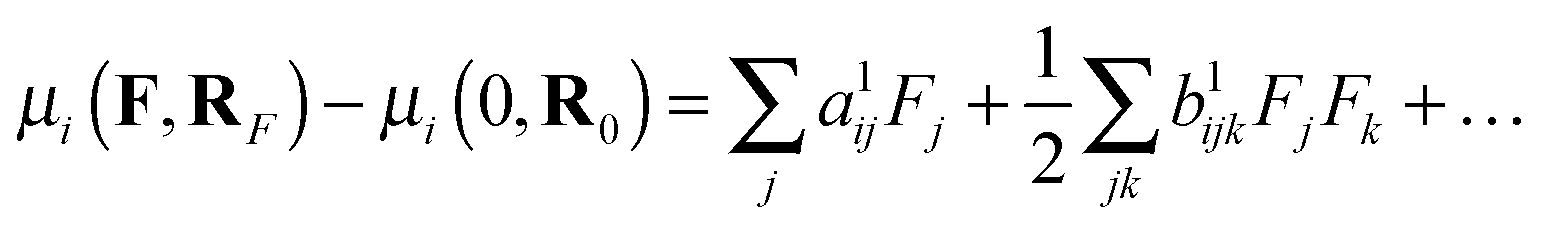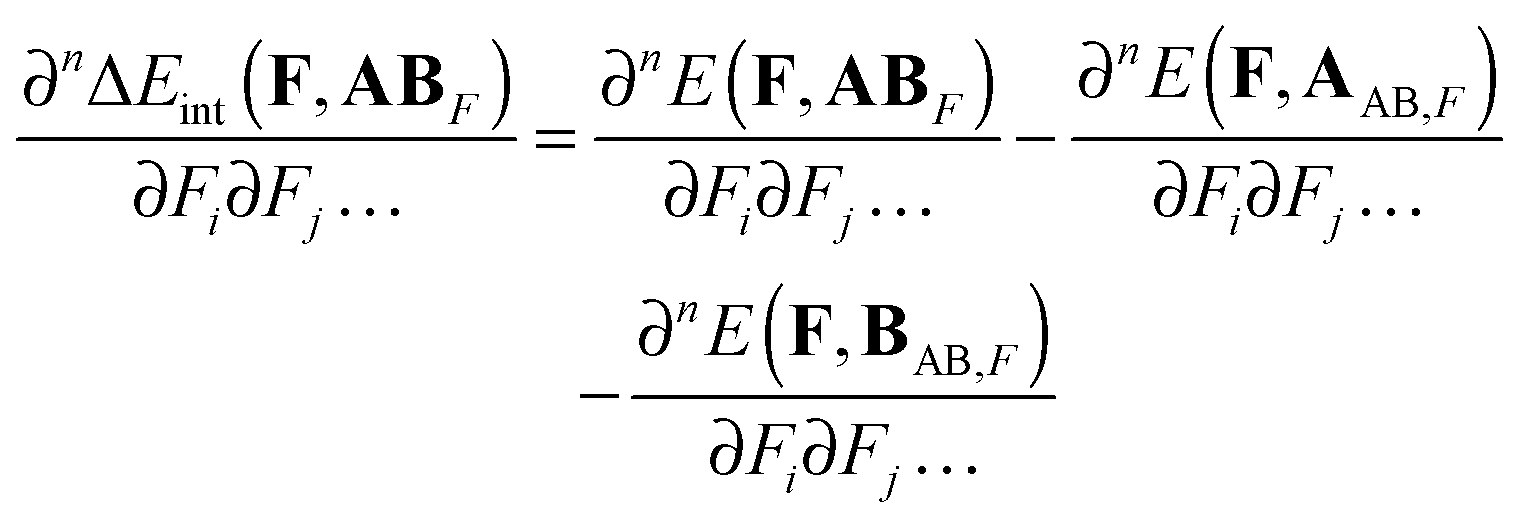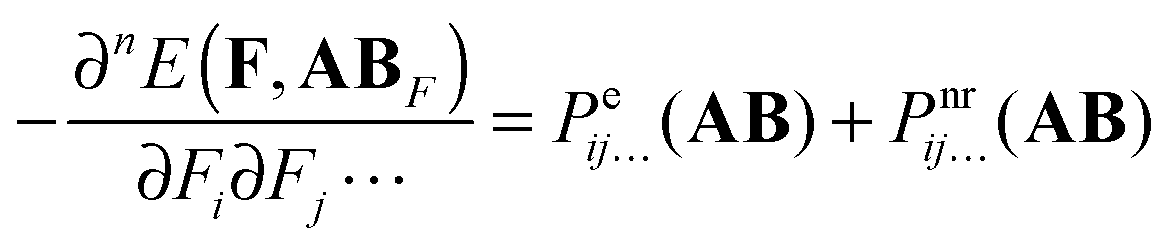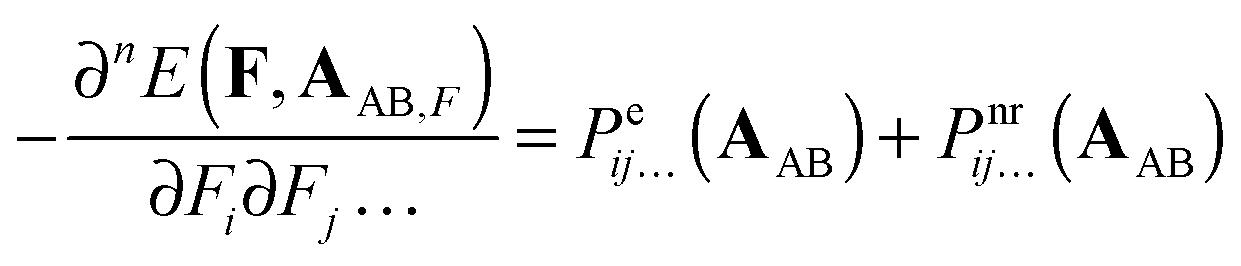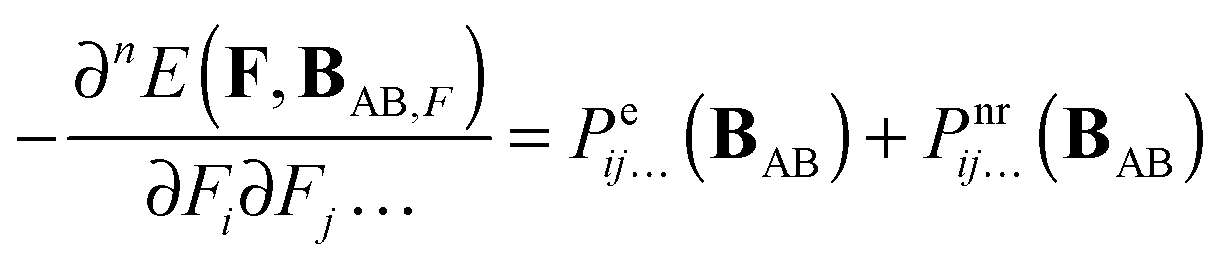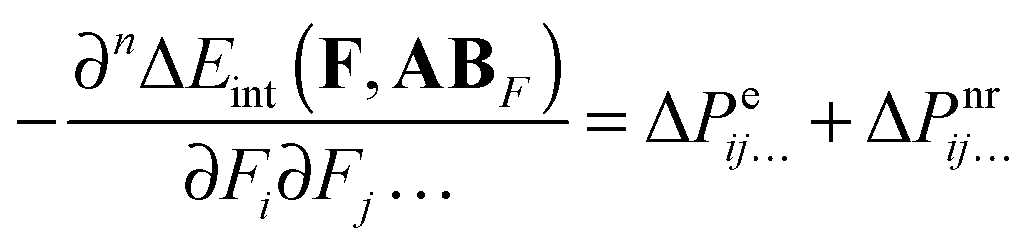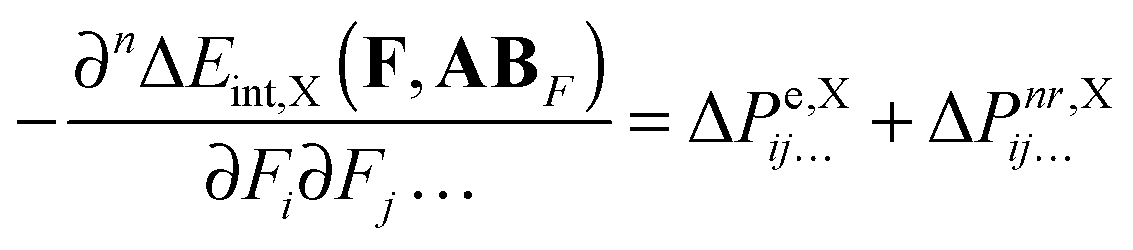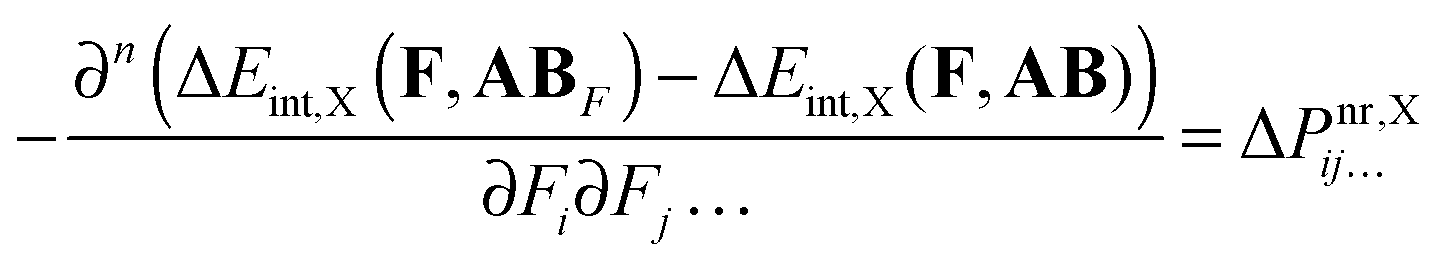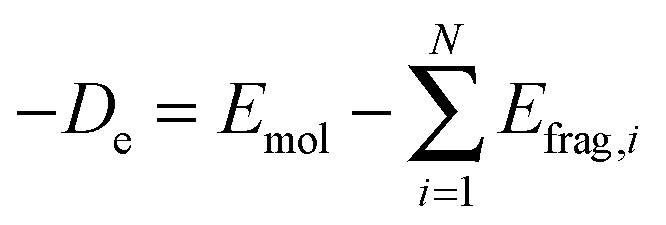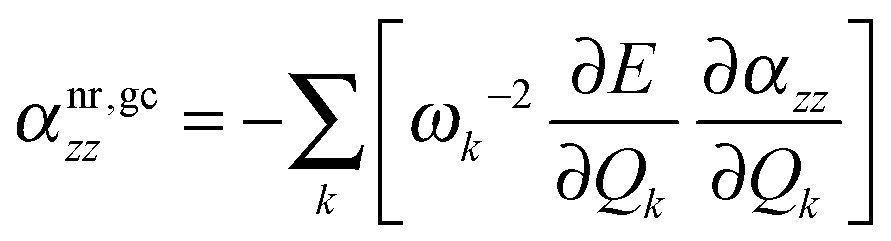On the physical origins of interaction-induced vibrational (hyper)polarizabilities†
Received
14th April 2016
, Accepted 18th July 2016
First published on 19th July 2016
Abstract
This paper presents the results of a pioneering exploration of the physical origins of vibrational contributions to the interaction-induced electric properties of molecular complexes. In order to analyze the excess nuclear relaxation (hyper)polarizabilities, a new scheme was proposed which relies on the computationally efficient Bishop–Hasan–Kirtman method for determining the nuclear relaxation contributions to electric properties. The extension presented herein is general and can be used with any interaction-energy partitioning method. As an example, in this study we employed the variational–perturbational interaction-energy decomposition scheme (at the MP2/aug-cc-pVQZ level) and the extended transition state method by employing three exchange–correlation functionals (BLYP, LC-BLYP, and LC-BLYP-dDsC) to study the excess properties of the HCN dimer. It was observed that the first-order electrostatic contribution to the excess nuclear relaxation polarizability cancels with the negative exchange repulsion term out to a large extent, resulting in a positive value of Δαnr due to the contributions from the delocalization and the dispersion terms. In the case of the excess nuclear relaxation first hyperpolarizability, the pattern of interaction contributions is very similar to that for Δαnr, both in terms of their sign as well as relative magnitude. Finally, our results show that the LC-BLYP and LC-BLYP-dDsC functionals, which yield smaller values of the orbital relaxation term than BLYP, are more successful in predicting excess properties.
1 Introduction
Great attention has been paid to the materials characterized by a large nonlinear optical response, as they are commonly used in optical rectification, modern waveguides, and optical communication technology and for optical switching and signal processing.1 At the molecular level, the linear and nonlinear optical properties are given by the linear polarizability (α) and first (β) and second (γ) hyperpolarizabilities, respectively. However, the molecular properties can be greatly influenced by the intermolecular interactions in the bulk material.2–5 The latter effect can be attributed to the interaction-induced (excess) properties, which are defined as the difference between a property of the complex and the sum of the properties of the noninteracting subsystems. The interest in excess properties and collision-induced spectra has evolved over the last two decades from pairs of interacting atoms6–20 to complexes involving molecules.21–43 An extensive body of literature exists on the purely electronic interaction-induced electric properties, including various aspects of their theoretical treatment. Also, several methods were proposed for partitioning the excess electric properties on the grounds of intermolecular perturbation theory, as it leads to a better understanding of their origins.7,10 The most comprehensive approach was proposed by Moszyński et al.,10 who formulated a symmetry-adapted perturbation theory of interaction-induced electric properties. A similar approach was followed by other authors, who carried out an exploration of the contributions to purely electronic excess properties.23,27,39,40 Below, we shall briefly summarize the most relevant findings.
Skwara et al.23 performed the decomposition of the excess electric properties (dipole moment, polarizability and first hyperpolarizability) of the linear HF dimer into interaction energy components. They highlighted a subtle balance of the electrostatic and exchange repulsion contributions. Góra et al. studied the excess electric properties of a set of hydrogen-bonded systems, encompassing quasi-linear dimers of hydrogen cyanide, urea, diformamide, 4-pyridone, 4-nitroaniline, and the complex of hydrogen fluoride with nitroacetylene.27,40 These authors found that the origins of the interaction-induced dipole moment are purely electrostatic in nature with only a minor contribution from the charge-delocalization term and negligible remaining components. On the other hand, the origins of interaction-induced polarizabilities were slightly more complicated