
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSearch for aromatic anions in the P2E3− (E = N, P, As, Sb, Bi) series†
Anton S.
Nizovtsev
 ab
ab
aNikolaev Institute of Inorganic Chemistry, Siberian Branch of the Russian Academy of Sciences, Academician Lavrentiev Avenue 3, Novosibirsk, 630090, Russian Federation. E-mail: anton.nizovtsev@gmail.com
bNovosibirsk State University, Pirogova Street 2, Novosibirsk, 630090, Russian Federation
First published on 24th May 2016
Abstract
We report a systematic computational study focused on the global minimum and low-lying isomer search for the P2E3− (E = N, P, As, Sb, Bi) series of anions. We found nine planar five-membered rings and proved their aromatic character by various quantum chemical techniques. The possible use of two different P2N3− aromatic anions as ligands in a number of sandwich complexes was also studied.
Aromatic species with delocalized (4n + 2)π electrons have increased stability compared to their non-aromatic analogues.1,2 Along with the well-known aromatic carbocyclic compounds (benzene (n = 1), naphthalene (n = 2), etc.), aromaticity was also detected in a number of species containing metals and main group elements.3,4 Special attention to this field leads to the novel aromatic compounds being intensively sought both experimentally and theoretically.5–8 Planar five-membered all-pnictogen cycles with 6π electrons are of particular interest among the main group aromatics because they are isovalent with the cyclopentadienyl anion (C5H5−) and can be used as inorganic building blocks.9–11 Specifically, P5− and As5− fragments were stabilized in several sandwich complexes,12 including a carbon-free metallocene [(η5-P5)2Ti]2−.13
Recently, Velian and Cummins reported the synthesis of P2N3− anion by an original “click” reaction between an anthracene-based source of P2, P2(C14H10)2,14 and the azide anion N3− in tetrahydrofuran solution and computationally confirmed the aromatic properties of P2N3−.15 The proposed P2 transfer reaction opens new opportunities for synthetic realization of unusual phosphorus-containing compounds. After that, Mandal et al. have predicted a number of all-pnictogen aromatic species with X2Y3− and X3Y2− formulae and explored the possibility of using a P3N2− anion as an η5-ligand in metallocenes by density functional theory calculations.16 Inspired by these two studies, here we report a systematic ab initio search for possible isomers with P2E3− (E = N, P, As, Sb, Bi) stoichiometry in order to characterize expected aromatic anions.
An unbiased quantum-chemical search17 for P2E3− species performed at the B3LYP-D3(BJ)/def2-SVP level of theory revealed that the global minimum for each E is a planar five-membered ring with a C2v-symmetrical (E = N, As, Sb, Bi) or D5h-symmetrical (E = P) structure (Fig. 1).18 It is important to note that two different kinds of such rings were found for P2E3− (E = N, As, Sb, Bi), both of them having increased stability over the rest of the corresponding isomers (Fig. 1 and Fig. S1–S5, ESI†). One structural motif has a pnictogen atom between two phosphorus centers (I.A-like), whereas another has no atoms between them (I.B-like). Bond lengths optimized at B3LYP-D3(BJ)/def2-QZVPD and CCSD/aug-cc-pVTZ(-PP) levels of theory (results obtained agree with each other within 0.06 Å) as well as natural population analysis (NPA)19 partial charges are presented in Fig. 1. Rather short P–P, E–P, and E–E interatomic distances suggest the presence of multiple bonding.
