
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnderstanding the thermal decomposition mechanism of a halogen-free chelated orthoborate-based ionic liquid: a combined computational and experimental study†
M.
Golets
*a,
M. R.
Shimpi
b,
Y.-L.
Wang
cd,
O. N.
Antzutkin
b,
S.
Glavatskih
de and
A.
Laaksonen
*af
aDepartment of Materials and Environmental Chemistry, Arrhenius Laboratory, Stockholm University, SE-106 91 Stockholm, Sweden. E-mail: aatto.laaksonen@mmk.su.se; mikhail.golets@yahoo.se
bChemistry of Interfaces, Luleå University of Technology, S-97187, Luleå, Sweden
cApplied Physical Chemistry, Department of Chemistry, KTH Royal Institute of Technology, SE-100 44 Stockholm, Sweden
dSystem and Component Design, KTH Royal Institute of Technology, SE-100 44 Stockholm, Sweden
eMechanical Construction and Production, Ghent University, B-9000 Ghent, Belgium
fStellenbosch Institute of Advanced Study (STIAS), Wallenberg Research Centre, Stellenbosch University, Marais Street, Stellenbosch 7600, South Africa
First published on 19th July 2016
Abstract
In the last few decades, ionic liquids (ILs) have gained significant attention as lubricants and lubricant additives due to their polar nature, low vapour pressure and tunable physicochemical properties. In this work, quantum chemistry calculations and atomistic Molecular Dynamics (MD) simulations were employed to predict thermal degradation mechanisms of a potential lubricating agent – the tributyloctylphosphonium bis(oxalato)borate ([P4,4,4,8][BOB]) IL. It was found that the onset of decomposition of the studied IL coincides with a cleavage of the B–O bonds in the [BOB]− anion. Consequently, a series of chemical reactions of the [P4,4,4,8]+ cation with the [BOB]− anion was triggered yielding alkylboranes, alkenes, trialkylphosphines, CO and CO2. Another ionic system, consisting of [P4,4,4,8][Cl], was also tested for a comparison. Thermogravimetric measurements have shown a higher thermal stability of [P4,4,4,8][BOB] compared to that of [P4,4,4,8][Cl] at least at the initial stage of decomposition, in accord with the presented calculations. Quantum chemical frequency calculations also agreed with the experimental Fourier Transform Infrared (FTIR) spectroscopy results.
1. Introduction
The unique chemistry of ionic liquids (ILs) allows many options for enhancing performance characteristics in existing applications and even enables new technologies. A temperature increase in industrial applications is not uncommon, which makes knowledge on thermal stability of ILs essential for proper IL selection. In some applications, such as lubrication, decomposition can be useful. Shear rates and contact pressures in lubricated contacts can be very high, 107 s−1 and 4 GPa, respectively. High thermo-mechanical loads may lead to a rupture of the hydrodynamic film causing a direct surface-to-surface contact. ILs should interact with the surfaces and build protective tribofilms to reduce friction and wear. Mechanisms of tribofilm formations by ILs are complex and not completely understood.1 They involve several stages possibly involving decomposition of the IL. The anionic moiety may promote the tribochemical reactions and tribofilm formation in the IL/solid interfacial region. However, the oxidative decomposition of ILs is promoted by the cationic moiety.2The thermal decomposition of various ammonium- and imidazolium-based ILs was extensively studied during the last decade. Experimental trials were performed referring to the tribo-tests3,4 and artificial degradation experiments, including oven-aging tests4 and thermo-gravimetric analysis (TGA).5 The obtained decomposition products were analyzed by means of X-ray photoelectron spectroscopy (XPS), mass spectrometry (MS) and white light confocal microscopy.3,4 The versatility of using the TGA-MS technique for studying IL systems was also verified by other research groups.6–9 An interesting method was reported by Anderson et al.10 for studying the thermal stability of ILs deposited in fused silica capillaries. The corresponding decomposition products were consequently detected using a flame ionization detector (FID) and MS.10,11 Kroon et al.12 performed DFT calculations in order to unveil decomposition mechanisms of imidazolium ILs. They observed a very small effect of changing the alkyl chain length of the cation on the onset temperature of decomposition. The activation energy barriers calculated for the decomposition of imidazolium ILs were compared with the onset temperatures of decomposition reported in other works.
The SN2 nucleophilic attack of anions or the so-called reverse Menschutkin-type dealkylation reaction occurs in almost all ILs studied and yields neutral products.4,13,14 The competitive decomposition path via E2 elimination was also observed.14 Interestingly, phosphonium salts tend to be the most stable in comparison with the corresponding ammonium and imidazolium salts.13,14 Higher thermal stability was claimed for the tetraalkylphosphonium cations containing double bonds in [P4,4,4,3][NTf2].15
Several publications on the decomposition of phosphonium- and boron-based ILs recently emerged, given that this family of ILs has attracted a broad interest in both academic and industrial communities. Oh et al.16 studied the electrochemical decomposition of lithium bis(oxalato)borate (BOB) and observed that the decomposition reactions were initiated when superoxide radicals promoted the nucleophilic substitution at the boron centre. In addition, a series of reduction and gas evolution reactions were observed. As in an older study by Zinigrad et al.,17 where the thermal decomposition of [Li][BOB] was studied, Oh et al. obtained similar decomposition products. An important computational study on oxidative stability and decomposition mechanism of [Li][BOB] was recently performed by Wang et al.18 Despite similar reaction products, the oxidative decomposition mechanism of [BOB]− differed from the one suggested in the experimental studies by Oh et al. and Zinigrad et al.
Trihexyl(tetradecyl)phosphonium chloride was studied by Ramnial et al.19 The authors expected to observe a deprotonation of the IL to a phosphorane product in a highly basic environment. Such reaction did not occur, demonstrating the stability of IL versus strongly basic and nucleophilic reagents. Tseng et al.20 studied the decomposition of tetraalkylphosphonium ILs under basic microwave irradiation conditions. A quick reaction of the tetraalkylphosphonium cation with sodium salts of substituted benzoates was observed via direct SN2 carboxylate alkylation resulting in ester formation. In the next step, these esters were consequently converted to aryl ketones by means of microwave irradiation. It was concluded that the tetraalkylphosphonium cation was highly acidic, electrophilic and reactive when combined with the benzoate anion. In another study, the [P6,6,6,14]+ was previously thermally degraded together with [CH3(CH2)8CO2]− and [NTf2]−. Mainly organophosphorous products with different carbon chain lengths were found; H3PO4 was also detected and was presumably formed in the reaction of P2O5 and water.7
Two interesting studies of the decomposition of carboxylate21 and imidazolium22 ILs were performed by combining experimental and computational approaches. MS, TGA, TGA-MS and differential scanning calorimetry (DSC) techniques were used. Density functional theory (DFT) was employed to provide more physical insights into the nanoscopic level.21,22 Still, the phenomenon of thermal decomposition of tetraalkylphosphonium borate ILs is not clear. Accordingly, the current work combines computational and experimental approaches to unveil main decomposition stages and to investigate the thermal stability of [P4,4,4,8][BOB], a potential lubricating agent. Phosphonium orthoborate ILs were previously studied by our group demonstrating promising lubrication properties.23 An ionic system of [P4,4,4,8][Cl] was also tested to observe the effect of the anion on the IL decomposition process. Quantum chemistry ab initio calculations provided optimized molecular geometries and potential energy surfaces of elongation and torsion of structural bonds. Systems consisting of neat ILs, [P4,4,4,8][BOB] and [P4,4,4,8][Cl], were further analyzed using the ab initio techniques in order to follow the whole reaction network. Infrared frequencies were also calculated and compared with experimental FTIR data. The MD simulations assisted in the understanding of the spatial orientation of ion pairs in the neat IL systems. Finally, the calculation results were compared with TGA and TGA-MS data for [P4,4,4,8][BOB] and [P4,4,4,8][Cl].