even though they also seem to be dominated by the first-order electrostatic term.40 In particular, in some of the studied complexes with bifurcated hydrogen bonds the exchange-repulsion effects were found to quench completely the associated first-order electrostatic term. Thus, the excess polarizabilities of smaller complexes were shown to be dominated by induction (polarization) and exchange-induction effects.40 In the case of excess first hyperpolarizability Góra and Błasiak reported that its origin was much more system-dependent than other electric properties. For example, in the case of the urea dimer, the excess first hyperpolarizability was found to be diminished due to induction and exchange-induction terms, whereas in diformamide and 4-pyridone dimers the observed reduction was primarily due to exchange-repulsion effects.40 Even the dispersion contribution, which is in most cases negligible, may sometimes become unusually large, e.g. it contributes more than 15% to the excess electronic first hyperpolarizability of the p-aminobenzoic acid⋯benzene complex.43 Excess electronic polarizabilities for a large set of nucleic acid base pair conformations were studied by Czyżnikowska et al.39 It turned out that the physical origins of this property are quite different for stacked and hydrogen-bonded base pairs.39 In the case of the stacked cytosine dimers the leading contribution to excess polarizability was due to the electrostatic contribution. The interaction pattern for Watson–Crick guanine–cytosine and adenine–thymine pairs was found to be different, i.e. the excess polarizability was dominated by exchange and induction interactions and electrostatics played only a minor role.39 The effects of many-body interactions on excess properties were also thoroughly examined.22,27
Finally, it should not be overlooked that several approaches to estimate partial contributions of atoms or molecular fragments to (hyper)polarizabilities have also been developed. Ángyán and co-workers have calculated distributed (hyper)polarizabilities using the atoms in molecules (AIM) partitioning scheme.44–46 Their approach allows the analysis of local induced polarizabilities and charge flow between different regions of a molecule. Ye and Autschbach have investigated local contributions to the first hyperpolarizability of donor–acceptor substituted molecules by means of the Mulliken type and natural bond orbital (NBO) analyses.47 Their results highlight the contributions from atoms and bonds on different functional groups to the total value of the first hyperpolarizability. Yang and coworkers proposed a decomposition scheme of the first hyperpolarizability by means of Hirshfeld partitioning analysis.48–50 In their analysis the hyperpolarizabilities are decomposed into local and charge-transfer parts. A quite different approach is the concept of the (hyper)polarizability densities developed by Nakano and coworkers,51–53 which are defined as the derivatives of the Mulliken charge densities with respect to the electric field. Their (hyper)polarizability density plots allow the visualization of the local contributions to the total properties.
At this point it is important to highlight that rather insufficient attention was paid to the vibrational contributions to interaction-induced electric properties. A preliminary treatment of this subject can be found in the paper of Avramopoulos et al.,54 who examined the influence of relativistic effects on the interaction-induced dipole moment and polarizability of the HF–AuH complex including the effect of molecular vibrations. These authors demonstrated, based on the low-order perturbation method of Bishop and Kirtman (BKPT),55 that the vibrational contribution to the induced polarizability is approximately an order of magnitude larger than its electronic counterpart.54 It should be noted, though, that they argued that this finding may significantly depend on approximations used in the low-order perturbation treatment.
Our aim in this work is to perform a pioneering exploration of the physical origins of nuclear relaxation contributions to the excess electric properties of molecular complexes. The HCN dimer is chosen as a model system for this purpose, mainly because its electronic excess properties were already thoroughly studied in our previous paper.27
2 Theory and computational details
This section is organized as follows. First, we outline the computational treatment of vibrational (hyper)polarizabilities. Next, we describe a framework for the partitioning of the excess vibrational (hyper)polarizabilities. Finally, we present the interaction energy decomposition schemes employed in this work.
