363, Casilla 40, Correo 33, Santiago, Chile
cDepartment of Chemistry, University of Helsinki, P.O. Box 55 (A.I. Virtanens plats 1), FIN-00014 University of Helsinki, Finland. E-mail: sundholm@chem.helsinki.fi
dCollege of Chemistry and Materials Science, Northwest University, 710127, Xi'an, China
eMillennium Nucleus of Molecular Engineering for Catalysis and Biosensors, ICM, Chile
First published on 28th September 2016
Abstract
Solar power is a strong alternative to the currently used fossil fuels in order to satisfy the world's energy needs. Among them, dye-sensitized solar cells (DSSC) represent a low-cost option. Efficient and cheap dyes are currently needed to make DSSCs competitive. Computational chemistry can be used to guide the design of new light-absorbing chromophores. Here, we have computationally studied the lowest excited states of ZnPBAT, which is a recently synthesized porphyrinoid chromophore with high light-absorption efficiency. The calculations have been performed at ab initio correlated levels of theory employing second-order coupled clusters (CC2) and algebraic diagrammatic construction using second order (ADC(2)) methods and by performing density functional theory (DFT) calculations using the time-dependent DFT (TDDFT) approach for excitation energies. The ultraviolet-visible (UV-vis) spectrum calculated at the ADC(2) and CC2 levels agrees well with the experimental one. The calculations show that ZnPBAT has six electronic transitions in the visible range of the absorption spectrum. The ab initio correlated calculations and previously reported experimental data have been used to assess the performance of several well-known density functionals that have been employed in the present TDDFT study. Solvent effects have been estimated by using the conductor-like screening model (COSMO). The influence of the addition of a TiO2 cluster to the chromophore systems has also been investigated. The results indicate that both CAM-B3LYP and Becke's “half-and-half” (BHLYP) density functionals are appropriate for the studies of excitation energies in the blue range of the visible spectrum for these kinds of porphyrinoid chromophores, whereas the excitation energies of the Q band calculated at the ab initio correlated level are more accurate than those obtained in the present TDDFT calculations. The inclusion of solvent effects has a modest influence on the spectrum of the protonated form of the studied chromophores, whereas solvent models are crucial when studying the absorption spectrum of the anionic chromophore. The calculated UV-vis spectrum for the chromophore anion is not significantly affected by attaching a TiO2 cluster to it.
1 Introduction
The energy-consumption rate of our civilization was 17.2 TW in 2014 and it is expected to increase by a factor of two in the coming decades.1,2 Solar energy is probably the most important sustainable energy source that has the potential to replace fossil energy, when the solar-energy capturing devices are efficient enough.1,3 Only 0.02% of the solar radiation has to be captured for satisfying future energy needs. Many different principles have been proposed for constructing efficient solar-energy capturing devices. However, since the discovery of the principles for dye-sensitized solar cells (DSSC) 25 years ago, DSSCs have attracted attention as a replacement for the traditional solar cells made of silicon.4,5The DSSCs originally proposed by Grätzel usually consist of a dye that is adsorbed on a nanocrystalline titanium dioxide (TiO2) electrode.4–9 More recently other semiconductors such as perovskites (CaTiO3) have been found to yield a higher efficiency than with TiO2.10 The light absorbing dyes often consist of metal complexes of, e.g., ruthenium or zinc, with efficiencies between 7% and 11% rendering technical applications feasible.5,11–16 Due to the high price of ruthenium, novel efficient DSSC dyes containing cheaper metals are desirable.5
Another class of potential DSSC dyes is inorganic complexes of organic molecules with extended π–π conjugation, such as metalloporphyrines (MPs), which can be designed to have specific functionalities and properties.17–20 Functionalized MPs can be deposited on semiconductor electrodes for use as dyes in solar energy harvesting devices.4,5 The MP-based DSSCs have good photostability and they are relatively cheap. The use of Zn as the central metal and molecules with metallo-supramolecular interactions has lead to a high efficiency of 6–11%.21–25
