
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Over 75% incident-photon-to-current efficiency without solid electrodes†
D.
Plana‡
a,
K. A.
Bradley
ab,
D.
Tiwari
a and
D. J.
Fermín
*a
aSchool of Chemistry, University of Bristol, Cantocks Close, Bristol BS8 1TS, UK. E-mail: david.fermin@bristol.ac.uk
bBristol Centre for Functional Nanomaterials, NSQI Building, University of Bristol, Tyndall Avenue, Bristol BS8 1FD, UK
First published on 15th April 2016
Abstract
The efficiency of photoelectrochemical reactions is conventionally defined in terms of the ratio between the current responses arising from the collection of carriers at electrical contacts and the incident photon flux at a given wavelength, i.e. the incident-photon-to-current-efficiency (IPCE). IPCE values are determined by a variety of factors such as the absorption constant of the active layer, bulk and surface recombination of photogenerated carriers, as well as their characteristic diffusion length. These parameters are particularly crucial in nanostructured photoelectrodes, which commonly display low carrier mobility. In this article, we examine the photoelectrochemical responses of a mesoporous TiO2 film in which the IPCE is enhanced by fast extraction of carriers via chemical reactions. TiO2 films are spontaneously formed by destabilisation of colloidal particles at the polarisable interface between two immiscible electrolyte solutions. The photocurrent arises from hole-transfer to redox species confined to the organic electrolyte, which is coupled to the transfer of electrons to oxygen in the aqueous electrolyte. The dynamic photocurrent responses demonstrate that no coupled ion transfer is involved in the process. The interplay of different interfacial length scales, molecularly sharp liquid/liquid boundary and mesoporous TiO2 film, promotes efficiencies above 75% (without correction for reflection losses). This is a significant step change in values reported for these interfaces (below 1%), which are usually limited to sub-monolayer coverage of photoactive molecular or nanoscopic materials.
Introduction
Nanostructured and mesoscopic photoactive materials have had a profound impact on the area of solar energy conversion.1–4 From dye-sensitised5–7 to organo-halide perovskite solar cells8,9 as well as photoelectrodes for water-splitting, the combination of high surface areas and light-capture cross sections allows the generation of devices with conversion efficiencies comparable to or even higher than mono or polycrystalline systems. However, these complex systems rely on certain boundary conditions mainly associated with the lifetime and collection rate of photoinduced carriers. The high density of defects in mesoporous layers often generates sites which can act as carrier trap states. These states effectively decrease carrier mobility and can promote recombination losses. In conventional dye-sensitised solar cells, device efficiencies are often limited by the collection of injected electrons into the mesoporous TiO2 film, particularly in the presence of water and oxygen.10–12 In this work, we shall introduce a new concept in which electron capture by species in solution increases the photoelectrochemical efficiency. In other words, photocurrent responses arise from redox species collecting carriers directly from a mesoporous titania network. This concept is based on polarisable interfaces between two immiscible electrolyte solutions (ITIES).Liquid/liquid interfaces provide very interesting platforms for assembling nanostructured materials,13–16 although few works have been devoted to probe the activity of the materials at the molecular interface.17–19 A number of studies have demonstrated that dye species and colloidal nanoparticles assembled at polarisable ITIES can generate photocurrent responses upon illumination in the presence of redox species.20–22 In these works, the photoactive component corresponds to a monolayer assembled at a molecularly flat surface, leading to incident-photon-to-current efficiencies (IPCE) below 1%. Herein, we demonstrate for the first time that films of coalesced TiO2 nanoparticles at the liquid/liquid boundary can act as photoactive layers featuring IPCE values above 75%, excluding reflection losses. These observations are remarkable in the sense that the ITIES show all of the electrochemical signatures of a molecularly sharp interface, despite the presence of a macroscopic assembly of non-sintered nanoparticles. Consequently, the high IPCE values is the result of a combination of charge transfer across a polarisable ITIES and carrier transport through a mesoporous nanoparticle layer, without the need of direct electrical wiring to the active interface.
