
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular polymorphism: microwave spectra, equilibrium structures, and an astronomical investigation of the HNCS isomeric family†
Brett A.
McGuire‡
 ab,
Marie-Aline
Martin-Drumel
b,
Sven
Thorwirth
c,
Sandra
Brünken
c,
Valerio
Lattanzi
d,
Justin L.
Neill
e,
Silvia
Spezzano
d,
Zhenhong
Yu
f,
Daniel P.
Zaleski
e,
Anthony J.
Remijan
a,
Brooks H.
Pate
e and
Michael C.
McCarthy
*ag
ab,
Marie-Aline
Martin-Drumel
b,
Sven
Thorwirth
c,
Sandra
Brünken
c,
Valerio
Lattanzi
d,
Justin L.
Neill
e,
Silvia
Spezzano
d,
Zhenhong
Yu
f,
Daniel P.
Zaleski
e,
Anthony J.
Remijan
a,
Brooks H.
Pate
e and
Michael C.
McCarthy
*ag
aNational Radio Astronomy Observatory, 520 Edgemont Rd, Charlottesville, VA 22903, USA
bHarvard-Smithsonian Center for Astrophysics, 60 Garden Street, Cambridge, MA 02138, USA
cI. Physikalisches Institut, Universität zu Köln, Zülpicher Str. 77, 50937 Köln, Germany
dMax-Planck Institut für Extraterrestrische Physik, Giessenbachstrasse 1, 85748 Garching, Germany
eDepartment of Chemistry, University of Virginia, McCormick Rd., Charlottesville, VA 22904, USA
fAerodyne Research, Inc, Billerica, MA, USA
gSchool of Engineering and Applied Sciences, Harvard University, 29 Oxford Street, Cambridge, MA 02138, USA. E-mail: mccarthy@cfa.harvard.edu
First published on 13th July 2016
Abstract
The rotational spectra of thioisocyanic acid (HNCS), and its three energetic isomers (HSCN, HCNS, and HSNC) have been observed at high spectral resolution by a combination of chirped-pulse and Fabry–Pérot Fourier-transform microwave spectroscopy between 6 and 40 GHz in a pulsed-jet discharge expansion. Two isomers, thiofulminic acid (HCNS) and isothiofulminic acid (HSNC), calculated here to be 35–37 kcal mol−1 less stable than the ground state isomer HNCS, have been detected for the first time. Precise rotational, centrifugal distortion, and nitrogen hyperfine coupling constants have been determined for the normal and rare isotopic species of both molecules; all are in good agreement with theoretical predictions obtained at the coupled cluster level of theory. On the basis of isotopic spectroscopy, precise molecular structures have been derived for all four isomers by correcting experimental rotational constants for the effects of rotation–vibration interaction calculated theoretically. Formation and isomerization pathways have also been investigated; the high abundance of HSCN relative to ground state HNCS, and the detection of strong lines of SH using CH3CN and H2S, suggest that HSCN is preferentially produced by the radical–radical reaction HS + CN. A radio astronomical search for HSCN and its isomers has been undertaken toward the high-mass star-forming region Sgr B2(N) in the Galactic Center with the 100 m Green Bank Telescope. While we find clear evidence for HSCN, only a tentative detection of HNCS is proposed, and there is no indication of HCNS or HSNC at the same rms noise level. HSCN, and tentatively HNCS, displays clear deviations from a single-excitation temperature model, suggesting weak masing may be occurring in some transitions in this source.
1 Introduction
Isomerism is one of the oldest and most important concepts in chemistry, dating back to the 1820s when Liebig and Wöhler first demonstrated that silver fulminate (AgCNO) and silver cyanate (AgOCN) – two compounds with the same elemental formula – have different physical properties. These findings led Berzelius to propose the concept of “isomer” in 1831.1 Because it is fundamentally linked to molecular structure and chemical bonding, isomerism – particularly of small molecules – has long fascinated experimentalists and theoreticians alike.Astrochemistry is one of the applied disciplines where structural isomers are of great importance because chemistry in the interstellar medium (ISM) is kinetically, rather than thermodynamically, controlled.2 Consequently, the abundances of isomers (e.g., HCN vs. HNC)3 in astronomical sources often provide a sensitive probe of the chemical evolution and physical conditions that are operative there. In many cases, isomeric abundance ratios deviate significantly from predictions based on thermodynamic considerations alone; in some astronomical sources, a higher-energy isomer may even be more abundant than the most stable conformer.4,5 A recent illustration of this point is provided by studies of three stable singlet H2C3O isomers in the Sagittarius B2(N), hereafter Sgr B2(N), star-forming region: Loomis et al.6 found no evidence for L-propadienone (H2CCCO) in this source, but rotational lines of the higher energy isomer, cyclopropenone (calculated to lie 6 kcal mol−1 above ground),7 are readily observed. Their results show that L-propadienone is at least an order of magnitude less abundant than cyclopropenone. Propynal [HC(O)CCH], a low-lying isomer of comparable stability to L-propadienone,7–11 is more than an order of magnitude more abundant than cyclopropenone. The authors suggest that these large variations in abundance could arise from different formation pathways on the surface of interstellar dust grains, for which some supporting laboratory evidence has been found from surface reaction experiments.7
Because nearly all well-known interstellar species have high-energy isomers with significant barriers to either isomerization or dissociation, there is much applied interest in identifying and precisely characterizing the rotational spectra of these metastable forms. Isocyanic acid (HNCO) was one of the first polyatomic molecules detected in space,12,13 but owing to the lack of laboratory rest frequencies, it was not until fairly recently that its higher energy isomers cyanic acid, HOCN (24.7 kcal mol−1 above ground),14 and fulminic acid, HCNO (70.7 kcal mol−1), were observed in interstellar molecular clouds.15,16 Soon after its initial astronomical identification,15 rotational lines of HOCN were reported in at least five other sources,17,18 findings which suggest that this isomer and HCNO are common constituents of the ISM. Abundance ratios and formation pathways of the HNCO isomers as a function of visual extinction, density, and temperature have now been the subject of several chemical modeling studies.18–20 Among the four singlet isomers, only isofulminic acid (HONC) has yet to be detected in space.
Apart from their astronomical interest, systematic studies of the structure, properties, and formation pathways of isomers are of fundamental importance. Such studies provide insight into a wide variety of bonding preferences (e.g., bond order), enable comparative studies of isovalent systems, and provide stringent tests for quantum chemical calculations. For example, rotational spectroscopy measurements of the elusive HONC (84 kcal mol−1) – the highest energy singlet isomer of HNCO21 – are consistent with a structure containing a polar C–N triple bond22 and a significant HOC bending angle (105°), in good agreement with a high-level coupled cluster calculation performed in conjunction with the experimental work. Furthermore, simultaneous detection of several isomers under similar experimental conditions may yield insight into isomerization pathways, and provide estimates of relative and absolute abundances, so follow-up experiments can be undertaken at other wavelengths.
Like their isovalent oxygen counterparts, thiocyanates (R-SCN) and isothiocyanates (R-NCS) have a rich history which is closely linked to isomerization. In one of the first investigations of these compounds, Hofmann established in 1868 that methyl thiocyanate rearranges to form methyl isothiocyanate at high temperature.23 Several years later, Billeter24 and Gerlich25 independently observed that allyl thiocyanate (H2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–CH2–NCS) thermally rearranges into allyl isothiocyanate, known as mustard oil,26 the compound largely responsible for the characteristic hot, pungent flavor of vegetables such as horseradish and mustard leaves. Today, the chemistry of thiocyanates and their derivatives is extensive and highly varied; compounds containing this chromophore are used in applications ranging from pharmaceuticals, dyes, and synthetic fiber to fungicides and photography, among others.
CH–CH2–NCS) thermally rearranges into allyl isothiocyanate, known as mustard oil,26 the compound largely responsible for the characteristic hot, pungent flavor of vegetables such as horseradish and mustard leaves. Today, the chemistry of thiocyanates and their derivatives is extensive and highly varied; compounds containing this chromophore are used in applications ranging from pharmaceuticals, dyes, and synthetic fiber to fungicides and photography, among others.
Isothiocyanic acid (HNCS), the simplest isothiocyanate, is calculated to be the most stable molecule in the [H, N, C, S] family, followed by thiocyanic acid (HSCN), lying about 6 kcal mol−1 above HNCS.27,28 These are followed by thiofulminic acid (HCNS) and isothiofulminic acid (HSNC), which are comparably stable to each other at 35.1 kcal mol−1 and 36.6 kcal mol−1 above HNCS, respectively (this work). Each isomer is calculated to possess a singlet ground state with a nearly linear heavy atom backbone, a planar equilibrium structure, and a large permanent electric dipole moment. Fig. 1 summarizes a number of the salient properties of the four singlet HNCS isomers.
