
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDecoupling strain and ligand effects in ternary nanoparticles for improved ORR electrocatalysis†
Paul C.
Jennings
 ,
Steen
Lysgaard
,
Heine A.
Hansen
and
Tejs
Vegge
*
,
Steen
Lysgaard
,
Heine A.
Hansen
and
Tejs
Vegge
*
Department of Energy Conversion and Storage, Technical University of Denmark, Lyngby, Denmark. E-mail: teve@dtu.dk; Fax: +45 4677 5758; Tel: +45 5164 1787
First published on 11th August 2016
Abstract
Ternary Pt–Au–M (M = 3d transition metal) nanoparticles show reduced OH adsorption energies and improved activity for the oxygen reduction reaction (ORR) compared to pure Pt nanoparticles, as obtained by density functional theory. The strain and ligand effects in nanoparticles are decoupled and correlated with the extended Pt(111) surface for benchmarking. The ternary metal in the core allows for tuning the catalytic activity through strain effects. Pt–Au–M for M = Cr, Mn, Co, Cu, Zn nanoparticles are of particular interest as they exhibit an optimal contribution of strain, ligand effects and stability. Good agreement is found with experimental studies showing increased activity of Pt–Au–Fe/Ni nanoparticles, and mid to late 3d transition metals are predicted to exhibit enhance activity and stability with respect to pure Pt nanoparticles.
1 Introduction
Proton exchange membrane fuel cells (PEMFC) have been earmarked for transport and stationary power applications1 but before becoming commercially viable for widespread deployment, the limited performance and durability of the cathode electrocatalyst needs to be addressed. To date, Pt-based nanoparticles and nanoalloys have been investigated extensively, primarily focusing on binary systems, with particular focus on improving cathodic performance.2–4 Recently, there has been growing interest in extending studies to ternary systems,5 in order to improve the stability of binary systems e.g. Pt3Y, Pt5La, Pt5Gd etc.6–8 Experimental studies utilising differing methodological procedures have been reported, generating a variety of trimetallic Pt-based nanoparticles.9–25 A number of theoretical studies have also been performed on similar bi- and trimetallic Pt-based systems.26–39Of particular interest to this study are Pt–Au–M (where M is a 3d transition metal) nanoparticles34,40 for catalysing the oxygen reduction reaction (ORR). The interest in these systems stems from work showing that the presence of Au in PEMFC electrocatalysts can significantly increase their durability. Zhang et al. showed that forming a binary Pt–Au system of Pt nanoparticles decorated with small Au clusters can enhance the durability of Pt-based electrocatalysts.41 While Stamenkovic et al. recently demonstrated the excellent stability of Pt–Au–Fe9 and Pt–Au–Ni19 nanoparticles. Computational work investigating Pt–Au–M slab systems, for a number of 3d transition metals, has also shown that these ternary extended surface are predicted to give improved reaction kinetics as a result of a reduction in the Pt–OH bond strength.38,42
The improved durability of these Au-containing electrocatalysts is largely attributed to the nobility of Au. In the case of the ternary Pt–Au–Ni nanoparticles, the authors suggest the formation of a Pt-skin on subsurface Au.19 The noble subsurface then acts as a passivation layer that provides a geometric barrier, preventing dissolution of other alloyed species, which gives rise to improved durability of the electrocatalyst. Though, it is noted that considering surface energies of Pt and Au, it would be expected that Au should preferentially occupy surface sites, which has indeed been found to be the case in a number of experimental and computational studies.43–45 It has also been shown possible to experimentally form metastable Au-core Pt-shell nanoparticles.46–49 Similar chemical ordering has also been shown possible with ternary Pt–Au–Pd nanoparticles, where theory predicted the preferential formation of a mixed Pd/Au shell on a Pt-rich core.34,37 However, experimental studies have successfully demonstrated the formation of particles with a Pt-rich shell on an alloyed Au-containing core.50–58 Adsorbate induced stabilisation has also been suggested to play a role in formation of Pt over an Au-containing core under ORR conditions. Oxygen and hydroxyl bond more strongly to Pt than to Au, thus when metastable structures are formed, the presence of an oxygen atmosphere stabilises this structure with a Pt-shell.42 It should also be noted that nanoparticles formed of a Pt–Au skin on an alloyed core can also exhibit improved durability.59
Further, C. Coutanceau et al. recently demonstrated the viability of preparing binary and ternary Pt–Au containing nanoparticles via the water in oil microemulsion method, noting the formation of Pt rich particle surfaces, for bulk Au atomic ratios up to 70%, which translates into the formation of a core–shell like structure with the Pt–Au alloy core of almost nominal composition surrounded by a Pt rich skin. Further, it was found that even if only the effect of the Pt–Au catalyst composition and structure on the kinetics current density is considered, in terms of mass activity (related to Pt mass in the catalyst), catalysts with up to 30 at% Au leads to higher ORR activity.60
In the following, Pt–Au–M nanoparticles are studied to decouple strain and ligand effects61 for ternary systems with select 3d transition metal cores. Mackay icosahedral nanoparticles of ∼2 nm (309 atoms) in size are utilised as a model for the experimentally relevant systems found in the literature that highlight the activity of icosahedral nanoparticles for the ORR.62–68 The choice to focus on the icosahedral structure was made to allow for screening of a greater range of ternary systems. Given the 5-fold symmetry, the icosahedral structure is only relevant for small nanoparticles, due to the large strain that otherwise develops in larger (nano)particles.66,69 These nanoparticles, therefore provide a unique possibility to design and balance strain, segregation and ligand effects to optimize the catalyst. Further, it is noted that strain effects for various icosahedral nanoparticles have been discussed in the literature, primarily focused on the effects of core compression. These studies have shown a preference for icosahedral nanoparticles forming off-centre core morphologies, or the preference for atomic vacancies in order to reduce strain effects.70,71
Considering previous studies, it is highly likely that forming nanoalloys with elements possessing a smaller atomic radius than Pt (or Au) will negate some core compression, thus improving overall stability of the nanoparticle.72 There also exists the possibility that the icosahedral structure may undergo some structural rearrangement in order to reduce strain. There exist examples in the literature where atomic substitution has occurred, resulting in a small amount of the core element being exchanged with surface atoms.73 It has also been found that a distortion to the anti-Mackay icosahedral structure may also reduce strain for certain systems.74 Each of these rearrangements are important though outside the scope of the current study, in which screening of a range of compositions is the ultimate aim. Thus, the following studies of strain effects will predominantly focus on changes to the shell, where there is greater effect on nanoparticle reactivity, focusing on idealised segregated core–shell particles.
2 Methodology
Density Functional Theory (DFT) calculations are performed using GPAW75,76 with a real space implementation of the projector-augmented wave (PAW) method.77 GPAW is run in the linear combination of atomic orbitals (LCAO) mode78 with a double zeta polarized basis set and RPBE exchange correlation functional.79 Calculations are run spin-polarised with a Fermi smearing of 0.05 eV in a non-periodic 32 × 32 × 32 Å unit cell. Atoms are relaxed by the FIRE optimisation algorithm80 until all atomic forces are below 0.05 eV Å−1. Further, adsorbate binding energies reported herein include the counterpoise correction to avoid basis set superposition error (BSSE).81 This general approach has previously been successfully used to study the stability of Cu–Ni and Au–Cu 309-atom nanoalloys and the adsorption reaction intermediates for the CO2 reduction reaction (CO2RR) and ORR.82,83To decouple strain from ligand effects, three models are compared for each system. For this methodology, all nanoparticles with differing compositions are initially relaxed to minimize any geometric strain. To investigate electronic effects, the relaxed ternary Pt162Au92M55 (shell, subsurface, core) nanoparticle is compared to that of the same (unrelaxed) geometric nanoparticle, i.e. the same atomic positions, but where all element types have been transposed to that of Pt, written as Pt309AuxyzMxyz henceforth. In this case, the two nanoparticles have the exact same geometric structure so any changes are due to electronic effects induced by forming multimetallic nanoparticles. Conversely, the strained Pt309AuxyzMxyz nanoparticle compared to that of the relaxed pure Pt309 nanoparticle will produce changes solely due to strain effects, as ligand effects through formation of a multimetallic nanoalloy have been negated.