2.1 Vibrational (hyper)polarizabilities
Within the Born–Oppenheimer approximation, one may define the total property P as the sum of electronic (Pe), nuclear relaxation (Pnr) and curvature (Pcurv) contributions:56Pnr and Pcurv arise from the change of the electronic and vibrational energies caused by the field-induced relaxation of the equilibrium geometry, respectively. The nuclear relaxation contributions contain all the leading terms of the BKPT formulas, including the anharmonic corrections of the lowest order. The curvature contributions, which include the rest of the high-order anharmonicity, are far more computationally expensive than Pnr and they are not computed here. Bishop, Hasan and Kirtman (BHK) demonstrated that one may employ a computationally efficient finite-field nuclear relaxation (FF-NR) formalism to evaluate the nuclear relaxation contribution to (hyper)polarizabilities.57,58 In the present study, we confine the analysis solely to static properties, including polarizability and first hyperpolarizability. As far as the nuclear relaxation contribution is concerned, following BHK we define:57| | 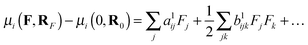 | (2) |
where μ(F,RF) is the dipole moment obtained at the field-relaxed geometry and μ(0,R0) is the same property for field-free conditions. The expansion coefficients yield the static properties:| | | a1ij = αeij(0;0) + αnrij(0;0) | (3) |
| | | b1ijk = βeijk(0;0,0) + βnrijk(0;0,0) | (4) |
When applying these FF-NR formulas, one should not overlook that the geometry relaxation must not include the rotations of the molecule through the alignment of the permanent and(or) induced dipole moment in the field direction (indeed this may be the easiest way for the system to lower its energy). Therefore, the field-dependent optimization must be performed strictly maintaining the Eckart conditions.59 Such optimizations can be carried out with the aid of the procedure developed by Luis et al.60
2.2 Excess vibrational (hyper)polarizabilities and their partitioning
Let the equilibrium geometry of a complex composed of A and B in an applied electric field, F, be denoted by ABF, while E(F,AAB,F) and E(F,BAB,F) are the field-dependent energy of A and B at the geometry corresponding to the field-relaxed geometry of ABF. AB stands for the equilibrium geometry of the complex without the field present. The field-dependent interaction energy at field-relaxed geometry ABF is thus given by:| | | ΔEint(F,ABF) = E(F,ABF) − E(F,AAB,F) − E(F,BAB,F) | (5) |
The derivative of ΔEint(F,ABF) with respect to electric field components along Cartesian directions i,j,… is given by:| | 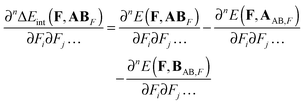 | (6) |
For n > 1 the first term on the r.h.s. of eqn (6) can be recognized as the sum of the static electronic (hyper)polarizability (Pe) and the nuclear relaxation (Pnr) of a complex AB:| | 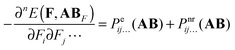 | (7) |
The two remaining terms on the r.h.s of eqn (6) can also be split in the electronic and nuclear-relaxation contributions to a property P of subsystems A and B, respectively. Henceforth, we will refer to Pnr(AAB) and Pnr(BAB) as pseudo-nuclear relaxation (pseudo-NR) contributions to monomer properties. We add the prefix pseudo because these monomeric contributions are computed at geometries corresponding to interacting AB, instead of the equilibrium geometry of A and B, respectively:| | 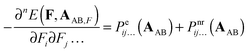 | (8) |
| | 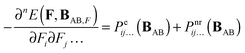 | (9) |
Combining eqn (6) with eqn (7–9) yields:| | 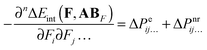 | (10) |
where| | | ΔPeij… = Peij…(AB) − Peij…(AAB) − Peij…(BAB) | (11) |
| | | ΔPnrij… = Pnrij…(AB) − Pnrij…(AAB) − Pnrij…(BAB) | (12) |
Since ΔEint can be represented as the sum of interaction energy components, Eint,X, it is possible to re-write eqn (10):| | 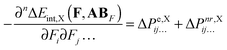 | (13) |
Finally, the above equation can be presented in a form that allows us to directly analyze the ΔPnr contribution in terms of interaction-energy components ΔEint,X:| | 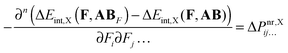 | (14) |
2.3 Interaction energy partitioning schemes
In the preceding section we have presented a framework for the partitioning of nuclear-relaxation (hyper)polarizabilities. However, such partitioning is general and it still requires a particular choice of interaction energy decomposition scheme. Since the seminal work of Morokuma, who was the first to introduce a robust scheme for the decomposition of supermolecular interaction energy into physically meaningful components based on analysis and interpretation of the Fock matrix elements,61 many similar approaches have been proposed including the extended transition state (ETS) method of Ziegler and Rauk,62 the improved Kitaura–Morokuma (KM) scheme,63 the constrained space orbital variation (CSOV),64 the reduced variational space self-consistent field (RVS-SCF),65 the natural energy decomposition analysis (NEDA),66 the block-localized wavefunction approach (BLW-ED)67 and the method recently proposed by Su and Li.68 There is also a variational-perturbational decomposition scheme (VP-EDS)69–72 which, like the above schemes, is based on the partitioning of the supermolecular interaction energy. However, in this scheme the interpretation of the obtained components refers to the intermolecular perturbation theory.73 Essentially, in this type of analysis the total interaction energy obtained in a supermolecular approach is partitioned into a selection of interaction energy terms analogous to the ones defined in state-of-the-art symmetry-adapted perturbation theory (SAPT).69–72
In this paper we decompose electronic and vibrational contributions to electric excess properties using the ab initio VP-EDS scheme and the ETS method within the Kohn–Sham density functional theory (KS-DFT) framework. Although the VP-EDS and KS-DFT schemes were previously used for the partitioning of interaction-induced properties,23,27,39,40 the reliability of the latter approach is largely determined by the choice of exchange–correlation potential. It can be argued that the excess property partitioning imposes additional requirements regarding the choice of density functional. Unfortunately, there is not much known about the performance of density functionals in computations of excess properties and available data are to some extent contradictory. Zawada et al. studied the purely electronic contributions to the interaction-induced electric properties by employing several exchange–correlation functionals, including B3LYP, LC-BLYP, PBE0, M06-2X and CAM-B3LYP, and concluded that their overall performance is satisfactory, at least for hydrogen-bonded chains consisting of HF, H2CO and NH3 molecules.29 More pessimistic conclusions regarding the reliability of the most popular functionals were drawn by Baranowska-Łączkowska et al., based on the results of computations of excess properties for CO– and N2–(HF)n complexes with n = 1,…,4.41 These authors reported that in the case of long chains, range-separated hybrid functionals or functionals with a large fraction of exact exchange are necessary to avoid errors exceeding 20%.41 Maroulis employed the B1LYP, B3LYP, B3PW91 and mPW1PW91 exchange–correlation functionals and determined the interaction-induced properties of the water dimer.36 Their comparison against the CCSD(T) reference values revealed that none of the functionals correctly predicts the sign (and magnitude) of the interaction-induced mean second hyperpolarizability.36 Moreover, a recent study by Bulik et al. demonstrates that computations of vibrational contributions by means of density functional theory still pose serious challenges.74 To bring the discussion to a close, let us conclude that an insufficient amount of data regarding the performance of DFT was one of the factors that motivated us to employ two decomposition schemes for the excess property partitioning. We thus decompose electronic and vibrational contributions to electric excess properties, using the ab initio VP-EDS and ETS schemes with a selection of exchange–correlation functionals.
2.3.1 VP-EDS scheme.
According to the VP-EDS scheme, the total interaction energy of a dimer, calculated by a supermolecular approach in the dimer-centered basis set (DCBS),75 using the second-order Møller–Plesset perturbation theory (MP2) is partitioned into the Hartree–Fock (HF) and the electron correlation interaction energy components:| | | ΔEMP2int = ΔEHFint + ΔEMP2corr | (15) |
The HF term can be further partitioned into the electrostatic interactions of unperturbed monomer charge densities, ε(10)el, as well as the associated exchange repulsion (ΔEHLex), and the charge delocalization (ΔEHFdel) term encompassing the induction and the associated exchange effects due to the Pauli exclusion principle:| | | ΔEHFint = ε(10)el + ΔEHLex + ΔEHFdel | (16) |
The second-order electron correlation term, ΔEMP2corr,| | | ΔEMP2corr = ε(12)el,r + ε(20)disp + ΔE(2)ex | (17) |
includes the second order dispersion interaction, ε(20)disp, as well as the electron correlation correction to the first order electrostatic interaction, ε(12)el,r, and the remaining electron correlation effects (ΔE(2)ex). The latter term accounts mainly for the uncorrelated exchange-dispersion and electron correlation corrections to the Hartree–Fock exchange repulsion.71,73 The ε(10)el and the ε(20)disp terms are obtained in the standard polarization perturbation theory, whereas the ε(12)el,r term is calculated using the formula proposed by Moszyński et al.76 The indices in parentheses denote perturbation orders in the intermolecular interaction operator and intramonomer correlation operator, respectively.
2.3.2 ETS scheme.
The ETS approach, which is usually carried out in the framework of density functional theory, can be employed to analyze chemical bonds between the fragments of a molecule. The energy of chemical bonds (De) is defined as the difference between the total energy of a molecule Emol and the sum of the energies of the fragments Efrag:| | 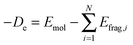 | (18) |
The dissociation energy may be split into two terms, the preparation energy (ΔEprep) and the interaction energy (ΔEint):The first one is given by two contributions: (i) the energy required to distort the equilibrium geometry of the isolated fragments to the geometry that they have in the molecule; and (ii) the energy necessary to excite the electronic ground states of the fragments to the states with the electronic configuration suitable to form the bonds. Obviously, if the fragments are atoms, ΔEprep = 0. The interaction energy, which is the difference between the total energy of the molecule and the sum of the energies of the prepared fragments, can be split into three different contributions: (i) the electrostatic interaction energy between the frozen charge densities of the fragments (ΔEel); (ii) the exchange repulsion energy due to the antisymmetrization and renormalization of the Hartree product of the fragment wavefunctions (ΔEPauli); and (iii) the orbital relaxation energy (ΔEorb):| | | ΔEint = ΔEel + ΔEPauli + ΔEorb | (20) |
It is interesting to note that if this scheme is applied to a weakly bound complex with the fragments chosen as interacting molecules, it becomes essentially equivalent to the partitioning of ΔEHFint in VP-EDS. However, if carried out within the KS-DFT framework, the obtained contributions also account for the intra-monomer correlation and to some extent the inter-monomer correlation effects (i.e. dispersion interaction), which in VP-EDS are included via the Møller–Plesset perturbation theory. In this paper we compare the ab initio VP-EDS and KS-DFT ETS partition of the electronic and vibrational hyperpolarizabilities to analyze the performance of BLYP, LC-BLYP and LC-BLYP-dDsC functionals.