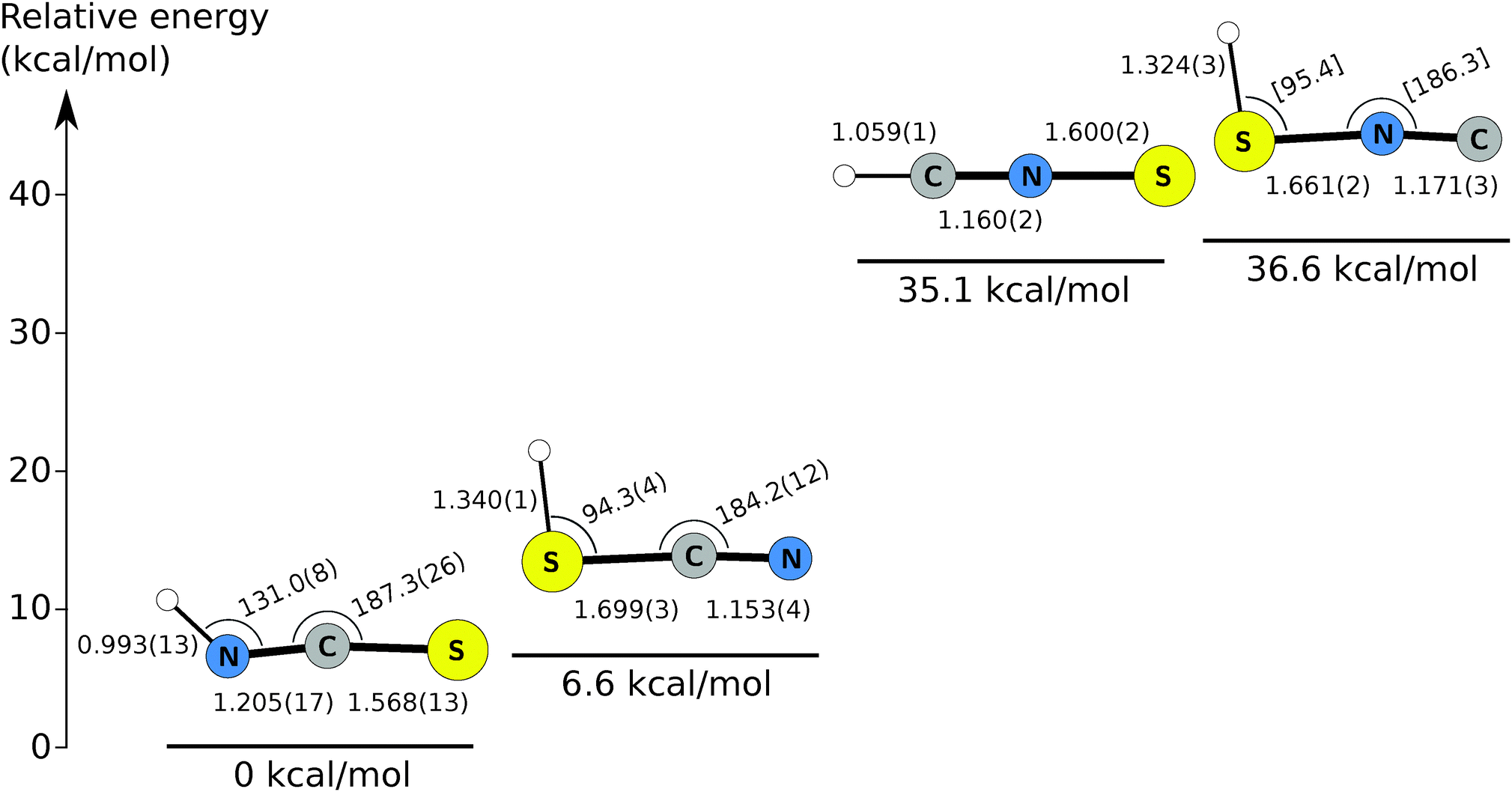 | ||
| Fig. 1 Relative energies and structures of the [H, N, C, S] isomer family. Energies are calculated at the ae-CCSD(T)/cc-pwCVQZ level and corrected for zero-point vibrational contributions calculated at the fc-CCSD(T)/cc-pV(Q+d)Z level. Semi-experimental equilibrium (rSEe) structures, obtained in this work, are indicated, along with associated uncertainties (1σ) derived from a least-squares optimization. Bond lengths are in Å, bond angles are in degrees. Square brackets indicate the structural parameter was fixed to the calculated value. | ||
With the exception of HNCS, our knowledge of the [H, N, C, S] isomeric system is limited. HNCS was first detected nearly 50 years ago by Beard and Dailey29 who determined its molecular structure and dipole moment from an analysis of the rotational spectrum and those of its more abundant isotopologues. Since then, HNCS and its isotopic species have been the subject of many high-resolution studies, from the microwave to the far infrared region;30,31 much of this work was aimed at understanding how large-amplitude bending vibrations affect structural rigidity. HNCS has also long been known as a constituent of the ISM; it was detected nearly 40 years ago via several a-type, Ka = 0 rotational transitions toward the high-mass star-forming region Sagittarius (Sgr) B2 region.32
Until quite recently, there was little spectroscopic information on HSCN, HCNS, or HSNC. HSCN and HSNC were first characterized experimentally at low spectral resolution by matrix-IR spectroscopy33 in which both isomers were formed by UV-photolysis of HNCS in solid argon and nitrogen matrices, but HCNS does not appear to have been studied at any wavelength. The microwave spectrum of HSCN was recently reported by several authors of this study,34 and soon after it was detected in the ISM.35,36 Owing to the absence of rotational spectra for HCNS and HSNC, astronomical searches have not been possible.
Since sulfur is less electronegative and has a larger atomic radius compared to oxygen, the energetics, structure, and bonding of analogous [H, N, C, S] and [H, N, C, O] isomers are predicted to differ. While the [H, N, C, S] isomers have the same energy ordering compared to their oxygen counterparts, the spread in energy is more than two times smaller (37 vs. 84 kcal mol−1).14,28 Because the reservoir of sulfur in space is not well established in dense molecular clouds,37 the abundance of higher energy [H, N, C, S] isomers may significantly differ from that found in the HNCO isomers.
In this paper we report a comprehensive laboratory study of the microwave spectra of HCNS and HSNC, the two remaining singlet isomers of HNCS, along with detection of their singly-substituted isotopic species and a number of rare isotopologues of HSCN, using both chirped-pulse Fourier-transform microwave (CP-FTMW) spectroscopy and cavity-FTMW spectroscopy. Because all four isomers can be simultaneously observed under the same experimental conditions, it has been possible to derive abundances relative to ground state HNCS, and consequently infer the dominant chemical reaction that yields HSCN. The relatively low abundance found for HSNC, with respect to isoenergetic HCNS, may indicate a low barrier to isomerization. By correcting the experimental rotational constants of each isotopic species for the effects of zero-point vibration calculated theoretically, precise semi-experimental equilibrium structures (rSEe) have been derived for each isomer. Finally, the results of an astronomical search using observations toward Sgr B2(N) from the Green Bank Telescope (GBT) Prebiotic Interstellar Molecular Survey (PRIMOS) project are reported. Although the recent millimeter study by Halfen et al.35 toward this source found that HNCS and HSCN are present in nearly equal column density, in the PRIMOS survey, firm evidence is found for HSCN alone, with only a tentative detection of HNCS. Lines of HSCN are observed in both absorption and emission, an indication that the excitation of this isomer is not well described by a single excitation temperature.
2 Quantum chemical calculations
Quantum chemical calculations guided the initial spectroscopic searches for HCNS and HSNC. These were performed using the CFOUR suite of programs,38,39 and follow the general strategies outlined in ref. 40. Briefly, calculations were performed at the coupled-cluster with singles, doubles, and perturbative triple excitations [CCSD(T)] level of theory,41 and Dunning's hierarchies of correlation-consistent polarized valence and polarized core valence basis sets. In the frozen core (fc) approach, the tight-d-augmented basis sets cc-pV(X+d)Z (X = T and Q) were used for the sulfur atom, and the corresponding cc-pVXZ basis sets for nitrogen, carbon, and hydrogen.42,43 The cc-pwCVXZ (X = T and Q) basis sets were used when considering all electrons (ae) in the correlation treatment.44Equilibrium geometries were calculated using analytic gradient techniques45 and basis sets as large as cc-pwCVQZ,46 a level that has been shown to yield highly accurate molecular equilibrium structures even for molecules containing second-row elements.47–50 Dipole moment components and nuclear quadrupole coupling constants were derived at the same level. Vibrational effects were calculated at the fc-CCSD(T)/cc-pV(Q+d)Z level using second-order vibrational perturbation theory (VPT2) based on the formulas given in ref. 51. Harmonic force-fields were computed analytically,52 while cubic and semi-diagonal quartic force fields were obtained via numerical differentiation of the analytically evaluated harmonic force fields.53 Overall, these calculations provided harmonic vibrational frequencies, centrifugal–distortion and vibration–rotation interaction constants (αi), zero-point vibrational corrections to rotational constants 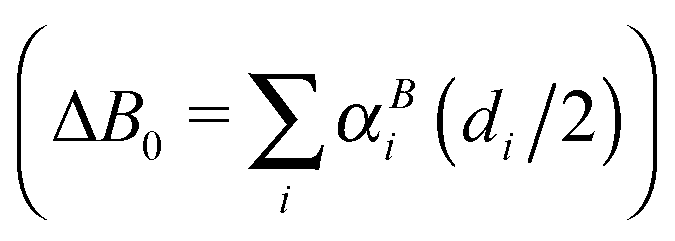 as well as fundamental vibrational frequencies (νi). Best estimates for the ground state rotational constants A0, B0, and C0 were then obtained using the relation B0 = Be − ΔB0 (with similar equations for A0 and C0) where the equilibrium rotational constants Ae, Be, and Ce are calculated from the ae-CCSD(T)/cc-pwCVQZ structure.
as well as fundamental vibrational frequencies (νi). Best estimates for the ground state rotational constants A0, B0, and C0 were then obtained using the relation B0 = Be − ΔB0 (with similar equations for A0 and C0) where the equilibrium rotational constants Ae, Be, and Ce are calculated from the ae-CCSD(T)/cc-pwCVQZ structure.
3 Experimental methods
Although metastable HCNS and HSNC are calculated to lie ∼35 kcal mol−1 above the HNCS global minimum (Fig. 1), prior work has shown that high-lying metastable isomers can be readily produced with an electrical discharge source and then probed in a rotationally-cold supersonic expansion.54 This unusual scenario arises because the reaction chemistry is closely coupled to the free energy of the electrons, which have an average kinetic energy of several eV,55 rather than to the extremely low (∼3 K) rotational excitation temperature of the expansion. The isovalent [H, C, N, O] isomers to those sought here, HCNO and HONC, lie substantially higher in energy relative to the ground state HNCO (70.7 and 84.1 kcal mol−1, respectively56), yet they were readily observed using essentially the same production and detection method as the one described here.14,56–583.1 Chirped-pulse FT microwave spectroscopy
Two configurations of the CP-FTMW spectrometer, one operating between 7.5 and 18.5 GHz (K-band) and the other between 25 and 40 GHz (Ka-band), were employed in this study. Their design and operation have been previously reported.59,60 Briefly, short (∼1 μs) frequency chirps are amplified by either a 300 W (7.5–18.5 GHz) or a 50 W (25–40 GHz) traveling wave tube amplifier and broadcast into a vacuum chamber by a standard gain horn antenna. Two or three pulsed nozzles, for the higher- and lower-frequency spectrometers, respectively, each equipped with an electrical discharge source,61 were positioned perpendicular to the axis of the microwave propagation in such a way that the resulting supersonic expansion of each intersects with the microwave radiation about 10 cm from the valve orifice. Because the transmission efficiency between the two horn antennas is poorer at Ka band compared to K band, the horns must be placed closer to each other in the high-band spectrometer, an arrangement which precludes using a third nozzle.Ten frequency chirps, separated by 30 s, are applied during each valve pulse. The resulting molecular free induction decay (FID) associated with each chirp is collected for 20 s by a second matched horn antenna, and the signal is amplified using a standard low-noise amplifier. The FID is then digitized at 100 GSa s−1 on a high speed (20 GHz) digital oscilloscope; the Fourier-transform of the FID yields the frequency-domain spectrum.
