
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe underlying mechanisms for self-healing of poly(disulfide)s
Sil
Nevejans
a,
Nicholas
Ballard
a,
José I.
Miranda
 b,
Bernd
Reck
c and
José M.
Asua
*a
b,
Bernd
Reck
c and
José M.
Asua
*a
aPOLYMAT, University of the Basque Country UPV/EHU, Joxe Mari Korta Center, Avenida Tolosa 72, 20018 Donostia-San Sebastián, Spain. E-mail: jm.asua@ehu.es
bSGIker, NMR Facility, University of the Basque Country UPV/EHU, Spain
cDispersions and Colloidal Materials, BASF SE, 67056 Ludwigshafen, Germany
First published on 9th September 2016
Abstract
Recently, self-healing polymers based on disulfide compounds have gained attention due to the versatile chemistry of disulfide bonds and easy implementation into polymeric materials. However, the underlying mechanisms of disulfide exchange which induce the self-healing effect in poly(disulfide)s remain unclear. In this work, we elucidate the process of disulfide exchange using a variety of spectroscopic techniques. Comparing a model exchange reaction of 4-aminophenyl disulfide and diphenyl disulfide with modified reactions in the presence of additional radical traps or radical sources confirmed that the exchange reaction between disulfide compounds occurred via a radical-mediated mechanism. Furthermore, when investigating the effect of catalysts on the model exchange reaction, it could be concluded that catalysts enhance the disulfide exchange reaction through the formation of S-based anions in addition to the radical-mediated mechanism.
Introduction
Many biological organisms have the ability to undergo autonomous repair upon physical damage. Taking inspiration from this, a considerable number of studies have been published concerning synthetic self-healing materials, and various research groups have reviewed this relatively new field by distinguishing different categories for the self-repair of polymeric materials.1–5 Depending on the mechanism of self-healing, Billiet et al. made a distinction between extrinsic and intrinsic self-healing materials.1 Extrinsic materials gain their self-healing properties by the insertion of healing agents into the bulk of the material, while intrinsic materials possess a self-healing functionality through the built-in material properties. Intrinsic self-healing materials typically make use of reversible chemical or physical bonds which allows them to undergo multiple healing events and makes them very interesting for long-term use. Examples of the different types of chemistry used in intrinsic materials are molecular interdiffusion,6 Diels–Alder/retro Diels–Alder reaction (DA),7,8 photo-reversible networks,9,10 radical fission/recombination,11 anionic reactions,12 hydrogen bonding,13,14 pH responsive systems,15 π–π interactions,16,17 metallo-supramolecular,18 host–guest interactions,19 and vitrimers.20Another important type of chemical moiety used to obtain intrinsic self-healing materials, which has not been mentioned yet in the examples above, is the disulfide bond. Disulfide bonds are of interest in self-healing systems because they are weaker than carbon bonds, consequently mechanical scission is much easier.21 The chemistry of disulfide bonds has gained attention due to its versatility and the relatively simple applicability for a large variety of polymers.22 Taking advantage of the reversible disulfide bond cleavage, various self-healing polymers based on disulfide compounds have been developed. One of the first examples of using a disulfide moiety for self-healing systems was reported in 2010 by Kamada et al. describing the use of redox exchange reactions to heal polymer star gels based on reversible covalent cross-linking through thiol/disulfide redox dynamic exchange reactions.23 Aside from exploiting the redox responsiveness of the disulfide bonds, other external stimuli have been used to introduce a self-healing functionality into disulfide-based polymers. For example, healing events were thermally triggered in rubber polydisulfide thermosets.24,25 In addition to thermal activation, light has also been utilized to induce self-healing in disulfide-containing polymers. Semicrystalline, covalently cross-linked networks that contained disulfide bonds could be healed by exposure to UV light,26 while networks containing thiuram disulfide units could self-heal under the stimulation of visible light.27 Furthermore, dynamic polymer hydrogels showed self-healing ability based on a change in pH by combining acylhydrazone and disulfide bonds together in the same system.28 However, considering the previous examples and their dependence on external stimuli (redox, heat, light or pH), healing events at room temperature without the need of these stimuli would be preferable. In this context, self-healing poly(urea-urethane) elastomers have recently been described in the literature which were capable of dynamic exchange in the absence of any additional stimulus. Bis(4-aminophenyl) disulfide was used as a dynamic cross-linker in order to obtain a material which showed quantitative healing efficiency at room temperature, without the need for any catalyst or external intervention.29
It is important to understand the underlying mechanism for the self-healing event in disulfide bonds because this opens up the possibility to develop a variety of self-healing poly(disulfide)s at room temperature without the need of external stimuli. However, both metathesis30–32 and a radical-mediated mechanism27,33 have been used in the literature to describe the mechanism for disulfide exchange. Moreover, some work has been published in which both mechanisms are used indistinctly to describe the exchange reaction,34–36 while other research describes the disulfide exchange without specifying the underlying mechanism.27,37 Therefore, it is obvious that further analysis of the mechanisms involved in this highly relevant reaction for self-healing is necessary in order to be able to provide a clarifying overview on this topic.
In this context, it is important to emphasize the difference between the [2 + 2] metathesis reaction mechanism and a [2 + 1] radical-mediated mechanism. As distinctively described by Ruipérez and co-workers, in the first case, the disulfide bonds would break and form simultaneously, while in the radical-mediated mechanism, the breaking of one disulfide bond would lead to the formation of sulfur-centered radicals that would eventually attack other disulfide bonds (Fig. 1).38 These authors theoretically predicted that the main reaction mechanism would go through the formation of sulfur-based radicals and three-membered transition states.38 In this work, we focus on the experimental validation of the underlying mechanisms of disulfide exchange by investigating the kinetics of the exchange of a mixture of aromatic disulfides.