2.4 Software and computational details
Geometry optimizations and vibrational structure calculations using MP2, CCSD and CCSD(T) methods were performed using the Gaussian suite of programs77 while property calculations were carried out using custom computer programs. VP-EDS calculations were carried out using a modified version of the Gamess (US) program.78,79 Finally, all DFT calculations were done using ADF program.80
3 Results and discussion
In a series of papers, Baranowska and collaborators carried out the assessment of basis sets on the excess electric properties24–26,38,41,42 and advocated the use of basis sets specifically tailored for such types of calculations (for the analysis of schemes for eliminating the basis set superposition error in calculations of the properties in question we refer to ref. 81 and 82). This is in line with the conclusions presented by Skwara et al.22 However, as demonstrated recently by some of us, property-oriented basis sets are not always the best choice to predict the nuclear relaxation (hyper)polarizabilities.83 On the other hand, it was reported in the same study that the average errors in these properties associated with the aug-cc-pVTZ basis set can be negligible (i.e. less than 1%) for small molecules. Considering these findings, in the present study we used Dunning's correlation-consistent basis sets aug-cc-pVXZ (X = D, T, Q)84–86 for VP-EDS calculations.
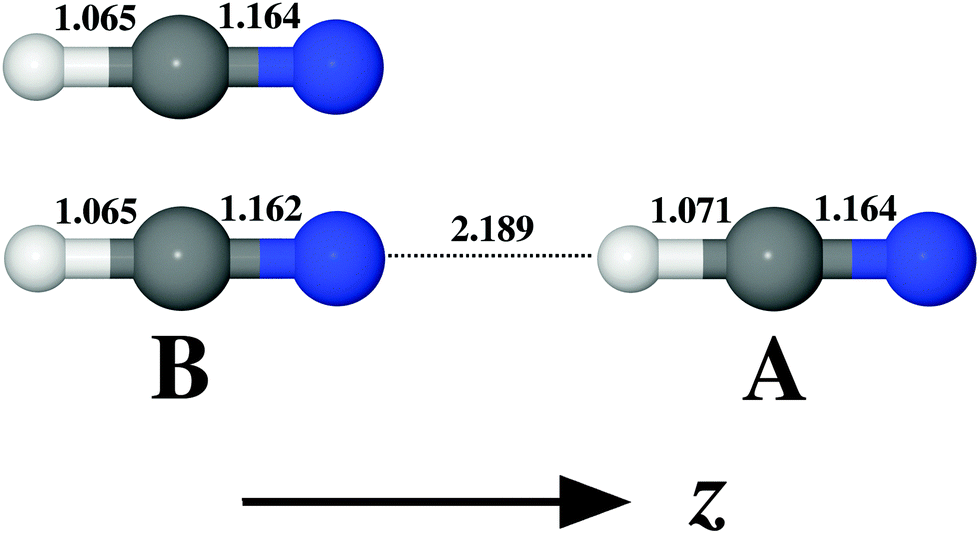
The structure and geometrical parameters for the HCN monomer and the dimer are shown in Fig. 1. The most remarkable change in the monomer geometry upon the complex formation is the increase in the C–H bond length from 1.065 Å (isolated monomer) to 1.071 Å (monomer A in the complex AB). There is also an accompanying shortening of the C–N bond length by 0.002 Å in the case of monomer B. In comparison with the isolated monomer, upon the complex formation one thus expects a much more significant perturbation to the vibrational structure and properties of A than to B. For the exhaustive analysis of the energetics of (HCN)2 isomer formation we refer to the study of Smith et al.87 and to the ESI in ref. 27 and references cited therein.
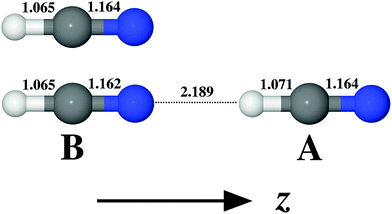 |
| | Fig. 1 Geometrical parameters (given in [Å] units) for the monomer and the dimer of HCN and the orientation of molecules in the Cartesian coordinate system. Equilibrium geometries were obtained at the MP2/aug-cc-pVQZ level of theory. | |
MP2 and coupled-cluster electronic and vibrational contributions to α and β of the HCN monomer and the dimer are shown in Table 1. The electronic electric-dipole properties of the monomer and the dimer were also studied by other authors and there is a good agreement between their results and the values reported in Table 1.26,27,88 It should be highlighted that there is a satisfactory convergence of monomer and dimer properties with respect to the basis set size, and the aug-cc-pVTZ basis set already delivers accurate estimates. Then, although in the following analysis we will focus on the data obtained at the MP2/aug-cc-pVQZ level, the same conclusions are obtained from aug-cc-pVDZ and aug-cc-pVTZ basis set results. We can also see that the MP2 results are in fairly good agreement with those provided by more sophisticated CCSD and CCSD(T) methods. This is in line with results obtained for the polarizabilities of long polyynes.89,90
Table 1 Electronic and nuclear relaxation polarizability and first hyperpolarizability of the HCN monomer and dimer (given in a.u.) at equilibrium geometries corresponding to the indicated methods
| Method |
α
e
zz
|
α
nr
zz
|
β
e
zzz
|
[μα](0,0)zzz |
[μ3](1,0)zzz |
[μ3](0,1)zzz |
β
nr
zzz
|
|
|
HCN |
| MP2/aug-cc-pVDZ |
22.80 |
0.17 |
3.40 |
−8.36 |
<0.01 |
−0.26 |
−8.62 |
| MP2/aug-cc-pVTZ |
22.20 |
0.18 |
0.43 |
−8.97 |
−0.02 |
−0.29 |
−9.28 |
| MP2/aug-cc-pVQZ |
22.09 |
0.18 |
−0.62 |
−9.14 |
−0.02 |
−0.29 |
−9.45 |
|
|
|
|
HCN⋯HCN |
| MP2/aug-cc-pVDZ |
50.60 |
4.84 |
−37.41 |
−163.75 |
−257.45 |
197.78 |
−223.42 |
| MP2/aug-cc-pVTZ |
49.15 |
4.71 |
−39.45 |
−157.04 |
−242.81 |
180.98 |
−218.87 |
| CCSD/aug-cc-pVTZ |
48.66 |
4.54 |
−29.91 |
— |
— |
— |
−210.50 |
| CCSD(T)/aug-cc-pVTZ |
49.92 |
4.71 |
−28.00 |
— |
— |
— |
−224.64 |
| MP2/aug-cc-pVQZ |
48.89 |
4.75 |
−39.84 |
−157.65 |
−246.61 |
189.39 |
−214.87 |
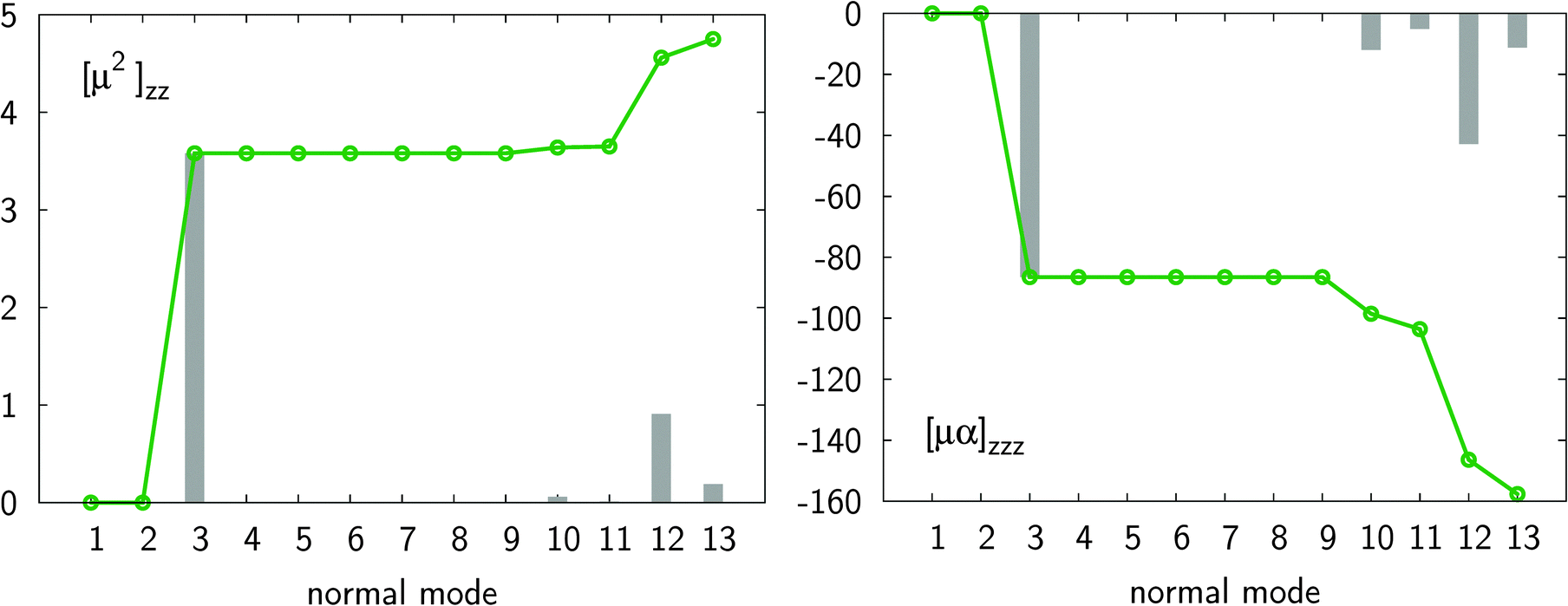
A comparison of the properties of the monomer with those of the dimer reveals a few interesting conclusions. For both systems the electronic polarizability is much larger than the nuclear relaxation polarizability, with the αnr/αe ratio increasing from 0.008 (monomer) up to 0.097 (dimer). In contrast to polarizability, βnr prevails over βe for both the monomer and the dimer, as the βnr/βe ratio is about 15 for the monomer and 5 for the dimer. Upon the complex formation there is a large increase in the nuclear relaxation contributions to polarizability (roughly 30-fold) and first hyperpolarizability (roughly 20-fold). In order to elucidate the origins of this enhancement we have performed normal mode analysis based on the Bishop–Kirtman perturbation theory. Fig. 2 shows that the major contribution of double harmonic diagonal terms (i.e. [μ2](0,0) and [μα](0,0)) is due to the vibrational normal mode of frequency ν3 = 123 cm−1, which corresponds to the intermolecular symmetric stretching along the principal symmetry axis (see also Fig. 3).