The discharge conditions were optimized to produce HSCN in high yield: a mixture of hydrogen sulfide (H2S, 0.15%) and methyl cyanide (CH3CN, 0.15%) heavily diluted in neon, a 1500 V dc discharge, and a stagnation pressure of 2.5 kTorr (3.2 atm) behind the 1 mm diameter supersonic nozzle were found to be optimal. Using these conditions, chirped spectra were collected over a 12 hour period in each frequency band. At the valve repetition rate of 5 Hz, nearly 2 million FIDs were recorded. The resulting spectrum has a high signal-to-noise ratio (SNR), and consequently the 34S and 13C isotopologues of HSCN were observed in natural abundance (see Fig. 2) in addition to the parent isotopologues of HNCS, HSCN, and HCNS. At this level of integration, about 500 molecular lines were detected in the two bands. In the high-band spectrum, for example, 24 molecules were readily identified: 18 are known interstellar molecules, a number which represents roughly 10% of known astronomical species (Table S1, ESI†). In addition to the work on the [H, N, C, S] family presented here, an analysis of this spectrum led to the first interstellar detection of E- and Z-ethanimine (CH3CHNH).62
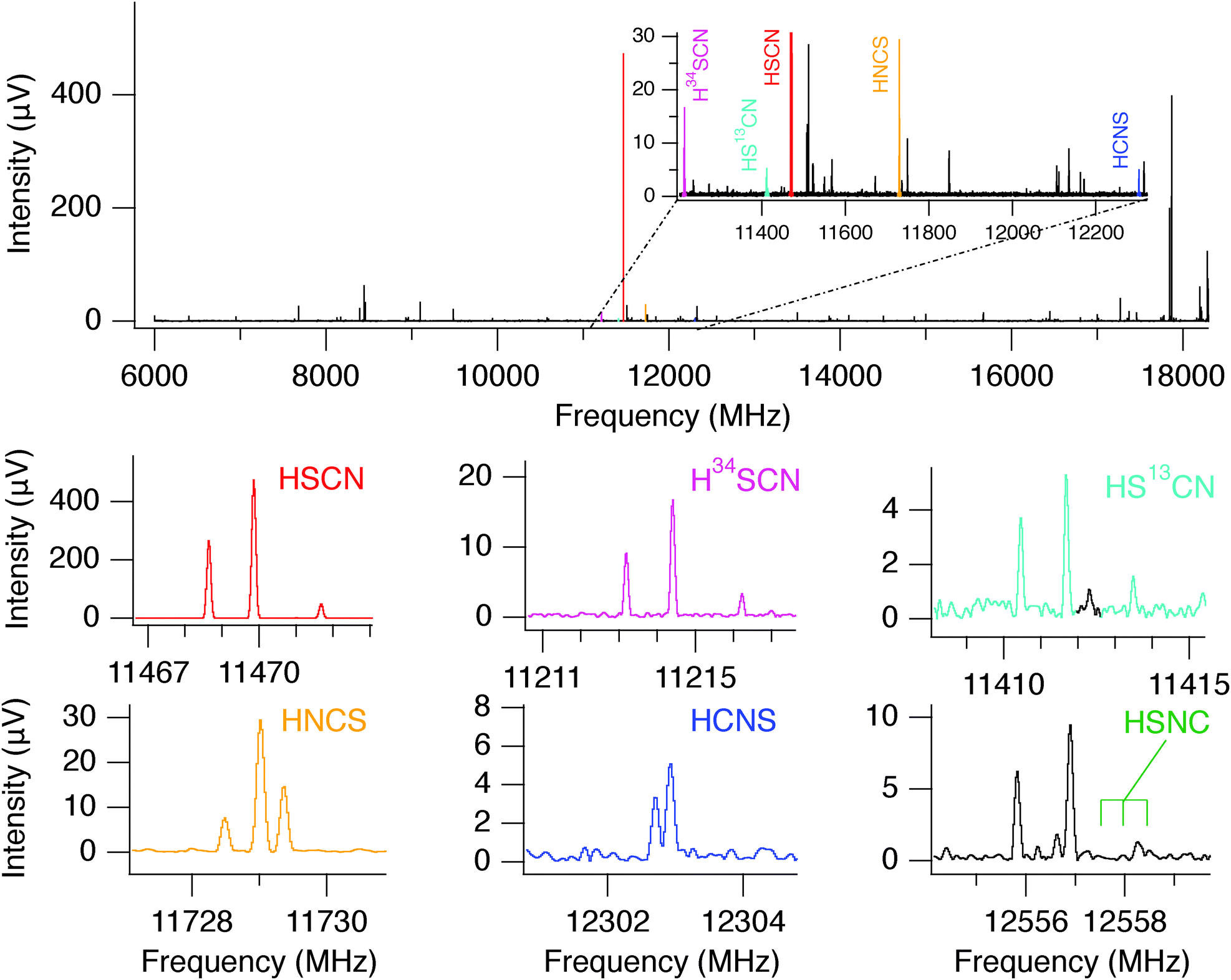 | ||
| Fig. 2 Chirped pulse spectrum between 7.5 and 18.5 GHz through a dilute discharge of H2S and CH3CN. The full bandwidth is shown, along with zoomed insets. The strongest feature in the spectrum arises from HSCN, and, owing to the high SNR, the 34S and 13C isotopologues are observed in natural abundance. The fundamental rotational transitions of each species are shown in colors. The hyperfine-split line of HSNC, not detected in this spectrum, is also indicated. The strong transitions above ∼17.9 GHz are from CH3CN and its isotopologues. | ||
Because HSCN and its isomers are relatively light molecules, a single rotational transition falls in each of the two CP-FTMW bands, although a third transition of HSNC falls near the lower edge of the Ka band. The CCSD(T) calculations performed here, however, are generally accurate to a few 0.1%, and zero-point energy corrections to the Be constants are generally small (∼0.4%) as well, meaning that the frequency uncertainty for the fundamental rotational transition of each isomer (10,1 → 00,0, predicted near 12 GHz) is expected to be less than ±75 MHz. Combined with the low line density and characteristic hyperfine splitting from the 14N nucleus, there were very few candidates lines for HCNS in the CP-FTMW spectrum. The lack of any viable candidate lines for HSNC, however, necessitated a follow-up study using more sensitive cavity FTMW spectroscopy.
3.2 Cavity FT microwave spectroscopy
Following the CP-FTMW measurements, a number of follow-up studies were performed using a cavity FTMW spectrometer operating between 5 and 43 GHz. These studies were needed to: (1) confirm the initial identifications with better SNR and at higher spectral resolution; (2) extend the rotational spectroscopy to higher-J transitions for the purpose of determining the leading centrifugal distortion terms; (3) detect rare isotopic species of isomers, both to provide overwhelming evidence for the identification and to allow precise determinations of molecular structures; and (4) detect HSNC, whose lines were not readily apparent in the CP-FTMW spectra.Details of the cavity FTMW spectrometer, which is based on the original design of Balle & Flygare,63 and its operation have been published previously.61,64,65 The nozzle source is identical to that used in the CP-FTMW experiment, except this source is mounted directly behind one of the cavity mirrors, and gas expands into the open resonator via a small hole in that mirror. As the gas passes through the beam waist of the cavity, molecules are excited by a short (1 μs) pulse of microwave radiation. The resulting FID is recorded using a sensitive microwave receiver; the Fourier-transform of the FID yields the frequency-domain spectrum. Owing to the high Q of the cavity, the instantaneous bandwidth is only about 1 MHz, but the sensitivity per unit MHz and time is substantially higher compared to the CP-FTMW spectrometer, by roughly a factor of 40.59 The spectral resolution, determined by the divergence of the molecular beam,66 is also quite high, approaching 0.1 ppm, or about a factor of 50 greater than that which can routinely be achieved in CP-FTMW spectra at the same frequency.
Owing to slight differences in nozzle geometry and expansion conditions with the chirped-pulse experiment, the optimum production conditions for HNCS and HSCN were similar but not identical between the two experiments. Although both molecules were observed using a mixture of H2S and CH3CN, a dilute mixture of H2S (0.035%) and cyanogen (NCCN, 0.03%) in neon and a discharge voltage of 800 V yielded somewhat stronger lines at the same backing pressure (2.5 kTorr) behind the nozzle.
To confirm the initial CP-FTMW assignments, searches were undertaken at higher frequencies to establish if additional lines were present at frequencies corresponding to ratios of integers, i.e. near 24 and 36 GHz for HSCN, as all four [H, N, C, S] isomers are either linear molecules or asymmetric tops very close to the prolate limit (κ ∼ −1). Two series of lines, both observed with good SNR, were quickly identified in this manner. Transitions of HSNC, undetected in the chirped-pulse experiment, were identified near 13, 25, and 38 GHz. Many additional cavity measurements were performed to detect isotopic species of HCNS, HSNC, and HSCN, either in natural abundance (34S) or using a variety of isotopically-enriched precursors (D2S, CH313CN, CH3C15N), in which searches were guided by the quantum chemical calculations described in Section 2.