One of the Zn porphyrins with the highest solar-light harvesting efficiencies is ZnPBAT, which is a Zn porphyrin with two electron-donating diphenylamine groups in two of the meso positions, a strong electron-withdrawing carboxyphenylethynyl moiety as the anchoring group in one of the meso positions, and a tBu substituted phenyl group in the fourth meso position. The effect of one diarylamino group in meso positions has previously been analyzed.22 The ethynyl group between the anchoring carboxyphenyl group and the porphyrin moiety leads to a slightly improved light-harvesting capability (η) of 10.1% as compared with the similar ZnPBA dye, which has the carboxyphenyl group directly connected to the meso position of the porphyrin ring. The ZnPBA chromophore reaches an η of 8.3%.22 ZnPBAT has broad Soret and Q bands, which enhance light absorption leading to a better performance of the solar cell.22
The interaction between the dye and TiO2 is important for the efficiency of DSSCs. Usually, the dyes have a carboxylate group that binds to TiO2 of the solid electrode. The interaction of the dyes with the (101) plane of TiO2 (anatase) or a TiO2 nanocluster has been studied computationally at quantum and classical levels of theory. The studies show that the dye is attached to titanium dioxide via carboxylate groups.26–34
In this work, we have studied the lowest excited states of the ZnPBAT and ZnPBA dyes at the second-order approximate coupled-cluster (CC2) and the algebraic diagrammatic construction of second order (ADC(2)) levels of theory. The results obtained using the ab initio correlated methods are compared with those calculated at the time-dependent density functional theory (TDDFT) level using a couple of functionals. TDDFT is widely used in excited state studies of large molecules, because of its good performance and its relatively low computational costs.35 However, since TDDFT calculations might suffer from charge transfer problems, the reliability of the obtained results should be assessed by comparing with the results calculated at higher levels of theory.36 Basis-set and solvent effects on the calculated vertical excitation energies have also been investigated. The present CC2 and ADC(2) calculations on the ZnPBAT and ZnPBA molecules and the dyes attached to a TiO2 cluster model are, to the best of our knowledge, the first ab initio study of the excited states of these important DSSC systems, for which experimental ultraviolet-visible (UV-vis) absorption data are available.22
2 Materials and methods
The molecular structures used in the time-dependent density functional theory (TDDFT)37 and ab initio correlated calculations were optimized at the density functional theory (DFT) level using the Becke–Perdew (BP86) functional38,39 and the Karlsruhe split-valence basis set augmented with polarization functions (def2-SVP).40 The molecular structures for a few selected molecules were also optimized using the Karlsruhe triple-ζ basis set augmented with polarization functions (def2-TZVP).40,41 Dispersion interactions were considered by using Grimme's D3 correction.42 The resolution of the identity approximation (RI) was used to speed up the calculations,43–45 the “chain of spheres” approximation was additionally used to speed up calculations using hybrid functionals,46 and the algorithm described by Kendall and Früchtl47 was used to speed up calculations using a long-range corrected functional.The calculations of the electronic excitation energies were performed at the TDDFT level using the long-range corrected CAM-B3LYP functional38,48,49 and the def2-SVP basis set augmented with diffuse basis functions (def2-SVPD) for the main group atoms.50 For Zn, the def2-SVP basis set was employed. Excited states were calculated using the B3LYP and BHLYP functionals.49,51–54 The CAM-B3LYP functional in combination with the def2-TZVP basis set was also used in the calculations of the excitation energies.
DFT calculations including solvent effects were performed using the conductor-like screening model (COSMO) with a dielectric constant of 24 representing ethanol.55,56 All DFT calculations were performed using the ORCA program package version 3.0.3.57
Excitation energies and oscillator strengths were calculated at the approximative second-order coupled cluster (CC2) and the algebraic diagrammatic construction through second order ADC(2) levels using the scaled opposite-spin approximation and the def2-TZVP basis set.58–60 The def2-SVPD basis set was also employed to assess the size of the basis-set truncation errors of the ab initio correlated calculations. The Laplace transformation (LT) algorithm and the reduced-virtual-space (RVS) approximation were used to speed up the computations.60–64 The RVS cut-off threshold was 60 eV. The COSMO was employed at the ADC(2) level to account for the solvent effects.65 The ab initio correlated calculations were performed using the TURBOMOLE 7.0 package.66–68
3 Results and discussion
The carboxylic acid group that anchors substituted porphyrin dyes to TiO2 in the solar cells is known to be protonated in solution.69 Protonated dyes were therefore initially analyzed in order to assess the different methodologies for the computational studies of the UV-vis spectra. The experimental reference data were reported by Kurotobi et al.22 The optimized geometry for the ZnPBAT molecule, studied in this work, is shown in Fig. 1. In the figure it can be seen that the triple-bond that connects benzoic acid with the porphyrinic ring allows coplanarity between both moieties.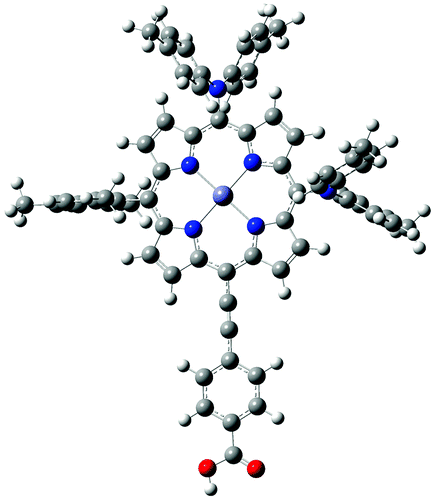 | ||