Results and discussion
The liquid/liquid interface (geometric area 2.27 cm2) is assembled in a custom made three-compartment all quartz electrochemical cell shown in Fig. 1a, with the aqueous and organic phase reference electrodes (RE) in separate compartments, connected to the main compartment through Luggin capillaries. Under potentiostatic control, the potential difference between the two REs is kept constant, while any charge transfer across the liquid/liquid boundary is balanced by redox reactions induced at the counter electrodes (CE). The interfacial TiO2 film was prepared by dispersing 25 nm TiO2 particles (Sigma-Aldrich) in the organic electrolyte (1 mg cm−3) by continuous sonication for 1 h. The organic phase solution contained 1,1′-dimethyl-ferrocene (DMFc) and bis(triphenylphosphoranylidene)-ammonium tetrakis(penta-fluorophenyl)borate (BTPPA-TPFB) as donor species and supporting electrolyte, respectively. BTPPA-TPFB was prepared following an established procedure.23 The aqueous electrolyte was added on top of the organic electrolyte and the system was sonicated for 4 additional minutes and allowed to stabilise during 3 hours. As illustrated in Fig. 1a, this procedure leads to a white film of TiO2 colloids suspended at the interface between two clear electrolyte solutions. Introducing a high concentration of LiCl in the aqueous phase (0.1 M) promotes a strong coalescence of the TiO2 particles (7 mg, ca. 2.5 × 1013 particles) mostly at the liquid/liquid boundary, with very few losses into the aqueous and organic electrolytes. Electrochemical cells generated by this approach will be denoted as Cell 1. This system will be compared to polarisable liquid/liquid interfaces with very similar composition but featuring a stable aqueous colloidal system of 5 nm TiO2 particles (Cell 2). The full compositions of the two electrochemical cells are described in the ESI.†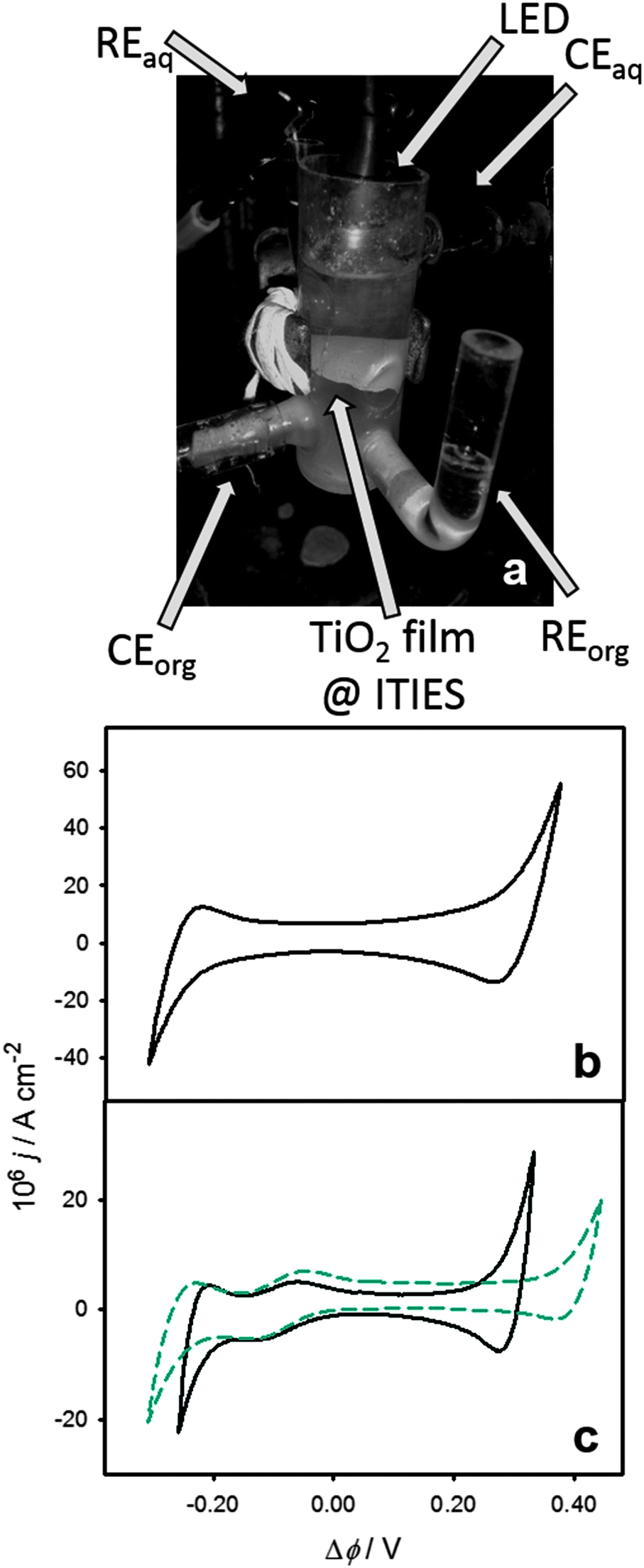 | ||
| Fig. 1 Three-compartment electrochemical cell featuring reference (RE) and counter electrodes (CE) in the aqueous (top) and organic electrolytes (bottom), as well as a layer of coalesced 25 nm TiO2 particles (a). The LED used for photoexcitation is located at the top of the cell. Cyclic voltammogram of Cell 1 in the presence of the coalesced TiO2 particle (25 nm) layer at the water/DCE interface at 0.05 V s−1 (b). Cyclic voltammograms of cells 1 (coalesced TiO2 nanoparticle layer) and 2 (5 nm TiO2 aqueous colloidal solution) at 0.05 V s−1 (c). The composition of the electrochemical cells can be found in the ESI.† Both cells show the characteristic ion transfer responses associated with the transfer of the 1,1′-dimethylferrocenyl cation (DMFc+) accumulated after prolonged illumination. The shorter potential window in Cell 1 is due to the higher concentration of LiCl in the aqueous electrolyte, in comparison to Cell 2. | ||