4 Experimental results and spectroscopic analysis
Spectroscopic constants of all species were determined by least-squares optimization using the CALPGM (SPFIT/SPCAT) suite of programs67 and either a standard asymmetric top (S-reduced) or linear molecule Hamiltonian, both of which include centrifugal distortion and hyperfine interactions, either nuclear quadrupole coupling from the I = 1 spin of nitrogen, deuterium, or both, or spin-rotation from the I = 1/2 13C nucleus. Frequencies derived from the higher resolution cavity FTMW data were used in each analysis.4.1 Isotopic HSCN (thiocyanic acid)
HSCN is an asymmetric rotor very close to the prolate limit (κ = −0.999),68 with an a-inertial axis almost coincident with the nearly-linear SCN heavy atom backbone, a large energy separation (E/k ∼ 15 K) between successive Ka levels, and a strong a-type spectrum. Because it is produced in high abundance in the discharge nozzle, it was possible to detect transitions in the Ka = 1 ladders for several isotopic species, despite the very low rotational excitation temperature. The Ka = 1 lines, however, are a factor of 1000 weaker in intensity than those from the same J in the Ka = 0 ladder.The rotational spectra of five isotopic species, H34SCN, HS13CN, HSC15N, D34SCN, and DSC15N, were observed in the centimeter-wave band (Tables S2 and S3, ESI†) and analyzed in this study. Because the present data set is limited to a-type transitions with Ka ≤ 1 below 40 GHz, it was not possible to determine the A rotational constant to better than ∼3% for the deuterated isotopic species, and even less so for H34SCN, HS13CN, and HSC15N. Consequently, the A rotational constant of these species was fixed to the value derived for the main isotopologue in the fits. Table 1 provides a summary of the derived constants for isotopic HSCN.
| Constanta | HSCNb | H34SCN | HS13CN | HSC15N | DSCNb | D34SCN | DSC15N |
|---|---|---|---|---|---|---|---|
| a Values in parentheses are 1σ uncertainties in units of the last significant digit. b Ref. 34. Additional higher-order constants, provided in ref. 34, are not reproduced here. c Fixed to value derived for normal HSCN. d Owing to the limited data set, B–C value constrained to value derived for normal HSCN. | |||||||
| A 0 | 289![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 737(64) 737(64) |
[289737]c |
[289737]c |
[289737]c |
151350(478) |
148302(568) |
150423(1051) |
| B 0 | 5794.71368(20) | 5664.4755(4) | 5765.0305(5) | 5585.5858(2) | 5705.09910(29) | 5583.8130(3) | 5495.9129(5) |
| C 0 | 5674.93940(20) | 5549.7261(3) | 5646.4473(3) | [5470.1341]d | 5489.24957(27) | 5376.0826(3) | 5295.2994(5) |
| 103DJ | 1.66557(21) | 1.54(2) | 1.62(2) | [1.66557]c | 1.560(20) | 1.36(3) | 1.37(4) |
| D JK | 0.15153(11) | 0.1453(2) | 0.1484(2) | [0.1515]c | 0.14152(34) | 0.1372(4) | 0.1331(6) |
| 106d1 | −35.91(21) | — | — | — | — | — | — |
| χ aa(N) | −4.0479(15) | −4.049(2) | −4.049(2) | — | −4.0530(11) | −4.051(1) | — |
| χ bb(N) | 2.8271(18) | 2.822(3) | 2.825(3) | — | 2.8292(18) | 2.829(4) | — |
| χ aa(D) | — | — | — | — | −0.0821(32) | −0.068(3) | −0.058(2) |
| χ bb(D) | — | — | — | — | 0.1445(26) | 0.134(3) | 0.136(3) |
| rms (kHz) | — | 2.2 | 2.8 | 4.8 | — | 1.9 | 1.6 |
| N lines | — | 27 | 24 | 3 | — | 66 | 25 |
4.2 HCNS (thiofulminic acid)
The three lowest rotational transitions of HCNS and its four singly-substituted species H13CNS, HC15NS, HCN34S, and DCNS have been measured between 12 and 36 GHz (see Tables S4–S6, ESI†). Because this isomer is predicted to be strictly linear, for the normal isotopic species, HC15NS, and HCN34S, a fit rms comparable to the experimental uncertainty (∼2 kHz) was achieved by optimizing only three parameters: the rotational constant B0, the centrifugal distortion constant D0, and the nitrogen quadrupole constant eQq(N) (except in the case of the 15N species). Owing to additional hyperfine structure from the deuteron and 13C for DCNS and H13CNS, one additional hyperfine term, eQq(D) and the spin-rotation constant C(13C), respectively, was required to achieve a similarly low rms. The best-fit constants are summarized in Table 2, along with results from the quantum-chemical calculations from Section 2. When comparison is possible, the agreement between the two sets of constants is excellent.| Constanta | HCNS | H13CNS | HC15NS | HCN34S | DCNS | |
|---|---|---|---|---|---|---|
| Measured | Calculatedb | |||||
| a Values in parentheses are 1σ uncertainties in units of the last significant digit. b See text for details. c Fixed to value derived for normal HCNS. | ||||||
| B 0 | 6151.4436(4) | 6153.961 | 5931.3801(4) | 6125.714(1) | 6008.8818(6) | 5617.8801(3) |
| 103D0 | 1.71(2) | 1.664 | 1.54(3) | [1.71]c | [1.71]c | 1.38(2) |
| eQq(N) | −0.723(2) | −0.711 | −0.728(2) | — | [−0.723]c | −0.725(3) |
| 103C(13C) | — | — | 11(1) | — | — | — |
| eQq(D) | — | — | — | — | — | 0.1958(4) |
| rms (kHz) | 1.7 | — | 2.4 | — | 1.1 | 2.5 |
| N lines | 13 | — | 19 | 3 | 3 | 29 |
4.3 HSNC (isothiofulminic acid)
Similarly to HSCN, isothiofulminic acid is also an asymmetric rotor very close to the prolate limit (κ = −0.999) with the least principal inertial axis almost coincident with the nearly-linear SNC heavy atom backbone (Fig. 1). Consequently, the molecule has a large A rotational constant (A = 290 GHz) and significant energy separation between successive Ka energy levels. At the very low rotational excitation temperatures that characterizes our supersonic jet, however, only rotational levels in the Ka = 0 ladder are significantly populated. As a consequence, only the effective constant Beff = (B + C)/2 was determinable. As with HCNS, the rotational spectra of the four singly-substituted isotopic species, H34SNC, HS15NC, HSN13C, and DSNC, have also been measured below 40 GHz (Tables S7 and S8, ESI†). Best-fit constants are given in Table 3.| Constanta | HSNC | H34SNC | HS15NC | HSN13C | DSNC | |
|---|---|---|---|---|---|---|
| Measured | Calculatedb | |||||
| a Values in parentheses are 1σ uncertainties in units of the last significant digit. b See text for details. c Fixed to value derived for normal HSNC. | ||||||
| B eff | 6279.0335(4) | 6274.1645 | 6141.6485(6) | 6242.520(1) | 6022.3790(3) | 6118.1242(4) |
| 103Deff | 4.51(2) | [4.51]c | 4.4(2) | 3.95(2) | 16.14(3) | |
| χ aa(N) | 1.213(2) | 1.204 | [1.21]c | — | 1.214(2) | [1.21]c |
| χ aa(D) | — | — | — | — | — | −0.05(1) |
| rms (kHz) | 1.6 | — | 1.6 | — | 2.5 | 2.2 |
| N lines | 13 | — | 3 | 8 | 19 | 19 |
4.4 Structural determinations
Purely experimental (r0) structures for HCNS, HSNC, and HSCN were derived by least-squares optimization of the structural parameters of each isomer to reproduce the measured moments of inertia of its normal and rare isotopic species using the STRFIT program.69 Because the moment of inertia is inversely proportional to the rotational constant along each axis, the three moments of inertia of each species are trivially calculated from constants in Tables 1–3. For HNCS, no attempt was made to derive an r0 structure because a substitution (rs) structure has been previously reported.31 For HCNS, only the three bond lengths were varied because of its linear geometry, but for the asymmetric top HSCN, the two bond angles were optimized as well. Owing to the limited data set for HSNC, the three bond lengths were varied, with the remaining two angles constrained to the calculated value. For isotopic species, poorly constrained spectroscopic constants in the fit were omitted in the structural determination. Tables S10–S13 (ESI†) summarize the geometries derived by this method.Semi-experimental equilibrium (rSEe) structures were determined by correcting the experimental rotational constants (A0, B0, C0) to account for zero-point vibrational effects as obtained by VPT2 (see Section 2). The semi-experimental equilibrium rotational constants (ASEe, BSEe, and CSEe) are used instead of the experimental constants in the structural optimization. The remainder of the procedure is otherwise identical to that employed for the r0 structures. For completeness, a rSEe structure has also been derived for ground state HNCS, in which the published constants are corrected for zero-point vibration.29,70 Vibrational corrections for each isotopic species of each HNCS isomer are summarized in Tables S14–S17 (ESI†), along with the corresponding BSEe constants. The best-fit rSEe structures are given in Tables S10–S13 (ESI†), and the values are reported in Fig. 1.
For most isomers, the most obvious difference between the r0 and rSEe structures is the higher accuracy of the derived parameters in the latter structure. For HSCN, the fractional precision of most parameters is improved by a factor of three or more, with bond lengths derived to a few mÅ, while the bond angles are accurate to 1° or better. The inertial defects derived for each isotopologue decrease by roughly a factor of 10 or more (e.g. from 0.0958 to 0.0045 amu Å2 for HSCN). Despite being the lowest energy and best studied of the isomers, the structural parameters for HNCS are the least well-determined of the four due to significant influence of large-amplitude motions on the structure.71
Compared to the analogous [H, N, C, O] isotopologues, the [H, N, C, S] species possess longer bond lengths, by ∼0.4–0.5 Å, for bonds involving sulfur rather than oxygen. CN, CH, and NH bond lengths are remarkably similar, usually within 0.01 Å. The bond angles for which sulfur is the central atom are smaller than their O-atom counterparts by 10–15°.71
4.5 Relative abundances and formation pathways
Table 4 summarizes the abundances of HSCN, HCNS, and HSNC relative to HNCS in the chirped-pulse spectrum, which have been derived from intensities of the fundamental a-type rotational transition of each species, accounting for differences in the hyperfine structure and dipole moment. This calculation also assumes that line intensity is proportional to the square of the a-type dipole moment, that all four isomers have the same rotational excitation temperature in the expansion, and that the instrument response function is uniform over the range of measurement (11.4–12.5 GHz).| Isomer | E rel | Dipole momenta | N X/NHNCSb | |
|---|---|---|---|---|
| μ a | μ b | |||
| a Calculated at the ae-CCSD(T)/cc-pwCVQZ level of theory, see text. b Measurements made by comparing observed intensities of the fundamental rotational transition for each species, and correcting for differences in dipole moment. Errors are estimated to be 10–15%. | ||||
| HNCS | 0 | 1.64 | 1.16 | 1 |
| HSCN | 6.6 | 3.29 | 0.89 | 4 |
| HCNS | 35.1 | 3.85 | 0.07 | |
| HSNC | 36.6 | 3.13 | 0.88 | <0.004 |
Under our discharge conditions, HSCN is the most abundant isomer by a factor of four, even though this metastable isomer lies 6.6 kcal mol−1 above HNCS. Because Λ-doublet transitions of the SH radical are readily observed near 8.4 GHz,72 and the CN radical is known to be a common fragment of CH3CN dissociation,73 HSCN is likely produced in high abundance via the radical–radical reaction HS (2Π) + CN (2Σ+).28 Although CN has no transitions in the frequency range of the CP-FTMW spectrum, detections of rotational lines of several other cyanides (e.g., HC3N, CH2CHCN, CH3CH2CN) and isocyanides (e.g., CH3NC, HCCNC, CH2CHNC; see Table S1, ESI†) strongly suggest that this radical plays an important role in the discharge chemistry.