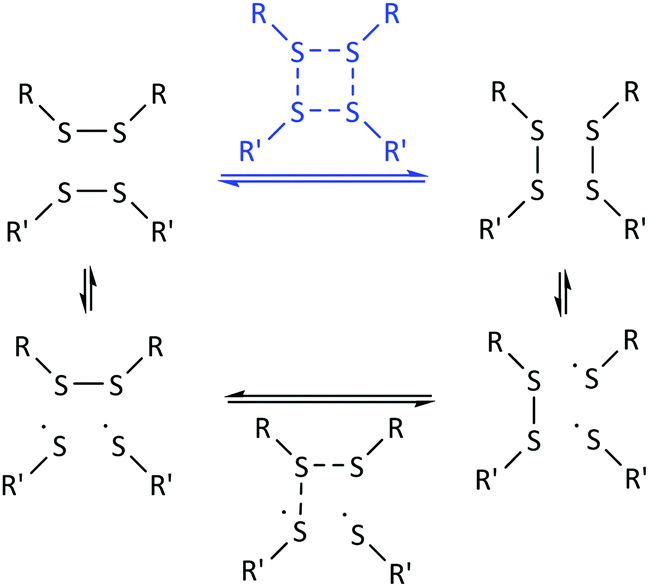 | ||
| Fig. 1 Schematic representation of the [2 + 2] metathesis (blue) and [2 + 1] radical-mediated (black) mechanisms. | ||
First, kinetic studies are performed by quantitative 13C Nuclear Magnetic Resonance (NMR) spectroscopy in order to gain more knowledge about the mechanism of the disulfide exchange reaction. A model exchange reaction of 4-aminophenyl disulfide with diphenyl disulfide is investigated and compared with modified reactions by adding or changing specific reagents (initiators, inhibitors, catalysts, monomers or disulfide compounds) or changing the reaction conditions (with or without UV radiation). Additionally, solid-state and solution Electron Paramagnetic Resonance (EPR) measurements are carried out to provide some insight into the presence of radicals in the disulfide bonds of different disulfide compounds and to check the influence of catalysts on the presence of these radicals. Finally, Ellman's reagent (5,5′-dithiobis-(2-nitrobenzoic acid), DTNB) is used to establish the presence of thiolate compounds when certain catalysts are added to the system.
Experimentals
Materials
2,2′-Azobis(4-methoxy-2,4-dimethyl valeronitrile) (V70, Wako Pure Chemical Industries, technical grade), 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB/Ellman's reagent, Sigma Aldrich, ≥98%), 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO, Sigma Aldrich, 98%), 4-aminophenyl disulfide (TCI, >98.0%), bis(4-hydroxyphenyl) disulfide (TCI, >98.0%), diethylamine (NHEt2, Sigma Aldrich, ≥99.5%), diphenyl disulfide (Sigma Aldrich, 99%), methyl methacrylate (MMA, Quimidroga, technical grade), triethylamine (NEt3, Sigma Aldrich, ≥99%) and tri-n-butylphosphine (TBP, Sigma Aldrich, 97%) were used as received.Methods
X-band EPR measurements were performed on a Bruker ELEXSYS 500 spectrometer equipped with a super-high-Q resonator ER-4123-SHQ and a maximum available microwave power of 200 mW. Solid samples and solutions were placed in quartz tubes and spectra were recorded at room temperature using typical modulation amplitudes of 0.05–0.1 mT at a frequency of 100 kHz. A NMR probe was used to calibrate the magnetic field and the frequency inside the cavity (∼9.4 GHz) was determined using an integrated MW-frequency counter. The liquid NMR spectra were recorded on a Bruker AVANCE 500 spectrometer equipped with a Z-gradient BBO probe. The kinetic studies of the reactions were conducted at 35 °C using 13C spectra. The 13C spectra were recorded every 5/15 min for 2/12 h using an inverse gated sequence at 125.13 MHz. A time domain of 64k and a spectral width of 31 kHz were considered, while the interpulse delay was 5 s and the acquisition time 1 s. In some experiments the interpulse delay was changed from 5 s to 20 s. An ultraviolet (UV) chamber (model BS 03, Dr Gröbel UV-Elektronik GmbH) was used equipped with 20 UV lamps of wavelength ranging from 315 to 400 nm with a maximum intensity at 368 nm. Ultraviolet-visible (UV-vis) spectra were obtained using a Cary-1 (Varian) spectrophotometer and analyzed using UVProbe as the appropriate software.Kinetic studies performed using 13C NMR spectroscopy
In a typical reaction, 24.8 mg of 4-aminophenyl disulfide was dissolved in 500 μL of DMSO and brought up to the temperature of 35 °C in the NMR spectrometer after which an initial scan was recorded. Subsequently, a solution of 21.8 mg of diphenyl disulfide in 200 μL of DMSO was added and the NMR tube was mixed briefly before inserting it into the spectrometer. Other reactions were performed and analyzed similarly after adding or varying certain reagents or reaction conditions as described in detail in Table 1. The degree of product formation for the different reactions was calculated using the following fraction of the integrals of a and b (Fig. 3):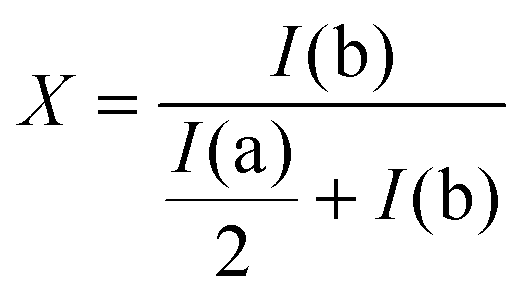
| Reaction | A (eq.) | B (eq.) | R | X | Solvent | Time (h) | Temperature (°C) |
|---|---|---|---|---|---|---|---|
| (i) | 1 | 1 | –NH2 | — | DMSO | 12 | 35 |
| (ii) | 1 | 1 | –NH2 | 0.1 eq. TEMPO | DMSO | 12 | 35 |
| (iii) | 1 | 1 | –NH2 | 0.5 eq. V70 | DMSO | 12 | 35 |
| (iv) | 1 | 1 | –NH2 | UV (10 min) | DMSO | 12 | 35 |
| (v) | 1 | 1 | –OH | — | DMSO | 12 | 35 |
| (vi) | 1 | 1 | –NH2 | 0.1 eq. NEt3 | DMSO | 2 | 35 |
| (vii) | 1 | 1 | –NH2 | 0.1 eq. NEt3 + 0.1 eq. TEMPO | DMSO | 12 | 35 |
| (viii) | 1 | 1 | –NH2 | 0.1 eq. TBP | DMSO | 2 | 35 |
| (ix) | 1 | 1 | –NH2 | 0.1 eq. TBP + 0.1 eq. TEMPO | DMSO | 12 | 35 |
The measured T1 values of a (1.14 s) and b (1.71 s) are low enough so that, with an interpulse delay of 5 s, the values obtained for the degree of product formation can be considered quantitative. To confirm this additional experiments with a delay time of 20 s were performed with quantitatively similar results. It should be noted that the carbons c and d as shown in Fig. 3 and 5 have significantly longer relaxation times (T1 = 4.01 and 3.22 s for carbons c and d respectively), therefore quantitative comparison is not possible for these signals. Analogously, also the reaction of 21.8 mg of diphenyl disulfide with 175 μL of MMA was followed using 13C NMR spectroscopy in 525 μL DMSO at room temperature. NMR measurements of the mixtures were taken after 2 days at room temperature and after additional exposure to UV radiation for 1 h 10 min also at room temperature. Furthermore, the reaction between MMA and diphenyl disulfide (1 eq.) in DMSO and in the presence of 1.4 μL (0.1 eq.) of NEt3 was followed using 13C NMR spectroscopy, measuring the reaction after 2 days at room temperature.