A characteristic cyclic voltammogram of the system described in Cell 1 recorded at 50 mV s−1 is shown in Fig. 1b. The voltammetric response exhibited the behaviour associated with a molecularly sharp interface between two immiscible electrolyte solutions, with the polarisable window limited by the transfer of Cl− and Li+ at negative and positive Galvani potential differences, respectively. The Galvani potential difference was estimated from the formal transfer potential of the DMFc+ cation observed after prolonged photoelectrolysis (cf.Fig. 1c).24 Impedance data in the dark recorded within the ideally polarisable window yielded interfacial capacitances of 5–50 μF cm−2, values consistent with a molecularly sharp interface. This point is further confirmed by the results in Fig. 1c, contrasting cyclic voltammograms of Cells 1 and 2. Very similar responses are observed for both cells in the Galvani potential difference range between −0.2 and 0.2 V. In this potential window, it can be clearly seen that the capacitive current density and the DMFc+ ion transfer responses are effectively unperturbed by the presence of the coalesced TiO2 interfacial layer. The only noticeable difference between the two voltammograms is the narrower polarisable window in Cell 1, which is due to the significantly higher concentration of LiCl in the aqueous electrolyte. These results show that, despite the macroscopic layer of TiO2 particles deposited at the liquid/liquid interface, the system retains the characteristic molecular sharpness, with no significant hindrance to ion transfer processes.
Photocurrent transient measurements under UV excitation in the presence of DMFc in the organic phase are exemplified in Fig. 2. Illumination was provided by a LED with a narrow spectral range centred at 311 nm, which was focused onto the water/1,2-dichloroethane either through the top (aqueous phase) or the bottom (organic phase) of the cell. This wavelength was chosen in order to use the same excitation source for Cells 1 and 2. The 5 nm TiO2 particles used in Cell 2 exhibit an absorption onset deeper in the UV region in comparison to bulk anatase.25 Information about the spectral emission of the LED and quantification of the photon flux are provided in the ESI.† The photoresponses were investigated as a function of the Galvani potential difference (Fig. 2a) and photon-flux (Fig. 2b), with illumination introduced through the bottom of the cell (i.e. through the organic electrolyte). A large fraction of the photoresponse arises in-phase with the light perturbation, which can be rationalised in terms of heterogeneous hole-transfer to DMFc across the liquid/liquid boundary. As formally described in previous studies, the time-dependence of this relatively fast photocurrent responses is determined by the RC time constant of the cell.26 Homogeneous photochemical reaction followed by transfer of photogenerated ions across the interface manifests itself by a slow rise of the photocurrent in the seconds to minutes time scale.23 However, a small component of the photocurrent after the instantaneous response shows a slower rise time, which is linked to the slow photocurrent decay in the off transient. The origin of the slow component of the photoresponses will be discussed further below.