The counterpart of HSCN, isothiofulminic acid HSNC is roughly 103 times less abundant in the same discharge, and underabundant by a factor of 40 compared to HCNS, its isoenergetic isomer. Previous theoretical work has explored the unimolecular isomerization of HNCS isomers at the MP2/aug-cc-pVTZ level of theory for structures, and CCSD(T)/aug-cc-pVTZ level of theory for single-point energy calculations.28 Although formation of both HSCN and HSNC from HS + CN is exothermic and barrierless, HSCN resides in a deeper potential well with larger barriers to isomerization compared to that of HSNC. The HSNC to HSCN isomerization proceeds through a cyclic intermediate, S(H)CN, with an energy barrier corrected for zero-point energy of only 23 kcal mol−1, while the reverse reaction must overcome a barrier of nearly 50 kcal mol−1; a similarly high barrier is also required to convert HSCN to HNCS (see Fig. 5 in ref. 28). For these reasons, energized HSCN is apparently easier to stabilize compared to energized HSNC and, once formed, HSCN probably undergoes relatively little isomerization. Because the exothermicity of the HS + NC → HSNC reaction is roughly three times the well depth of the HSNC minimum, however, considerable rearrangement to the thermodynamically-favored HSCN may occur prior to stabilization of HSNC.
Guided by the theoretical work of Wierzejewska and Moc28, it appears likely that HNCS and HCNS are formed through reactions involving sequential reactions of CN with atomic S and H, either H (2S) + NCS (2Π), which can produce both HNCS and HSCN, or H (2S) + CNS (2Π), which only yields HCNS. Once formed, like HSCN, considerable barriers must be overcome to isomerize HNCS, the lowest of which is about 60 kcal mol−1 to form HSCN. The preferential production of HCNS relative to HSNC, despite their nearly identical energetics, is a likely indication of much greater stability of the former to isomerization. HCNS isomerization to either HSCN or HNCS is energetically unfavorable; both involve barriers of about 60 kcal mol−1.
5 Astronomical search
We have conducted a search for each of the four [H, N, C, S] isomers in the publicly-available PRIMOS survey§ toward Sgr B2(N)74 where HSCN and HNCS have been previously detected.32,35,75 Based on a purely thermodynamically-driven treatment, a detectable column density of HSNC or HCNS in this source is not expected, given their much lower calculated stabilities. However, prior observations of Sgr B2(N) have shown that chemistry in this star-forming region is kinetically-controlled, rather than thermodynamically-controlled,¶ and abundances of higher-energy isomers commonly defy thermodynamic predictions.6,74 Sgr B2(N) is a spatially- and radiatively-complex source, and careful treatment is required to obtain accurate column densities and excitation temperatures. Here, we follow the convention of Hollis et al.76 to derive these values; a detailed description of the techniques is provided in the ESI.†5.1 HNCS
HNCS has been previously detected in Sgr B2(N) with a column density NT = 1.3(5) × 1013 cm−2 and a rotational excitation temperature Tex = 19(2) K.35 Only the J = 20,2–10,1 transition of HNCS at 23.5 GHz is accessible with PRIMOS since the next two Ka = 0 lines, at 35187 and 46916 MHz, fall within small gaps in the survey coverage, while the 10,1–00,0 at 11729 MHz is contaminated with radio frequency interference (RFI). While a simulation based on the previously-observed column density and excitation temperature predicts a weak absorption at the frequency of the 2–1 line, no absorption feature is visible (Fig. 3). Instead, a weak emission feature could potentially be assigned to HNCS. While this assignment is only tentative, if correct it would indicate that HNCS is not described by a single excitation temperature, and in fact may be experiencing weak maser action in this low-J transition. Such behavior has been reported previously in PRIMOS data toward Sgr B2(N): both carbodiimide (HNCNH) and methyl formate (CH3OCHO) display weak masing.77,78
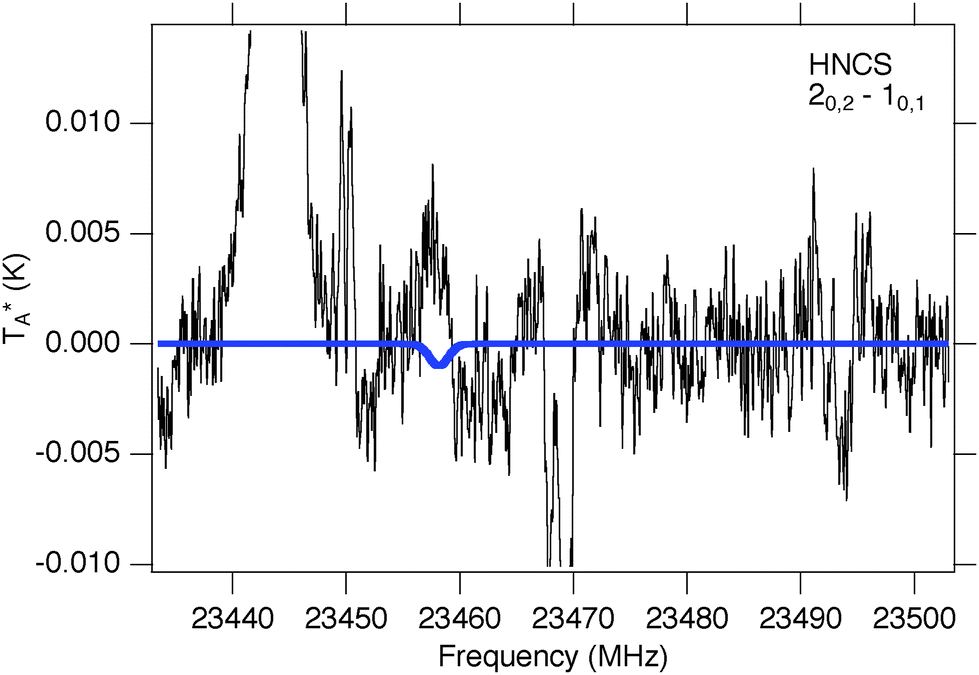 | ||
| Fig. 3 Simulation of HNCS at previously-observed35 column density and excitation temperature (blue trace, ΔV = 10 km s−1, VLSR = +64 km s−1) overlaid on PRIMOS observations at 23.5 GHz. | ||
5.2 HSCN
HSCN has been previously detected in Sgr B2(N) with a column density NT = 3.3(2) × 1013 cm−2 and a rotational excitation temperature Tex = 18(3) K.35 Unlike HNCS, four transitions of HSCN (40,4–30,3, 30,3–20,2, 20,2–10,1, and 10,1–00,0) are covered by the available PRIMOS observations. Using eqn (S1) (ESI†), and the previously-derived column density and temperature, these transitions are predicted to be detectable above the noise threshold of the PRIMOS observations. As indicated in Fig. 4, lines of HSCN are indeed detected at the expected frequencies, some with resolved hyperfine structure. Because the hyperfine structure is known exactly from experiment, and is partially resolved in the astronomical data, our analysis explicitly treats this structure. We find that a linewidth of 6.6 km s−1, a column density of NT = 1.0 × 1013 cm−2, and a rotational excitation temperature of Tex = 5.0 K best reproduce the PRIMOS observations (Fig. 4; red). This linewidth is significantly narrower than the previously-determined value of ∼25 km s−1. The discrepancy may be because Halfen et al.35 do not appear to explicitly treat the hyperfine components of the transitions, as the components do not appear to be resolved in their higher-frequency observations, which could yield a much broader derived linewidth.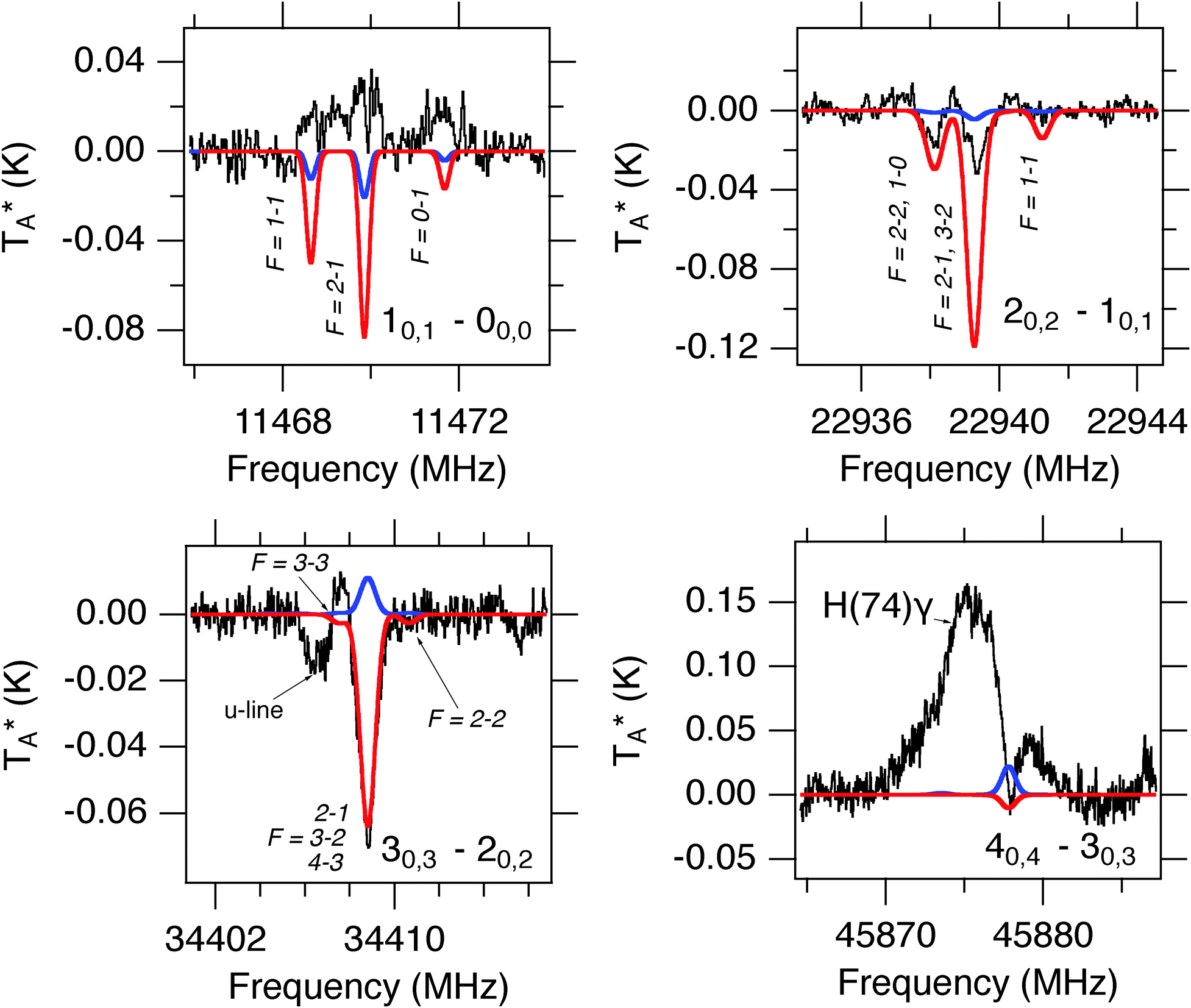 | ||