EPR measurements
4-Aminophenyl disulfide and bis(4-hydroxyphenyl) disulfide were analyzed for the presence of radicals using EPR spectroscopy. An amount of 70 mg of each disulfide compound was used to be able to carry out solid-state measurements. Furthermore, to analyze the effect of NEt3 on the disulfide bond and the presence of radicals in this bond, also measurements in solutions were performed. In 1.5 mL eppendorf tubes, a solution was made of 2 mg of the solid disulfide compound in the required amount of NEt3 as a solvent to reach a total volume of 1.5 mL. Eventually 0.5 mL of the mixture was used to perform the solution EPR measurements.UV-vis measurements in the presence of Ellman's reagent
Quantitative experiments were performed by using UV-vis spectroscopy. In these experiments NEt3, NHEt2 and TBP were tested by adding 50 μL of a DTNB solution (4 mg DTNB per mL DMSO) to a 14.3 mM solution of the reagents in DMSO. These reaction mixtures were then measured using UV-vis spectroscopy in a spectral range varying form 190–700 cm−1.Results and discussion
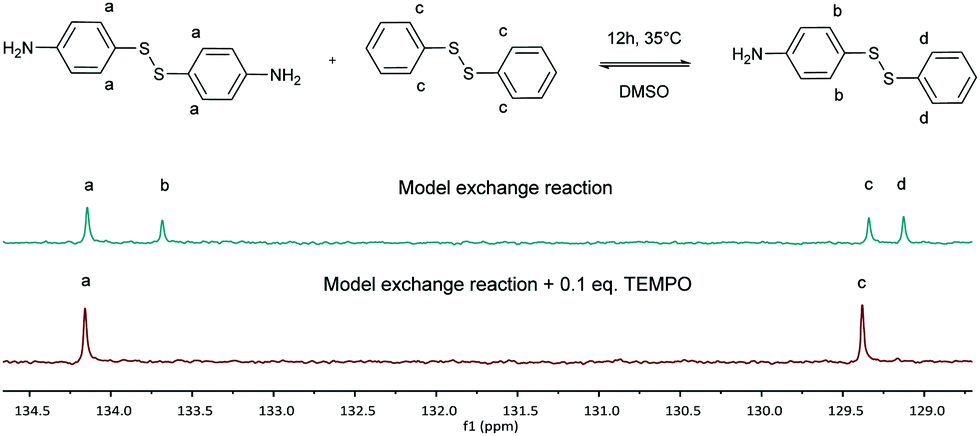
First, the model exchange reaction of 4-aminophenyl disulfide with diphenyl disulfide forming the mixed product (Fig. 2) was followed kinetically for 12 h at 35 °C using NMR spectroscopy. The product formation was analyzed through the change of signals in the 13C NMR spectra as depicted in Fig. 3. The signals a and c in Fig. 3 were both assigned to the reactants, while b and d were ascribed to the formed product. The degree of product formation for the model exchange reaction (i) (Table 1) was calculated by monitoring the change in the relative integrals of the 13C NMR spectra and is shown in Fig. 4. It can be seen that, after 12 h, the exchange reaction reached a conversion of almost 60%. When experiments were performed under inert conditions (N2-atmosphere), similar results were obtained which indicated that oxygen has little to no influence on the disulfide exchange reaction. Addition of the radical scavenger TEMPO (0.1 eq.) resulted in a complete inhibition of the disulfide exchange (Fig. 5). This inhibition is possible since TEMPO combines with sulfur-based radicals forming an adduct which can also decompose to give inert species.39 It is particularly important to highlight that complete inhibition of the exchange would not be possible using such a low amount of TEMPO if it is assumed that the exchange proceeds via a simple cleavage and recombination of the disulfide bond.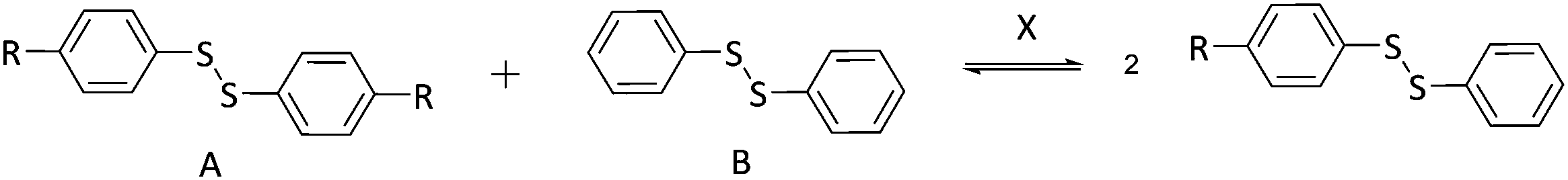 | ||
| Fig. 2 Exchange reactions studied by 13C NMR. | ||
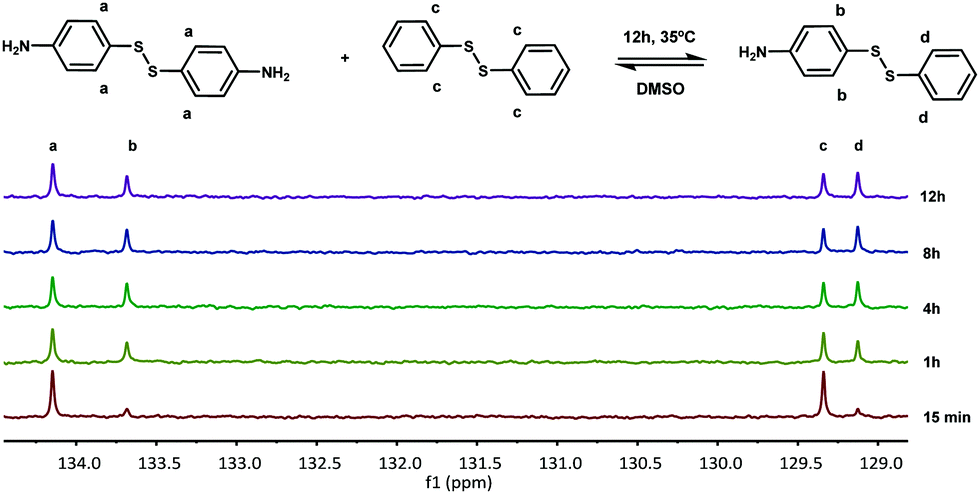 | ||
| Fig. 3 13C NMR spectra of the exchange reaction of 4-aminophenyl disulfide with diphenyl disulfide (delay time of 20 s). | ||
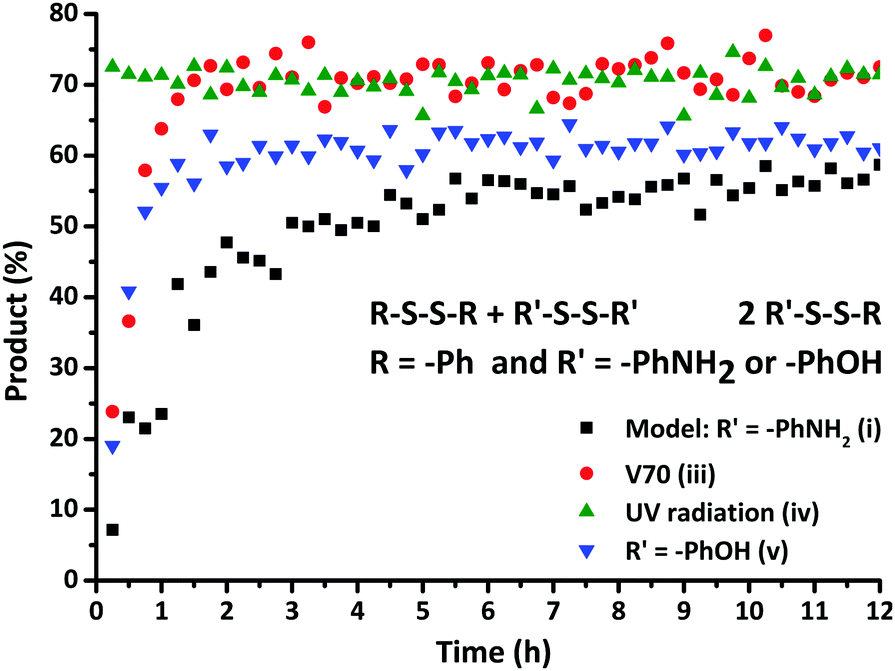 | ||
| Fig. 4 Formation of the exchange product as a function of time for the exchange reactions (i), (iii), (iv) and (v). | ||
 | ||
| Fig. 5 Formation of the exchange product via the model exchange reaction (i) and in the presence of TEMPO (ii) (delay time of 20 s). | ||
When an initiator with a fast decomposition at low temperature (t1/2 = 10 h at 30 °C), V70 (0.5 eq.) was introduced into the model reaction mixture, a fast increase to a maximum product formation (70%) at 2 h was observed (Fig. 4(iii)). Based on the change in kinetics of the disulfide exchange in the presence of a radical trap and a radical source, it can be presumed that the mechanism for the disulfide exchange is radical-mediated as proposed by theoretical studies.
To further confirm the radical mechanism, the reaction conditions of the model exchange experiment were modified. When the reaction mixture was exposed to UV radiation, the exchange reaction was rapid. After exposure for 10 min to UV radiation (iv), the reaction mixture was kinetically studied and the corresponding data obtained by 13C NMR spectroscopy are illustrated in Fig. 4. This result demonstrates that the exchange reaction had already reached product formation of 70% at the first measurement of the kinetic study (15 min). This drastic increase in the rate of product formation when conducted under UV radiation further confirmed the proposed mechanism.