| Fig. 1 Optimized geometry for the ZnPBAT–H molecule. | ||
3.1 Ab initio calculations
The electronic excitation spectra of the molecule have been calculated at the ADC(2) and CC2 levels of theory for the protonated form of the ZnPBAT molecule (ZnPBAT–H). ADC(2) results are reported with and without the use of the COSMO implicit solvation model (ε = 24). The calculated absorption and wavelengths are compared to the experimental data in Table 1. The excitation energies calculated at the ADC(2) level using the COSMO are in excellent agreement with the experimental ones with deviations of less than 0.1 eV. However, due to the band broadening of the experimental spectrum, it is hard to determine the exact values for the vertical excitation energies and the number of transitions that contribute to each band. The present calculations suggest that each of the experimental B (Soret) bands at 460 nm and 433 nm comprises two electronic transitions. The results show that the ZnPBAT–H molecule has six excited states in the visible range, which is two more than predicted from Gouterman's four-orbital model.70 The plot of the exciton densities shown in Fig. 2 shows that transitions of the B bands lead to a charge-transfer from the meso substituents to the porphyrin ring, whereas the Q bands are more local excitations.| Band character | Experimental | ADC(2) (vacuum) | ADC(2) (ethanol) | CC2 (vacuum) | ||||
|---|---|---|---|---|---|---|---|---|
| λ | Intensity | λ | f | λ | f | λ | f | |
| Q | 661 | 0.15 | 639 | 0.17 | 647 | 0.22 | 621 | 0.16 |
| Q | 596 | 0.08 | 615 | 0.01 | 618 | 0.01 | 602 | 0.01 |
| B | 460 | 0.60 | 463 | 0.16 | 472 | 0.22 | 452 | 0.20 |
| B | 448 | 0.42 | 457 | 0.62 | 439 | 0.40 | ||
| B | 433 | 0.75 | 435 | 0.51 | 443 | 0.67 | 430 | 0.48 |
| B | 418 | 0.15 | 424 | 0.15 | 413 | 0.08 | ||
| 398 | 1.90 | |||||||
 | ||
| Fig. 2 The calculated exciton density of the selected excited states of ZnPBAT–H. The excited states were obtained at the ADC(2)/def2-TZVP/COSMO (ε = 24) level of theory. The exciton densities are labelled with its absorption wavelength. The exciton density of the strong band at 370 nm reported in the ESI† is also shown. The plots are made using a 0.003 isosurface. Negative exciton densities (holes) are depicted in red and positive in blue. | ||
The excitation energies calculated at the ADC(2) and CC2 levels are also in excellent agreement with one another. The largest deviation is only 0.07 eV. The solvent effects at the ADC(2) level lead to a redshift of the bands by 3–9 nm (0.01–0.05 eV).
3.2 TDDFT calculations
The calculated absorption wavelengths are compared to the experimental data in Table 2. The values can also be compared with equivalent ab initio results in Table 1. In Table 2 we can see that CAM-B3LYP and BHLYP calculations yield similar excitation energies and that the calculated spectra agree qualitatively with the experimental one. The Q band at ∼600 nm calculated at the CAM-B3LYP and BHLYP levels is slightly blue shifted as compared to the measured Q band at ∼660 nm. The trend of solvent shifts in CAM-B3LYP is similar to that observed at the ADC(2) level, although the shifts observed are larger for the B band at the CAM-B3LYP level, and leads to a better reproduction of the experimental bands. A similar behavior is observed for the BHLYP functional, as shown in the ESI.†| Experiment22 | B3LYP | CAM-B3LYP | CAM-B3LYP (vacuum) | BHLYP | |||||
|---|---|---|---|---|---|---|---|---|---|
| λ | Intensity | λ | f | λ | f | λ | f | λ | f |
| 661 | 0.15 | 780 | 0.30 | 610 | 0.33 | 601 | 0.32 | 594 | 0.32 |
| 596 | 0.08 | 727 | 0.07 | 583 | 0.02 | 579 | 0.02 | 570 | 0.01 |
| 460 | 0.6 | 691 | 0.08 | 492 | 0.07 | 456 | 0.09 | 487 | 0.06 |
| 433 | 0.75 | 636 | 0.03 | 463 | 0.21 | 439 | 0.25 | 454 | 0.21 |
| 526 | 0.04 | 450 | 0.10 | 418 | 0.13 | 450 | 0.14 | ||
| 526 | 0.10 | 430 | 0.22 | 394 | 0.26 | 432 | 0.21 | ||
| 516 | 0.18 | ||||||||
| 474 | 0.04 | ||||||||
| 452 | 0.20 | ||||||||
| 407 | 1.78 | ||||||||
At the B3LYP level, the first two transitions corresponding to the Q band appear at 780 nm and 727 nm. The B band is also significantly redshifted as compared to the experiment. The first strong transition corresponding to the B band appears at 526 nm. In addition, several spurious transitions with small oscillator strengths are obtained in the B3LYP calculations but not at any other level of theory that is employed in this work. The B3LYP calculations of the excitation energies suffer from charge transfer problems, which lead to slightly too low excitation energies and spurious excited states. Thus, the UV-vis spectrum calculated at the B3LYP level is not reliable and is therefore not discussed further in this work.