| Fig. 4 Observed transitions of HSCN in the PRIMOS spectra in black, with simulations of HSCN based on the derived column density and excitation temperature from Halfen et al.35 overlaid in blue, and the derived best-fit model from the PRIMOS observations (Tex = 5 K) overlaid in red. | ||
The 40,4–30,3 transition of HSCN is seen in absorption superimposed on an emission feature from H(74)γ, while the remaining three transitions are unblended. The 30,3–20,2 is well-fit by our derived parameters, however the intensity of the 20,2–10,1 is over-predicted, and the 10,1–00,0 is observed in emission, rather than absorption. Because the three higher-frequency transitions (at correspondingly lower continuum levels) are seen in absorption, the 10,1–00,0 line must be described by a separate excitation temperature greater than the continuum level at that frequency (Tc > 125 K). This behavior is strongly indicative of a weak maser, and indeed weak masing in the 20,2–10,1 transition would also account for the over prediction of the absorption depth in our single-excitation model. We note that in the 10,1–00,0 emission line, narrow self-absorption features appear to be present. If real, these features could be indicative of a spatially-distinct masing population embedded or behind colder, absorbing gas. Owing to the limited data set, however, a detailed analysis is not feasible.
5.3 HCNS and HSNC
No compelling evidence is found for either HCNS or HSNC in the PRIMOS data (Fig. 5), yielding the upper limits given in Table 5 for a set of common conditions in the Sgr B2(N) environment. In both cases, the J = 1–0 transitions of each fall within regions contaminated with RFI. The PRIMOS observations are largely sensitive to cold, extended material, and thus the upper limits are correspondingly larger for warm, compact components. If these isomers are present, they will likely only be detected if their ground-state transitions display masing enhancement like that of HSCN, or if high-sensitivity mm/sub-mm observations with instruments such as the atacama large millimeter/sub-millimeter array (ALMA) are undertaken.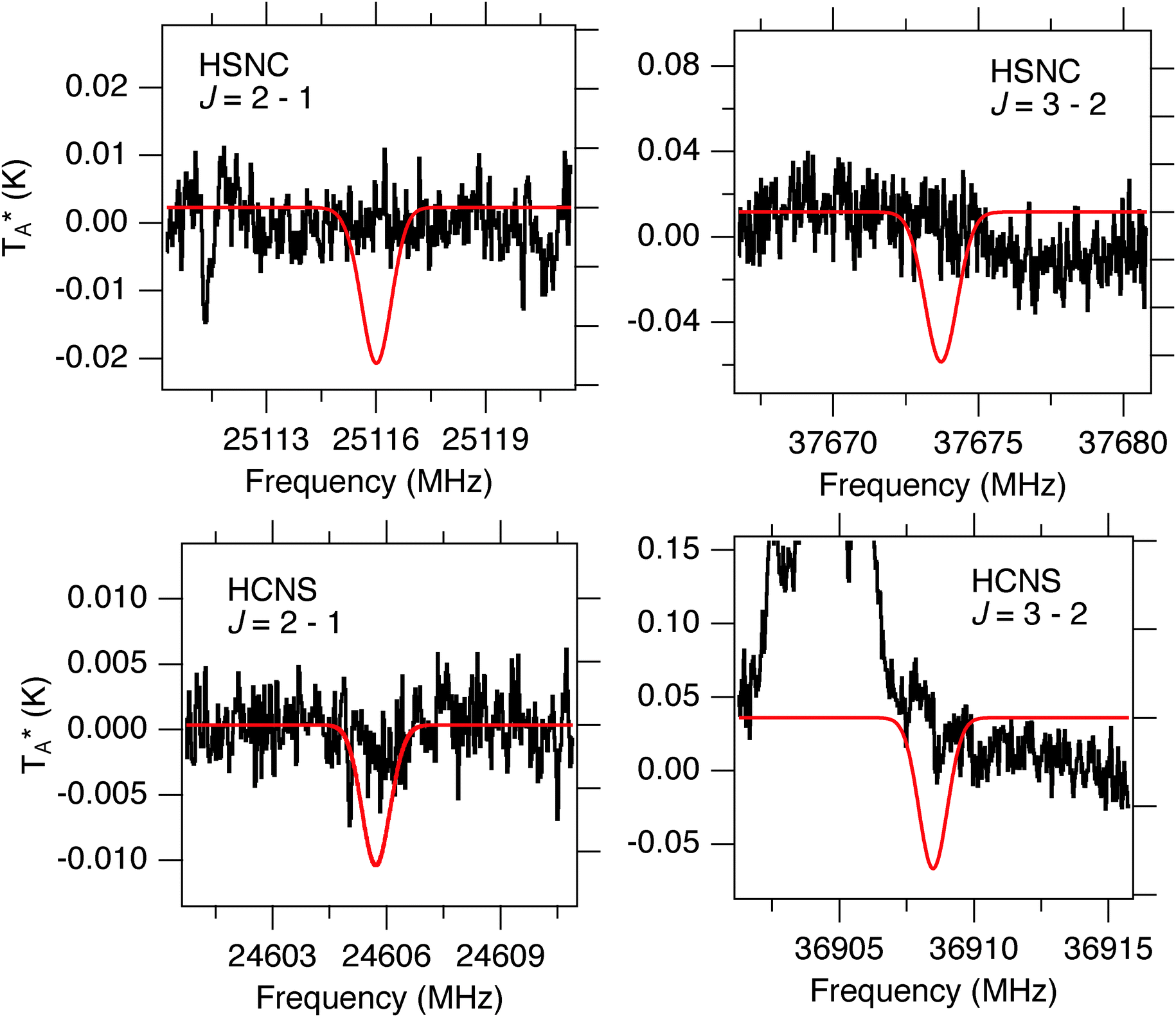 | ||
| Fig. 5 Non-detections of HSNC and HCNS in PRIMOS spectra (black). For illustration, red traces show simulations of the intensity profile of the transitions, in arbitrary units, for a linewidth of 10 km s−1. | ||
| T ex (K) | Source size (′′) | Upper limit column density (cm−2) | |
|---|---|---|---|
| HCNS | HSNC | ||
| Note: we estimate the uncertainties in the upper limits to be ∼30%, largely arising from absolute flux calibration and pointing uncertainties. | |||
| 8 | 20 | 1.79 × 1011 | 1.43 × 1012 |
| 18 | 20 | 1.12 × 1012 | 2.07 × 1013 |
| 80 | 5 | 1.74 × 1013 | 2.13 × 1014 |
| 140 | 5 | 2.67 × 1013 | 3.02 × 1014 |
5.4 Astrophysical implications
The derived excitation temperature for HSCN agrees extremely well with other cold, extended molecules observed in PRIMOS, which are well described by Tex = 5–10 K.62,74,79 We note that these excitation temperatures are dependent on the assumed continuum temperature, Tc, which in all these studies has been derived from the observations of Hollis et al.80 The continuum structure in Sgr B2 is extremely complex, and observations with different beam sizes|| (using other telescopes) and/or at higher-frequencies can be sensitive to different background continuum values.The previously-reported column density and excitation temperature for HSCN systematically underpredict the observed PRIMOS features. In the cases of the 40,4–30,3 and 30,3–20,2 transitions, they predict emission features, rather than the absorption features which are observed. The difference is likely exclusively due to large variations in the continuum levels between the PRIMOS and ARO observations. Nevertheless, a Tex = 19 K is too high to completely account for the absorption seen in PRIMOS given the Tc observed.80 Conversely, the Tex = 5 K derived here is likely too low to completely account for the previously-observed emission at higher frequencies; the lack of continuum information in the previous work prevents a rigorous comparison. A full, quantitative radiative-transfer model** would be required to properly reconcile the emission observed at mm-wavelengths with the absorption and potential masing seen in PRIMOS, but the necessary collisional cross-section information is not available. Regardless, the PRIMOS observations are consistent with a cold, spatially-extended population of molecules distinct from species observed toward the hot core.