In order to investigate the influence of the molecular structure of the disulfide compounds on the exchange reaction, more specifically the effect of alternative substituents on the aromatic ring, the reactant 4-aminophenyl disulfide was replaced by bis(4-hydroxyphenyl) disulfide in the model reaction. As visible in Fig. 4, this exchange reaction (v) reached a conversion of 60% after approximately 2 h in comparison to 12 h in the case of 4-aminophenyl disulfide. The increase in the rate of conversion suggests that the radical generation of bis(4-hydroxyphenyl) disulfide is faster than that of 4-aminophenyl disulfide. Further confirmation of the radical-mediated exchange mechanism was obtained via EPR spectroscopy. Solid-state EPR measurements showed an EPR signal, visible in Fig. 6, for bis(4-hydroxyphenyl) disulfide while no signal could be observed for 4-aminophenyl disulfide. The isotropic EPR signal with a g-value of 2.0025 affirmed the presence of a (sulfur-based) radical in bis(4-hydroxyphenyl) disulfide which is in strong accordance with the g-value of a sulfur-based radical in a thiuram disulfide diol unit (g = 2.004).27 An explanation for the absence of an EPR signal in the analysis of the 4-aminophenyl disulfide would be the lower radical concentration present in this disulfide compound which is in agreement with the slower rate of product formation as depicted above in Fig. 4 and the limited sensitivity of solid-state EPR spectroscopy which probably prevents the detection of this lower concentration.
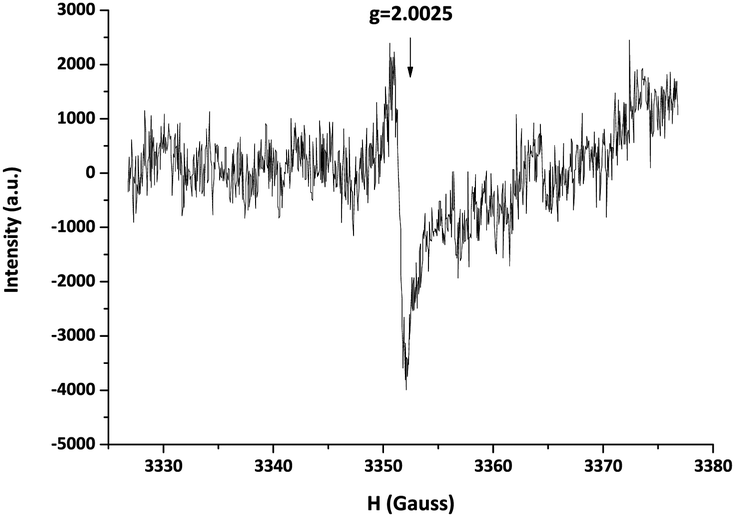 | ||
| Fig. 6 EPR signal of bis(4-hydroxyphenyl) disulfide. | ||
As a final experiment to validate the presence of radicals in the underlying mechanism of disulfide exchange, diphenyl disulfide was reacted with MMA at room temperature. Since disulfides can act simultaneously as an initiator, a transfer agent and a terminator (iniferter) in radical polymerization, poly(methyl methacrylate) (PMMA) is expected to be formed with initiator fragments of the disulfide compound as end groups.40,41 The 13C NMR spectra of the reaction of diphenyl disulfide with MMA after 2 days at room temperature (r.t.) and after an additional exposure to UV radiation for 10 min and 1 h 10 min at r.t. are depicted in Fig. 7. These spectra show that after 2 days a small amount of PMMA could be observed at the region of 1–0.5 ppm. However, when UV radiation was applied to increase the cleavage of the disulfide compound, larger signals for the formed product appear. This proves that UV radiation cleaves the disulfide bond forming sulfur-based radicals which can induce radical polymerization of MMA, since a blank of MMA in DMSO under UV radiation for the same time interval did not show any polymer product. The suppression of the exchange reaction by the inhibitor, the enhancement by the initiator, by UV radiation and by changing the disulfide and the role of the disulfide as iniferter in the radical polymerization of MMA all suggested that the underlying mechanism for the disulfide exchange reaction is radical-mediated. However, the experimental results of the model reaction in the presence of TEMPO showed that a mechanism which only forms radicals via cleavage and recombination could be ruled out, because only 0.1 equivalent of TEMPO was necessary to suppress the formation of the product. Therefore a radical-mediated mechanism in which cleavage leads to the formation of two radicals which can recombine or undergo a chain transfer reaction can be proposed (Scheme 1). According to this mechanism, only a small amount of radicals have to be formed to achieve extensive disulfide exchange, and hence even a small amount of inhibitor (0.1 eq. TEMPO) can kill the possibility of forming any product.