To decouple strain and ligand effects, each of the aforementioned nanoparticles will need to be geometrically constrained prior to performing any study. This is a necessity of the methodology as otherwise the Pt309AuxyzMxyz nanoparticle would simply re-optimize to the geometry of the relaxed Pt309 nanoparticle. For the constrained systems, the geometry of the locally relaxed nanoparticle is fixed, so no minimisation of the metal atoms occurs. However, the adsorbate is able to fully relax on the surface of these geometrically fixed nanoparticles. Constrained nanoparticles will be denoted (*). The final binding energy of the reaction intermediates is calculated for the fully relaxed Pt162Au92M55 nanoparticle, where all metal and adsorbate atoms are allowed to relax. This is imperative for the consideration of potential adsorbate induced structural changes of the nanoparticles. Comparing the constrained multimetallic Pt162Au92M55* nanoparticle with the fully relaxed counterpart gives an indication of geometric flexibility related to the effect of changing the core metal.
3 Results and discussion
3.1 RMSD analysis
Root Mean Squared Displacement (RMSD) values are calculated based on absolute atomic positions, giving the magnitude of the atomic displacement induced upon formation of the ternary nanoparticles relative to the pure Pt309 nanoparticle. All nanoparticles are locally minimised to the icosahedral geometry. Thus the displacements discussed herein are small changes as a result of strain due to the formation of multimetallic nanoparticles compared to the pure Pt nanoparticle. These small strain effects do not result in significant distortion of the nanoparticles away from the starting icosahedral structure, but can have a significant effect on the activity of the electrocatalyst.It is to be expected that greater displacements are likely to result in more significant strain effects. The results in Table 1 show there is a general reduction in displacement of Pt-shell atoms when comparing early 3d transition metals with late 3d transition metals. This coincides with a reduction in the 3d lattice parameter, where M–M bulk bond distances are also shown, with Sc having the longest bond of 3.21 Å and Fe the shortest at 2.48 Å. The % change of the Au–Au subsurface and Pt–Pt shell bond lengths are also calculated relative to the Au–Au and Pt–Pt bulk bond lengths, respectively. This shows expansion of the Au–Au and Pt–Pt bond lengths when alloyed with the Sc or Ti core, which each have longer bulk M–M bond lengths. Conversely, there is a contraction when alloyed with the other 3d transition metal cores, which have shorter bulk M–M bonds. There is greater variation in the RMSD values calculated for the M-core and Au-subsurface, with no discernible trend. It should be noted, RMSD gives an indication of how the overall structure of the nanoparticle changes for varying compositions, where large values could be the result of contraction or expansion.
| 3d | RMSD (Å) | M–M | % Strain rel. bulk | ||||
|---|---|---|---|---|---|---|---|
| Core | Particle | M | Au | Pt | Bulk (Å) | Au | Pt |
| Sc | 0.36 | 0.34 | 0.36 | 0.36 | 3.21 | 11.46 | 15.47 |
| Ti | 0.13 | 0.07 | 0.11 | 0.15 | 2.90 | 0.69 | 4.32 |
| V | 0.16 | 0.31 | 0.12 | 0.09 | 2.62 | −9.03 | −5.76 |
| Cr | 0.16 | 0.31 | 0.12 | 0.09 | 2.50 | −13.19 | −10.07 |
| Mn | 0.09 | 0.17 | 0.07 | 0.04 | 2.73 | −5.21 | −1.80 |
| Fe | 0.17 | 0.34 | 0.14 | 0.04 | 2.48 | −13.89 | −10.79 |
| Co | 0.23 | 0.44 | 0.22 | 0.08 | 2.51 | −12.85 | −9.71 |
| Ni | 0.20 | 0.37 | 0.21 | 0.07 | 2.49 | −13.54 | −10.43 |
| Cu | 0.14 | 0.27 | 0.15 | 0.04 | 2.56 | −11.11 | −7.91 |
| Zn | 0.06 | 0.04 | 0.04 | 0.07 | 2.67 | −7.29 | −3.96 |
| Pt | 0.20 | 0.24 | 0.19 | 0.07 | 2.78 | −3.47 | 0.00 |
Plots showing RMSD through a cross section of the nanoparticle are in Fig. 1, where in each shell there are 1, 12, 42, 92, 162 atoms, from the inner to outer shell, respectively. Visualising the RMSD in this way allows for site specific differentiation not apparent from the averaged values in Table 1. The Pt162Au92Sc55 nanoparticle is an outlier with rather significant displacement for the majority of atoms. For nanoparticles formed with Ti, Mn, and Zn cores, there is very little displacement of atoms throughout the cross section. For other nanoparticles, it is generally found that there is greatest displacement of the outer 42-atoms of the shell, with marginally less displacement of the inner 13-atoms. Interestingly, when comparing the cross-sections in Fig. 1, it is obvious that there is less displacement of the surface Pt atoms for the majority of Pt162Au92M55 nanoparticles compared to the Pt162Au147 and Pt162Au92Au55 nanoparticles. This shows that the presence of the 3d transition metal core significantly reduces Pt shell strain induced from formation of the subsurface layer of Au.
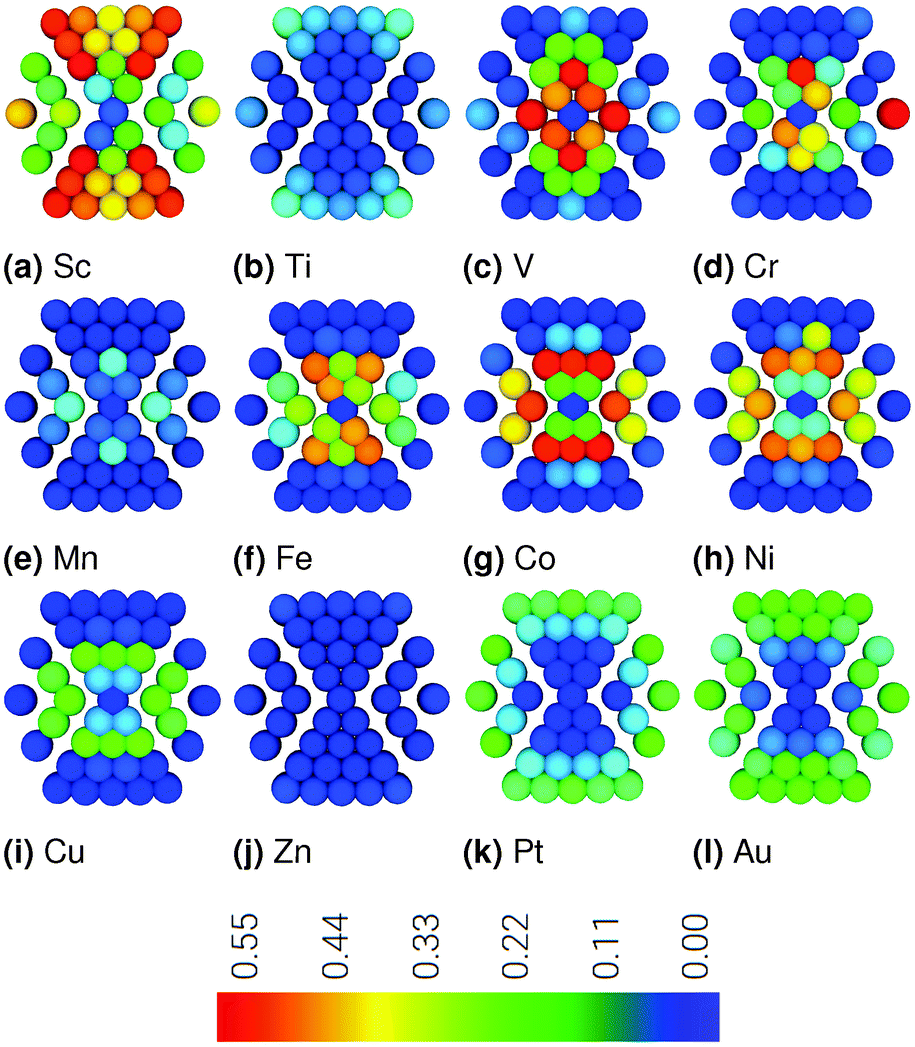 | ||
| Fig. 1 RMSD plots for a cross-section of the ternary nanoparticles relative to the Pt nanoparticle. Each atom is coloured according to displacement from the range 0.00 to 0.55 Å, coloured blue to red, respectively. | ||
The exact reason behind the changes in the RMSD values are the result of a complex relationship between the three elements and the positions within the particle that they occupy. In order to gain a better understanding of this, dimers have been studied for M–M and M–Au elemental pairs, the results of which are in the ESI,† Table S1. It is found that there is a general reduction in M–M and M–Au bond lengths compared to the Au–Au dimer. This reduction in bond length relative to forming an Au–Au core will likely be the reason for the reduced Pt-surface strain when forming the ternary particles. It is more difficult to utilise the dimer data to form reasons for the Ti, Mn and Zn containing particles having particularly small RMSD. This is ultimately the result of changes to Pt–Pt, Pt–Au, Au–Au, Au–M and M–M bond lengths as a result of forming the multimetallic nanoparticles. Though these changes are not easily separated, as changes to one bonding parameter may enhance or cancel out changes to another.