2. Experimental
2.1. Chemicals and synthetic procedure of [P4,4,4,8][BOB]
Boric acid (99.999%) and oxalic acid anhydrous (99.999%) were purchased form Aldrich Chemicals and sodium carbonate (99.999%) was purchased from Merck suppliers. Acetonitrile, ethanol (97.5%) and dichloromethane were purchased from Sigma-Aldrich and used as supplied. Tributyloctylphosphonium chloride [P4,4,4,8][Cl] was purchased from Cytec Canada, and used as received. Milli-Q water was used in all experiments through the work (conductivity less than 0.1 μS cm−1).Oxalic acid (3 mol) and boric acid (1 mol) were separately dissolved in water and then mixed together under constant stirring. Na2CO3 (0.5 mol) was added slowly to the mixture with vigorous stirring. The turbid solution was heated in an oil bath at 120 °C until dry. The crude white product was isolated at room temperature. The crude product was redispersed in a hot acetonitrile under one hour of stirring and the white product precipitate was isolated by vacuum filtration. The product material was further washed with cold ethanol and the powder was dried in an oven at 60 °C for 12 hours. Thereafter, the obtained Na[BOB] product was mixed with tributyloctylphosphonium chloride with equimolar ratio in dichloromethane. The reaction mixture was stirred overnight at room temperature. Water was added to the reaction mixture with constant stirring at room temperature. The reaction product was extracted with 80 mL of dichloromethane. The dichloromethane organic layer was washed three times with 100 mL of water. The dichloromethane was rotary evaporated at reduced pressure and the product was dried in a vacuum oven at 60 °C for 2 days. A viscous colourless ionic liquid of tributyloctylphosphonium bis(oxalato)borate [P4,4,4,8][BOB] was obtained. The purity of the [P4,4,4,8][BOB] and [P4,4,4,8][Cl] was confirmed by nuclear magnetic resonance (NMR) spectroscopy and FTIR techniques (ESI†).
2.2. Physicochemical characterization
Thermogravimetric analysis (TGA) was performed by means of TA Instruments 2950 thermogravimetric analyzer at a rate of 10 °C min−1. Tested samples were placed in a platinum 100 μL sample pan. The samples were then inserted into the TG furnace under nitrogen gas with a flow rate of 10.0 mL min−1. Nickel and AlumelTM were used as the calibration standards. The TA Universal Analysis software determined the Tonset, temperatures corresponding to the loss of 2, 5 and 10 wt%. The differential thermal analysis (DTA) was performed as a complementary technique (ESI†).The TGA-MS test was performed following an analogous thermogravimetric procedure in an identical TGA apparatus, which was connected to the Thermo Star MS (Pfeiffer Vacuum, Germany). The trigger mode was applied for the MS ion currents at the start of a TGA run. MS: 0–70 amu at 15 s cycle speed; TGA: 20 °C to 500 °C in 2 °C min−1 in air with a total purging rate of 100 mL min−1.
2.3. Computational details
Quantum chemistry calculations of [P4,4,4,8][BOB] decomposition were performed using Gaussian 09 software.24 The DFT method was applied with B3LYP functional and 6-31G(d,p) basis set for optimizing molecular geometries of [P4,4,4,8]+ and [BOB]−. The B3LYP hybrid functional was chosen for computations as a suitable tool, which was widely used for various molecular systems and, particularly, to study the thermal decomposition of ILs.6,12 Relaxed energy scans of structural bond lengths in [P4,4,4,8]+ and [BOB]− were performed for 10 steps with a step interval of 0.4 Å. Furthermore, torsion angles were scanned for alkyl chains in [P4,4,4,8]+ for 12 steps with an angle interval of 30°. Finally, quantum chemistry calculations were performed to determine activation energy barriers and transition states for an isolated [BOB]−.A less computationally demanding system consisting of [P4,4,4,8]+ and [Cl]− anion was studied first. The weakest structural bonds in [P4,4,4,8][Cl] were scanned with 10 steps of 0.4 Å and reactions with [Cl]− were observed. The intermediates previously identified for [BOB]− were optimized together with [P4,4,4,8]+. Consequently, a series of calculations was performed for scanning the weakest bonds in the [P4,4,4,8]+ with 10 steps of 0.4 Å in order to observe the potential reactions between [P4,4,4,8]+ and [BOB]− fragments. Furthermore, the vibration analysis of the above ion pair structures was applied in order to compute the infrared (IR) spectra of the separate species in neat IL. Also, the calculated IR spectra of ILs were similar to those obtained with the FTIR (ESI†).
Standard three-dimensional periodic boundary conditions were used for liquid-state atomistic simulations using M.Dynamix software package.25 Simulations on the neat [P4,4,4,8][BOB] were performed in our previous study.26 The Tuckerman–Berne double time step algorithm27 was applied to integrate equations of motion. A short time step of 0.2 fs represented fast intramolecular vibrations and non-bonded interactions, whereas van der Waals and electrostatic interactions were integrated with a long time step of 2.0 fs. We applied the standard Ewald summation method to treat electrostatic interactions between atom-centred point charges. The MD simulations were performed for 10 ns in an isothermal–isobaric ensemble. A Nosé–Hoover thermostat (333 K) and a barostat (1 atm) were employed with time coupling constants of 1000 and 30 fs, respectively. The computation followed with 10 ns canonical ensemble simulations with consequent recording of simulation trajectories at an interval of 100 fs. Further, the microscopic liquid structures in IL were illustrated by spatial distribution functions (SDFs), which provide the probability of finding an atom around a specific molecule in three-dimensional space. In this study the gOpenMol software package28,29 was employed for SDF visualization.
3. Results and discussion
3.1. Geometry optimization and vibration analysis
The optimized molecular geometries of the [P4,4,4,8]+ and [BOB]− are presented in Fig. 1. The optimized molecular geometry of [BOB]− is characterized by uniform O–B–O angles of 111.7° and equal lengths of 1.47 Å for B–O bonds, indicating a high symmetry in [BOB]−. The optimized structure of [P4,4,4,8]+ has three short alkyl chains (labeled as B, T and R in Fig. 1) and one long alkyl chain (labeled as L in Fig. 1), respectively. In [P4,4,4,8]+, the C–C bond lengths in all alkyl chains continuously increase from 1.53 up to 1.54 Å from the last carbon atom to the central one. Additionally, the C–P bond lengths of 1.81 and 1.84 Å were observed at C8 and C4 chains, respectively.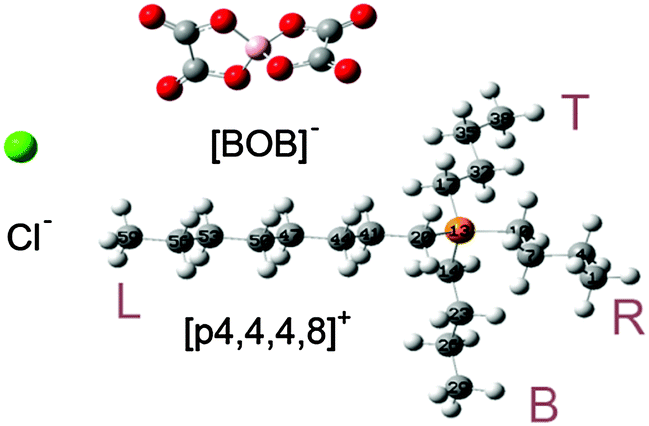 | ||
| Fig. 1 Molecular structures of IL species studied herein: [P4,4,4,8]+, [BOB]− and [Cl]−. Oxygen, boron, phosphorus, chlorine and carbon atoms are shown in red, pink, orange, green and gray, respectively. The letters L, R, B and T stand for left, right, bottom and top alkyl chains, respectively. | ||
The IR spectra of the [P4,4,4,8]+ and [BOB]− were obtained by vibrational frequency analysis and presented in Fig. 2 and 3, respectively. In the [P4,4,4,8]+, strong asymmetric stretching vibrations were observed in C–H bonds as follows: ν[C(56–59)–H] at 3106 cm−1; ν[C(41–44–47–50–53–56)–H] at 3026–3106 cm−1; ν[C(17–32–35–38)–H] at 3048 cm−1; ν[C(14–23–26–29)–H], ν[C(1–4–7–10)–H] at 3055–3115 cm−1, respectively. Clearly, these IR bands demonstrated the presence of strong intramolecular van der Waals interactions promoted by high mobility of hydrogens. Further, less intensive asymmetric bending vibrations were detected at 756–1528 cm−1 for all C–H bonds. Interestingly, the vibrations in C–C (195–492 cm−1) and C–P (407 cm−1) skeletal bonds were negligible indicating high rigidity of the cation molecule. While in the [BOB]−, strongest vibrations referred to asymmetric stretching of B–O (1040 cm−1) and C–C (1315 cm−1) bonds, respectively. Strong asymmetric stretching of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds (1871 cm−1) was also observed, as shown in Fig. 3.