 | ||
| Fig. 1 Representative isomers of (I) P2N3−, (II) P5−, (III) P2As3−, (IV) P2Sb3−, and (V) P2Bi3−, their point group symmetries, spectroscopic states, bond lengths (Å), NPA partial charges (|e|, in italics), and relative energies (kcal mol−1). The energies are given at the CCSD(T)/aug-cc-pVTZ(-PP)//B3LYP-D3(BJ)/def2-QZVPD + ΔSR (E = N, P, As) + ΔSO + ΔZPE level of theory. Bond lengths are given at B3LYP-D3(BJ)/def2-QZVPD and CCSD/aug-cc-pVTZ(-PP) (in bold) theoretical levels. | ||
It should be stressed that, in contrast to the recently reported anion I.B,15 anionic heterocycle I.A has been unknown to date. Moreover, I.A is a global minimum structure, which lies 16.1 kcal mol−1 below I.B in energy (here and elsewhere results on the total energy differences for P2E3− are given at the CCSD(T)/aug-cc-pVTZ(-PP)//B3LYP-D3(BJ)/def2-QZVPD + ΔSR (E = N, P, As) + ΔSO + ΔZPE theoretical level; ΔSR, ΔSO, and ΔZPE denote scalar relativistic, spin–orbit, and zero-point energy corrections, respectively).18 However, isomers with I.B-like structures are the most preferred ones in the P2E3− (E = As, Sb, Bi) series so that the relative energy of the second lowest-lying isomer (I.A-like structures) changes from 1.1 kcal mol−1 (P2As3−) and 3.6 kcal mol−1 (P2Sb3−) to 10.5 kcal mol−1 (P2Bi3−), as our calculations predict (Fig. 1). The energy difference between the first two lowest-lying isomers was calculated to be 7.0 kcal mol−1 and 32.6 kcal mol−1 in the case of P2N3− and P5−, respectively (Fig. S1 and S2, ESI†).
Thus, our computational findings suggest that P2E3− (E = N, As, Sb, Bi) anions with I.A-like and I.B-like structures, most of which were characterized for the first time (see Tables S1 and S2, ESI,† for predicted IR and Raman spectra and 31P chemical shifts), possess increased thermodynamic stability and can be considered as potential targets for chemical synthesis.
To check whether these P2E3− anions exhibit aromatic properties, we performed nucleus-independent chemical shift (NICS),20 natural resonance theory (NRT),21 and adaptive natural density partitioning (AdNDP)22 calculations.18
All NICSzz profiles calculated for both the planar P2E3− and C5H5− anions consist of negative NICS values and show a minimum (Fig. 2, Fig. S6, S7 and Table S3, ESI†), which is typical for aromatic compounds.23 A decrease of the maximum absolute NICSzz value is accompanied by the displacement of the profile’s minimum to higher distances above the ring critical point at the electron density gradient field as the atomic weight of E is increased, indicating weakening of the π-aromaticity. It is worth noting that absolute NICSzz values of I.B-like structures are higher than those of I.A-like ones.
 | ||
| Fig. 2 NICSzz profiles for P2E3− anions with (a) I.A-like and (b) I.B-like structures computed at the B3LYP/def2-QZVPD level of theory. Results for C5H5− anions are also presented for comparison. | ||
NRT analysis reveals five canonical resonance structures with the contribution ranging from 11% to 17% for each system (Fig. S8 and S9, ESI†), reflecting electron delocalization.
In contrast to NRT analysis, the AdNDP method allows one to describe electron delocalization, being an important feature of aromatic compounds, via multicenter bonds without invoking the concept of resonance. As the picture of chemical bonding in P2E3− species of similar structure slightly depends on the pnictogen E, here we discuss in detail the results of AdNDP calculations only for the I.A and I.B anions with E = N (Fig. 3). The complete AdNDP results are given in Fig. S10 and S11 (ESI†).
 | ||
| Fig. 3 Results of AdNDP analysis for the (a) I.A and (b) I.B structures. | ||
The chemical bonding pattern in the global minimum of P2N3− (I.A, Fig. 3a) represents five s-type lone pairs (two P lone pairs with occupation numbers (ON) equal to 2.0 |e| and three N lone pairs with ON = 1.9 |e|), five covalent two-center two-electron (2c-2e) σ-bonds with ON = 2.0 |e| (four P–N and one N–N bonds), and three 5c-2e π-bonds (ON = 2.0 |e|) between all atoms of the ring. The same picture of chemical bonding is observed in structure I.B (Fig. 3b), except for the presence of three different types of 2c-2e σ-bonds (two P–N, two N–N, and one P–P bonds with ON = 2.0 |e|).24 It is important to note that the set of three 5c-2e π-bonds found in P2E3− species is also the key element of the chemical bonding pattern for C5H5− (Fig. S12, ESI†), which is a well-known 6π-electron aromatic system.