In order to quantify changes in stability resulting from the formation of these multimetallic nanoparticles the mixing energy is calculated. Mixing energies indicate a measured change in the energy of the multimetallic nanopartilcle that is compositionally proportional to the pure nanoparticles. As discussed in the introduction, Au will preferentially occupy surface sites over core positions, though this has not prevented the formation of metastable Pt-shell, Au-core nanoparticles from being produced experimentally. Indeed, the calculated mixing energy for the Pt162Au147 nanoparticle is 0.12 eV per atom. This positive mixing energy suggest that the Pt162Au147 nanoparticle is indeed metastable, though encouragingly the formation of the multimetallic nanoparticles reduces the destabilization attributable to the presence of subsurface Au. Nanoparticles with the ternary core, particularly those of interest, typically have mixing energies of below 0.04 eV per atom. This would typically suggest that if these metastable nanoparticles were produced, they would be more stable than their binary PtAu counterparts. Interestingly, Fe has a slightly negative mixing energy of −0.01 eV per atom, though for earlier d-metals such as Ti, this is increased up to −0.05 eV per atom. The mixing energies are shown in the ESI,† Fig. S1.
3.2 Adsorbate studies
For the 309-atom icosahedral nanoparticle there are a large number of possible symmetry inequivalent sites at which to bind adsorbates. Due to the computational expense of studying all sites for all compositions, two representative surface sites have been selected for detailed investigation. These best represent two distinct and important sites of particular interest which are numbered 1 and 2 in Fig. 2, for the edge-bridge and atop sites, respectively. The edge-bridge position provides a favourable site of strong binding, due to the under-coordinated edge Pt atoms. At this site, the adsorbates form bonds with two Pt atoms located at the edge of the facet, each atom is denoted “1” in Fig. 2. These positions give particularly stable binding allowing for study of phenomena related to this, though it is not expected that these sites will dominate the catalytic activity due to the overly strong adsorbate binding energies. The second site considered is the atop-facet site, which will bind adsorbates more weakly, though as nanoparticle size increases these sites will become more abundant compared to edge-bridge sites. The adsorbate forms a bond to a single Pt atom located towards the centre of the facet at the atop position, the relevant atom is denoted with a “2” in Fig. 2. It is these atop positions that are likely to be some of the more catalytically active sites on the nanoparticle.84,85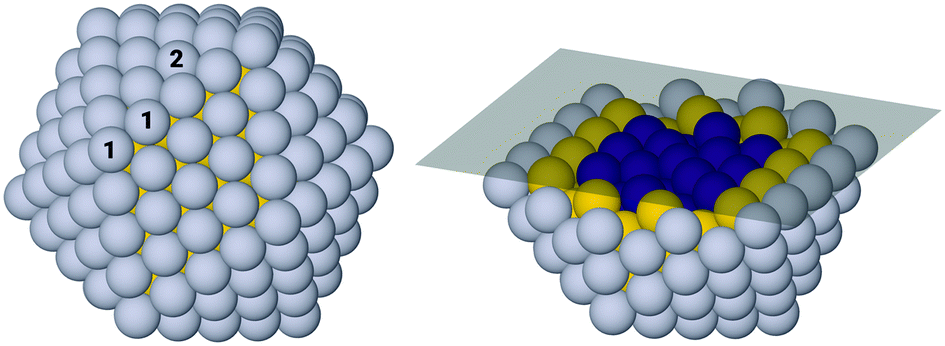 | ||
| Fig. 2 Schematic showing the complete icosahedral nanoparticle (left) and the position of the cross-section at which RMSD values are projected (right). The atoms are coloured showing the Pt shell (grey), Au subsurface (yellow) and M core (purple). Adsorption sites are also denoted on the complete particle, showing the edge-bridge position (1) and the atop position (2), investigated later in the article. | ||
To improve ORR kinetics, it is preferable that alloying effects result in a weakening of the Pt–OH binding energy by ∼0.1 eV relative to the atop site on the Pt(111) extended surface.6 For the purposes of this study, we firstly investigate Pt–OH binding with respect to comparisons between the pure Pt309 and alloyed Pt162Au92M55 nanoparticles for the atop and edge-bridge sites. This allows for an understanding of OH binding trends across the particles, though these trends are largely dependent on the size and shape effects of the particles. Thus it is difficult to compare activity of the alloyed 309-atom icosahedral nanoparticles with other nanoparticles in this size range without a better benchmark. Therefore, further comparisons are made between the particles and the more widely studied extended surface to allow for a broader benchmark with other systems.
Edge-bridge OH binding energies relative to the Pt309 nanoparticle are shown in Table 2 and plotted in Fig. 3. For all the Pt162Au92M55* constrained multimetallic nanoparticles, OH binding is reduced with respect to the pure Pt309* nanoparticle, on the edge-bridge site. The weakening of Pt–OH binding is not significant for the Pt162Au92Pt55* or Pt162Au92Au55* nanoparticles, though all other ternary systems exhibit weakening of the Pt–OH bond by at least 0.1 eV. Comparing multimetallic Pt162Au92M55* nanoparticles to the strained Pt309AuxyzMxyz* nanoparticles, the weakened OH binding is dominated by electronic effects, where OH binding on Pt309AuxyzMxyz* nanoparticles is strengthened compared to the Pt309 nanoparticle in every case. Thus the OH weakening found for the Pt162Au92M55 * nanoparticles is a result of electronic effects whilst strain has the inverse effect, strengthening OH binding. There is generally little deviation from the Pt309* Pt–OH binding energy as a result of strain effects. The Pt309AuxyzAuxyz* nanoparticle gives the greatest strengthening of the Pt–OH bond, followed by the Pt309AuxyzPtxyz* nanoparticle. This correlates well with the somewhat muted weakening of the Pt–OH binding for the multimetallic Pt162Au92Pt55* and Pt162Au92Au55* nanoparticles.