Such spatially-extended, low-excitation material in Sgr B2(N) is thought to be the result of bulk liberation of molecular ices from the surfaces of dust grains by the passage of low-velocity shocks.76,81,82 Indeed, several of the complex species detected toward this source are found exclusively to be cold and extended, and have not yet been detected in the compact hot core (e.g., trans-methyl formate, propanal, propenal, propynal, and ethanimine).62,74,79 Previous work on HNCS and HSCN ratios in the ISM has suggested several gas-phase formation routes, proceeding through the cation precursor HNCSH+.36 Within pre-shock molecular ices, however, radical–radical reactions of simple species may be an important pathway for the evolution of more complex species.83,84 HSCN and HSNC could also conceivably be formed in the condensed phase by the reaction of the SH radical with the CN radical, both known constituents of the ISM, before being liberated into the gas phase.85–88 Owing to its greater stability and larger barriers to isomerization, however, HSCN may be preferentially formed in space. Radical–radical recombination reactions on grain surfaces are relatively energetic by interstellar standards, with the excess energy often stabilized by the grain surface acting as a third body. Some of this initial energy, however, could easily drive population from HSNC to HSCN, where it is then trapped in the deeper potential well.
6 Conclusions
In the present investigation, the pure rotational spectra of HSCN, HCNS, and HSNC were recorded by a combination of chirped-pulse and cavity FTMW spectroscopy. Rotational constants were obtained from fits to these spectra, and experimental r0 structures were derived. Using high-level quantum-chemical calculations, these structures were corrected for zero-point vibrational energy effects, and semi-experimental equilibrium rSEe structures were determined for these three species, as well as for HNCS.Now that four isomers of the [H, N, C, S] system up to 37 kcal mol−1 above ground have been characterized experimentally, even more energetic isomers may be within reach. On the singlet potential energy surface, Wierzejewska and Moc28 calculate three ring molecules, c-C(H)NS, c-S(H)CN, and c-N(H)CS (i.e., where the hydrogen atom is bound to either of the heavy atoms forming a three-membered ring) to energetically follow the four chains characterized here at roughly 45, 54 and 72 kcal mol−1 above ground, respectively. Given that isomers of [H, N, C, O] as high as 84 kcal mol−1 above ground have been observed already,21 any of the three [H, N, C, S] ring isomers seem plausible candidates for future laboratory microwave searches. Even triplet species might be amenable to detection: the lowest triplet species, branched C(H)NS, is predicted at 63 kcal mol−1 followed by the bent chains HNCS and HCNS at 67 and 80 kcal mol−1, respectively.28
In the course of the present study, we have also searched the publicly-available PRIMOS centimeter-wave survey of Sgr B2(N), and find no evidence for a cold population of HCNS or HSNC, and only a tentative detection of weak emission from HNCS. Lines of HSCN are clearly observed, and evidence is found for weak maser activity in its 10,1–00,0 transition near 11469 MHz. Future astronomical searches for HCNS and HSNC in molecule rich sources, however, are clearly warranted in the millimeter-wave regime. While the data obtained here are not sufficient to predict the millimeter wave spectra of these two species accurately enough for astronomical searches, they are indispensable in guiding laboratory searches at still higher frequencies.
To further explore structural isomerism in analogous systems to the [H, N, C, O] family, comprehensive studies of molecules in which carbon and/or nitrogen are replaced with their heavier counterparts such as those of the [H, Si, N, O] and [H, C, P, O] families may be worthwhile.89,90 As a first step in this direction, the corresponding lowest-energy silicon and phosphorus analogs HNSiO and HPCO were recently detected by their pure rotational spectra.91 Owing to the relatively small energy separation between isomers, and that there are very few experimental studies of these heavier analogs, members of these (seemingly simple) four-atom molecular systems should provide a fertile testbed for further experimental study of molecular isomerism.
Acknowledgements
This work was supported by NSF CHE 1213200 and NASA grant NNX13AE59G. B. A. M. is grateful to G. A. Blake for access to computing resources. S. T. acknowledges support from the Deutsche Forschungsgemeinschaft (DFG) through grants TH 1301/3-2 and SFB 956 and the Regional Computing Center of the Universität zu Köln (RRZK) for providing computing time on the DFG-funded High Performance Computing (HPC) system CHEOPS. The National Radio Astronomy Observatory is a facility of the National Science Foundation operated under cooperative agreement by Associated Universities, Inc.References
- S. Esteban, J. Chem. Educ., 2008, 85, 1201–1203 CrossRef CAS.
- T. Hirota, S. Yamamoto, H. Mikami and M. Ohishi, Astrophys. J., 1998, 503, 717–728 CrossRef CAS.
- J. C. Loison, V. Wakelam and K. M. Hickson, Mon. Not. R. Astron. Soc., 2014, 443, 398–410 CrossRef CAS.
- F. J. Lovas, D. F. Plusquellic, S. L. W. Weaver, B. A. McGuire and G. A. Blake, J. Mol. Spectrosc., 2010, 264, 10–18 CrossRef CAS.
- D. P. Zaleski, N. A. Seifert, A. L. Steber, M. T. Muckle, R. A. Loomis, J. F. Corby, O. Martinez Jr., K. N. Crabtree, P. R. Jewell, J. M. Hollis, F. J. Lovas, D. Vasquez, J. Nyiramahirwe, N. Sciortino, K. Johnson, M. C. McCarthy, A. J. Remijan and B. H. Pate, Astrophys. J., 2013, 765, L10 CrossRef.
- R. A. Loomis, B. A. McGuire, C. Shingledecker, C. H. Johnson, S. Blair, A. Robertson and A. J. Remijan, Astrophys. J., 2015, 799, 34–38 CrossRef.
- L. Zhou, R. I. Kaiser, L. G. Gao, A. H. H. Chang, M.-C. Liang and Y. L. Yung, Astrophys. J., 2008, 686, 1493–1502 CrossRef CAS.
- A. Komornicki, C. E. Dykstra, M. A. Vincent and L. Radom, J. Am. Chem. Soc., 1981, 103, 1652–1656 CrossRef CAS.
- R. G. Maclagan, M. J. McEwan and G. Scott, Chem. Phys. Lett., 1995, 240, 185–192 CrossRef CAS.
- S. Ekern, J. Szczepanski and M. Vala, J. Phys. Chem., 1996, 100, 16109–16115 CrossRef CAS.
- A. Karton and D. Talbi, Chem. Phys., 2014, 436–437, 22–28 CrossRef CAS.
- L. E. Snyder and D. Buhl, Astrophys. J., 1972, 177, 619–623 CrossRef CAS.
- D. Buhl, L. E. Snyder and J. Edrich, Astrophys. J., 1972, 177, 625–628 CrossRef CAS.
- M. S. Schuurman, S. R. Muir, W. D. Allen and H. F. Schaefer, J. Chem. Phys., 2004, 120, 11586–11599 CrossRef CAS PubMed.
- S. Brünken, C. A. Gottlieb, M. C. McCarthy and P. Thaddeus, Astrophys. J., 2009, 697, 880–885 CrossRef.
- N. Marcelino, J. Cernicharo, B. Tercero and E. Roueff, Astrophys. J., Lett., 2009, 690, L27–L30 CrossRef CAS.
- S. Brünken, A. Belloche, S. Martín, L. Verheyen and K. M. Menten, Astron. Astrophys., 2010, 516, A109 CrossRef.
- N. Marcelino, S. Brünken, J. Cernicharo, D. Quan, E. Roueff, E. Herbst and P. Thaddeus, Astron. Astrophys., 2010, 516, A105 CrossRef.
- D. Quan, E. Herbst, Y. Osamura and E. Roueff, Astrophys. J., 2010, 725, 2101–2109 CrossRef CAS.
- A. Jiménez-Escobar, B. M. Giuliano, G. M. M. Caro, J. Cernicharo and N. Marcelino, Astrophys. J., 2014, 788, 19 CrossRef.
- M. Mladenović, M. Lewerenz, M. C. McCarthy and P. Thaddeus, J. Chem. Phys., 2009, 131, 174308 CrossRef PubMed.
- D. Poppinger, L. Radom and J. A. Pople, J. Am. Chem. Soc., 1977, 99, 7806–7816 CrossRef CAS.
- A. W. Hofmann, Chem. Ber., 1868, 1, 201–202 CrossRef.
- O. Billeter, Ber. Dtsch. Chem. Ges., 1875, 8, 462 CrossRef.
- G. Gerlich, Liebigs Ann. Chem., 1875, 178, 80 CrossRef.
- P. A. S. Smith and D. W. Emerson, J. Am. Chem. Soc., 1960, 82, 3076–3082 CrossRef CAS.
- B. Bak, J. Christiansen, O. Nielsen and H. Svanholt, Acta Agric. Scand., Sect. A, 1977, 31, 666–668 CrossRef CAS.
- M. Wierzejewska and J. Moc, J. Phys. Chem. A, 2003, 107, 11209–11216 CrossRef CAS.
- C. I. Beard and B. P. Dailey, J. Chem. Phys., 1950, 18, 1437–1441 CrossRef CAS.
- K. Yamada, M. Winnewisser, G. Winnewisser, L. Szalanski and M. Gerry, J. Mol. Spectrosc., 1979, 78, 189–202 CrossRef CAS.
- K. Yamada, M. Winnewisser, G. Winnewisser, L. Szalanski and M. Gerry, J. Mol. Spectrosc., 1980, 79, 295–313 CrossRef.
- M. A. Frerking, R. A. Linke and P. Thaddeus, Astrophys. J., 1979, 234, L143–L145 CrossRef CAS.
- M. Wierzejewska and Z. Mielke, Chem. Phys. Lett., 2001, 349, 227–234 CrossRef CAS.
- S. Brünken, Z. Yu, C. A. Gottlieb, M. C. McCarthy and P. Thaddeus, Astrophys. J., 2009, 706, 1588–1593 CrossRef.
- D. T. Halfen, L. M. Ziurys, S. Brünken, C. A. Gottlieb, M. C. McCarthy and P. Thaddeus, Astrophys. J., 2009, 702, L124–L127 CrossRef CAS.
- G. R. Adande, D. T. Halfen, L. M. Ziurys, D. Quan and E. Herbst, Astrophys. J., 2010, 725, 561–570 CrossRef CAS.
- V. Wakelam, P. Caselli, C. Ceccarelli, E. Herbst and A. Catets, Astron. Astrophys., 2004, 422, 159–169 CrossRef CAS.