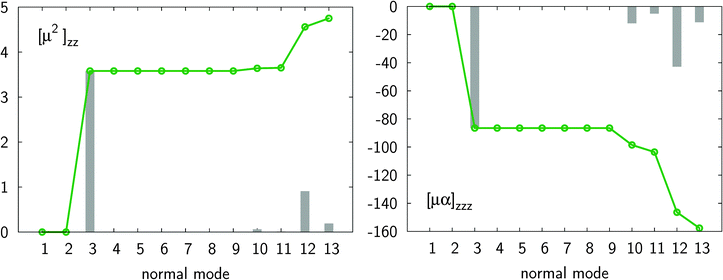 |
| | Fig. 2 Convergence of longitudinal components of [μ2](0,0) and [μα](0,0) for the HCN dimer with respect to the vibrational normal modes (dotted curves). Individual contributions are represented by gray bars. All results were obtained at the MP2/aug-cc-pVQZ level of theory. | |
 |
| | Fig. 3 Atomic displacements for vibrational normal mode of frequency ν3 = 123 cm−1 for the HCN dimer. See the text for more details. | |
There is only a negligible net anharmonic contribution to nuclear hyperpolarizability of the monomer (3.28%). On the contrary, although the electrical and mechanical anharmonic contributions to βnr of the dimer cancel each other out to a large extent, their net contribution is still substantial (26.63%). In contrast to other problematic hydrogen-bonded systems, like for instance the water dimer or the hydrogen fluoride dimer studied by Eckart and Sadlej,91 the data in Table 1 do not indicate that the BKPT expansion is likely to diverge.
We will now turn to the excess properties. Table 2 contains the values of electronic and nuclear relaxation interaction-induced polarizability and hyperpolarizability computed based on eqn (11) and (12). Although we noted earlier that αe is far larger than αnr for both the monomer and the dimer, the excess electronic and nuclear relaxation polarizabilities are similar in magnitude and they amount to 4.63 and 5.10 a.u., respectively. On the contrary, the excess Δβnr is much larger than its electronic counterpart and the Δβnr/Δβe ratio is nearly 7. Table 2 also contains the values of pseudo-NR contributions, i.e. Pnr(AAB) and Pnr(BAB). These quantities arise from the definition of interaction energy which involves geometries of monomers corresponding to a dimer. Table 2 shows that there are large differences between βnr(AAB) and βnr(BAB). The comparison of αnr and βnr values listed in Tables 1 and 2 reveals that Pnr(AAB) deviates more substantially from the values for the isolated monomer than Pnr(BAB). Both αnr(AAB) and βnr(AAB) have opposite signs compared to the corresponding properties of the monomer at its equilibrium geometry. Presumably, a major factor contributing to observed discrepancies is the formerly mentioned elongation of the C–H bond in monomer A upon complex formation.
Table 2 Electronic and nuclear relaxation contributions to the polarizability and first polarizability of the HCN monomers and the HCN dimer. All data in this table correspond to MCBS calculations and were obtained at the MP2 level
|
|
aug-cc-pVDZ |
aug-cc-pVTZ |
aug-cc-pVQZ |
|
α
e
zz
(AB) |
50.60 |
49.15 |
48.89 |
|
α
e
zz
(AAB) |
22.90 |
22.30 |
22.20 |
|
α
e
zz
(BAB) |
22.75 |
22.18 |
22.06 |
| Δαezz |
4.95 |
4.67 |
4.63 |
|
α
nr
zz
(AB) |
4.84 |
4.71 |
4.75 |
|
α
nr
zz
(AAB) |
−0.55 |
−0.52 |
−0.56 |
|
α
nr
zz
(BAB) |
0.22 |
0.19 |
0.21 |
| Δαnrzz |
5.17 |
5.04 |
5.10 |
| Δαezz + Δαnrzz |
10.12 |
9.71 |
9.73 |
|
|
|
β
e
zzz
(AB) |
−37.41 |
−39.45 |
−39.84 |
|
β
e
zzz
(AAB) |
3.80 |
0.84 |
−0.20 |
|
β
e
zzz
(BAB) |
3.35 |
0.47 |
−0.62 |
| Δβezzz |
−44.56 |
−40.76 |
−39.02 |
|
β
nr
zzz
(AB) |
223.42 |
−218.87 |
−214.87 |
|
β
nr
zzz
(AAB) |
65.77 |
62.88 |
62.86 |
|
β
nr
zzz
(BAB) |
−8.57 |
−8.26 |
−9.04 |
| Δβnrzzz |
−280.62 |
−273.49 |
−268.69 |
| Δβezzz + Δβnrzzz |
−325.18 |
−314.25 |
−307.71 |
In order to shed more light on the pseudo-NR contributions, we employed an approach proposed by Ingamells et al. who studied vibrational corrections to electric properties at non-equilibrium geometries.92,93 We shall focus solely on αnr as the extension of this approach to nuclear relaxation hyperpolarizability is not straightforward. Following their treatment, we define the geometry correction (αnr,gczz) to the nuclear relaxation polarizability as:
| | 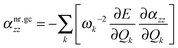 | (21) |
where
k runs over all vibrational normal modes
Q and
E is the total energy at non-optimum geometry. The largest normal-mode contributions to
αnr,gczz are shown in
Table 3. The major contributions to
αnr,gc are due to two vibrational modes (
k = 3 and
k = 4). The latter vibrational mode contribution prevails in the case of monomer A and it corresponds to C–H stretching. It comes as no surprise that this vibrational mode does not make a significant contribution to geometry correction for monomer B. As pointed out by Ingamells
et al. the geometry correction is approximately equal to the difference between the electronic polarizability at the non-optimum (
αe(
Rneq)) and optimum geometries (
αe(
Req)):
| | | αnr,gczz ≈ αezz(Rneq) − αezz(Req) | (22) |
The results in
Table 3 show that there is an excellent agreement between
αnr,gc and the difference given on the r.h.s. of the above equation. Finally, we briefly comment on the fact that the values reported in
Tables 2 and 3 were obtained, for the sake of consistent analysis, employing the monomer-centered basis sets (MCBSs)
75 in the case of pseudo-NR contributions. The differences in excess properties between MCBS and DCBS results at the MP2/aug-cc-pVQZ level are not substantial as Δ
αe + Δ
αnr is 9.73 a.u. for MCBS and 9.66 a.u. for DCBS, while Δ
βe + Δ
βnr is −307.71 a.u. for MCBS and −301.58 a.u. for DCBS.
Table 3 The geometry correction (αnr,gczz) to the diagonal nuclear relaxation polarizability together with non-vanishing normal mode (k) contributions. All results are given in a.u. and were obtained at the MP2/aug-cc-pVQZ level
| Monomer geometry |
α
nr,gc
zz
(k = 3) |
α
nr,gc
zz
(k = 4) |
α
nr,gc
zz
(total) |
α
e
zz
(Rneq) − αezz (Req) |
|
A
AB
|
0.04 |
0.07 |
0.11 |
0.11 |
|
B
AB
|
−0.04 |
0.01 |
−0.03 |
−0.03 |
We shall now analyze the results of electronic and vibrational excess property decomposition in terms of interaction energy components. Table 4 shows the results obtained based on the VP-EDS scheme. The excess electronic (hyper)polarizabilities of HCN aggregates have been thoroughly studied by some of us.27 Note, however, that in the present study, electronic (hyper)polarizabilities and equilibrium geometries are determined at the very same level of theory. This is the factor contributing to small differences between the present values and those given in ref. 27. With this in mind, we only briefly comment on our results of calculations of electronic properties. Electronic excess polarizability is dominated by the electrostatic contribution which prevails over the remaining components. Exchange-repulsion and delocalization components are similar in magnitude but of opposite signs while the sum of electron correlation corrections is negligible.