| M | Pt162Au92M55* | Pt309AuxyzMxyz* | Pt–Pt* % strain | Pt162Au92M55 |
|---|---|---|---|---|
| *Denotes a constrained nanoparticle.a Local relaxation of structure results in significant distortion. | ||||
| Sc | 0.10 | −0.09 | 2.73 | —a |
| Ti | 0.21 | −0.07 | 1.92 | —a |
| V | 0.12 | −0.09 | 0.14 | 0.21 |
| Cr | 0.10 | −0.03 | 0.57 | 0.16 |
| Mn | 0.14 | −0.03 | 0.71 | 0.20 |
| Fe | 0.15 | −0.01 | 0.04 | 0.23 |
| Co | 0.17 | −0.06 | −0.14 | 0.20 |
| Ni | 0.17 | −0.01 | 0.07 | 0.17 |
| Cu | 0.14 | −0.08 | 0.14 | 0.13 |
| Zn | 0.14 | −0.06 | 1.10 | 0.16 |
| Pt | 0.06 | −0.11 | 1.35 | 0.08 |
| Au | 0.01 | −0.37 | 1.60 | −0.02 |
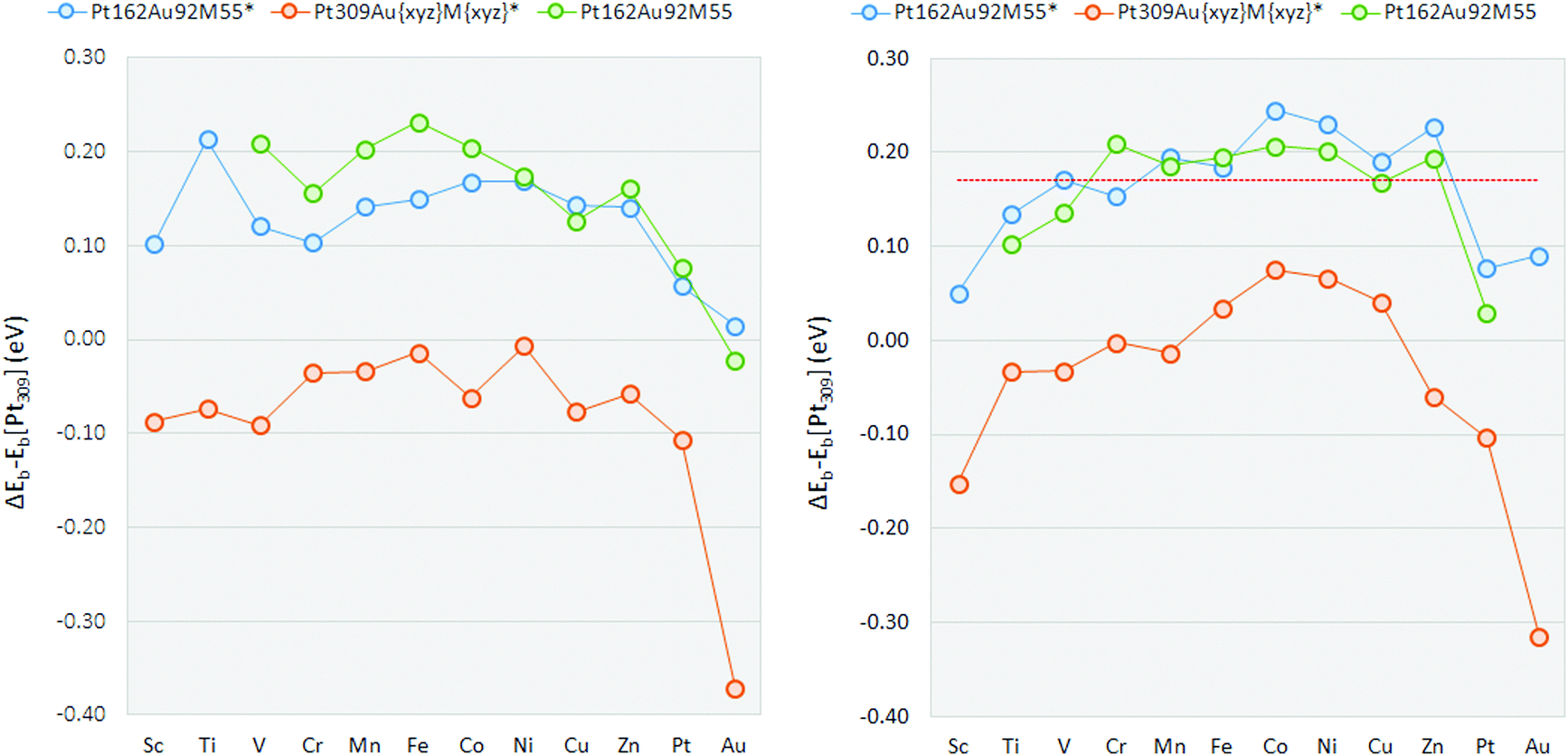 | ||
| Fig. 3 Pt–OH binding energies on the Pt162Au92M55 nanoparticles plotted relative to the Pt309 nanoparticle for binding at the edge-bridge (left) and atop (right) sites. Missing data points indicate that significant distortion of the icosahedral nanoparticle was found during local relaxation, thus reasonable comparisons could not be made. The dashed red line indicates the comparable OH binding energy on the Pt(111) extended surface. | ||
For the Pt162Au92Au55* nanoparticle, the tensile strain induced upon the Pt-shell largely negates any positive effects from subsurface Au by strengthening Pt–OH binding by 0.37 eV. However, while the Pt–OH binding for the Pt162Au92Au55* nanoparticle is only 0.01 eV weaker than that for the pure Pt309* nanoparticle, if tensile strain effects were reduced, this would suggests electronic effects resulting from subsurface Au accounted for a weakening of the Pt–OH bond by 0.38 eV. When this is compared, particularly to the ternary nanoparticles, the presence of a non-Au core results in electronic effects overcoming the increased binding from the strain effects to give a more significant weakening of the Pt–OH binding. For a number of the ternary Pt162Au92M55* nanoparticles studied here, there is a weakening of Pt–OH binding by 0.14–0.21 eV, i.e. the experimentally studied Fe and Ni containing nanoparticles, as well as Ti, V, Mn, Co, Cu and Zn containing nanoparticles. Again, this is particularly encouraging for potential improvements to ORR kinetics, where a weakening of Pt–OH binding by ∼0.1 eV relative to Pt(111) is suggested to be optimal.
When full relaxations are performed on the edge-bridge systems, the Pt162Au92Sc55 and Pt162Au92Ti55 nanoparticles with a single adsorbate deform during the geometric minimisation. This results in the Pt-shell expanding, and ultimately breaking, with Au exposed at the surface of the nanoparticle. This is likely attributed to two phenomena, firstly the large atomic radii of both Sc and Ti could induce excessive tensile strain in the Pt shell. Though, the nanoparticle locally minimised to the icosahedral structure prior to the addition of the adsorbate, it is likely that slight electronic changes in the Pt shell, induced by adsorbate interaction, result in a weakening of Pt–Pt bonding around the adsorption site, allowing the nanoparticle to distort. Pt–Pt bond elongation is commonly observed when relaxing the structure of the other nanoparticles in the presence of the adsorbate, though the strain induced upon the shell is not enough to distort the nanoparticle resulting in the core being exposed at the surface. For the majority of the remaining Pt162Au92M55 nanoparticles, there is generally a slight weakening of the Pt–OH binding when compared to the constrained Pt162Au92M55* nanoparticle. This is likely the result of electronic effects caused by slight atomic rearrangement around the adsorbate, where interaction with the adsorbate concurrently results in a slightly stronger interaction of the affected Pt with the subsurface Au. Though this has little effect on the more promising nanoparticles which still give Pt–OH weakening in the range of ∼0.1–0.2 eV when compared to the Pt309 nanoparticle.