Additionally, vertical detachment energies (VDEs) from the highest occupied molecular orbital range from 3.1 eV (V.B) to 4.3 eV (I.B), as calculated by the OVGF/aug-cc-pVTZ(-PP) method (Table S4, ESI†). The VDEs are similar to those for E5− (E = P, As, Sb, Bi) aromatic cluster anions reported by Zhai and coworkers (2.9–4.1 eV),4 but they are higher than VDE for C5H5− (2.0 eV, Table S5, ESI†).
Thus, one can conclude that all P2E3− anions shown in Fig. 1 are aromatic.
Taking into account the resembling aromatic properties of C5H5− and P2N3− anions, we also studied hypothetical M(η5-P2N3)2, M(η5-C5H5)(η5-P2N3), and M(η5-C5Me5)(η5-P2N3) sandwich complexes (M = Fe, Ru, Os; Me = CH3) to draw a parallel with classical metallocenes M(η5-C5H5)2. Compounds with one P2N3− (I.B) moiety were found to be ca. 20 kcal mol−1 higher in energy (M06-L/def2-TZVP level of theory) than their P2N3− (I.A)-substituted isomers (Fig. 4, Fig. S14 and S15, ESI†), which is in line with the results obtained for P2N3− anions (Fig. 1). Dissociation energies of the P2N3-containing complexes into the M2+ and two negatively charged ligands (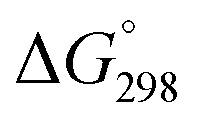 = 610–740 kcal mol−1) are comparable with those of M(η5-C5H5)2 (
= 610–740 kcal mol−1) are comparable with those of M(η5-C5H5)2 ( = 720–780 kcal mol−1), indicating their thermodynamic stability (Tables S6 and S7, ESI†).
= 720–780 kcal mol−1), indicating their thermodynamic stability (Tables S6 and S7, ESI†).
 | ||
| Fig. 4 Representative isomers of (I) Fe(η5-P2N3)2, (II) Ru(η5-P2N3)2, and (III) Os(η5-P2N3)2 complexes with P2N3− (I.A and I.B) ligands, their point group symmetries, spectroscopic states and relative energies (kcal mol−1). The energies are given at the M06-L/def2-TZVP//M06-L/def2-TZVP + ΔZPE level of theory. | ||
According to an energy decomposition analysis (EDA; Tables S8 and S9, ESI†), the energy of electrostatic interactions (ΔEelstat, 50–55%) between ML+ and P2N3− fragments gives slightly higher contribution to the total attractive interactions compared to the orbital interaction energy (ΔEorb, 45–50%). EDA results for M(η5-C5H5)2 compounds show that M(η5-C5H5)+ and C5H5− fragments interact more strongly than ML+ and P2N3− ones (ΔΔEint = 24–58 kcal mol−1) because of the gain in electrostatics (ΔEelstat, 57–58%).
In summary, exploring the potential energy surfaces of P2E3− (E = N, P, As, Sb, Bi) anions revealed planar five-membered cyclic structures for each E under study. An increased stability of the structures over the corresponding isomers can be explained by their aromatic properties. Two different structural motifs were found for aromatic anions (E = N, As, Sb, and Bi), one of them being theoretically characterized for the first time. Remarkably, the recently synthesized P2N3− aromatic anion turned out to lie ca. 16 kcal mol−1 higher in energy than the global minimum, which is the aromatic anion predicted in this work. Substitution of the C5H5− or C5Me5− ligand by the P2N3− moiety can lead to sandwich complexes with bonding properties, similar to the classical metallocene ones. Recent progress in the synthesis of aromatic pnictogen-containing heterocycles15,25–27 allows us to expect that the present contribution will stimulate further development of this research field.
The Siberian Supercomputer Center is gratefully acknowledged for providing computational resources.
Notes and references
- P. V. R. Schleyer, Chem. Rev., 2001, 101, 1115–1118 CrossRef CAS PubMed.