The shoulder in the experimental spectra at 596 nm is reproduced at the CAM-B3LYP and BHLYP levels. However, the calculated band strength is 4–8 times weaker than the experimental intensity. This weakening is also observed at the ab initio levels. The absorption energies of the strong B bands (Soret) calculated at the CAM-B3LYP and BHLYP levels agree well with the experimental values. The CAM-B3LYP and BHLYP calculations suggest that the B band consists of four transitions, whereas only two peaks are observed in the experimental spectrum.22 It cannot be ruled out that a third strong transition contributes to the strong and broad Soret band in the experimental spectrum. Thus, the experimental UV-vis spectrum is largely reproduced by TDDFT calculations employing the CAM-B3LYP and BHLYP functionals. Since the results obtained at the CAM-B3LYP and BHLYP levels are qualitatively the same, the rest of the BHLYP results are reported only in the ESI.†
The absorption wavelengths and oscillator strengths for ZnPBAT–H calculated at the CAM-B3LYP level are presented in Table 2. Comparing CAM-B3LYP wavelengths with those calculated using the COSMO in Table 2 shows that the solvent contribution is rather small but not negligible especially for the transitions of the B band, which are redshifted by 24–36 nm (0.15–0.26 eV) in the COSMO calculation. For the two transitions of the Q band the solvent shifts are 9 and 4 nm (0.02–0.03 eV), respectively. The inclusion of the solvent effect in the CAM-B3LYP calculation leads to better agreement with experiment for the B band, whereas the Q band is almost unaffected by solvent effects. In the CAM-B3LYP calculation using the COSMO, the lowest transition appears 0.15 eV higher in energy than the maximum of the first peak in the experimental spectrum.
The excitation energies calculated at the CC2 and ADC(2) levels are in excellent agreement. The largest deviation is only 0.07 eV. The calculations at the ab initio levels confirm that ZnPBAT–H has six excited states in the visible range, which is two more than predicted from Gouterman's four-orbital model.70 The trend of the solvent shifts of the ADC(2) excitation energies is similar to the one obtained at the CAM-B3LYP level. However, a solvent redshift of 3–9 nm (0.01–0.05 eV) at the ADC(2) level is observed, although smaller than that obtained in the CAM-B3LYP calculations. The excitation energies calculated at the ADC(2) level using the COSMO are in excellent agreement with the experimental ones with deviations of less than 0.1 eV. However, due to the band broadening of the experimental spectrum, it is hard to determine the exact values for the vertical excitation energies and the number of transitions that contribute to each band. The present calculations suggest that each of the experimental bands at 460 nm and 433 nm comprises two electronic transitions.
3.3 Basis-set study
The excitation energies of ZnPBAT–H calculated at the CAM-B3LYP/COSMO (ε = 24) and ADC(2)/COSMO (ε = 24) levels using the def2-SVPD and def2-TZVP basis sets are almost identical. The absorption wavelengths reported in Tables 3 and 4 differ at most by 1–3 nm. Thus, the def2-SVPD basis set is sufficiently large for the present purpose.| TZVP | SVPD | ||
|---|---|---|---|
| λ | f | λ | f |
| 609 | 0.32 | 610 | 0.32 |
| 583 | 0.02 | 583 | 0.02 |
| 491 | 0.08 | 492 | 0.08 |
| 462 | 0.22 | 463 | 0.22 |
| 450 | 0.12 | 450 | 0.12 |
| 430 | 0.22 | 430 | 0.22 |
| TZVP | SVPD | ||
|---|---|---|---|
| λ | f | λ | f |
| 647 | 0.22 | 644 | 0.21 |
| 618 | 0.01 | 616 | 0.01 |
| 472 | 0.22 | 474 | 0.22 |
| 457 | 0.62 | 457 | 0.60 |
| 443 | 0.67 | 443 | 0.64 |
| 424 | 0.15 | 425 | 0.16 |
3.4 Chromophores attached to TiO2
The studied molecules can be used as light absorbing chromophores in DSSCs,5 where the molecules are attached to a semiconductor electrode such as TiO2. The carboxylate group of porphyrinoids is deprotonated and coordinated to one or two Ti atoms.22,71–74 The optimized geometries of the ZnPBAT molecule, and that of the ZnPBA, also studied in this section, are shown in Fig. 3. The excitation energies of the chromophore attached to TiO2 were calculated at the CAM-B3LYP/def2-SVPD/COSMO (ε = 24) level to assess how TiO2 affects the light-absorption wavelengths. Since the anchoring carboxylate group deprotonates upon coordination to TiO2, the spectra were calculated for the deprotonated ZnPBAT and ZnPBA dyes attached to TiO2. The obtained spectra are compared to the excitation energies calculated for the ZnPBAT− and ZnPBA− anions at the same level of theory in order to estimate the effect of