 | ||
| Fig. 2 Photocurrent transients responses for Cell 1 under illumination through the organic phase (I0 = 7.4 × 1013 cm−2 s−1) as a function of the Galvani potential difference (a). The organic electrolyte contains 1 mM DMFc. Photon flux dependence of the photocurrent density (b) and incident photon-to-current-conversion efficiency (c) at Δϕ = 0.2 V. | ||
As shown in Fig. 2a, the initial photocurrent density is little affected by the Galvani potential difference, while a slight increase in the photostationary value is observed with increasing applied potentials. The origin of this behaviour is yet to be fully rationalised. The photocurrent decay at 0.1 V could be in principle linked to (1) a slow back electron transfer to DMFc+ in the organic phase, (2) the transfer of DMFc+ from the organic to the aqueous phase or (3) the diffusion of the redox species in the organic electrolyte.26 Options 2 and 3 can be disregarded based on the facts that the formal transfer potential of DMFc+ is located at significantly more negative potentials (see Fig. 1c) and that the shape of the photocurrent transient is independent of the photon-flux (Fig. 2b). It could be argued that the potential dependence of the photostationary current could be linked to slow charge transfer processes involving deep electron trap states at the TiO2 surface,27 however, this possibility would require further studies. With regards to the initial photocurrent, the weak potential dependence is in stark contrast to the behaviour observed in dye-sensitised ITIES, where field induced changes in the adsorbed dye coverage and rate of heterogeneous redox quenching are significant.28 Photocurrent responses involving electrostatically stabilised TiO2 colloids also showed a strong potential dependence due to changes in the nanoparticle coverage at the water/DCE interface.25,29
Photocurrent transients and the corresponding IPCE at Δϕ = 0.2 V are displayed in Fig. 2b and c, respectively. Increasing photon-fluxes (corrected for absorption of the organic phase, see ESI†) lead to an increase in the photocurrent density, although the relationship is not linear. This is clearly seen in the IPCE trends which actually decrease as the photon-flux increases. This behaviour is associated with second-order recombination of charge carriers in the TiO2 nanostructured films. Photogenerated holes are swiftly trapped at the particle surface to generate OH radicals, while electrons will be in a dynamic trapping/detrapping process between the conduction band edge and deep trap states.27 The oxygen reduction reaction (ORR) is the key reaction pathway coupled to the oxidation of DMFc mediated by the surface OH radicals. The trend in Fig. 2c shows that as the photostationary concentration of carriers in the TiO2 film decreases, i.e. lower photon-fluxes, the probability for surface OH radical reacting with DMFc through heterogeneous electron transfer increases.
Photoresponses in the absence of DMFc in the organic phase were more than an order of magnitude smaller. Experiments carried out under 12 h of constant illumination revealed that the organic electrolyte (BTPPA+TPFB−) can be partially decomposed in the presence of TiO2. 1H, 19F and 31P NMR measurements (data not shown) confirm that trace amounts of the electrolyte undergo degradation after long term exposure, although the exact nature of the products is yet to be determined. No evidence of DCE decomposition was observed in these experiments. However, the contribution of these photochemical processes is negligible within the time scale of the photocurrent transients discussed here.