Atop relative OH binding energies are in Table 3 and plotted in Fig. 3. For all Pt162Au92M55* constrained multimetallic nanoparticles, as with the edge-bridge site, atop-OH binding is reduced with respect to the pure Pt309* nanoparticle. In general there is a more prominent weakening of the Pt–OH binding for OH binding at the atop site compared to the edge-bridge on the Pt162Au92M55* nanoparticles. The V, Cr, Mn, Fe, Co, Ni, Cu, and Zn containing constrained nanoparticles exhibit a weakening of Pt–OH binding by 0.14–0.24 eV. For strained Pt309AuxyzMxyz* nanoparticles, there is once again little deviation from the Pt309* binding energy. As with the edge-bridge position, the strained Pt309AuxyzAuxyz* nanoparticle results in the strongest Pt–OH binding energy. For the atop position, formation of a number of Pt309AuxyzMxyz* nanoparticles results in a weakening of the Pt–OH binding energy relative to Pt309*.
| M | Pt162Au92M55* | Pt309AuxyzMxyz* | Pt162Au92M55 | ΔEslab |
|---|---|---|---|---|
| *Denotes a constrained nanoparticle.a Local relaxation of structure results in significant distortion. | ||||
| Sc | 0.05 | −0.15 | —a | —a |
| Ti | 0.13 | −0.03 | 0.10 | −0.07 |
| V | 0.17 | −0.03 | 0.14 | −0.03 |
| Cr | 0.15 | 0.00 | 0.21 | 0.04 |
| Mn | 0.19 | −0.01 | 0.19 | 0.02 |
| Fe | 0.18 | 0.03 | 0.19 | 0.03 |
| Co | 0.24 | 0.07 | 0.21 | 0.04 |
| Ni | 0.23 | 0.07 | 0.20 | 0.03 |
| Cu | 0.19 | 0.04 | 0.17 | 0.00 |
| Zn | 0.23 | −0.06 | 0.19 | 0.03 |
| Pt | 0.08 | −0.10 | 0.03 | −0.14 |
| Au | 0.09 | −0.31 | —a | —a |
The Sc and Au-rich nanoparticles both distort away from the initial icosahedral structures, though unlike the edge-bridge site, the Pt162Au92Ti55 nanoparticle does not distort. Though this is still likely to be due to the same effects as discussed previously. As with the studies on the edge-bridge site, the mid to late 3d transition metal containing nanoparticles all give promising results for the atop position with OH binding energies weakened in the range of ∼0.2 eV. Again, there is general agreement with the experimental studies showing Pt–Au–Fe/Ni are promising candidates.9,19 OH binding on the atop site is correlated with the Pt(111) slab, ΔEslab values are calculated comparing the OH binding energy on the slab to the relaxed Pt162Au92M55 nanoparticles. The Pt309 nanoparticle binds OH 0.17 eV stronger than the Pt(111) slab, in good agreement with expectations due to the increased Pt–Pt bond distance for the nanoparticle surface. The surface of the pure Pt309 nanoparticle is strained by 3.57% with respect to the extended surface due to inherent strain effects for the icosahedral particle in which core bonds are generally compressed and shell bonds elongated.86 When this effect is accounted for, the shift in binding energy is such that those nanoparticles showing most promise have approximately the same binding energy as the Pt(111) slab. For this model, Pt–OH weakening on the nanoparticle system should therefore be approximately 0.27 eV weaker than the Pt309 nanoparticle to be optimal.
Chorkendorff et al. showed the size dependence of Pt nanoparticles with reference to the bulk.87 They found the mass-activity peaked at around 3 nm nanoparticle size, showing the advantages of the nanoparticle systems over the bulk. Though they also found that the activity per surface area decreased with decreasing nanoparticle size. Nørskov et al. presented theory results showing strong OH binding energies for small clusters, but these tended towards the bulk for large nanoparticle sizes.88,89 This supports the observations above, where stronger OH binding is found on the Pt309 nanoparticle than extended Pt(111) surface. This suggests that whilst the OH binding energy of the ternary nanoparticles may not be significantly reduced with respect to the Pt(111) slab, the mass-activity of the nanoparticles is likely to be significantly improved. Further, it should be emphasised that the reason for forming Au containing alloys is to improve the stability of the nanoparticle, an additional benefit over the expected improved mass activity. Utilising core atoms with preferential bonding to Au will also improve the stability of the nanoparticle and by selecting materials like Cu, it is also possible to stabilise the icosahedral particle structure.83
3.3 Density of states analysis
The d-band model by Hammer and Nørskov is often a highly useful way to ascribe variation in the electron structure of the nanoparticle to changes in adsorbate binding energies.90 As a general expectation, a decrease in the average d-band energy (termed d-centre), away from the Fermi energy, suggests the system will exhibit weaker adsorbate binding. Conversely, an increase in the d-centre, toward the Fermi energy, suggest there will be stronger adsorbate binding. Though, there have been noted exceptions to this approximation.91 Data from the analysis of the d-band for the edge-bridge and atop sites are in the ESI,† Table S2 and S3, respectively. The first moment (μ1) of the d-band gives the d-centre. The square root of the second moment (μ21/2) gives the width of the d-band relative to the d-centre. While values for skewness and kurtosis are represented by the third (μ3/μ23/2) and fourth moments (μ4/μ22). Further, occupation numbers and fractional filling values are also given. Finally, the upper d-band edge, defined by F. Abild-Pedersen et al. as the highest peak position of the Hilbert transform of the d-band is also identified.92For the trimetallic nanoparticles, there is an increase in the d-centre towards the Fermi energy for all systems relative to the pure Pt309 nanoparticle. This is contrary to the expected trend as in the majority of cases, the trimetallic nanoparticles bind OH more weakly than the Pt309 nanoparticle. There is a notable change in the shape of the projected d-band, with a prominent peak close to the Fermi energy present for all trimetallic nanoparticles, as well as the Pt162Au147 nanoparticle, but absent for the pure Pt309 nanoparticle. This change in d-band shape makes direct comparisons between the pure and trimetallic nanoparticles difficult. The significant variation in band shape, with the prominent peak close to the Fermi energy for the Au containing nanoparticles means that the weighting of the average of the d-band will be skewed close to the Fermi energy, irrespective of band filling. Plots of the pDOS for the various nanoparticles are shown in Fig. 4, where greater d-band weight can be seen near the Fermi energy, set at 0 eV. Indeed, when comparing the skewness of the d-band, there is a positive skew present for all Au containing nanoparticles and a negative skew for the pure Pt309.