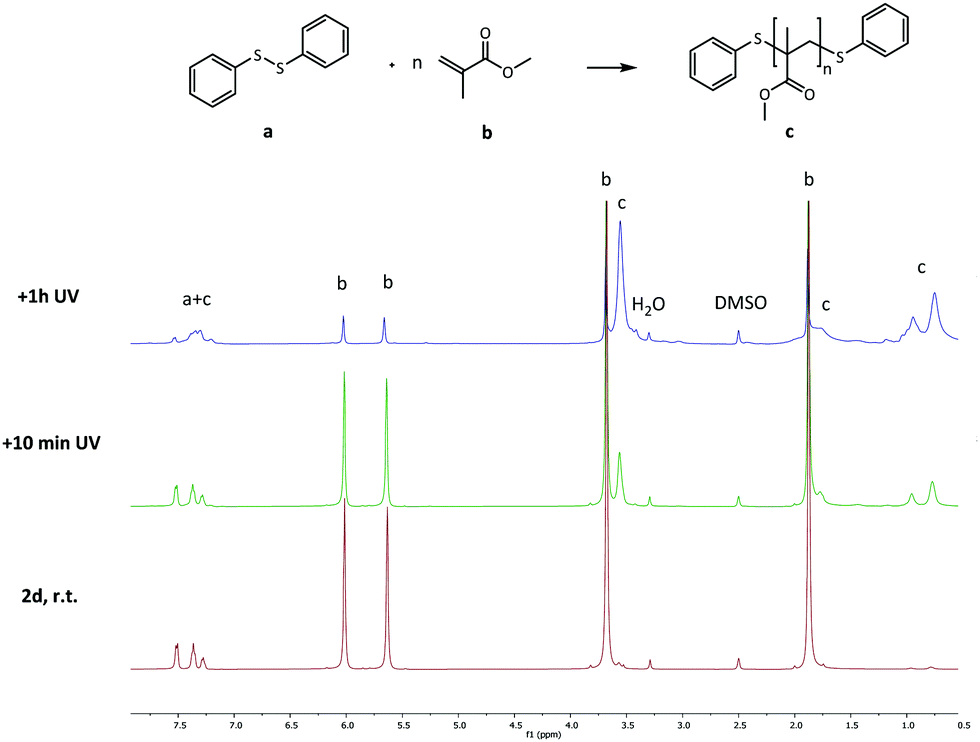 | ||
| Fig. 7 13C NMR spectra of the reaction of diphenyl disulfide with MMA. | ||
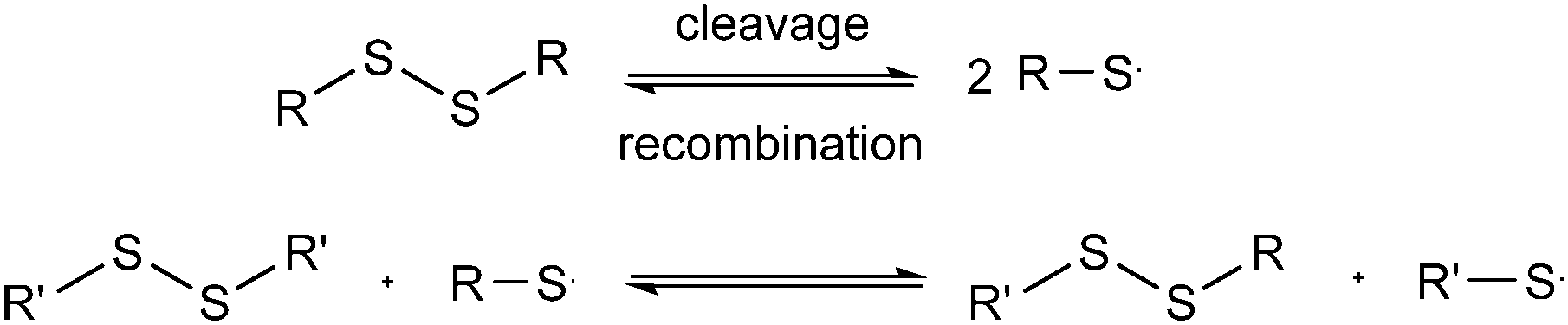 | ||
| Scheme 1 Probable radical-mediated mechanism for the disulfide exchange reaction. | ||
While the aromatic disulfides undergo spontaneous exchange, other disulfide compounds require the addition of catalysts, such as triethylamine (NEt3)29 and tri-n-butylphosphine (TBP),32 to enhance the disulfide exchange reaction. Kinetic studies were carried out using 13C NMR spectroscopy to verify the enhancing influence of NEt3 as a catalyst on the exchange reaction as described by Rekondo et al.29Fig. 8 demonstrates that the exchange reaction with the addition of NEt3 (vi) had already reached a conversion of 70% after approximately 1 h. Similar results were obtained adding TBP (viii) as a catalyst to the model exchange reaction. In order to verify the underlying mechanism of this increase, additional solution EPR measurements were carried out in order to examine the effect of a catalyst (NEt3) on the presence of radicals in the disulfide bonds of both disulfide compounds. Since the rate enhanced substantially, a corresponding large increase in the EPR signal would be expected when a radical mechanism is assumed. However, no increase in the EPR signal was observed in the presence of the catalyst, indicating that the increase in the rate could not be associated with a radical-mediated mechanism. Furthermore, addition of NEt3 had no impact on the extent of polymerization of MMA in the presence of diphenyl disulfide after 2 days at room temperature, proving that adding a catalyst does not lead to an increase in the radical concentration.