O bonds (1871 cm−1) was also observed, as shown in Fig. 3.
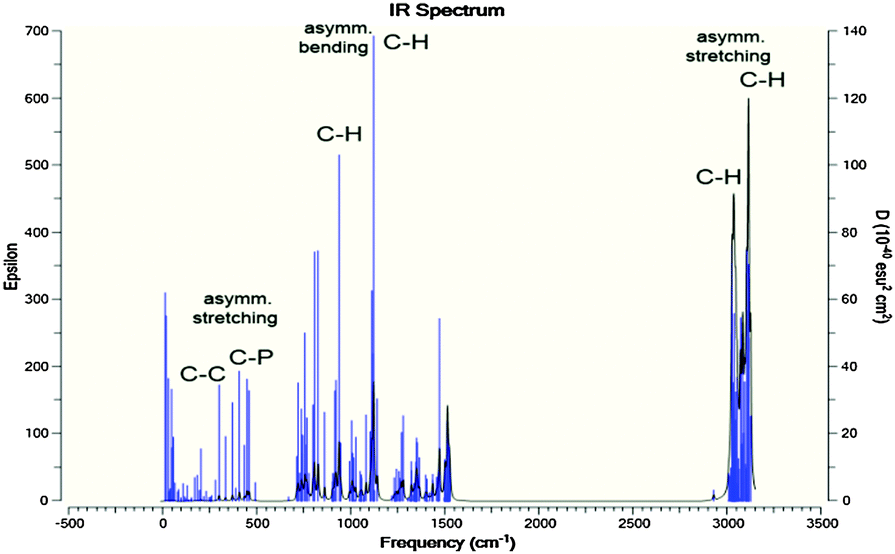 | ||
| Fig. 2 Calculated IR spectra for [P4,4,4,8]+. Highest IR frequencies attributed to the asymmetric stretching and bending of C–H bonds in alkyl chains; C–P and C–C vibrations are negligible. | ||
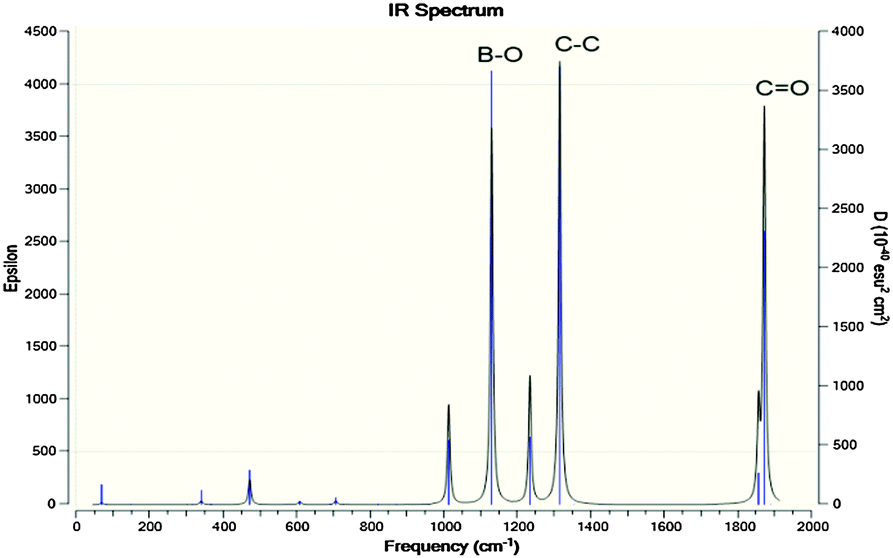 | ||
| Fig. 3 Calculated IR spectra for [BOB]−. Highest IR frequencies attributed to asymmetric stretching in C–C, CO and B–O bonds. | ||
3.2. Scanning of the potential energy surface in isolated [BOB]−
The potential energy surface (PES) was studied at the B3LYP/6-31G(d,p) level of theory. Chemical bonds in [BOB]− were scanned first and the [BOB]− was treated separately from the heavy cation to get a quick insight into its reactivity. It is shown that the decomposition of [BOB]− should start with the cleavage of one of the four B–O bonds given that [BOB]− possesses a negative charge equally spread among four oxygens around the central boron atom. Consequently, C–O, B–O and C–C bonds were scanned taking into account high symmetry of this molecule. Transformations observed throughout the cleavage of these bonds are described below. According to calculations, small [BOB]− undergoes rather simple decomposition pathways. The results of the PES scan are presented in Fig. 4. The optimized geometries of the transition states and stationary points observed throughout the reaction network are presented in Fig. 5.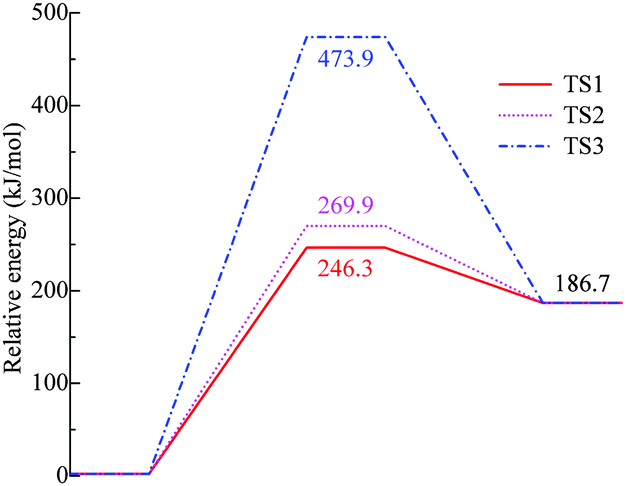 | ||
| Fig. 4 Relative energies of stationary points and transition states of decomposition of isolated [BOB]−. TS1 is B–O, TS2 is C–O and TS3 is the C–C bond, respectively. | ||
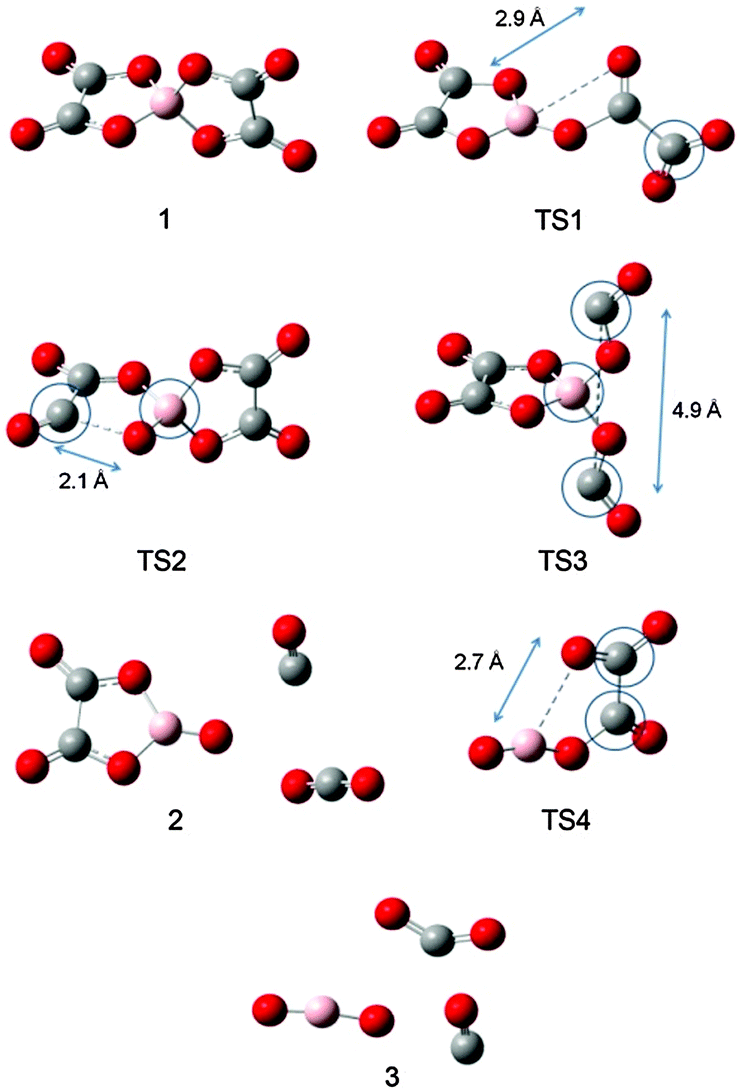 | ||
| Fig. 5 Optimized structures of stationary points and transition states observed for the decomposition of isolated [BOB]−. Scanned coordinates in transition states are marked with the dotted lines. Atomic distances are presented in Ångstroms. Blue circles are attributed to imaginary frequencies: TS1 (−1381.61 cm−1), TS2 (−270.30 cm−1), TS3 (−494.31 cm−1) and TS4 (−1924.23 cm−1) as asymmetric stretching around circled atoms. | ||
As observed in Fig. 4, the cleavage of either B–O (TS1) or C–O (TS2) bonds required 246.3 and 269.9 kJ mol−1, respectively. On the contrary, the decomposition via the cleavage of C–C bonds (TS3) was more energy demanding with an energy barrier of 473.9 kJ mol−1. In any of these three cases, the product mixtures consisting of a monocyclic anion 2, CO and CO2 were generated. Further decomposition of the monocyclic anion 2 proceeded via the scission of B–O bonds (Ea = 157.4 kJ mol−1, TS4) and resulted in the formation of a boron-containing anion 3, CO and CO2, respectively. At the final decomposition stage, the boron trioxide molecule could be produced from two [BOB]− anions. As stated by Lu et al.,3 such stable inorganic products of anion decomposition may interact with solid surfaces forming thick tribofilms. By such tribofilms the capture of the gaseous decomposition products was promoted thus increasing the tribofilm stability for these particular ILs. In analogy with a previous study,7 boric acid could also be originated from the reaction of B2O3 with water. The overall reaction network of the [BOB]− decomposition is depicted in Scheme 1.