Table 4 Results of decomposition of electronic and nuclear relaxation polarizability and first hyperpolarizability (given in a.u.) of the HCN dimer, performed at the MP2 level of theory. Note that each term, including ΔPHF and ΔPMP2, was independently computed by numerical differentiation (corresponding numerical errors are given in the ESI)
|
P
|
ΔPHF |
ΔPHF |
ΔPMP2corr |
ΔPMP2 |
| ΔP(10)el |
ΔPHLex |
ΔPHFdel |
ΔP(12)el,r |
ΔP(20)disp |
ΔP(2)ex |
|
|
aug-cc-pVTZ |
|
α
e
zz
|
5.04 |
−1.84 |
1.41 |
4.61 |
0.24 |
0.35 |
−0.55 |
4.65 |
|
α
nr
zz
|
10.02 |
−12.75 |
7.28 |
4.55 |
−0.30 |
2.05 |
−1.37 |
4.93 |
|
β
e
zzz
|
−14.9 |
13.9 |
−23.2 |
−24.2 |
−10.5 |
−0.2 |
−3.6 |
−38.5 |
|
β
nr
zzz
|
−448 |
672 |
−468 |
−245 |
3 |
−93 |
65 |
−264 |
|
|
|
|
aug-cc-pVQZ |
|
α
e
zz
|
5.01 |
−1.83 |
1.40 |
4.58 |
0.21 |
0.35 |
−0.54 |
4.61 |
|
α
nr
zz
|
9.82 |
−12.44 |
7.22 |
4.59 |
−0.30 |
2.03 |
−1.26 |
5.06 |
|
β
e
zzz
|
−15.0 |
14.0 |
−23.2 |
−24.2 |
−10.4 |
−0.2 |
−3.6 |
−38.4 |
|
β
nr
zzz
|
−461 |
697 |
−489 |
−275 |
3 |
−108 |
75 |
−263 |
Much more complicated interplay of interaction energy terms is found in the case of electronic contributions to excess first hyperpolarizability, i.e. one finds that the first-order electrostatic component is quenched by the associated exchange repulsion, and the delocalization component makes the major contribution to excess properties. Except a significant contribution of electron correlation correction to the electrostatic component, the remaining terms computed at the correlated level are not substantial.
We now turn to the decomposition of excess nuclear relaxation polarizability and hyperpolarizability. There are several interaction energy components which are large in magnitude and make a substantial contribution to Δαnr. These include: first-order electrostatic (+194%), exchange repulsion (−246%) and delocalization (+143%) components. As indicated by their signs given in parentheses, the first two terms cancel each other out to a large extent. The positive Hartree–Fock component is further slightly increased by the sum of dispersion contribution (+40%) and the electron correlation correction to exchange energy (−25%). In the case of excess nuclear relaxation first hyperpolarizability the pattern of interactions is similar (i.e. first-order electrostatic (+175%), exchange repulsion (−265%), delocalization (+143%), dispersion (+40%), and electron correlation correction to exchange repulsion (−25%)). Interestingly, there is an identical interference pattern between the negative and positive interaction components for Δαnr and Δβnr. Indeed, even the relative magnitude of the different components of Δαnr and their counterparts for Δβnr are strikingly similar.
Finally, we will discuss the results of calculations of electronic and nuclear relaxation contributions to α and β performed at the DFT/aug-TZ2P level of theory (cf.Table 5). The aug-TZ2P basis set is composed of Slater-type orbitals (STOs), which have the correct nuclear cusp and long-range behavior, as opposed to Gaussian-type orbitals (GTOs). Therefore, the properties converge much quicker with STOs than with GTOs with respect to the basis set size.94
Table 5 Results of decomposition of electronic and nuclear relaxation polarizability and first hyperpolarizability (given in a.u.) of the HCN dimer, performed at the DFT/aug-TZ2P level of theory. Corresponding numerical errors are given in the ESI
| |
P(AB) |
P(A) |
P(B) |
ΔP |
ΔPPauli |
ΔPel |
ΔPorb |
ΔPorb-σ |
ΔPorb-π |
|
|
α
e
zz
|
| BLYP |
52.98 |
23.85 |
23.69 |
5.43 |
−2.81 |
5.75 |
2.50 |
2.02 |
0.46 |
| LC-BLYP |
48.61 |
22.01 |
21.84 |
4.82 |
−2.70 |
5.49 |
2.02 |
1.67 |
0.35 |
| LC-BLYP-dDsC |
48.96 |
21.99 |
21.99 |
4.98 |
−2.99 |
5.74 |
2.18 |
1.80 |
0.38 |
|
|
|
|
α
nr
zz
|
| BLYP |
6.28 |
−0.87 |
0.18 |
7.03 |
−17.12 |
12.09 |
12.00 |
10.79 |
1.21 |
| LC-BLYP |
5.21 |
−0.78 |
0.23 |
5.84 |
−13.78 |
10.24 |
9.38 |
8.30 |
1.08 |
| LC-BLYP-dDsC |
5.02 |
−0.89 |
0.28 |
5.63 |
−11.62 |
8.81 |
8.67 |
7.69 |
0.98 |
|
|
|
|
β
e
zzz
|
| BLYP |
−38.5 |
9.0 |
8.2 |
−55.8 |
19.1 |
−21.9 |
−53.0 |
−54.9 |
0.5 |
| LC-BLYP |
−35.1 |
0.7 |
0.2 |
−36.2 |
22.2 |
−22.7 |
−35.4 |
−32.7 |
−2.7 |
| LC-BLYP-dDsC |
−35.7 |
0.4 |
1.2 |
−37.2 |
23.0 |
−22.4 |
−39.7 |
−34.0 |
−2.5 |
|
|
|
|
β
nr
zzz
|
| BLYP |
−293.9 |
107.6 |
−11.3 |
−390.5 |
1000.7 |
−584.7 |
−813.1 |
−675.2 |
−92.3 |
| LC-BLYP |
−224.6 |
55.2 |
−19.2 |
−269.5 |
1092.6 |
−683.6 |
−678.8 |
−585.1 |
−93.4 |
The calculations were performed employing three functionals, namely: BLYP, LC-BLYP and dispersion corrected LC-BLYP-dDsC. As seen, there are large differences in the αe value for the complex (AB) between BLYP and its long-range-corrected counterparts. The relative error with respect to the CCSD(T)/aug-cc-pVTZ value of αe is 6.13%, 2.62% and 1.92%, for BLYP, LC-BLYP and LC-BLYP-dDsC, respectively. Note the excellent performance of the latter two functionals in predicting electronic polarizability. In the case of electronic first hyperpolarizability, the corresponding relative errors with respect to CCSD(T)/aug-cc-pVTZ are much larger at the DFT/aug-TZ2P level of theory and they amount to 37.50%, 25.36% and 27.50% for BLYP, LC-BLYP and LC-BLYP-dDsC, respectively. Also for αnr, the relative errors with respect to the CCSD(T)/aug-cc-pVTZ value are larger than for αe, i.e. 33.33%, 10.62% and 6.58% for BLYP, LC-BLYP and LC-BLYP-dDsC, respectively. Similar discrepancies are found in the case of βnr, i.e. the errors are equal to 30.83% and 0.02% for BLYP and LC-BLYP. Due to large numerical instabilities, the value of this property is not shown for the dispersion corrected LC-BLYP-dDsC functional. Therefore, for the HCN dimer, the LC-BLYP functional not only delivers much more accurate estimates in comparison with the BLYP for the electronic contributions of α and β, but also for their nuclear relaxation counterparts.
Table 5 also contains the results of decomposition of electronic and nuclear relaxation interaction-induced properties based on the ETS scheme. As already highlighted in the previous section, when the chosen fragments are interacting molecules, the ETS scheme is essentially equivalent to the partitioning of ΔEHF in VP-EDS. However, the DFT-ETS terms also partly account for electron correlation effects. Therefore, the DFT-ETS electrostatic term should be compared with VP-EDS ΔP(10)el + ΔP(12)el,r; DFT-ETS exchange repulsions with VP-EDS ΔPHLex + ΔP(2)ex; and DFT-ETS orbital relaxation with VP-EDS ΔPHFdel. In what follows we will compare the VP-EDS data determined at the MP2/aug-cc-pVQZ (GTOs) level of theory basis set with the results obtained based on the DFT-ETS scheme and using the aug-TZ2P (STOs) basis set. The absolute values of the DFT-ETS electrostatic, exchange repulsion and orbital relaxation terms contributing to Δαe, Δαnr, Δβe and Δβnr are larger than their VP-EDS counterparts. The only exceptions are the electrostatic DFT-ETS terms of Δβe and the electrostatic and exchange terms of Δαnr determined by employing the LC-BLYP-dDsC functional. The discrepancy is larger for the orbital relaxation term than for the other two terms, with the exception of LC-BLYP and LC-BLYPdDsC Δβe. Since the ΔP(2)disp term was not explicitly taken into account in the above comparison between VP-EDS and DFT-ETS terms, at least for BLYP and LC-BLYP, the observed discrepancies could partly be attributed to the missing (or non-representative) dispersion effects in DFT calculations. Our results show that the ability of DFT methods to reproduce the ab initio results is directly related to the magnitude of the orbital relaxation terms. That is, the origin of the too large DFT NLOP properties is the overshooting of the orbital relaxation term. Finally, we focus on the effect of the dispersion correction of LC-BLYP-dDsC results. The dDsC corrections to Δα, and Δβ contributions are far smaller than the ΔP(2)disp term, and do not systematically improve LC-BLYP values. Thus, although dDsC correction improves the accuracy of the interaction energy, the same is not true for the excess properties.