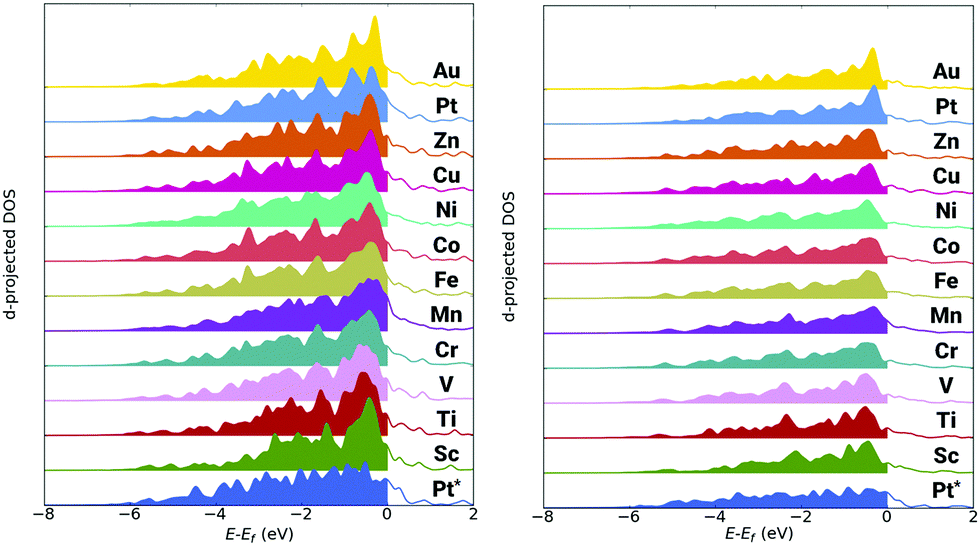 | ||
| Fig. 4 Projected d-band DOS for the edge-bridge sites (left) and atop sites (right). The Pt162Au92M55 nanoparticles are compared to the pure Pt309 nanoparticle, denoted Pt*. | ||
The d-band difference is plotted in the ESI† Fig. S2, to better show this difference in shape. The d-band character of the Pt309 nanoparticle is subtracted from that of the Pt162Au92M55 nanoparticle in each case. Thus negative intensity shows where the intensity of the Pt309 d-band is greater than the intensity of the Pt162Au92M55 nanoparticle d-band. This is found to be the case at negative energies away from the Fermi energy. Conversely, positive intensities indicate where the Pt162 Au92M55 nanoparticle d-band is greater in intensity than the Pt309 d-band. This is found close to the Fermi energy where there is often a sharp peak. Significant charge transfer can also result in problems when trying to compare d-band character between systems. A Bader analysis of the various nanoparticles has also been performed and is discussed in the ESI.† Though it is found that there is not a substantial variation in the charge transfer to the Pt-shell at which the d-band analysis is performed.
For comparative purposes, the character of the d-band for the bimetallic (A162B147) nanoparticles has also been investigated for a number of compositions. The bimetallic nanoparticles display a similar d-band character to the Pt309 nanoparticle, with a similar negative skew as in Table S2 (ESI†), while the ternary Au containing nanoparticles had positive skew. Further, investigating the d-centre of the bimetallic nanoparticles reveals a decrease relative to the pure Pt309 nanoparticle. This suggests that the prominent peak is likely attributable to the subsurface Au layer, as it is present for all Pt162Au92M55 nanoparticles as well as the Pt162Au147 nanoparticle. However, the relative increase in the d-centre is clearly not indicative of strengthening of adsorbate interactions as would normally be expected.
This suggests that the standard d-band model is not a reliable measure for the complex systems being considered in this study. It should be noted that a similar trend has been observed before, where Pt occupation is skewed close to the Fermi energy when alloyed with subsurface Au.38 The pDOS for the atop sites give a similar up-shift in the d-centre relative to the Fermi energy as found for all multimetallic systems when comparing to the pure Pt309 nanoparticle. Again, there is a characteristic peak close to the Fermi energy, most prominent for the Pt162Au147 nanoparticle, but clearly present for all the ternary nanoparticles. For the atop site, a number of the multimetallic nanoparticles also exhibit a negative skew of the d-band, though the skew for these nanoparticles is significantly less than for the Pt309 nanoparticle.
4 Conclusions
We have identified a number of new ternary nanoparticles based on Pt–Au–M for M = Cr, Mn, Co, Cu, Zn that are predicted to be promising electrocatalysts for the ORR. This is due to an improvement in ORR kinetics as a result of weakening of Pt–OH binding with respect to a pure Pt nanoparticle. This is further enhanced by an improvement in the stability of the nanoparticles in harsh operating conditions at the cathode by forming alloys with the more noble Au. Our other results are in broad agreement with previous experimental studies by Stamenkovic et al. for the ternary Pt–Au–Fe/Ni systems. Further, it is proposed that formation of a Cr, Co or Cu-rich core will likely sustain the active icosahedral structure due to the small lattice constants. While the pure icosahedral nanoparticle has been suggested to be highly active due to the interplay between strain and ligand effects, these effects can be further amplified by formation of the ternary system. The trimetallic Pt162Au92M55 nanoparticles indeed exhibit a number of advantageous properties over pure Pt or bimetallic Pt/M nanoparticles.Initially, a number of experimental studies have already shown increased durability induced through formation of Pt nanoalloys with subsurface Au. Preferential mixing of M/Au for a number of the systems further suggests stabilisation of the ternary Pt162Au92M55 nanoparticle compared to the binary Pt162Au147 nanoparticle. Of particular interest are the trimetallic nanoparticles formed with late 3d transition metals. The compressive strain exhibited on the Au subsurface by the 3d transition metal core ultimately leads to a favourable weakening of the Pt–OH binding energy for a number of the trimetallic nanoparticles. The current study has investigated nanoparticle systems in the experimentally relevant size range of ∼2 nm showing particularly promising weakening of Pt–OH binding by 0.1–0.2 eV for a number of late 3d transition metals compared to the pure Pt nanoparticle. When benchmarked against the extended Pt(111) surface, stronger Pt–OH binding on nanoparticles compared to extended surfaces means that there is only a small weakening of binding energy when comparing the nanoparticles to the Pt(111) extended surface, though it is expected that the nanoparticles will provide better mass-activity. Based on the results presented in this study, experimental investigation of the highlighted alloys has been undertaken, where Pt–Au–M nanoparticles are prepared via the water in oil microemulsion method, as discussed in the introduction.60
Acknowledgements
The authors thank Prof. Christophe Coutanceau for valuable discussions and insight. The financial support of the European Commission under the FP7 Fuel Cells and Hydrogen Joint Technology Initiative grant agreement FP7-2012-JTI-FCH-325327 for the SMARTCat project is gratefully acknowledged. The calculations were carried out on the resources provided on the Niflheim compute facilities at the Technical University of Denmark, as well as resources on Gardar funded through the Nordic High Performance Computing (NHPC) project.References
- A. Chandan, M. Hattenberger, A. El-kharouf, S. Du, A. Dhir, V. Self, B. G. Pollet, A. Ingram and W. Bujalski, J. Power Sources, 2013, 231, 264–278 CrossRef CAS.
- N. M. Marković, T. J. Schmidt, V. Stamenković and P. N. Ross, Fuel Cells, 2001, 1, 105–116 CrossRef.
- Z. Liu, L. Ma, J. Zhang, K. Hongsirikarn and J. G. Goodwin, Catal. Rev., 2013, 55, 255–288 CAS.
- P. C. Jennings, H. A. Aleksandrov, K. M. Neyman and R. L. Johnston, J. Phys. Chem. C, 2015, 119, 11031–11041 CAS.
- M. K. Jeon, C. H. Lee, G. I. Park and K. H. Kang, J. Power Sources, 2012, 216, 400–408 CrossRef CAS.
- J. Greeley, I. E. L. Stephens, A. S. Bondarenko, T. P. Johansson, H. A. Hansen, T. F. Jaramillo, J. Rossmeisl, I. Chorkendorff and J. K. Nørskov, Nat. Chem., 2009, 1, 552–556 CrossRef CAS PubMed.
- M. Escudero-Escribano, A. Verdaguer-Casadevall, P. Malacrida, U. Grønbjerg, B. P. Knudsen, A. K. Jepsen, J. Rossmeisl, I. E. L. Stephens and I. Chorkendorff, J. Am. Chem. Soc., 2012, 134, 16476–16479 CrossRef CAS PubMed.
- P. Malacrida, M. Escudero-Escribano, A. Verdaguer-Casadevall, I. E. L. Stephens and I. Chorkendorff, J. Mater. Chem. A, 2014, 2, 4234 CAS.
- C. Wang, D. van der Vliet, K. L. More, N. J. Zaluzec, S. Peng, S. Sun, H. Daimon, G. Wang, J. Greeley, J. Pearson, A. P. Paulikas, G. Karapetrov, D. Strmcnik, N. M. Markovic and V. R. Stamenkovic, Nano Lett., 2011, 11, 919–926 CrossRef CAS PubMed.