TiO2 on the absorption spectra. The TiO2 semiconductor is simulated by a TiO2 cluster consisting of 27 atoms, which has been derived from the crystal structure of anatase. The dangling bonds are saturated with hydrogens yielding a cluster consisting of 45 atoms. The calculations were performed at the CAM-B3LYP/def2-SVPD/COSMO (ε = 24) level, because the calculated UV-vis spectra calculated at that level largely agree with the ones obtained in the ADC(2) calculations. The calculated excitation energies and oscillator strengths reported in Table 5 show that the UV-vis spectra of the deprotonated porphyrinoids and the spectra for the chromophores attached to the TiO2 cluster are practically identical with differences of 1–2 nm. Thus, TiO2 does not significantly affect the UV-vis spectra of the chromophore attached to the semiconductor. The calculated UV-vis spectra for ZnBAT–H and for ZnBAT–(Ti6O21H18) also agree well having the largest deviation of 7 nm (0.02 eV) for the lowest transition. For a solvent such as ethanol, the UV-vis absorption spectra of porphyrinoid chromophores attached to TiO2 can be modeled by performing calculations on the deprotonated or neutral chromophore employing a continuum solvent model. | ||
| Fig. 3 Optimized geometry for the ZnPBAT and ZnPBA chromophores bound to a TiO2 cluster. | ||
| ZnPBAT–TiO2 | ZnPBAT–TiO2 (vacuum) | ZnPBAT− | ZnPBA–TiO2 | ZnPBA− | |||||
|---|---|---|---|---|---|---|---|---|---|
| λ | f | λ | f | λ | f | λ | f | λ | f |
| 603 | 0.33 | 594 | 0.37 | 602 | 0.28 | 570 | 0.06 | 569 | 0.06 |
| 583 | 0.03 | 586 | 0.04 | 582 | 0.03 | 568 | 0.05 | 567 | 0.05 |
| 481 | 0.07 | 421 | 0.25 | 481 | 0.07 | 452 | 0.20 | 454 | 0.18 |
| 462 | 0.20 | 420 | 0.01 | 462 | 0.20 | 451 | 0.19 | 454 | 0.19 |
| 448 | 0.07 | 399 | 1.68 | 448 | 0.07 | 414 | 0.18 | 412 | 0.16 |
| 429 | 0.20 | 392 | 0.01 | 429 | 0.20 | 411 | 0.24 | 409 | 0.23 |
| 391 | 0.85 | ||||||||
3.5 Anionic porphyrinoid chromophores
For the neutral ZnPBAT–H chromophore, the solvent did not introduce any large changes in the calculated UV-vis spectrum. The differences in the solvent shifts calculated at the CAM-B3LYP and ADC(2) levels are even larger than the solvent shift calculated at the ADC(2) level. However, the solvent effects are significant for the anionic (deprotonated) chromophores as seen in Table 6. In the vacuum calculations at the CAM-B3LYP, ADC(2), and CC2 levels, a few extra states with very small oscillator strengths are obtained in the visible region. For a single anionic chromophore in the gas phase, five states with low oscillator strength appear in the TDDFT calculations using the CAM-B3LYP and BHLYP functionals. Four of the extra states are also present at the ADC(2) and CC2 levels indicating that most of them are true states and not artificial charge transfer states of the employed TDDFT method. See the ESI.†| CAM-B3LYP (vacuum)a | ADC(2) (vacuum)a | ADC(2) (ethanol) | CC2 (vacuum)a | ||||
|---|---|---|---|---|---|---|---|
| λ | f | λ | f | λ | f | λ | f |
| a States obtained in the vacuum calculations with very small oscillator strengths are omitted. The wavelengths omitted in the table are 543, 473, 472, 467, 423, 415 nm at the CAM-B3LYP level; 551, 484, 476, 472, 422 nm at the ADC(2) level; and 493, 445, 440, 432 nm at the CC2 level. | |||||||
| 597 | 0.40 | 631 | 0.19 | 641 | 0.19 | 618 | 0.03 |
| 594 | 0.06 | 630 | 0.04 | 616 | 0.01 | 616 | 0.21 |
| 421 | 0.64 | 427 | 1.44 | 462 | 0.30 | 430 | 1.36 |
| 415 | 0.47 | 427 | 0.06 | 452 | 0.70 | 416 | 0.72 |
| 421 | 0.74 | 442 | 0.58 | 410 | 0.44 | ||
| 422 | 0.14 | 404 | 0.18 | ||||
| 396 | 1.80 | ||||||
A solvation model such as the COSMO is not needed when studying neutral porphyrinoid dyes in solution, whereas it is necessary to enclose anionic chromophores with dielectric media in order to avoid the extra states with small oscillator strengths. Ethanol, which is the solvent used in this work, is rather polar and can form hydrogen bonds with the dye and the semiconductor, which are not considered in the COSMO calculations. We have used the dielectric constant of ethanol in the COSMO calculations, because ethanol was used as solvent in the experimental study by Kurotobi et al.22 Since the solvent effects are small, considering the solvent effects is less crucial when investigating neutral chromophores in non-polar solvents. The inclusion of a solvent model is important for a proper treatment of chromophores bound to TiO2 when simulating the light absorption spectra of DSSCs. In the absence of the dielectricum, the calculated UV-vis spectrum for ZnBAT–(Ti6O21H18) differs