4 Summary and conclusions
The concept of excess properties is a convenient framework to gain insight into the effect of intermolecular interactions on the electric properties of molecular complexes and clusters, which can also be relevant to set up appropriate models for calculations of bulk properties. Moreover, in combination with an appropriate interaction energy decomposition scheme this concept enables us to understand the physical origins of the excess properties. This paper reports the results of a pioneering investigation of nuclear relaxation contributions to the excess electric properties of a model molecular complex. Due to the presence of the hydrogen bond, which plays a pivotal role in supramolecular chemistry, the HCN dimer was chosen as a model system in our study. In order to analyze the interaction-induced nuclear relaxation (hyper)polarizabilities, the approach for the evaluation of purely electronic excess properties has been extended to the total (i.e. electronic and vibrational) excess properties. The new scheme relies on the computationally efficient Bishop–Hasan–Kirtman method for the partitioning of the total value into the electronic and nuclear relaxation contributions. Since in our extension both contributions to electric properties can be expressed in terms of the interaction energy derivatives with respect to the external field, it is straightforward to use an appropriate interaction-energy decomposition scheme and express the excess properties as the sums of physically meaningful contributions. In our study, we apply the VP-EDS scheme at the MP2/aug-cc-pVQZ level and the ETS scheme by employing selected DFT functionals (BLYP, LC-BLYP, and LC-BLYP-dDsC) with the aug-TZ2P basis set.
A significant increase of both electronic and vibrational electric properties upon the complex formation is reflected in the magnitude of the corresponding excess quantities. Moreover, let us highlight the particularly large value of the excess nuclear relaxation first hyperpolarizability, which is roughly 7 times larger than its electronic counterpart. From the VP-EDS analysis it follows that: (i) while the electronic excess polarizability is strongly dominated by the electrostatic contribution, in the case of the electronic excess first hyperpolarizability, the electrostatic component is practically quenched by the exchange repulsion contribution, and then it is the delocalization term which finally plays the major role; (ii) there are several interaction energy components which are large in magnitude and make a substantial contribution to nuclear relaxation excess polarizability. It was observed that the first-order electrostatic contribution cancels out with the negative exchange repulsion term to a large extent resulting in a positive value of Δαnr due to the contribution from the delocalization and dispersion terms. In the case of excess nuclear relaxation first hyperpolarizability, the pattern of interaction contributions is very similar to that for Δαnr, both in terms of their sign as well as relative magnitude.
For the functionals explored in this work all three interaction energy terms within the DFT-ETS scheme contribute to the overestimation of excess properties. Nevertheless, the largest overshooting is always given by the orbital relaxation term. Finally, one may conclude that the LC-BLYP and LC-BLYP-dDsC functionals, which have a smaller orbital relaxation term than BLYP, are much more successful in predicting excess properties. Thus, comparison between VP-EDS and DFT-ETS partition schemes can be applied as a useful tool to analyze in depth the performance of DFT functionals for computing NLO properties and obtain helpful hints to pursue in the future design of more accurate functionals.
Acknowledgements
R. Z. gratefully acknowledges support from the National Science Centre, Poland (Grant No. 2015/19/B/ST4/01881). M. G.-B. and J. M. L. thank the Spanish Ministerio de Economía y competividad (MINECO, CTQ2014-52525-P) and the Generalitat de Catalunya (project number 2014SGR931). M. M. acknowledges the financial support of the Slovak Research and Development Agency (project No. APVV-15-0105). All calculations reported herein were performed at the Wroclaw Center for Networking and Supercomputing. The authors declare no competing financial interest.
References
- P. A. Sullivan and L. R. Dalton, Acc. Chem. Res., 2010, 43, 10–18 CrossRef CAS PubMed.
- S. D. Bella, M. A. Ratner and T. J. Marks, J. Am. Chem. Soc., 1992, 114, 5842–5849 CrossRef.
-
B. Champagne and D. M. Bishop, Adv. Chem. Phys., John Wiley & Sons, Inc., 2003, vol. 126, pp. 41–92 Search PubMed.
- A. Facchetti, E. Annoni, L. Beverina, M. Morone, P. Zhu, T. J. Marks and G. A. Pagani, Nat. Mater., 2004, 3, 910–917 CrossRef CAS PubMed.
- A. Datta and S. K. Pati, Chem. Soc. Rev., 2006, 35, 1305–1323 RSC.
- A. Buckingham and K. Clarke, Chem. Phys. Lett., 1978, 57, 321–325 CrossRef CAS.
- P. Fowler and A. Sadlej, Mol. Phys., 1992, 77, 709–725 CrossRef CAS.
- P. W. Fowler, K. L. C. Hunt, H. M. Kelly and A. J. Sadlej, J. Chem. Phys., 1994, 100, 2932–2935 CrossRef CAS.
- R. Moszyński, T. G. A. Heijmen, P. E. S. Wormer and A. van der Avoird, J. Chem. Phys., 1996, 104, 6997–7007 CrossRef.
- T. G. A. Heijmen, R. Moszyński, P. E. S. Wormer and A. van der Avoird, Mol. Phys., 1996, 89, 81–110 CrossRef CAS.
- C. Hättig, H. Larsen, J. Olsen, P. Jørgensen, H. Koch, B. Fernandez and A. Rizzo, J. Chem. Phys., 1999, 111, 10099–10107 CrossRef.
- H. Koch, C. Hättig, H. Larsen, J. Olsen, P. Jørgensen, B. Fernandez and A. Rizzo, J. Chem. Phys., 1999, 111, 10108–10118 CrossRef CAS.
- M. Jaszuński, A. Rizzo and P. Jørgensen, Theor. Chem. Acc., 2001, 106, 251–258 CrossRef.
- A. Rizzo, C. Hättig, B. Fernandez and H. Koch, J. Chem. Phys., 2002, 117, 2609–2618 CrossRef CAS.
- J. L. Cacheiro, B. Fernandez, D. Marchesan, S. Coriani, C. Hättig and A. Rizzo, Mol. Phys., 2004, 102, 101–110 CrossRef CAS.
- A. Antušek, M. Jaszuński and A. Rizzo, J. Chem. Phys., 2007, 126, 074303 CrossRef PubMed.
- T. Bancewicz and G. Maroulis, Chem. Phys. Lett., 2010, 498, 349–352 CrossRef CAS.
- A. Chantzis and G. Maroulis, Chem. Phys. Lett., 2011, 507, 42–47 CrossRef CAS.
- D. Xenides, A. Hantzis and G. Maroulis, Chem. Phys., 2011, 382, 80–87 CrossRef CAS.
- G. Maroulis, A. Haskopoulos, W. Glaz, T. Bancewicz and J. Godet, Chem. Phys. Lett., 2006, 428, 28–33 CrossRef CAS.
- B.-Q. Wang, Z.-R. Li, D. Wu, X.-Y. Hao, R.-J. Li and C.-C. Sun, J. Phys. Chem. A, 2004, 108, 2464–2468 CrossRef CAS.
- B. Skwara, W. Bartkowiak, A. Zawada, R. W. Góra and J. Leszczynski, Chem. Phys. Lett., 2007, 436, 116–123 CrossRef CAS.
- B. Skwara, A. Kaczmarek, R. W. Góra and W. Bartkowiak, Chem. Phys. Lett., 2008, 461, 203–206 CrossRef CAS.
- A. Baranowska, B. Fernandez, A. Rizzo and B. Jansik, Phys. Chem. Chem. Phys., 2009, 11, 9871–9883 RSC.
- A. Baranowska, A. Zawada, B. Fernandez, W. Bartkowiak, D. Kedziera and A. Kaczmarek-Kedziera, Phys. Chem. Chem. Phys., 2010, 12, 852–862 RSC.
- A. Baranowska, B. Fernandez and A. J. Sadlej, Theor. Chem. Acc., 2011, 128, 555–561 CrossRef CAS.
- R. W. Góra, R. Zaleśny, A. Zawada, W. Bartkowiak, B. Skwara, M. G. Papadopoulos and D. L. Silva, J. Phys. Chem. A, 2011, 115, 4691–4700 CrossRef PubMed.
- A. Zawada, R. W. Góra, M. M. Mikołajczyk and W. Bartkowiak, J. Phys. Chem. A, 2012, 116, 4409–4416 CrossRef CAS PubMed.
- A. Zawada, A. Kaczmarek-Kedziera and W. Bartkowiak, J. Mol. Model., 2012, 18, 3073–3086 CrossRef CAS PubMed.
- M. H. Champagne, X. Li and K. L. C. Hunt, J. Chem. Phys., 2000, 112, 1893–1906 CrossRef CAS.
- X. Li, C. Ahuja, J. F. Harrison and K. L. C. Hunt, J. Chem. Phys., 2007, 126, 214302 CrossRef CAS PubMed.
- T. Karman, E. Miliordos, K. L. C. Hunt, G. C. Groenenboom and A. van der Avoird, J. Chem. Phys., 2015, 142, 084306 CrossRef PubMed.
- M. Abel, L. Frommhold, X. Li and K. L. C. Hunt, J. Phys. Chem. A, 2011, 115, 6805–6812 CrossRef CAS PubMed.
- M. Abel, L. Frommhold, X. Li and K. L. C. Hunt, J. Chem. Phys., 2012, 136, 044319 CrossRef PubMed.
- C. D. Zeinalipour-Yazdi and D. P. Pullman, J. Phys. Chem. B, 2006, 110, 24260–24265 CrossRef CAS PubMed.
- G. Maroulis, Int. J. Quantum Chem., 2012, 112, 2231–2241 CrossRef CAS.
- J.-L. Godet, T. Bancewicz, W. Glaz, G. Maroulis and A. Haskopoulos, J. Chem. Phys., 2009, 131, 204305 CrossRef PubMed.
- A. Baranowska-Łączkowska, B. Fernandez, A. Rizzo and B. Jansik, Mol. Phys., 2012, 110, 2503–2512 CrossRef.
- Ż. Czyżnikowska, R. W. Góra, R. Zaleśny, W. Bartkowiak, A. Baranowska-Łączkowska and J. Leszczynski, Chem. Phys. Lett., 2013, 555, 230–234 CrossRef.