The effect of illumination through the top (aqueous electrolyte) or bottom (organic electrolyte) in the photocurrent responses at porous thin films and electrostatically stabilised TiO2 colloids are contrasted in Fig. 3. The composition of the electrochemical cell used in the presence of the colloid is described as Cell 2 in the ESI.†
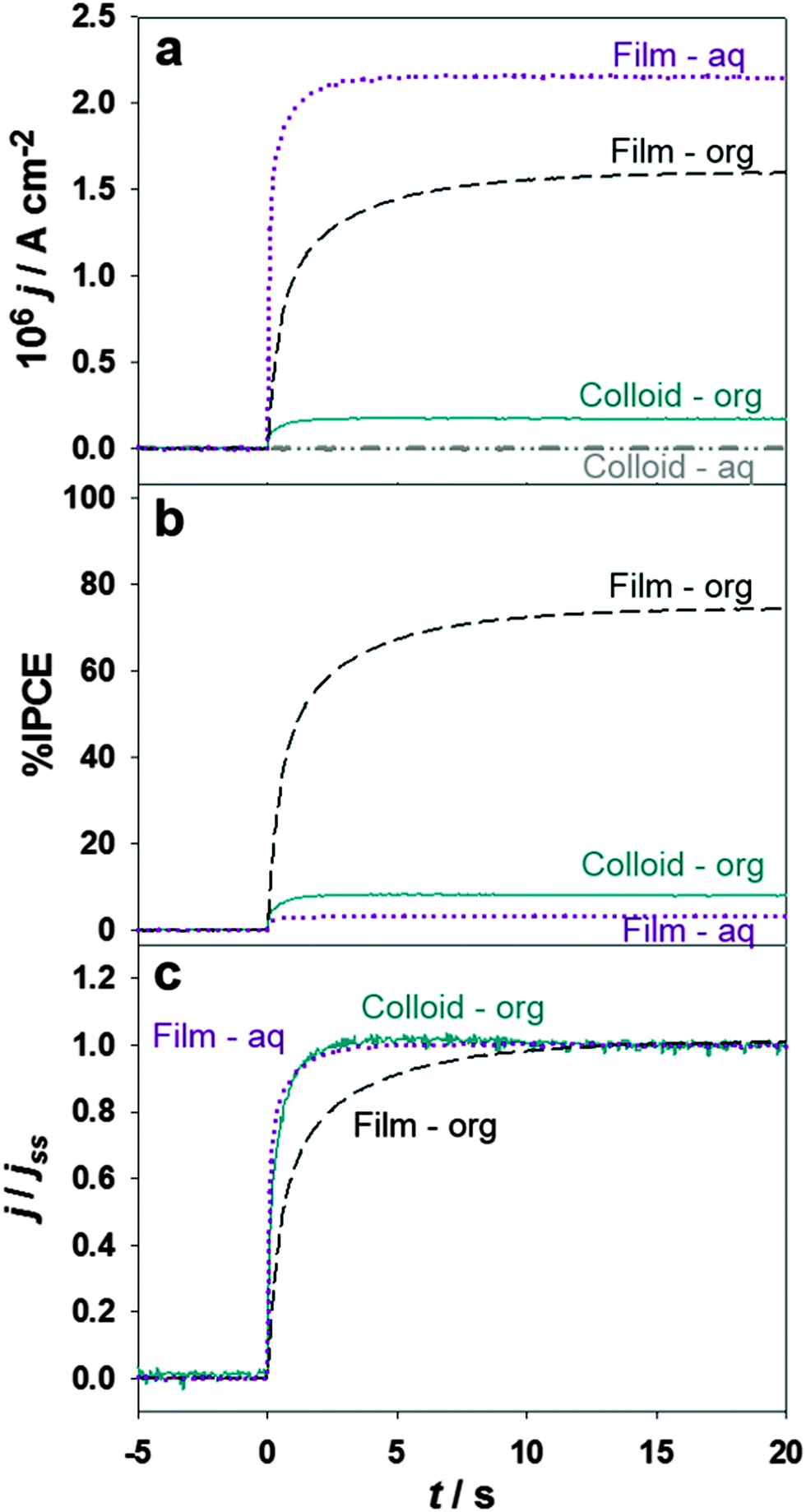 | ||
| Fig. 3 Time dependent photocurrent responses (a) and their corresponding values after normalisation by the incident photon flux (IPCE, b) and photostationary currents (j/jss, c) at Δϕ = 0.2 V in the presence of 5 mM DMFc in the organic phase. Data for colloidal (solid cyan and dashed-dotted grey) and titania film (dashed black and dotted purple) is presented, with illumination from the organic phase (I0 = 1.3 × 1013 cm−2 s−1, solid cyan and dashed black) and aqueous phase (I0 = 4.2 × 1014 cm−2 s−1, dashed-dotted grey and dotted purple). | ||
The photon-flux impinging onto the electrochemical cell (4.2 × 1014 cm−2 s−1) and the Galvani potential difference (0.2 V) were identical in all measurements. As discussed below, the effective photon flux at the liquid/liquid boundary depends on whether the cell is illuminated through either the top or the bottom. The photocurrent density values for the thin porous TiO2 film were significantly higher than in the case of stabilised nanoparticles under all conditions (Fig. 3a), confirming that the photocurrent responses are determined by the effective number of particles at the liquid/liquid boundary. Quantifying the interfacial particle number density is a rather complex issue, in particular considering that the nominal particle size in the thin film is larger than the stabilised colloids. It is interesting to note that measurable photocurrents can be seen in the presence of colloid nanoparticles under bottom illumination as opposed to top illumination. This is due to the fact that light absorption occurs throughout the whole of the TiO2-containing aqueous phase and the effective photon-flux reaching the liquid/liquid boundary is very small. In the case of the thin film, the concentration of TiO2 in the bulk of the aqueous phase is negligible and significant amount of photons are able to strike the ITIES under top illumination.