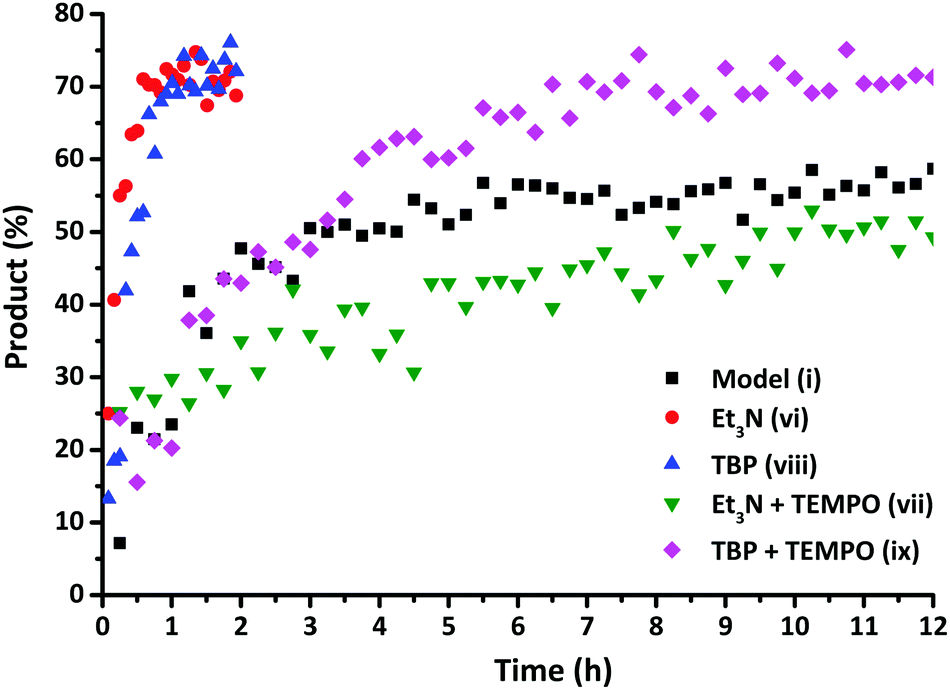 | ||
| Fig. 8 Formation of the exchange product as a function of time for the exchange reactions (i), (vi), (vii), (viii) and (ix). | ||
Since both catalysts, NEt3 and TBP, are nucleophiles and are known to reduce disulfide compounds, forming thiols, the most probable effect of these catalysts is to enhance the disulfide exchange reaction through the formation of sulfur-based anions.42 In order to prove this, Ellman's reagent (DTNB), an aromatic disulfide similar in structure to the compounds used in this work, was used as a reactant. Upon reduction, 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB) yields a highly coloured thiolate species (TNB) which can be detected by UV-vis spectroscopy.43,44 Thus, the disulfide DTNB was reacted with NEt3, TBP and NHEt2 in similar concentrations as used in the previous NMR studies and UV-vis spectroscopy was used to measure the formation of the coloured TNB. As can be derived from Fig. 9, the UV-vis signal increased with the nucleophilicity of the catalyst (TBP > NHEt2 > NEt3),45,46 starting from small in the case of NEt3, to bigger in the case of NHEt2 and the biggest for TBP as a catalyst, confirming that in the presence of nucleophiles aromatic disulfide compounds fragment to yield a thiolate anion.
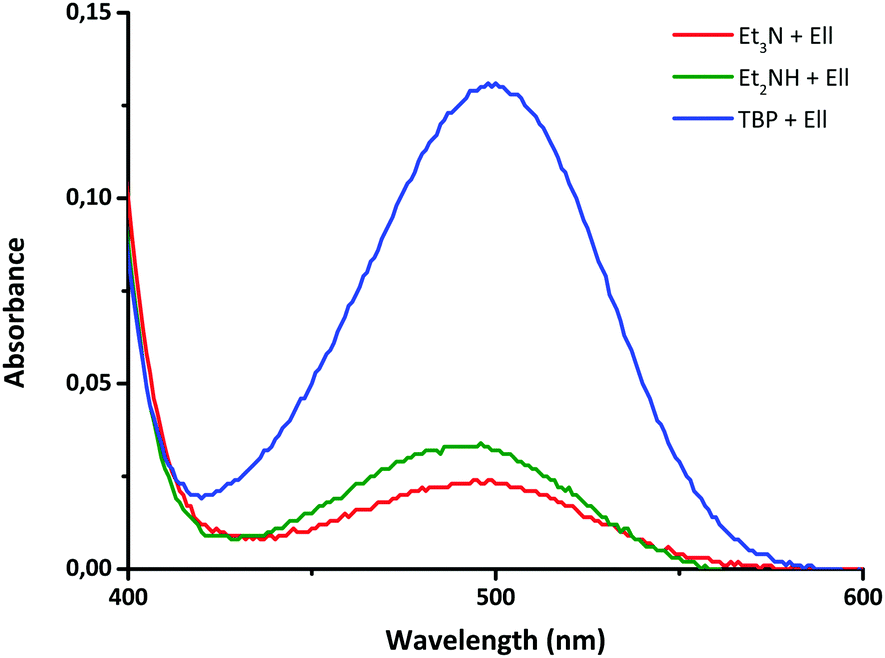 | ||
| Fig. 9 UV-vis measurements of different catalysts (TBP, NHEt2, NEt3) with DTNB. | ||
Based on these experiments, it would be suggested that in the presence of a catalyst both radical-mediated and thiol-mediated exchanges occur simultaneously as shown in Scheme 2. In order to confirm this, reactions were conducted in the presence of both the catalyst and the radical scavenger TEMPO. Whilst TEMPO was capable of completely preventing the radical-mediated exchange in the absence of a catalyst, when a catalyst (NEt3 or TBP) is added the disulfide exchange still occurs, although at a slower rate than when no TEMPO is present (Fig. 8). The faster relative rate of conversion in the presence of TBP compared to NEt3 is due to the higher nucleophilicity of the TBP in comparison to NEt3, since a higher nucleophilicity enhances the formation of sulfur-based anions and therefore promotes the disulfide exchange.43 In the presence of TEMPO and a catalyst, the radical