- R. W. Góra and B. Błasiak, J. Phys. Chem. A, 2013, 117, 6859–6866 CrossRef PubMed.
- A. Baranowska-Łączkowska, B. Fernandez and R. Zaleśny, J. Comput. Chem., 2013, 34, 275–283 CrossRef PubMed.
- A. Baranowska-Łączkowska and B. Fernandez, Mol. Phys., 2015, 113, 3362–3369 CrossRef.
- M. Medved', Š. Budzak, A. D. Laurent and D. Jacquemin, J. Phys. Chem. A, 2015, 119, 3112–3124 CrossRef PubMed.
- J. G. Ángyán, G. Jansen, M. Loss, C. Hättig and B. A. Heß, Chem. Phys. Lett., 1994, 219, 267–273 CrossRef.
- H. Reis, M. G. Papadopoulos, C. Hättig, J. G. Ángyán and R. W. Munn, J. Chem. Phys., 2000, 112, 6161–6172 CrossRef CAS.
- M. in het Panhuis, P. L. A. Popelier, R. W. Munn and J. G. Ángyán, J. Chem. Phys., 2001, 114, 7951–7961 CrossRef CAS.
- A. Ye and J. Autschbach, J. Chem. Phys., 2006, 125, 234101 CrossRef PubMed.
- F. Yang, X. Wang, M. Yang, A. Krishtal, C. v. Alsenoy, P. Delarue and P. Senet, Phys. Chem. Chem. Phys., 2010, 12, 9239–9248 RSC.
- X. Chu, M. Yang and K. A. Jackson, J. Chem. Phys., 2011, 134, 234505 CrossRef PubMed.
- Q. Zeng, L. Liu, W. Zhu and M. Yang, J. Chem. Phys., 2012, 136, 224304 CrossRef PubMed.
- M. Nakano, I. Shigemoto, S. Yamada and K. Yamaguchi, J. Chem. Phys., 1995, 103, 4175–4191 CrossRef CAS.
- M. Nakano, S. Ohta, K. Tokushima, R. Kishi, T. Kubo, K. Kamada, K. Ohta, B. Champagne, E. Botek and H. Takahashi, Chem. Phys. Lett., 2007, 443, 95–101 CrossRef CAS.
- M. Nakano, A. Takebe, R. Kishi, H. Fukui, T. Minami, K. Kubota, H. Takahashi, T. Kubo, K. Kamada, K. Ohta, B. Champagne and E. Botek, Chem. Phys. Lett., 2008, 454, 97–104 CrossRef CAS.
- A. Avramopoulos, M. G. Papadopoulos and A. J. Sadlej, J. Chem. Phys., 2002, 117, 10026–10038 CrossRef CAS.
- D. Bishop and B. Kirtman, J. Chem. Phys., 1991, 95, 2646–2658 CrossRef CAS.
- D. Bishop, Adv. Chem. Phys., 1998, 104, 1–40 CrossRef CAS.
- D. M. Bishop, M. Hasan and B. Kirtman, J. Chem. Phys., 1995, 103, 4157 CrossRef CAS.
- B. Kirtman, J. M. Luis and D. M. Bishop, J. Chem. Phys., 1998, 108, 10008–10012 CrossRef CAS.
- C. Eckart, Phys. Rev., 1926, 28, 711 CrossRef.
- J. M. Luis, M. Duran, J. L. Andrés, B. Champagne and B. Kirtman, J. Chem. Phys., 1999, 111, 875 CrossRef CAS.
- K. Morokuma, J. Chem. Phys., 1971, 55, 1236–1244 CrossRef CAS.
- T. Ziegler and A. Rauk, Theor. Chim. Acta, 1977, 46, 1–10 CrossRef CAS.
- K. Kitaura and K. Morokuma, Int. J. Quantum Chem., 1976, 10, 325–340 CrossRef CAS.
- P. S. Bagus, K. Hermann and C. W. Bauschlicher, J. Chem. Phys., 1984, 80, 4378 CrossRef CAS.
- W. J. Stevens and W. H. Fink, Chem. Phys. Lett., 1987, 139, 15–22 CrossRef CAS.
- E. D. Glendening and A. Streitwieser, J. Chem. Phys., 1994, 100, 2900 CrossRef CAS.
- Y. Mo, J. Gao and S. D. Peyerimhoff, J. Chem. Phys., 2000, 112, 5530 CrossRef CAS.
- P. Su and H. Li, J. Chem. Phys., 2009, 131, 014102 CrossRef PubMed.
- M. Gutowski, F. V. Duijneveldt, G. Chałasiński and L. Piela, Mol. Phys., 1987, 61, 233–247 CrossRef CAS.
- W. A. Sokalski, S. Roszak and K. Pecul, Chem. Phys. Lett., 1988, 153, 153–159 CrossRef CAS.
- S. M. Cybulski, G. Chałasiński and R. Moszyński, J. Chem. Phys., 1990, 92, 4357–4363 CrossRef CAS.
- G. Chałasiński and M. M. Szczȩśniak, Chem. Rev., 1994, 94, 1723–1765 CrossRef.
- G. Chałasiński and M. M. Szczȩśniak, Mol. Phys., 1988, 63, 205–224 CrossRef.
- I. W. Bulik, R. Zaleśny, W. Bartkowiak, J. M. Luis, B. Kirtman, G. E. Scuseria, A. Avramopoulos, H. Reis and M. G. Papadopoulos, J. Comput. Chem., 2013, 34, 1775–1784 CrossRef CAS PubMed.
- F. B. van Duijneveldt, J. G. C. M. van Duijneveldt-van de Rijdt and J. H. van Lenthe, Chem. Rev., 1994, 94, 1873–1885 CrossRef CAS.
- R. Moszyński, S. Rybak, S. Cybulski and G. Chałasiński, Chem. Phys. Lett., 1990, 166, 609–614 CrossRef.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 Revision D.01, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis and J. A. Montgomery, J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS.
-
R. W. Góra, EDS package, Revision 2.8.3, Wrocław, Poland, 1998–2008.
-
E. J. Baerends, T. Ziegler, J. Autschbach, D. Bashford, A. Bérces, F. Bickelhaupt, C. Bo, P. M. Boerrigter, L. Cavallo, D. P. Chong, L. Deng, R. M. Dickson, D. E. Ellis, M. van Faassen, L. Fan, T. H. Fischer, C. F. Guerra, M. Franchini, A. Ghysels, A. Giammona, S. J. A. van Gisbergen, A. W. Götz, J. A. Groeneveld, O. V. Gritsenko, M. Grüning, S. Gusarov, F. E. Harris, P. van den Hoek, C. R. Jacob, H. Jacobsen, L. Jensen, J. W. Kaminski, G. van Kessel, F. Kootstra, A. Kovalenko, M. V. Krykunov, E. van Lenthe, D. A. McCormack, A. Michalak, M. Mitoraj, S. Morton, J. Neugebauer, V. Nicu, L. Noodleman, V. P. Osinga, S. Patchkovskii, M. Pavanello, P. H. T. Philipsen, D. Post, C. C. Pye, W. Ravenek, J. I. Rodríguez, P. Ros, P. R. T. Schipper, G. Schreckenbach, J. S. Seldenthuis, M. Seth, J. G. Snijders, M. Solà, M. Swart, D. Swerhone, G. te Velde, P. Vernooijs, L. Versluis, L. Visscher, O. Visser, F. Wang, T. A. Wesolowski, E. M. van Wezenbeek, G. Wiesenekker, S. K. Wolff, T. K. Woo and A. L. Yakovlev, ADF 2010.01, SCM, Amsterdam, 2010 Search PubMed.
- B. Skwara, W. Bartkowiak and D. L. Da Silva, Theor. Chem. Acc., 2009, 122, 127–136 CrossRef CAS.
- A. Roztoczyńska, A. Kaczmarek-Kedziera, R. W. Góra and W. Bartkowiak, Chem. Phys. Lett., 2013, 571, 28–33 CrossRef.
- R. Zaleśny, A. Baranowska-Łączkowska, M. Medved and J. M. Luis, J. Chem. Theory Comput., 2015, 11, 4119–4128 CrossRef PubMed.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- R. A. Kendall, T. H. Dunning and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- D. E. Woon and T. H. Dunning, J. Chem. Phys., 1994, 100, 2975–2988 CrossRef CAS.
- I. W. M. Smith, D. Talbi and E. Herbst, Astron. Astrophys., 2001, 369, 611–615 CrossRef CAS.
- G. Maroulis and C. Pouchan, Phys. Rev. A: At., Mol., Opt. Phys., 1998, 57, 2440–2447 CrossRef CAS.
- C. D. Zeinalipour-Yazdi and D. P. Pullman, J. Phys. Chem. B, 2008, 112, 7377–7386 CrossRef CAS PubMed.
- C. D. Zeinalipour-Yazdi and D. P. Pullman, J. Phys. Chem. B, 2009, 113, 9628 CrossRef CAS.
- U. Eckart and A. J. Sadlej, Mol. Phys., 2001, 99, 735–743 CrossRef CAS.
- V. E. Ingamells, M. G. Papadopoulos and A. J. Sadlej, Chem. Phys., 2000, 260, 1–10 CrossRef CAS.
- V. E. Ingamells, M. G. Papadopoulos and A. J. Sadlej, J. Chem. Phys., 2000, 112, 1645–1654 CrossRef CAS.
- M. Güell, J. M. Luis, M. Sola and M. Swart, J. Phys. Chem. A, 2008, 112, 6384–6391 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp02500e |
|
| This journal is © the Owner Societies 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Miroslav
Medved'
c and
Josep M.
Luis
*d
a,
Miroslav
Medved'
c and
Josep M.
Luis
*d