- N. Martín and L. T. Scott, Chem. Soc. Rev., 2015, 44, 6397–6400 RSC.
- X. Li, A. E. Kuznetsov, H.-F. Zhang, A. I. Boldyrev and L.-S. Wang, Science, 2001, 291, 859–861 CrossRef CAS PubMed.
- H.-J. Zhai, L.-S. Wang, A. E. Kuznetsov and A. I. Boldyrev, J. Phys. Chem. A, 2002, 106, 5600–5606 CrossRef CAS.
- A. I. Boldyrev and L.-S. Wang, Chem. Rev., 2005, 105, 3716–3757 CrossRef CAS PubMed.
- J. M. Mercero, A. I. Boldyrev, G. Merino and J. M. Ugalde, Chem. Soc. Rev., 2015, 44, 6519–6534 RSC.
- I. Fernández, G. Frenking and G. Merino, Chem. Soc. Rev., 2015, 44, 6452–6463 RSC.
- A. I. Boldyrev and L.-S. Wang, Phys. Chem. Chem. Phys., 2016, 18, 11589–11605 RSC.
- J. Bai, A. V. Virovets and M. Scheer, Science, 2003, 300, 781–783 CrossRef CAS PubMed.
- C. Heindl, E. V. Peresypkina, A. V. Virovets, W. Kremer and M. Scheer, J. Am. Chem. Soc., 2015, 137, 10938–10941 CrossRef CAS PubMed.
- M. Fleischmann, J. S. Jones, F. P. Gabbaï and M. Scheer, Chem. Sci., 2015, 6, 132–139 RSC.
- O. J. Scherer, Angew. Chem., Int. Ed. Engl., 1990, 29, 1104–1122 CrossRef.
- E. Urnezius, W. W. Brennessel, C. J. Cramer, J. E. Ellis and P. V. R. Schleyer, Science, 2002, 295, 832–834 CrossRef CAS PubMed.
- A. Velian, M. Nava, M. Temprado, Y. Zhou, R. W. Field and C. C. Cummins, J. Am. Chem. Soc., 2014, 136, 13586–13589 CrossRef CAS PubMed.
- A. Velian and C. C. Cummins, Science, 2015, 348, 1001–1004 CrossRef CAS PubMed.
- S. Mandal, S. Nandi, A. Anoop and P. K. Chattaraj, Phys. Chem. Chem. Phys., 2016, 18, 11738–11745 RSC.
- A. P. Sergeeva, B. B. Averkiev, H.-J. Zhai, A. I. Boldyrev and L.-S. Wang, J. Chem. Phys., 2011, 134, 224304 CrossRef PubMed.
- Complete computational details are given in the ESI†.
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS.
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. v. R. Schleyer, Chem. Rev., 2005, 105, 3842–3888 CrossRef CAS PubMed.
- E. D. Glendening and F. Weinhold, J. Comput. Chem., 1998, 19, 593–609 CrossRef CAS.
- D. Y. Zubarev and A. I. Boldyrev, Phys. Chem. Chem. Phys., 2008, 10, 5207–5217 RSC.
- J. O. C. Jiménez-Halla, E. Matito, J. Robles and M. Solà, J. Organomet. Chem., 2006, 691, 4359–4366 CrossRef.
- The third 5e-2c π-bond of the I.B structure (Fig. 3b) has a contribution from all atoms at lower isovalues (see Fig. S13, ESI†).
- G. He, O. Shynkaruk, M. W. Lui and E. Rivard, Chem. Rev., 2014, 114, 7815–7880 CrossRef CAS PubMed.
- C. Hering-Junghans and E. Rivard, Angew. Chem., Int. Ed., 2015, 54, 10077–10079 CrossRef CAS PubMed.
- C. Heindl, E. V. Peresypkina, A. V. Virovets, G. Balázs and M. Scheer, Chem. – Eur. J., 2016, 22, 1944–1948 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Computational details, complete results of calculations performed, including IR and Raman spectra and 31P chemical shifts for aromatic P2E3− anions, and Cartesian coordinates of the most relevant species. See DOI: 10.1039/c6cp02241c |
| This journal is © the Owner Societies 2016 |