pathway is blocked, but the exchange can still occur due to the generation of thiols by the catalyst. However, it is noteworthy to mention that when an excess of TEMPO (1 eq.) is added to the model reaction in the presence of NEt3 (0.1 eq.) no product formation was visible. This indicates that TEMPO may also retard the formation of sulfur-based anions formed by the addition of catalysts.47
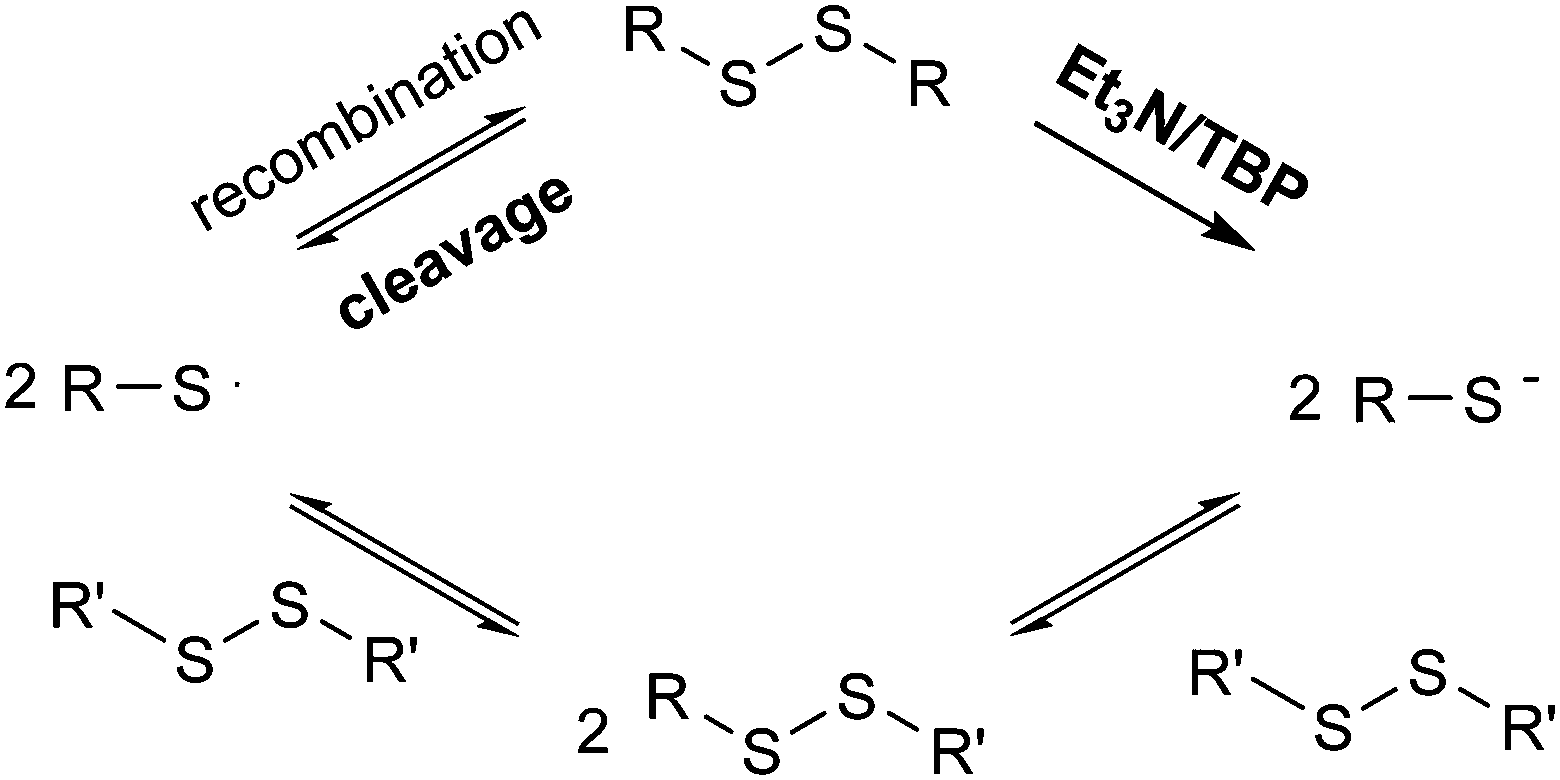 | ||
| Scheme 2 Disulfide exchange through the formation of sulfur-based radicals and sulfur-based anions. | ||
Conclusion
In summary, the results presented herein demonstrate that the general mechanism responsible for the self-healing event of disulfide bonds is radical-mediated and involves homolytic cleavage of the disulfide bond followed by subsequent radical transfer of sulfur-based radicals. In the presence of nucleophiles, such as NEt3 and TBP, disulfides yield thiolate anions which can similarly undergo a series of transfer reactions and enhance the rate of the disulfide exchange. It is hoped that this mechanistic picture of the disulfide exchange reaction will help in the synthesis of new self-healing materials based on the dynamic exchange of the disulfide bond.Acknowledgements
The European Union's Horizon 2020 research and innovation programme is accredited for the financial support through Project TRACKWAY-ITN 642514 under the Marie Sklodowska-Curie grant agreement. Prof. Luis Lezama is acknowledged for the EPR measurements.Notes and references
- S. Billiet, X. K. D. Hillewaere, R. F. A. Teixeira and F. E. Du Prez, Macromol. Rapid Commun., 2013, 34, 290–309 CrossRef CAS PubMed.
- J. A. Syrett, C. R. Becer and D. M. Haddleton, Polym. Chem., 2010, 1, 978–987 RSC.
- P. Michael, D. Döhler and W. H. Binder, Polymer, 2015, 69, 216–227 CrossRef CAS.
- S. Y. An, D. Arunbabu, S. M. Noh, Y. K. Song and J. K. Oh, Chem. Commun., 2015, 51, 13058–13070 RSC.
- N. Roy, B. Bruchmann and J. M. Lehn, Chem. Soc. Rev., 2015, 44, 3786–3807 RSC.
- N. A. Yufa, J. Li and S. J. Sibener, Polymer, 2009, 50, 2630–2634 CrossRef CAS.
- S. R. White, N. R. Sottos, P. H. Geubelle, J. S. Moore, M. R. Kessler, S. R. Sriram, E. N. Brown and S. Viswanathan, Nature, 2001, 409, 794–797 CrossRef CAS PubMed.
- Y. Heo and H. A. Sodano, Adv. Funct. Mater., 2014, 24, 5261–5268 CrossRef CAS.