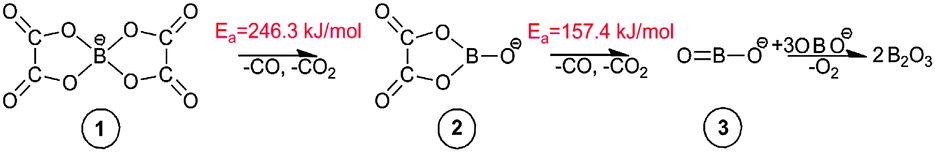 | ||
| Scheme 1 Reaction network for decomposition of [BOB]−. | ||
Oh et al.16 have studied [Li][BOB] and observed deoxygenative formation of boron trioxide (B2O3) from two identical molecules of the boron-containing anion 3, which was previously supported by the literature.17 Moreover, in an earlier study, Zinigrad et al. have also observed the formation of CO and CO2 from [Li][BOB].17 The formation of B2O3 from the decomposition of an alkylimidazolium-BF4 IL on steel surfaces is also a well-studied phenomenon.30 Wang et al. proposed an alternative decomposition pathway for the isolated [BOB]−via a consecutive breakage of C–C and B–O bonds.18 The combined activation energy of reaction barriers observed in their work (ca. 124 kJ mol−1) was much smaller than the energy obtained in the current study. A direct comparison of these studies is not straightforward as Wang et al. investigated an oxidation mechanism by applying PCM calculations with acetone dielectric constant while our study is mainly focused on thermal decomposition of [BOB]−.
3.3. Scanning of the potential energy surface in isolated [P4,4,4,8]+
Activation barriers were also searched for an isolated [P4,4,4,8]+. In contrast with the highly reactive [BOB]−, no barriers were observed when scanning structural bonds in the [P4,4,4,8]+. Short and long carbon chains were altogether scanned and only a continuous increment in the potential energy was observed. It is presented in panels (a) and (b) in Fig. 6. The elongation of C–P bonds requires less energy in comparison with that of C–C bonds. The scanning of C–C bonds in alkyl chain tails was the most energy demanding. Meanwhile, equal energies were required to elongate the C–P bonds in various alkyl chains. As mentioned,21 IL anions with long or branched alkyl chains had a minimal effect on decomposition temperatures. Analogous observations for IL cations were reported elsewhere.12 As shown in panel (b) in Fig. 6, the two last C–C bonds in the C8 chain (labeled as L chain in Fig. 1) were the most rigid and energetically stable as compared to other C–C bonds in the short alkyl chains.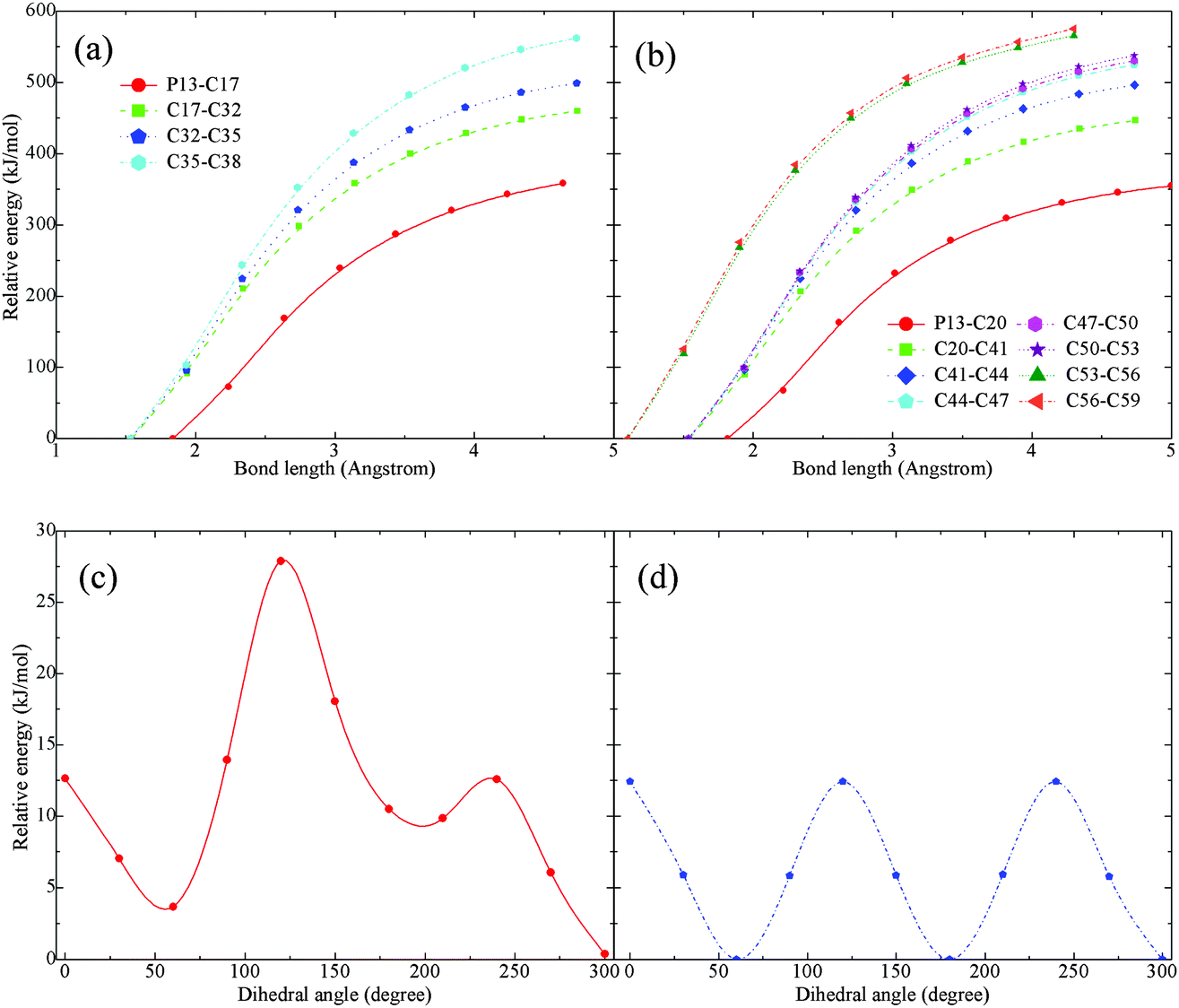 | ||
| Fig. 6 Potential energy surface scans for isolated [P4,4,4,8]+: (a) bond elongation in C4 alkyl chain; (b) bond elongation in C8 alkyl chain; (c) dihedral rotation in the middle of C8 alkyl chain; and (d) dihedral rotation in the tail of C8 alkyl chain. | ||
Rotational barriers were also calculated and are shown in panels (c) and (d) in Fig. 6. The rotation of H–C–C–H dihedral angles in the tails of alkyl chains resulted in equal harmonic barriers indicating a low possibility for transformations. However, fluctuations were observed while shifting the rotated dihedral from the middle of alkyl chains to the centre of the cation. Although no clear reaction barriers were detected, the above-mentioned calculations, when combined with structural and vibration analyses as well as published data,3,13,14 indicated a preferable cleavage of a C–P bond at the initial stage of decomposition of an isolated [P4,4,4,8]+.