- CFOUR, a quantum chemical program package written by J. F. Stanton, J. Gauss, M. E. Harding, P. G. Szalay with contributions from A. A. Auer, R. J. Bartlett, U. Benedikt, C. Berger, D. E. Bernholdt, Y. J. Bomble, L. Cheng, O. Christiansen, M. Heckert, O. Heun, C. Huber, T.-C. Jagau, D. onsson, J. Jusélius, K. Klein, W. J. Lauderdale, D. A. Matthews, T. Metzroth, L. A. Mück, D. P. O'Neill, D. R. Price, E. Prochnow, C. Puzzarini, K. Ruud, F. Schiffmann, W. Schwalbach, S. Stopkowicz, A. Tajti, J. Visázquez, F. Wang, J. D. Watts and the integral packages MOLECULE (J. Almlöf and P. R. Taylor), PROPS (P. R. Taylor), ABACUS (T. Helgaker, H. J. Aa Jensen, P. Jørgensen and J. Olsen), and ECP routines by A. V. Mitin and C. van Wüllen. For the current version, see http://www.cfour.de.
- M. E. Harding, T. Metzroth, J. Gauss and A. A. Auer, J. Chem. Theory Comput., 2008, 4, 64–74 CrossRef CAS PubMed.
- C. Puzzarini, J. F. Stanton and J. Gauss, Int. Rev. Phys. Chem., 2010, 29, 273–367 CrossRef CAS.
- K. Raghavachari, G. W. Trucks, J. A. Pople and M. Head-Gordon, Chem. Phys. Lett., 1989, 157, 479–483 CrossRef CAS.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- T. H. Dunning, K. A. Peterson and A. K. Wilson, J. Chem. Phys., 2001, 114, 9244–9253 CrossRef CAS.
- K. A. Peterson and T. H. Dunning, J. Chem. Phys., 2002, 117, 10548–10560 CrossRef CAS.
- J. D. Watts, J. Gauss and R. J. Bartlett, Chem. Phys. Lett., 1992, 200, 1–7 CrossRef CAS.
- K. A. Peterson and T. H. Dunning, J. Chem. Phys., 2002, 117, 10548–10560 CrossRef CAS.
- S. Coriani, D. Marchesan, J. Gauss, C. Hättig, T. Helgaker and P. Jørgensen, J. Chem. Phys., 2005, 123, 184107 CrossRef PubMed.
- S. Thorwirth and M. E. Harding, J. Chem. Phys., 2009, 130, 214303 CrossRef PubMed.
- L. A. Mück, S. Thorwirth and J. Gauss, J. Mol. Spectrosc., 2015, 311, 49–53 CrossRef.
- M. A. Martin-Drumel, A. Roucou, G. G. Brown, S. Thorwirth, O. Pirali, G. Mouret, F. Hindle, M. C. Mccarthy and A. Cuisset, J. Chem. Phys., 2016, 144, 084305 CrossRef CAS PubMed.
- I. M. Mills, in Vibration-Rotation Structure in Asymmetric- and Symmetric-Top Molecules, ed. K. N. Rao and C. W. Mathews, Academic Press, New York, 1972, pp. 115–140 Search PubMed.
- J. Gauss and J. F. Stanton, Chem. Phys. Lett., 1997, 276, 70–77 CrossRef CAS.
- J. F. Stanton, C. L. Lopreore and J. Gauss, J. Chem. Phys., 1998, 108, 7190–7196 CrossRef CAS.
- M. C. McCarthy and J. Gauss, J. Phys. Chem. Lett., 2016, 7, 1895–1900 CrossRef CAS PubMed.
- M. E. Sanz, M. C. McCarthy and P. Thaddeus, J. Chem. Phys., 2005, 122, 194319 CrossRef PubMed.
- M. Mladenović, M. Elhiyani and M. Lewerenz, J. Chem. Phys., 2009, 131, 034302 CrossRef PubMed.
- S. Brünken, C. A. Gottlieb, M. C. McCarthy and P. Thaddeus, Astrophys. J., 2009, 697, 880–885 CrossRef.
- M. Mladenović, M. Lewerenz, M. C. McCarthy and P. Thaddeus, J. Chem. Phys., 2009, 131, 174308 CrossRef PubMed.
- G. G. Brown, B. C. Dian, K. O. Douglass, S. M. Geyer, S. T. Shipman and B. H. Pate, Rev. Sci. Instrum., 2008, 79, 053103 CrossRef PubMed.
- D. P. Zaleski, J. L. Neill, M. T. Muckle, N. A. Seifert, P. Brandon Carroll, S. L. Widicus Weaver and B. H. Pate, J. Mol. Spectrosc., 2012, 280, 68–76 CrossRef CAS.
- M. C. McCarthy, W. Chen, M. J. Travers and P. Thaddeus, Astrophys. J., Suppl. Ser., 2000, 129, 611–623 CrossRef CAS.
- R. A. Loomis, D. P. Zaleski, A. L. Steber, J. L. Neill, M. T. Muckle, B. J. Harris, J. M. Hollis, P. R. Jewell, V. Lattanzi, F. J. Lovas, O. Martinez Jr, M. C. McCarthy, A. J. Remijan, B. H. Pate and J. F. Corby, Astrophys. J., 2013, 765, L9 CrossRef.
- T. J. Balle and W. H. Flygare, Rev. Sci. Instrum., 1981, 52, 33–45 CrossRef CAS.
- K. N. Crabtree, O. Martinez Jr, L. Barreau, S. Thorwirth and M. C. McCarthy, J. Phys. Chem. A, 2013, 117, 3608–3613 CrossRef CAS PubMed.
- J.-U. Grabow, E. S. Palmer, M. C. McCarthy and P. Thaddeus, Rev. Sci. Instrum., 2005, 76, 093106 CrossRef.
- C. A. Gottlieb, A. J. Apponi, M. C. McCarthy, P. Thaddeus and H. Linnartz, J. Chem. Phys., 2000, 113, 1910–1915 CrossRef CAS.
- H. M. Pickett, J. Mol. Spectrosc., 1991, 148, 371–377 CrossRef CAS.
- C. H. Townes and A. L. Schawlow, Microwave Spectroscopy, Dover Publications, New York, 1975 Search PubMed.
- Z. Kisiel, Spectroscopy from Space, Springer Netherlands, Dordrecht, 2001, pp. 91–106 Search PubMed.
- M. Niedenhoff, G. Winnewisser, K. Yamada and S. P. Belov, J. Mol. Spectrosc., 1995, 169, 224–242 CrossRef CAS.
- M. Mladenović, M. Elhiyani and M. Lewerenz, J. Chem. Phys., 2009, 130, 154109 CrossRef PubMed.
- W. L. Meerts and A. Dymanus, Can. J. Phys., 1975, 53, 2123–2141 CrossRef CAS.
- C. A. McDowell and J. W. Warren, Trans. Faraday Soc., 1952, 48, 1084–1093 RSC.
- J. L. Neill, M. T. Muckle, D. P. Zaleski, A. L. Steber, B. H. Pate, V. Lattanzi, S. Spezzano, M. C. McCarthy and A. J. Remijan, Astrophys. J., 2012, 755, 153 CrossRef.
- A. Belloche, H. S. P. Müller, K. M. Menten, P. Schilke and C. Comito, Astron. Astrophys., 2013, 559, A47–A187 CrossRef.
- J. M. Hollis, P. R. Jewell, F. J. Lovas and A. Remijan, Astrophys. J., 2004, 613, L45–L48 CrossRef CAS.
- B. A. McGuire, R. A. Loomis, C. M. Charness, J. F. Corby, G. A. Blake, J. M. Hollis, F. J. Lovas, P. R. Jewell and A. J. Remijan, Astrophys. J., 2012, 758, L33 CrossRef.
- A. Faure, A. J. Remijan, K. Szalewicz and L. Wiesenfeld, Astrophys. J., 2014, 783, 72–78 CrossRef.
- J. M. Hollis, P. R. Jewell, F. J. Lovas, A. Remijan and H. Møllendal, Astrophys. J., 2004, 610, L21–L24 CrossRef CAS.
- J. M. Hollis, P. R. Jewell, A. J. Remijan and F. J. Lovas, Astrophys. J., 2007, 660, L125–L128 CrossRef CAS.
- J. N. Chengalur and N. Kanekar, Astron. Astrophys., 2003, 403, L43–L46 CrossRef CAS.
- M. A. Requena-Torres, J. Martín-Pintado, A. Rodríguez-Franco, S. Martín, N. J. Rodríguez-Fernández and P. De Vicente, Astron. Astrophys., 2006, 455, 971–985 CrossRef CAS.
- R. T. Garrod, S. L. Widicus Weaver and E. Herbst, Astrophys. J., 2008, 682, 283–302 CrossRef CAS.
- R. T. Garrod, Astrophys. J., 2013, 765, 60 CrossRef.
- A. McKellar, Publ. Astron. Soc. Pac., 1940, 52, 187–192 CrossRef CAS.
- K. B. Jefferts, A. A. Penzias and R. W. Wilson, Astrophys. J., 1970, 161, L87–L89 CrossRef.
- W. S. Adams, Astrophys. J., 1941, 93, 11–23 CrossRef CAS.
- D. A. Neufeld, E. Falgarone, M. Gerin, B. Godard, E. Herbst, G. Pineau des Forêts, A. I. Vasyunin, R. Güsten, H. Wiesemeyer and O. Ricken, Astron. Astrophys., 2012, 542, L6 CrossRef.
- P. Raghunath, S. Sitha, K. Bhanuprakash and B. Choudary, J. Phys. Chem. A, 2003, 107, 11497–11504 CrossRef CAS.
- H. Fu, H. Yu, Y. Chi, Z. Li, X. Huang and C. Sun, Chem. Phys. Lett., 2002, 361, 62–70 CrossRef CAS.
- S. Thorwirth, V. Lattanzi and M. C. McCarthy, J. Mol. Spectrosc., 2015, 310, 119–125 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp03871a |
| ‡ B.A.M. is a Jansky Fellow of the National Radio Astronomy Observatory. |
| § The entire PRIMOS dataset, as well as further information on the project, is accessible at http://www.cv.nrao.edu/PRIMOS/ |
| ¶ The reaction rates and barriers, both to formation and destruction, of molecules almost always dominate over differences in their relative energies (stabilities). |
| || The solid angle on the sky probed by the observations. |
| ** Such models explicitly treat the relative energy level population of molecules using both radiative and collisional excitation mechanisms. |
| This journal is © the Owner Societies 2016 |