- B. N. Wanjala, J. Luo, B. Fang, D. Mott and C.-J. Zhong, J. Mater. Chem., 2011, 21, 4012 RSC.
- P.-P. Fang, S. Duan, X.-D. Lin, J. R. Anema, J.-F. Li, O. Buriez, Y. Ding, F.-R. Fan, D.-Y. Wu, B. Ren, Z. L. Wang, C. Amatore and Z.-Q. Tian, Chem. Sci., 2011, 2, 531 RSC.
- W. Chanmanee, N. R. de Tacconi, K. Rajeshwar, W.-Y. Lin, L. Nikiel and W. A. Wampler, J. Electrochem. Soc., 2012, 159, F226–F233 CrossRef CAS.
- G.-R. Zhang, J. Wu and B.-Q. Xu, J. Phys. Chem. C, 2012, 116, 20839–20847 CAS.
- S. W. Kang, Y. W. Lee, Y. Park, B.-S. Choi, J. W. Hong, K.-H. Park and S. W. Han, ACS Nano, 2013, 7, 7945–7955 CrossRef CAS PubMed.
- Q. He, P. J. Miedziak, L. Kesavan, N. Dimitratos, M. Sankar, J. A. Lopez-Sanchez, M. M. Forde, J. K. Edwards, D. W. Knight, S. H. Taylor, C. J. Kiely and G. J. Hutchings, Faraday Discuss., 2013, 162, 365–378 RSC.
- L. Wang, Y. Zhang and Z. Li, Mater. Lett., 2013, 94, 179–182 CrossRef CAS.
- B. Loganathan and B. Karthikeyan, Colloids Surf., A, 2013, 436, 944–952 CrossRef CAS.
- Y. Song and S. Chen, Nanoscale, 2013, 5, 7284–7289 RSC.
- Y. Kang, J. D. Snyder, M. Chi, D. Li, K. L. More, N. M. Markovic and V. R. Stamenkovic, Nano Lett., 2014, 14, 6361–6367 CrossRef CAS PubMed.
- S. A. Kondrat, P. J. Miedziak, M. Douthwaite, G. L. Brett, T. E. Davies, D. J. Morgan, J. K. Edwards, D. W. Knight, C. J. Kiely, S. H. Taylor and G. J. Hutchings, ChemSusChem, 2014, 7, 1326–1334 CrossRef CAS PubMed.
- H. Zhang, L. Lu, Y. Cao, S. Du, Z. Cheng and S. Zhang, Mater. Res. Bull., 2014, 49, 393–398 CrossRef CAS.
- S.-I. Choi, M. Shao, N. Lu, A. Ruditskiy, H.-C. Peng, J. Park, S. Guerrero, J. Wang, M. J. Kim and Y. Xia, ACS Nano, 2014, 8, 10363–10371 CrossRef CAS PubMed.
- X. Huang, Z. Zhao, Y. Chen, E. Zhu, M. Li, X. Duan and Y. Huang, Energy Environ. Sci., 2014, 7, 2957 CAS.
- X. Liu, E. H. Yu and K. Scott, Appl. Catal., B, 2015, 162, 593–601 CrossRef CAS.
- X. Tan, S. Prabhudev, A. Kohandehghan, D. Karpuzov, G. A. Botton and D. Mitlin, ACS Catal., 2015, 150127154142005 Search PubMed.
- L. Rubinovich and M. Polak, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 155405 CrossRef.
- R. Callejas-Tovar and P. B. Balbuena, J. Phys. Chem. C, 2012, 116, 14414–14422 CAS.
- C. Wang, D. Li, M. Chi, J. Pearson, R. B. Rankin, J. Greeley, Z. Duan, G. Wang, D. van der Vliet, K. L. More, N. M. Markovic and V. R. Stamenkovic, J. Phys. Chem. Lett., 2012, 3, 1668–1673 CrossRef CAS PubMed.
- P. C. Jennings, B. G. Pollet and R. L. Johnston, Phys. Chem. Chem. Phys., 2012, 14, 3134–3139 RSC.
- P. C. Jennings, B. G. Pollet and R. L. Johnston, J. Phys. Chem. C, 2012, 116, 15241–15250 CAS.
- L. Zhang, R. Iyyamperumal, D. F. Yancey, R. M. Crooks and G. Henkelman, ACS Nano, 2013, 7, 9168–9172 CrossRef CAS PubMed.
- R. Huang, G.-F. Shao, Y.-H. Wen and S.-G. Sun, Phys. Chem. Chem. Phys., 2014, 16, 22754–22761 RSC.
- X. Wu and Y. Dong, New J. Chem., 2014, 38, 4893–4900 RSC.
- Z. Zhao, M. Li, D. Cheng and J. Zhu, Chem. Phys., 2014, 441, 152–158 CrossRef CAS.
- P. C. Jennings, H. A. Aleksandrov, K. M. Neyman and R. L. Johnston, Nanoscale, 2014, 6, 1153–1165 RSC.
- P. C. Jennings, H. A. Aleksandrov, K. M. Neyman and R. L. Johnston, Phys. Chem. Chem. Phys., 2014, 16, 26539–26545 RSC.
- G.-H. Wu, Q.-M. Liu and X. Wu, Chem. Phys. Lett., 2015, 620, 92–97 CrossRef CAS.
- C.-E. Kim, D.-H. Lim, J. H. Jang, H. J. Kim, S. P. Yoon, J. Han, S. W. Nam, S.-A. Hong, A. Soon and H. C. Ham, J. Chem. Phys., 2015, 142, 034707 CrossRef PubMed.
- L. Zhang and G. Henkelman, ACS Catal., 2015, 5, 655–660 CrossRef CAS.
- N. Toshima, R. Ito, T. Matsushita and Y. Shiraishi, Catal. Today, 2007, 122, 239–244 CrossRef CAS.
- J. Zhang, K. Sasaki, E. Sutter and R. R. Adzic, Science, 2007, 315, 220–222 CrossRef CAS PubMed.
- V. Tripković, H. A. Hansen, J. Rossmeisl and T. Vegge, Phys. Chem. Chem. Phys., 2015, 17, 11647–11657 RSC.
- W. B. Pearson, The crystal chemistry and physics of metals and alloys, 1972 Search PubMed.
- A. Logsdail, L. O. Paz-Borbón and R. L. Johnston, J. Comput. Theor. Nanosci., 2009, 6, 857–866 CrossRef CAS.
- D. T. Tran and R. L. Johnston, Proc. R. Soc. A, 2011, 467, 2004–2019 CrossRef CAS.
- N. Kristian and X. Wang, Electrochem. Commun., 2008, 10, 12–15 CrossRef CAS.
- J. Luo, L. Wang, D. Mott, P. N. Njoki, Y. Lin, T. He, Z. Xu, B. N. Wanjana, I.-I. S. Lim and C.-J. Zhong, Adv. Mater., 2008, 20, 4342–4347 CrossRef CAS.
- G.-R. Zhang, D. Zhao, Y.-Y. Feng, B. Zhang, D. S. Su, G. Liu and B.-Q. Xu, ACS Nano, 2012, 6, 2226–2236 CrossRef CAS PubMed.
- E. Higuchi, K. Okada, M. Chiku and H. Inoue, Electrochim. Acta, 2015, 179, 100–107 CrossRef CAS.
- L. Wang and Y. Yamauchi, J. Am. Chem. Soc., 2010, 132, 13636–13638 CrossRef CAS PubMed.
- Y. Xing, Y. Cai, M. B. Vukmirovic, W.-P. Zhou, H. Karan, J. X. Wang and R. R. Adzic, J. Phys. Chem. Lett., 2010, 1, 3238–3242 CrossRef CAS.