3.4. Decomposition of [P4,4,4,8][Cl]
After the geometry optimization of the ionic system, the [Cl]− anion was oriented along the cavity formed by L, R, and T alkyl chains and near the central phosphonium atom in [P4,4,4,8]+, as shown in Fig. 7. Identical activation barriers were observed for decomposition reactions with T and R chains of the cation, given that they are symmetrically oriented along [Cl]−. A series of relaxed scans were performed for C–P bond lengths: 13–14, 13–17 and 13–20 C–P bonds, as labeled in Fig. 1, were scissored resulting in a separation of B, T and L chains, respectively. A general reaction network is presented in Scheme 2.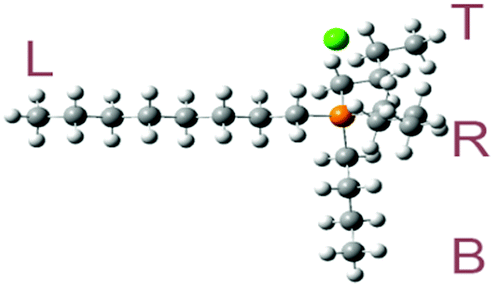 | ||
| Fig. 7 Spatial orientation of a [P4,4,4,8][Cl] ion pair. | ||
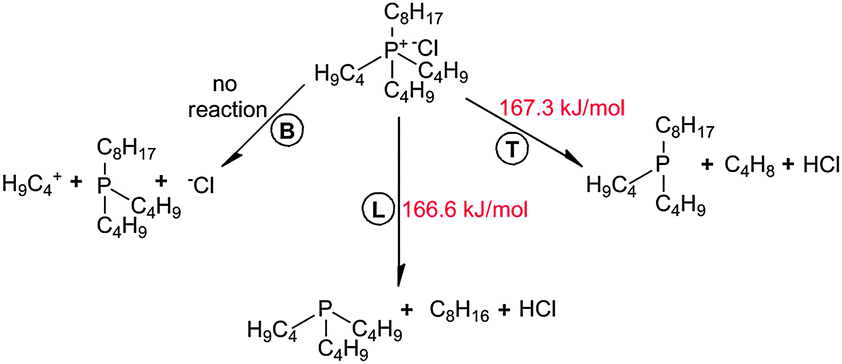 | ||
| Scheme 2 General reaction network for decomposition of [P4,4,4,8][Cl]. | ||
Quantum chemistry calculations showed two deprotonation reactions of [Cl]− with linear hydrocarbons of T and L alkyl chains, in which linear alkenes and neutral [P4,4,4] or [P4,4,8] trialkylphosphine molecules were generated. Nearly identical activation barriers of 166.6 and 167.3 kJ mol−1 were found for L and T alkyl chains, respectively, indicating similar reactivity of these alkyl chains in the [P4,4,4,8]+. The C–P bond reactivity does not depend on the bond length even if the C–P bond in the L alkyl chain is shorter. 1-Octene and 1-butene with similar activation energies of decomposition reactions of 107.3 and 109.3 kJ mol−1 were formed. In fact, such a reaction was not observed when separating the bottom C4 chain (B). Presumably, the nucleophilicity of the [Cl]− anion was not high enough for it to react with the neutral [P4,4,8] trialkylphosphine, whereas the separated C4H9 linear hydrocarbon was inaccessible. A low nucleophilic character of an anion promotes the alkene formation during a thermal cleavage.14 Furthermore, the size of the anion has an influence. If compared with the decomposition of an isolated [BOB]− anion, [P4,4,4,8][Cl] IL decomposes with a lower energy barrier, and it is hence less stable compared with an isolated [BOB]− anion.
3.5. Decomposition of [P4,4,4,8][BOB]
Similar to the Cl-containing phosphonium IL, four parallel decomposition pathways involving the corresponding four alkyl chains in the [P4,4,4,8]+ cation, L, T, R and B as shown in Fig. 8, were evaluated. No reaction was initially observed between [BOB]− and alkyl chains in [P4,4,4,8]+. Hence, the initial decomposition step of [BOB]−should occur prior to the onset of decomposition of [P4,4,4,8]+. Product 2, which was previously observed in the decomposition of an isolated [BOB]−, was introduced to the current system. The spatial orientation of this anion around [P4,4,4,8]+ was identical to that observed in [P4,4,4,8][Cl] IL, as shown in Fig. 7, i.e. product 2 was oriented towards L, T and R alkyl chains in [P4,4,4,8]+ while the B alkyl chain was isolated. As mentioned above, the decomposition reaction was triggered with a scission of one of the C–P bonds in the [P4,4,4,8]+ cation. The generalized reaction network for the anionic species 2 is presented in Scheme 3.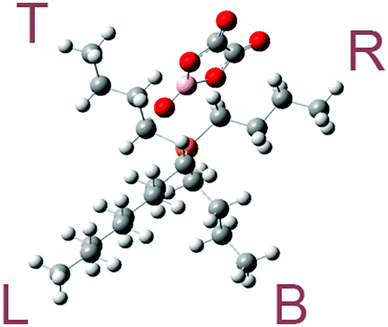 | ||
| Fig. 8 Spatial orientation of a [P4,4,4,8][BOB fragment] ion pair. | ||
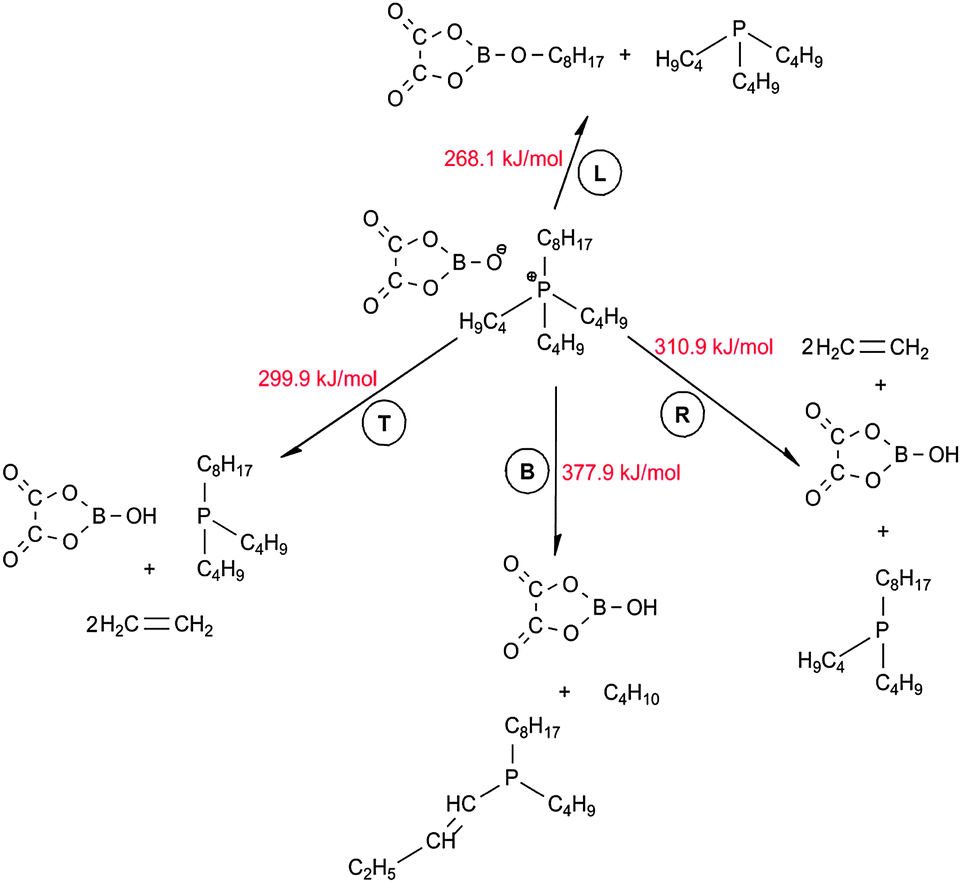 | ||
| Scheme 3 General reaction network for decomposition of [P4,4,4,8][BOB]. | ||
Elimination reactions were observed for T and R alkyl chains with the consequent cleavage of the C–P bond of the cation, proton abstraction and formation of CC bonds. Similar activation energies were demonstrated for reactions of T and R alkyl chains, as shown in Scheme 3. Reactions with these two alkyl chains are highly reversible as reaction energies of 294.2 and 299.5 kJ mol−1 were observed for T and R alkyl chains, respectively. For shorter C4 chains (T and R) cracking reactions occurred resulting in the formation of ethylene. In contrast, alkylboranes were obtained from the irreversible reaction of 2 with the longer C8 hydrocarbon (L), which occurred with the activation barrier of 268.1 kJ mol−1. At this point, the long L alkyl chain exhibited a higher decomposition stability. A slightly lower energy barrier was observed for the reaction with the chain L. The L chain decomposed via a classical SN2 substitution reaction, which is common for most of studied ILs. Through these reactions neutral trialkylphosphines were also produced. This is in line with an observation of the formation of alkylphosphites for high performance lubricants reported in the literature.31 Hence, the decomposition of [P4,4,4,8][BOB] may produce species that enhance lubrication.