from the one calculated for the same system when it is surrounded by a solvent model. The UV-vis spectra of ZnBAT–(Ti6O21H18) and ZnBAT–H calculated in vacuo also differ significantly. For ZnBAT–(Ti6O21H18), states with very small oscillator strengths appear as for the anionic chromophore when the dielectricum is not considered in the calculation. Another important change in the calculated spectra is that one of the strong transitions in the Soret band disappears in the vacuum calculation on ZnBAT–(Ti6O21H18). Thus, in the vacuum calculation ZnBAT–(Ti6O21H18) has only one strong transition in the Soret region with an absorption wavelength of 421 nm. The present study shows that the interaction between the porphyrinoid dye and the TiO2 cluster is altered by the surrounding dielectricum.
4 Conclusions
We have employed different computational methodologies for investigating the light absorption spectra of substituted porphyrinoid molecules and of the same molecules attached to a TiO2 cluster that simulates the semiconductor. The study focuses on the recently synthesized ZnPBAT chromophores that are efficient dyes in dye-sensitized solar cells (DSSCs).22 The computational studies performed on the second-order coupled-cluster (CC2) and algebraic diagrammatic construction at the second order (ADC(2)) levels of theory yield excitation energies and band strengths that agree with the experimental data. Four transitions have been assigned in the experimental spectrum.22 However, the ADC(2) and CC2 calculations show that ZnPBAT–H has six electronic excitation energies in the visible range. Two electronic transitions form the strong B band at 460 nm and two excited states also contribute to the strong B transition at 433 nm.Calculations at the time-dependent density functional theory (TDDFT) level show that a significant share of Hartree–Fock (HF) exchange has to be included in the functional in order to avoid spurious charge-transfer states. Calculations using the B3LYP functional with 20% HF exchange result in too small excitation energies and spurious charge-transfer states in the visible range, which disappear when the BHLYP functional with 50% HF exchange, or the long-range corrected CAM-B3LYP is used. The excitation energies calculated using the BHLYP and CAM-B3LYP functionals also agree with the experimental data. Our results suggest that these functionals can be employed in the studies of the light absorption properties of these kinds of porphyrinoids. However, the present study shows that TDDFT calculations on large chromophores should, due to charge-transfer problems, be benchmarked against ab initio data whenever possible. Our results indicate that the def2-SVPD basis set can be employed in TDDFT and ab initio correlated studies of the light-absorption properties of porphyrin-based DSSC chromophores. However, at the ab initio correlated levels the computational time does not increase much when using the larger basis set, because most of the virtual orbitals will be omitted in the calculation of excitation energies when the reduced virtual space (RVS) approximation is employed.64
For the neutral porphyrinoid chromophores, the inclusion of solvent effects via the COSMO55 approach has a modest effect on the calculated light-absorption wavelengths and oscillator strengths. For the anionic (deprotonated) chromophores, it is crucial to consider solvent effects because unphysical diffuse excited states are obtained for the anion when solvent effects are omitted. The diffuse states with small oscillator strengths disappear when the chromophore is surrounded by a dielectricum simulating the solvent.
Acknowledgements
The authors gratefully acknowledge financial support under Conicyt-Aka-ERNC-001 and for computational resources from CSC – IT Center for Science, Finland. FM acknowledges financial support from Fondecyt under project 1140503 and from project RC120001 of the Iniciativa Científica Milenio (ICM) del Ministerio de Economía, Fomento y Turismo del Gobierno de Chile. RM-A acknowledges financial support from Proyecto de Atracción e Inserción de Capital Humano Avanzado en la Academia No. 79150043. W-HX thanks the support from China Scholarship Council. This work has also been supported by the Academy of Finland through projects (266227 and 275845) and the Magnus Ehrnrooth Foundation. DS thanks the Swedish Cultural Foundation in Finland, the Alexander von Humboldt Stiftung, and the Fulbright Foundation for financial support during his sabbatical leave.References
- N. Armaroli and V. Balzani, Chem. – Eur. J., 2016, 22, 32–57 CrossRef CAS PubMed.