- K. Gong, D. Su and R. R. Adzic, J. Am. Chem. Soc., 2010, 132, 14364–14366 CrossRef CAS PubMed.
- G. Wang, B. Huang, L. Xiao, Z. Ren, H. Chen, D. Wang, H. D. Abruña, J. Lu and L. Zhuang, J. Am. Chem. Soc., 2014, 136, 9643–9649 CrossRef CAS PubMed.
- X. Sun, D. Li, Y. Ding, W. Zhu, S. Guo, Z. L. Wang and S. Sun, J. Am. Chem. Soc., 2014, 136, 5745–5749 CrossRef CAS PubMed.
- H. Li, R. Yao, D. Wang, J. He, M. Li and Y. Song, J. Phys. Chem. C, 2015, 119, 4052–4061 CAS.
- L. B. Venarusso, J. Bettini and G. Maia, J. Solid State Electrochem., 2016, 20, 1753–1764 CrossRef CAS.
- L.-L. Shen, G.-R. Zhang, S. Miao, J. J. Liu and B.-Q. Xu, ACS Catal., 2016, 6, 1680–1690 CrossRef CAS.
- X. Sun, D. Li, S. Guo, W. Zhu and S. Sun, Nanoscale, 2016, 8, 2626–2631 RSC.
- M. Gatalo, P. Jovanovič, G. Polymeros, J.-P. Grote, A. Pavlišič, F. Ruiz- Zepeda, V. S. Šelih, M. Šala, S. Hočevar, M. Bele, K. J. Mayrhofer, N. Hodnik and M. Gaberšček, ACS Catal., 2016, 6, 1630–1634 CrossRef CAS.
- S. Lankiang, M. Chiwata, S. Baranton, H. Uchida and C. Coutanceau, Electrochim. Acta, 2015, 182, 131–142 CrossRef CAS.
- J. Kitchin, J. Nørskov, M. Barteau and J. Chen, Phys. Rev. Lett., 2004, 93, 156801 CrossRef CAS PubMed.
- J. Wu, L. Qi, H. You, A. Gross, J. Li and H. Yang, J. Am. Chem. Soc., 2012, 134, 11880–11883 CrossRef CAS PubMed.
- D. S. He, D. He, J. Wang, Y. Lin, P. Yin, X. Hong, Y. Wu and Y. Li, J. Am. Chem. Soc., 2016, 138, 1494–1497 CrossRef CAS PubMed.
- N. V. Long, T. Asaka, T. Matsubara and M. Nogami, Acta Mater., 2011, 59, 2901–2907 CrossRef.
- R. Choi, S.-I. Choi, C. H. Choi, K. M. Nam, S. I. Woo, J. T. Park and S. W. Han, Chemistry, 2013, 19, 8190–8198 CrossRef CAS PubMed.
- W. Zhou, J. Wu and H. Yang, Nano Lett., 2013, 13, 2870–2874 CrossRef CAS PubMed.
- X. Wang, M. Vara, M. Luo, H. Huang, A. Ruditskiy, J. Park, S. Bao, J. Liu, J. Howe, M. Chi, Z. Xie and Y. Xia, J. Am. Chem. Soc., 2015, 137, 15036–15042 CrossRef CAS PubMed.
- X. Wang, S.-I. Choi, L. T. Roling, M. Luo, C. Ma, L. Zhang, M. Chi, J. Liu, Z. Xie, J. A. Herron, M. Mavrikakis and Y. Xia, Nat. Commun., 2015, 6, 7594 CrossRef PubMed.
- S. Patala, L. D. Marks and M. Olvera de la Cruz, J. Phys. Chem. Lett., 2013, 4, 3089–3094 CrossRef CAS.
- K. Laasonen, E. Panizon, D. Bochicchio and R. Ferrando, J. Phys. Chem. C, 2013, 117, 26405–26413 CAS.
- B. Zhu, H. Guesmi, J. Creuze, B. Legrand and C. Mottet, Phys. Chem. Chem. Phys., 2015, 17, 28129–28136 RSC.
- C. Mottet, G. Rossi, F. Baletto and R. Ferrando, Phys. Rev. Lett., 2005, 95, 035501 CrossRef CAS PubMed.
- R. Ferrando, J. Phys.: Condens. Matter, 2015, 27, 013003 CrossRef PubMed.
- D. Bochicchio and R. Ferrando, Nano Lett., 2010, 10, 4211–4216 CrossRef CAS PubMed.
- J. Mortensen, L. Hansen and K. Jacobsen, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 035109 CrossRef.
- J. Enkovaara, C. Rostgaard, J. J. Mortensen, J. Chen, M. Dułak, L. Ferrighi, J. Gavnholt, C. Glinsvad, V. Haikola, H. A. Hansen, H. H. Kristoffersen, M. Kuisma, A. H. Larsen, L. Lehtovaara, M. Ljungberg, O. Lopez-Acevedo, P. G. Moses, J. Ojanen, T. Olsen, V. Petzold, N. A. Romero, J. Stausholm-Møller, M. Strange, G. A. Tritsaris, M. Vanin, M. Walter, B. Hammer, H. Häkkinen, G. K. H. Madsen, R. M. Nieminen, J. K. Nørskov, M. Puska, T. T. Rantala, J. Schiøtz, K. S. Thygesen and K. W. Jacobsen, J. Phys.: Condens. Matter, 2010, 22, 253202 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- A. H. Larsen, M. Vanin, J. J. Mortensen, K. S. Thygesen and K. W. Jacobsen, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 195112 CrossRef.
- B. Hammer, L. Hansen and J. Nørskov, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 7413–7421 CrossRef.
- E. Bitzek, P. Koskinen, F. Gähler, M. Moseler and P. Gumbsch, Phys. Rev. Lett., 2006, 97, 170201 CrossRef PubMed.
- S. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- S. Lysgaard, D. D. Landis, T. Bligaard and T. Vegge, Top. Catal., 2013, 57, 33–39 CrossRef.
- S. Lysgaard, J. S. G. Mýrdal, H. A. Hansen and T. Vegge, Phys. Chem. Chem. Phys., 2015, 17, 28270–28276 RSC.
- J. X. Wang, H. Inada, L. Wu, Y. Zhu, Y. Choi, P. Liu, W.-P. Zhou and R. R. Adzic, J. Am. Chem. Soc., 2009, 131, 17298–17302 CrossRef CAS PubMed.
- C. di Paola and F. Baletto, Phys. Chem. Chem. Phys., 2011, 13, 7701–7707 RSC.
- R. Ferrando, J. Jellinek and R. L. Johnston, Chem. Rev., 2008, 108, 845–910 CrossRef CAS PubMed.
- F. J. Perez-Alonso, D. N. McCarthy, A. Nierhoff, P. Hernandez-Fernandez, C. Strebel, I. E. L. Stephens, J. H. Nielsen and I. Chorkendorff, Angew. Chem., Int. Ed., 2012, 51, 4641–4643 CrossRef CAS PubMed.
- A. H. Larsen, J. Kleis, K. S. Thygesen, J. K. Nørskov and K. W. Jacobsen, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 245429 CrossRef.
- G. A. Tritsaris, J. Greeley, J. Rossmeisl and J. K. Nørskov, Catal. Lett., 2011, 141, 909–913 CrossRef CAS.
- B. Hammer and J. Nørskov, Surf. Sci., 1995, 343, 211–220 CrossRef CAS.
- H. Xin and S. Linic, J. Chem. Phys., 2010, 132, 221101 CrossRef PubMed.
- H. Xin, A. Vojvodic, J. Voss, J. K. Nørskov and F. Abild-Pedersen, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 115114 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: In-depth analysis of the d-band density of states as well as bader analysis of the various nanoparticles investigated in the study. See DOI: 10.1039/c6cp04194a |
| This journal is © the Owner Societies 2016 |