Another situation occurred for the reaction of B alkyl chain. Given that this chain was initially isolated from anion 2, simply the deprotonation reaction was initiated between 2 and neutral trialkylphosphine when scissing of the C–P bond at B alkyl chain. As a result, a double bond on the short T alkyl chain in trialkylphosphine molecule was formed. A linear alkane was also formed via this reaction. In fact, no reaction was previously observed between the [Cl]− and B alkyl chain. Hence, the nucleophilic properties of 2 are stronger compared to [Cl]− and anionic sizes also have a minor effect.
Furthermore, the formation of phosphates was not observed at this level of decomposition, as oxygen is required to form phosphates. As presented elsewhere,32 oxygen-containing species were mainly found outside the wear scar, i.e. outside the main tribo-reaction zone, formed during a tribo-test with ILs as lubricants. The formation of oxygen-containing phosphate molecules during the decomposition of ILs could occur at the final stages, which are not considered in this work, but were observed on a worn metal surface using XPS.33
In fact, [P4,4,4,8][BOB] is initially more stable when compared with [P4,4,4,8][Cl]. For example, only 167.3 kJ mol−1 of energy is required to decompose the T alkyl chain in [P4,4,4,8][Cl] compared to 246.3 kJ mol−1 in [BOB]−. The decomposition of [P4,4,4,8][BOB] is a two-stage reaction. During calculations with product 3 even a higher activation barrier will be required for this three-stage reaction, i.e. (a) formation of product 2, (b) formation of product 3, and (c) the reaction of 3 with [P4,4,4,8]+.
3.6. MD simulations of [P4,4,4,8][BOB]
Fig. 9 shows spatial distribution of boron in [BOB]− around [P4,4,4,8]+ calculated from the neat IL. A particular coordination of boron to the central polar segment of [P4,4,4,8]+ was observed in a tetrahedral region around [P4,4,4,8]+. Such observation could be explained by strong electrostatic interactions between central polar segments of ionic species. Moreover, a strong steric effect of the four linear alkyl substituents in [P4,4,4,8]+ imposed a strong impact on the observed coordination environment. Similar phenomena are also observed for different anions around tetraalkylphosphonium cations in a tetrahedral arrangement.34–36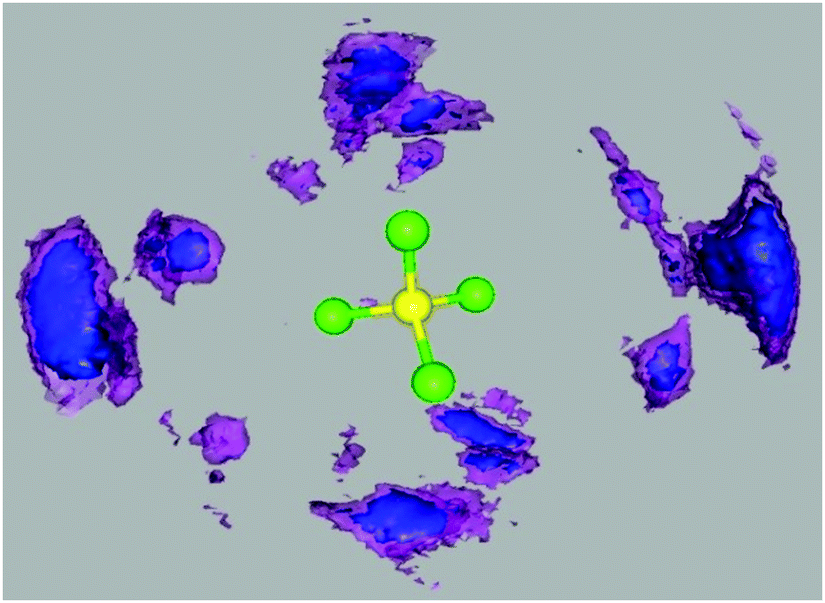 | ||
| Fig. 9 Three-dimensional probability distributions of boron atoms (blue/purple meshed surface) around a simplified representation of [P4,4,4,8]+ in neat [P4,4,4,8][BOB]. Purple and blue bounded contour surfaces are drawn at 5.0 and 6.0 times the average density of the corresponding boron atoms. The phosphorous atom and its directly connected four carbon atoms in [P4,4,4,8]+ are represented by yellow and green beads, respectively. | ||
3.7. TGA
The corresponding TGA curves for [P4,4,4,8][BOB] and [P4,4,4,8][Cl] are presented in Fig. 10. Both ILs were thermally stable up to ca. 300 °C with a further dramatic decomposition. Temperatures corresponding to the weight loss of 2, 5 and 10 wt%, as well as the percentage of mass loss at Tonset, are presented in Table 1.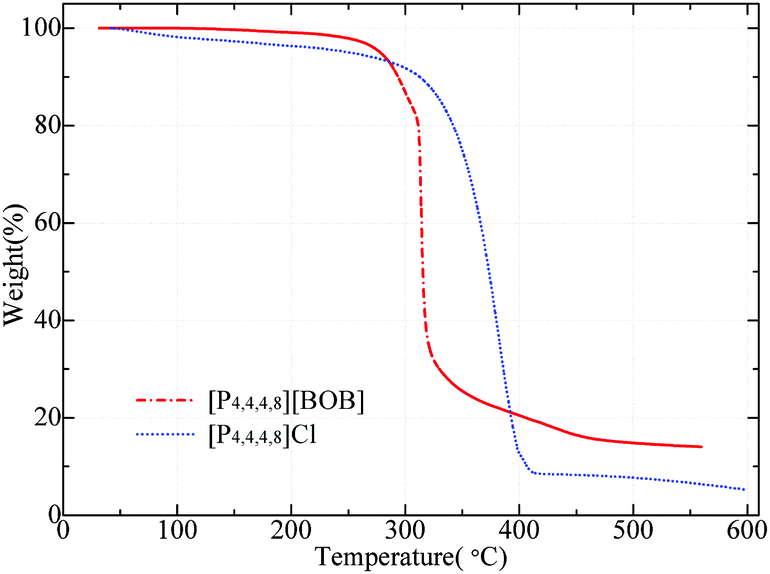 | ||
| Fig. 10 TGA curves for [P4,4,4,8][Cl] and [P4,4,4,8][BOB]. | ||
| T onset, °C | T 2%, °C | T 5%, °C | T 10%, °C | Δmonset, wt% | |
|---|---|---|---|---|---|
| [P4,4,4,8][Cl] | 325.85 | 109.23 | 251.02 | 312.60 | 13.93 |
| [P4,4,4,8][BOB] | 309.98 | 247.71 | 278.0 | 293.63 | 17.09 |
[P4,4,4,8][Cl] decomposed gradually between 300 and 400 °C. In contrast, the decomposition of [BOB]-containing IL was instant at ca. 320 °C. As claimed, the thermal stability of ILs containing symmetric or small ions (i.e. Cl−) is obviously higher due to a closer packing.37 The initial decomposition rate of [P4,4,4,8][BOB] was lower and in agreement with the simulated results. At the final stage [P4,4,4,8][Cl] demonstrated a slightly higher thermal stability and a loss of 10% by weight at 312.6 °C; whereas a similar weight loss was observed for [P4,4,4,8][BOB] at 293.6 °C (Table 1). [P4,4,4,8][BOB] was initially more stable losing 2 and 5 wt% at higher temperatures. ILs with oxygen-containing anions generally exhibit a two-step decomposition.38 The decomposition of phosphonium ILs consequently started from the anion and the thermal stability depended on the number of degradation steps.39 The decomposition of [P4,4,4,8][BOB] proceeded in two stages. It started with the cleavage of the B–O bond in [BOB]−. Presumably, the B–O bond in the [BOB]− anion was stable up to ca. 250–280 °C when the significant weight loss was detected. Meanwhile, in the [Cl]− salt the decomposition started from the cation and proceeded gradually. The DTA analysis results obtained for [P4,4,4,8][BOB] were in line with the TGA plot and the IL structure degraded at around 260 °C (ESI†).