- K. S. Gallagher, Daedalus, 2013, 142, 59–77 CrossRef.
- G. Singh, Energy, 2013, 53, 1–13 CrossRef.
- B. O'regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef.
- M. Grätzel, Acc. Chem. Res., 2009, 42, 1788–1798 CrossRef PubMed.
- H. Imahori, T. Umeyama and S. Ito, Acc. Chem. Res., 2009, 42, 1809–1818 CrossRef CAS PubMed.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed.
- M. Pastore, E. Mosconi, F. D. Angelis and M. Grätzel, J. Phys. Chem. B, 2010, 114, 7205–7212 CAS.
- F. D. Angelis, S. Fantacci, E. Mosconi, M. K. Nazeeruddin and M. Grätzel, J. Phys. Chem. B, 2011, 115, 8825–8831 Search PubMed.
- J. Burschka, N. Pellet, S. J. Moon, R. Humphry-Baker, P. Gao, M. K. Nazeeruddin and M. Grätzel, Nature, 2013, 316–319 CrossRef CAS PubMed.
- F. Gajardo, M. Barrera, R. Vargas, I. Crivelli and B. Loeb, Inorg. Chem., 2011, 50, 5910–5924 CrossRef CAS PubMed.
- F. Gajardo, A. M. Leiva, B. Loeb, A. Delgadillo, J. R. Stromberg and G. J. Meyer, Inorg. Chim. Acta, 2008, 361, 613–619 CrossRef CAS.
- D. Casanova, F. P. Rotzinger and M. Grätzel, J. Chem. Theory Comput., 2010, 6, 1219–1227 CrossRef CAS.
- J. Feng, Y. Jiao, W. Ma, M. K. Nazeeruddin, M. Grätzel and S. Meng, J. Phys. Chem. B, 2013, 117, 3772–3778 CAS.
- J. Warnan, Y. Pellegrin, E. Blart and F. Odobel, Chem. Commun., 2012, 48, 675–677 RSC.
- A. Yella, H.-W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. Nazeeruddin, E. W.-G. Diau, C.-Y. Yeh, S. M. Zakeeruddin and M. Grätzel, Science, 2011, 334, 629–634 CrossRef CAS PubMed.
- G. R. Desiraju, J. Chem. Sci., 2010, 122, 667–675 CrossRef CAS.
- S. Kinge, M. Crego-Calama and D. N. Reinhoudt, ChemPhysChem, 2008, 9, 20–42 CrossRef CAS PubMed.
- R. Otero, J. M. Gallego, A. L. V. de Parga, N. Martín and R. Miranda, Adv. Mater., 2011, 23, 5148–5176 CrossRef CAS PubMed.
- D. Bonifazi, S. Mohnani and A. Llanes-Pallas, Chem. – Eur. J., 2009, 15, 7004–7025 CrossRef CAS PubMed.
- L.-L. Li and E. W.-G. Diau, Chem. Soc. Rev., 2013, 42, 291–304 RSC.
- K. Kurotobi, Y. Toude, K. Kawamoto, Y. Fujimori, S. Ito, P. Chabera, V. Sundström and H. Imahori, Chem. – Eur. J., 2013, 19, 17075–17081 CrossRef CAS PubMed.
- S.-H. Liu, H. Fu, Y.-M. Cheng, K.-L. Wu, S.-T. Ho, Y. Chi and P.-T. Chou, J. Phys. Chem. B, 2012, 116, 163380 Search PubMed.
- H. Kusama and K. Sayama, J. Phys. Chem. B, 2012, 116, 23906–23914 CAS.
- T. Higashino and H. Imahori, Dalton Trans., 2015, 44, 448–463 RSC.
- F. D. Angelis, S. Fantacci, A. Selloni, M. K. Nazeeruddin and M. Grätzel, J. Phys. Chem. B, 2010, 114, 6054–6061 CrossRef PubMed.
- E. Jakubikova, R. C. Snoeberger III, V. S. Batista, R. L. Martin and E. R. Batista, J. Phys. Chem. A, 2009, 113, 12532–12540 CrossRef CAS PubMed.
- T. L. Bahers, T. Pauporté, P. P. Lainé, F. Labat, C. Adamo and I. Ciofini, J. Phys. Chem. Lett., 2013, 4, 1044–1050 CrossRef PubMed.
- F. Risplendi, G. Cicero, G. Mallia and N. M. Harrison, Phys. Chem. Chem. Phys., 2013, 15, 235–243 RSC.