- M. Burnworth, L. Tang, J. R. Kumpfer, A. J. Duncan, F. L. Beyer, G. L. Fiore, S. J. Rowan and C. Weder, Nature, 2011, 472, 334–337 CrossRef CAS PubMed.
- G. L. Fiore, S. J. Rowan and C. Weder, Chem. Soc. Rev., 2013, 42, 7278–7288 RSC.
- C. Yuan, M. Z. Rong, M. Q. Zhang, Z. P. Zhang and Y. C. Yuan, Chem. Mater., 2011, 23, 5076–5081 CrossRef CAS.
- P. Zheng and T. J. McCarthy, J. Am. Chem. Soc., 2012, 134, 2024–2027 CrossRef CAS PubMed.
- P. Cordier, F. Tournilhac, C. Soulie-Ziakovic and L. Leibler, Nature, 2008, 451, 977–980 CrossRef CAS PubMed.
- A. Faghihnejad, K. E. Feldman, J. Yu, M. V Tirrell, J. N. Israelachvili, C. J. Hawker, E. J. Kramer and H. Zeng, Adv. Funct. Mater., 2014, 24, 2322–2333 CrossRef CAS.
- G. Deng, C. Tang, F. Li, H. Jiang and Y. Chen, Macromolecules, 2010, 43, 1191–1194 CrossRef CAS.
- S. Burattini, H. M. Colquhoun, J. D. Fox, D. Friedmann, B. W. Greenland, P. J. F. Harris, W. Hayes, M. E. Mackay and S. J. Rowan, Chem. Commun., 2009, 6717–6719 RSC.
- L. R. Hart, N. A. Nguyen, J. L. Harries, M. E. Mackay, H. M. Colquhoun and W. Hayes, Polymer, 2015, 69, 293–300 CrossRef CAS.
- G. N. Hong, H. Zhang, Y. J. Lin, Y. J. Chen, Y. Z. Xu, W. G. Weng and H. P. Xia, Macromolecules, 2013, 46, 8649–8656 CrossRef CAS.
- M. Nakahata, Y. Takashima and A. Harada, Macromol. Rapid Commun., 2016, 37, 86–92 CrossRef CAS PubMed.
- M. Capelot, D. Montarnal, F. Tournilhac and L. Leibler, J. Am. Chem. Soc., 2012, 134, 7664–7667 CrossRef CAS PubMed.
- I. Park, S. S. Sheiko, A. Nese and K. Matyjaszewski, Macromolecules, 2009, 42, 1805–1807 CrossRef CAS.
- S. J. Garcia, Eur. Polym. J., 2014, 53, 118–125 CrossRef CAS.
- J. Kamada, K. Koynov, C. Corten, A. Juhari, J. A. Yoon, M. W. Urban, A. C. Balazs and K. Matyjaszewski, Macromolecules, 2010, 43, 4133–4139 CrossRef CAS.
- U. Lafont, H. van Zeijl and S. van der Zwaag, ACS Appl. Mater. Interfaces, 2012, 4, 6280–6288 CAS.
- J. Canadell, H. Goossens and B. Klumperman, Macromolecules, 2011, 44, 2536–2541 CrossRef CAS.
- B. T. Michal, C. A. Jaye, E. J. Spencer and S. J. Rowan, ACS Macro Lett., 2013, 2, 694–699 CrossRef CAS.
- Y. Amamoto, H. Otsuka, A. Takahara and K. Matyjaszewski, Adv. Mater., 2012, 24, 3975–3980 CrossRef CAS PubMed.
- G. H. Deng, F. Y. Li, H. X. Yu, F. Y. Liu, C. Y. Liu, W. X. Sun, H. F. Jiang and Y. M. Chen, ACS Macro Lett., 2012, 1, 275–279 CrossRef CAS.
- A. Rekondo, R. Martin, A. Ruiz de Luzuriaga, G. Cabanero, H. J. Grande and I. Odriozola, Mater. Horiz., 2014, 1, 237–240 RSC.
- A. M. Belenguer, T. Friščić, G. M. Day and J. K. M. Sanders, Chem. Sci., 2011, 2, 696 RSC.
- R. Caraballo, PhD thesis, KTH Royal Institute of Technology, 2010.
- Z. Q. Lei, H. P. Xiang, Y. J. Yuan, M. Z. Rong and M. Q. Zhang, Chem. Mater., 2014, 26, 2038–2046 CrossRef CAS.
- B. D. Fairbanks, S. P. Singh, C. N. Bowman and K. S. Anseth, Macromolecules, 2011, 44, 2444–2450 CrossRef CAS PubMed.
- T. Ohishi, Y. Iki, K. Imato, Y. Higaki, A. Takahara and H. Otsuka, Chem. Lett., 2013, 42, 1346–1348 CrossRef CAS.
- H. Otsuka, S. Nagano, Y. Kobashi, T. Maeda and A. Takahara, Chem. Commun., 2010, 46, 1150–1152 RSC.
- U. F. Fritze and M. von Delius, Chem. Commun., 2016, 52, 6363–6366 RSC.
- R. J. Sarma, S. Otto and J. R. Nitschke, Chem. – Eur. J., 2007, 13, 9542–9546 CrossRef CAS PubMed.
- J. M. Matxain, J. M. Asua and F. Ruipérez, Phys. Chem. Chem. Phys., 2016, 18, 1758–1770 RSC.
- S. Goldstein, A. Samuni and G. Merenyi, J. Phys. Chem. A, 2008, 112, 8600–8605 CrossRef CAS PubMed.
- K. Matyjaszewski, Macromol. Rapid Commun., 2005, 26, 135–142 CrossRef CAS.
- T. Otsu, Y. Kinoshita and M. Imoto, Makromol. Chem., 1964, 73, 225–230 CrossRef.
- I. V Koval, Russ. Chem. Rev., 1994, 63, 735–750 CrossRef.
- G. L. Ellman, Arch. Biochem. Biophys., 1959, 82, 70–77 CrossRef CAS PubMed.
- P. W. Riddles, R. L. Blakeley and B. Zerner, Methods Enzymol., Academic Press, 1983, vol. 91, pp. 49–60 Search PubMed.
- J. L. Methot and W. R. Roush, Adv. Synth. Catal., 2004, 346, 1035–1050 CrossRef CAS.
- W. A. Henderson and C. J. Schultz, J. Org. Chem., 1962, 27, 4643–4646 CrossRef CAS.
- S. Antonello, K. Daasbjerg, H. Jensen, F. Taddei and F. Maran, J. Am. Chem. Soc., 2003, 125, 14905–14916 CrossRef CAS PubMed.
| This journal is © the Owner Societies 2016 |