With an increase in temperature, anionic products 2 and 3 were continuously accumulated in [P4,4,4,8][BOB] promoting more concurrent reactions with the cation. As a result, at a critical concentration of these species the rapid decomposition of [P4,4,4,8][BOB] was initiated at a temperature of around 320 °C. At this point, higher nucleophilicity of 2 than of [Cl]− should be also taken into account. Evidently, the decomposition process of [P4,4,4,8][BOB] was more complex and correlated with calculations. As shown by initial mass loss of two tested ILs and by calculations, [BOB]− started to decompose first. Hence, the thermal stability of tetraalkylphosphonium ILs could be promoted by improving the stability of the anion.
The results of TGA-MS tests of [P4,4,4,8][BOB] are presented in Fig. 11. The mass to charge ratios (m/z) (amu) for characteristic decomposition products were detected as follows:40 fragmented charged species (m/z: below 16); ethylene and CO (m/z: 28); oxygen and methanol (m/z: 32); and acetylene and allene (m/z: 40). The signal, which was assigned to ethylene and CO, was continuous and intensive. Meanwhile, as the temperature reached 400 °C the intensity of the peak assigned to the oxygen species decreased, indicating the consequent decomposition mechanism of [BOB]−, as shown by simulations. More complex decomposition products, acetylene and allene, were also detected.
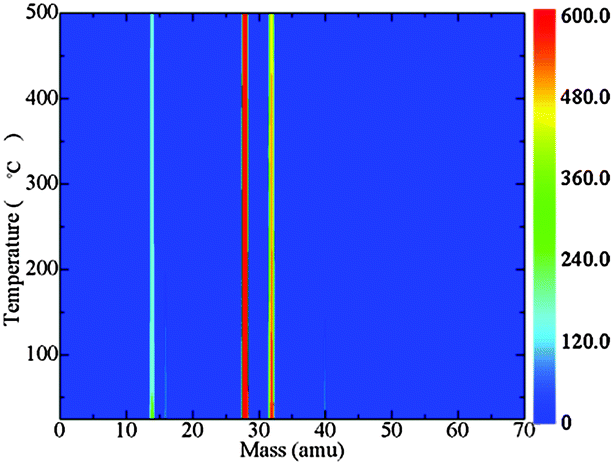 | ||
| Fig. 11 A TGA-MS diagram for [P4,4,4,8][BOB]: fragmented charged species (m/z: below 16); ethylene and CO (m/z: 28); oxygen and methanol (m/z: 32); acetylene and allene (m/z: 40). | ||
Our computational and experimental results along with the published data23,39 show the dominating role of the anion in the IL decomposition during both artificial aging and tribological tests. The decomposition process may be affected by the presence of water, which is one of the main impurities in IL samples originating from the preparation environment. Some ILs (i.e. [C4mim][NTf2]) are less thermally stable in the presence of water whilst the others (i.e. [C4mim][PF6] and [C4mim][Cl]) are more stable.41,42 Clearly, water may interact with the anion species. The water impact on the decomposition of [C2mim][BF4] was reported by Van Valkenburg et al. Up to 31 wt% of water was introduced into [C2mim][BF4], which was then subjected to TGA tests. The results showed no effect of the increasing water content on the thermal decomposition of [C2mim][BF4]. However, the authors observed the formation of HF from the [BF4]− in the presence of water.43 Depending of the cation type, [BOB]− may form highly acidic oxalates when in contact with water.44 Our further studies will unveil how water and the structure of the boron based anions ([BOB]−vs. bis(mandelato)borate, bis(salicylato)borate, and bis(malonato)borate) influence the decomposition mechanisms of phosphonium ILs.
4. Conclusions
The calculation procedure reported clearly explains the phenomena of thermal decomposition of [P4,4,4,8][BOB] and [P4,4,4,8][Cl]. Structural features of [P4,4,4,8][BOB] were evaluated and described by means of quantum chemistry calculations. In contrast to symmetric [BOB]−, the [P4,4,4,8]+ demonstrated a rigid and unstable central region, which was detected by vibration analysis and potential energy scans. No reaction with [BOB]− occurred during the decomposition of [P4,4,4,8]+. In contrast, the decomposition of neat [P4,4,4,8][BOB] started with a cleavage of a B–O bond in [BOB]− with an activation barrier of 246.3 kJ mol−1. Monocyclic anionic species were formed and then reacted with [P4,4,4,8]+, in which C–P bonds were least stable. The spatial orientation of ionic species is crucial as three alkyl chains in [P4,4,4,8]+ were oriented towards [BOB]− and reacted with it with an activation barriers from 268.1 to 310.9 kJ mol−1. The forth alkyl chain was isolated and a reaction between [BOB]− and a neutral trialkylphosphine molecule occurred at a higher activation barrier of 377.9 kJ mol−1. In all cases, trialkylphosphines, alkenes, CO and CO2 were identified from the reaction products. Alkylboranes were identified from the reaction of the anionic fragment with the C8 carbon chain. The product distribution suggested by computations was supported by TGA-MS results.[P4,4,4,8][Cl] showed a lower calculated stability as reactions with accessible alkyl chains occurred at barriers of 167.0 kJ mol−1. In the case of an isolated alkyl chain no reaction with [Cl]− anion was observed. Dominating effects of nucleophilicity and anion size were visible, i.e. [BOB]− decomposition fragments possessed stronger nucleophilic properties. On the other hand, TGA measurements indicated a higher thermal stability of [P4,4,4,8][Cl] at elevated temperatures. However, the initial weight loss, as shown by the tests, was less for [P4,4,4,8][BOB]. Hence, at the initial stage of the reaction the boron-containing IL was more stable, which correlated with simulated results. The thermal stability of [P4,4,4,8][BOB] can be improved by exchanging [BOB]− to a more stable anion.