- X. Zarate, E. Schott, T. Gomez and R. Arratia-Pérez, J. Phys. Chem. A, 2013, 117, 430–438 CrossRef CAS PubMed.
- E. Mosconi, J.-H. Yum, F. Kessler, C. J. G. García, C. Zuccaccia, A. Cinti, M. K. Nazeeruddin, M. Grätzel and F. D. Angelis, J. Am. Chem. Soc., 2012, 134, 19438–19453 CrossRef CAS PubMed.
- E. Maggio, N. Martsinovich and A. Troisi, J. Phys. Chem. B, 2012, 116, 7638–7649 CAS.
- L. Yang, L. Guo, Q. Chen, H. Sun, H. Yan, Q. Zeng, X. Zhang, X. Pan and S. Dai, J. Mol. Graphics Modell., 2012, 38, 82–89 CrossRef CAS PubMed.
- S. Ikäläinen and K. Laasonen, Phys. Chem. Chem. Phys., 2013, 15, 11673–11678 RSC.
- L. Goerigk and S. Grimme, Phys. Chem. Chem. Phys., 2011, 13, 6670–6688 RSC.
- F. Neese, JBIC, J. Biol. Inorg. Chem., 2006, 11, 702–711 CrossRef CAS PubMed.
- M. Casida and M. Huix-Rotllant, Annu. Rev. Phys. Chem., 2012, 63, 287–323 CrossRef CAS PubMed.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- K. Eichkorn, O. Treutler, H. Öhm, M. Häser and R. Ahlrichs, Chem. Phys. Lett., 1995, 242, 652–660 CrossRef CAS.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- K. Eichkorn, F. Weigend, O. Treutler and R. Ahlrichs, Theor. Chim. Acta, 1997, 97, 119–124 CAS.
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys., 2009, 356, 98–109 CrossRef CAS.
- A. R. Kendall and A. H. Früchtl, Theor. Chim. Acta, 1997, 97, 158–163 Search PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- D. Rappoport and F. Furche, J. Chem. Phys., 2010, 133, 134105 CrossRef PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
- P. J. Stevens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- S. Sinnecker, A. Rajendran, A. Klamt, M. Diedenhofen and F. Neese, J. Phys. Chem. A, 2006, 110, 2235–2245 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS.
- J. Schirmer, Phys. Rev. A: At., Mol., Opt. Phys., 1982, 26, 2395–2416 CrossRef CAS.
- O. Christiansen, H. Koch and P. Jørgensen, Chem. Phys. Lett., 1995, 243, 409–418 CrossRef CAS.
- N. O. Winter and C. Hättig, J. Chem. Phys., 2011, 134, 184101 CrossRef PubMed.
- Y. Jung, R. C. Lochan, A. D. Dutoi and M. Head-Gordon, J. Chem. Phys., 2004, 121, 9793–9802 CrossRef CAS PubMed.
- Y. M. Rhee and M. Head-Gordon, J. Chem. Phys., 2007, 111, 5314–5326 CrossRef CAS PubMed.
- N. O. C. Winter and C. Hättig, J. Chem. Phys., 2012, 401, 217–227 CAS.
- R. Send, V. R. I. Kaila and D. Sundholm, J. Chem. Phys., 2011, 134, 214114 CrossRef PubMed.
- B. Lunkenheimer and A. Köhn, J. Chem. Theory Comput., 2013, 9, 977–994 CrossRef CAS PubMed.
- TURBOMOLE V7.0 2015, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007; available from http://www.turbomole.com.
- F. Furche, R. Ahlrichs, C. Hättig, W. Klopper, M. Sierka and F. Weigend, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 91–100 CrossRef CAS.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- B. Völker, F. Wölzl, T. Bürgi and D. Lingenfelser, Langmuir, 2012, 28, 11354–11363 CrossRef PubMed.
- M. Gouterman, in The Porphyrins, ed. D. Dolphin, Academic Press, London and New York, 1978, vol. 3, p. 1 Search PubMed.
- K. Syres, A. Thomas, F. Bondino, M. Malvestuto and M. Grätzel, Langmuir, 2010, 26, 14548–14555 CrossRef CAS PubMed.
- T. Gomez, X. Zarate, E. Schott and R. Arratia-Perez, RSC Adv., 2014, 4, 9639–9646 RSC.
- K. B. Ørnsø, C. S. Pedersen, J. M. Garcia-Lastra and K. S. Thygesen, Phys. Chem. Chem. Phys., 2014, 16, 16246–16254 RSC.
- K. Chaitanya, X.-H. Ju and B. M. Heron, RSC Adv., 2014, 4, 26621–26634 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Coordinates for the systems studied and additional computationally obtained excitation energies and oscillator strengths. See DOI: 10.1039/c6cp04627d |
| ‡ These authors contributed equally to this work. |
| This journal is © the Owner Societies 2016 |