Acknowledgements
The computations were performed on resources provided by the Swedish National Infrastructure for Computing (SNIC) at HPC2N. The Swedish Research Council, COST Action CM1206 and the Knut and Alice Wallenberg Foundation are acknowledged for the financial support. Alexander Lyubartsev and Inna Ermilova from the Department of Materials and Environmental Chemistry (Stockholm University) are personally acknowledged for their valuable advices. Jekabs Grins and Zoltan Bacsik from the Department of Materials and Environmental Chemistry (Stockholm University) are also acknowledged for their support during the TGA-MS and FTIR analyses.References
- A. E. Somers, P. C. Howlett, D. R. MacFarlane and M. Forsyth, Lubricants, 2013, 1, 3 CrossRef.
- I. Minami, Molecules, 2009, 14, 2286 CrossRef CAS PubMed.
- R. Lu, S. Mori, K. Kobayashi and H. Nanao, Appl. Surf. Sci., 2009, 255, 8965 CrossRef CAS.
- L. Pisarova, V. Totolin, C. Gabler, N. Dorr, E. Pittenauer, G. Allmaier and I. Minami, Tribol. Int., 2013, 65, 13 CrossRef CAS.
- R. Alamdrari, F. G. Zamani and M. Shekarriz, Orient. J. Chem., 2015, 31, 1127 CrossRef.
- Y. Hao, J. Peng, S. Hu, J. Li and M. Zhai, Thermochim. Acta, 2010, 501, 78 CrossRef CAS.
- M. Y. Keating, F. Gao and J. B. Ramsey, J. Therm. Anal. Calorim., 2011, 106, 207 CrossRef CAS.
- Y. Chen, Y. Cao, Y. Shi, Z. Xue and T. Mu, Ind. Eng. Chem. Res., 2012, 51, 7418 CrossRef CAS.
- S. D. Chambreau, J. A. Boatz, G. L. Vaghjiani, C. Koh, O. Kostko, A. Golan and S. R. Leone, J. Phys. Chem. A, 2012, 116, 5867 CrossRef CAS PubMed.
- J. L. Anderson, R. F. Ding, A. Ellern and D. W. Armstrong, J. Am. Chem. Soc., 2005, 127, 593 CrossRef CAS PubMed.
- J. Alvarez, D. Gomis, P. Abrodo, D. A. Llorents, E. Busto, N. Lombardia, V. G. Fernandez and M. D. G. Alvarez, Anal. Bioanal. Chem., 2011, 400, 1209 CrossRef CAS PubMed.
- M. C. Kroon, W. Buijs, C. J. Peters and H.-J. Witkamp, Thermochim. Acta, 2007, 465, 40 CrossRef CAS.
- S. Sowmiah, V. Srinivasadesikan, M.-C. Tseng and Y.-H. Chu, Molecules, 2009, 14, 3780 CrossRef CAS PubMed.
- C. Maton, N. De Vos and V. Stevens, Chem. Soc. Rev., 2013, 42, 5963 RSC.
- K. Tsunashima, Y. Ono and M. Sugiya, Electrochim. Acta, 2011, 56, 4351 CrossRef CAS.
- S. H. Oh, T. Yim, E. Pomerantseva and L. F. Nazar, Electrochem. Solid. St., 2011, 14, 185 CrossRef.
- E. Zinigrad, L. Larush-Asraf, G. Salitra, M. Sprecher and D. Aurbach, Thermochim. Acta, 2007, 457, 64 CrossRef CAS.
- Y. Wang, L. Xing, X. Tang, X. Li, W. Li, B. Li, W. Huang, H. Zhou and X. Li, RSC Adv., 2014, 4, 33301 RSC.
- T. Ramnial, D. I. Daisuke and J. A. C. Clyburne, Chem. Commun., 2005, 325 RSC.
- M. C. Tseng, H. C. Kan and Y. H. Chu, Tetrahedron Lett., 2007, 48, 9085 CrossRef CAS.
- M. T. Clough, K. Geyer, P. A. Hunt, J. Mertes and T. Welton, Phys. Chem. Chem. Phys., 2013, 15, 20480 RSC.
- E. Thomas, D. Thomas, K. P. Vijayalakshmi and B. K. George, RSC Adv., 2016, 6, 9421 RSC.
- F. U. Shah, S. Glavatskih, D. R. MacFarlane, A. Somers, M. Forsyth and O. N. Antzutkin, Phys. Chem. Chem. Phys., 2011, 13, 12865 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 (Revision E.01), Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- A. Lyubartsev and A. Laaksonen, Comput. Phys. Commun., 2000, 12, 565 CrossRef.
- Y.-L. Wang, F. U. Shah, S. Glavatskih, O. N. Antzutkin and A. Laaksonen, J. Phys. Chem. B, 2014, 118, 8711 CrossRef CAS PubMed.
- M. Tuckerman, B. J. Berne and G. J. Martyna, J. Chem. Phys., 1992, 97, 1990 CrossRef CAS.
- L. A. Laaksonen, J. Mol. Graphics, 1992, 10, 33 CrossRef CAS PubMed.
- D. L. Bergman, L. Laaksonen and A. Laaksonen, J. Mol. Graphics Modell., 1997, 15, 301 CrossRef CAS PubMed.
- W. Liu, C. Ye, Q. Gong, H. Wang and P. Wang, Tribol. Lett., 2002, 13, 81 CrossRef CAS.
- D. J. Degonia and P. G. Griffin, E.P., 0395258B1, 1994 Search PubMed.
- V. Pejakovic, C. Tomastik, N. Dorr and M. Kalin, Tribol. Int., 2016, 97, 234 CrossRef CAS.
- V. Totolin, I. Minami, C. Gaber and N. Dorr, Tribol. Int., 2013, 67, 191 CrossRef CAS.
- Y.-L. Wang, S. Sarman, B. Li and A. Laaksonen, Phys. Chem. Chem. Phys., 2015, 17, 22125 RSC.
- Y.-L. Wang, S. Sarman, S. Glavatskih, O. N. Antzutkin, M. W. Rutland and A. Laaksonen, J. Phys. Chem. B, 2015, 119, 5251 CrossRef CAS PubMed.
- Y.-L. Wang, M. Shimpi, S. Sarman, O. N. Antzutkin, S. Glavatskih, L. Kloo and A. Laaksonen, J. Phys. Chem. B, 2016 DOI:10.1021/acs.jpcb.6b02921.
- W. C. Barnhill, J. Qu, H. Luo, H. M. Meyer, C. Ma, M. Chi and B. L. Papke, ACS Appl. Mater. Interfaces, 2014, 6, 22585 CAS.
- B. Yu, D. G. Bansal, J. Qu, X. Sun, H. Luo, S. Dai, P. J. Blau, B. G. Bunting, G. Mordukhovich and D. J. Smolenski, Wear, 2012, 289, 58 CrossRef CAS.
- A. Hernandez Battez, M. Bartolome, D. Blanco, J. L. Viesca, A. Fernandez-Gonzalez and R. Gonzalez, Tribol. Int., 2016, 95, 118 CrossRef CAS.
- Search for Species Data by Molecular Weight, National Institute of Standards and Technology, http://webbook.nist.gov/chemistry/mw-ser.html, accessed January 2016.
- J. G. Huddleston, A. E. Visser, W. M. Reichert, H. D. Willauer, G. A. Broker and R. D. Rogers, Green Chem., 2001, 3, 156 RSC.
- F. Wendler, L.-N. Todi and F. Meister, Thermochim. Acta, 2012, 528, 76 CrossRef CAS.
- M. E. Van Valkenburg, R. L. Vaughn, M. Williams and J. S. Wilkes, Thermochim. Acta, 2005, 425, 181 CrossRef CAS.
- R. Gusain and O. P. Khatri, RSC Adv., 2015, 5, 25287 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp03191a |
| This journal is © the Owner Societies 2016 |