Estimation of the IPCE shows a surprising trend as displayed in Fig. 3b. Although the photocurrent density for bottom and top illumination are comparable in the case of thin films, the IPCE values (corrected for light absorption in the organic electrolyte) are significantly different. The presence of 5 mM DMFc in the organic phase attenuates the total photon-flux by a factor of 30 (see ESI,† for details about estimation of the photon flux), resulting in IPCE values close to 80% under bottom illumination. Considering that reflection losses are not included in this estimation, these observations lead to the conclusion that the effective IPCE is close to 100% under these conditions. In our view, this is the first observation of high photocurrent yields in systems where charge carriers are not collected by solid electrodes. The relatively low IPCE values observed under top illumination of the TiO2 thin film reveal that only a fraction carriers are able to react at the liquid/liquid boundary. Under these conditions, the efficiency of the photoelectrochemical reaction is highly sensitive to the thickness of the TiO2 layer. This issue will be examined in a separate work.
Another key observation is the difference in photocurrent rise time between top and bottom illumination of the thin film, which is more easily discerned after normalisation of the time dependent photocurrents by the photostationary values (j/jss) as shown in Fig. 3c. The transient photocurrent responses show a complex multi-exponential rise associated with the convolution of the RC time constant of the cell (<10 ms) and a slower component associated with carrier transport in the nanostructured film. The transient response in the case of stabilised colloidal particles is also shown for comparison. Remarkably, the colloidal particles exhibits a similar rise time than the one observed for the TiO2 film under top illumination. Photocurrent responses obtained under bottom illumination of the cell are characterised by a slower rise time, suggesting the contribution of carrier diffusion away from the interface.
In order to rationalise the differences in the photocurrent transient responses under top and bottom illumination, it should be considered that the molecular liquid/liquid boundary is located within the coalesced layer of TiO2 nanoparticles. If illumination is introduced from the top, photocurrent responses mainly arise from hole-transfer involving nanoparticles located in the immediate vicinity to organic electrolyte. The photon flux, and the carrier generation rate, will be strongly attenuated towards the liquid/liquid boundary. This is the key reason why the IPCE is lower and the photocurrent responses are essentially in phase with the light perturbation (as in the case of stable colloidal solution). In the case of bottom illumination, most of the carrier generation occurs in the nanoparticles located in the organic side of the interface. Hole-capture is carried out by DMFc, but the photocurrent response takes place once electrons diffuse to the aqueous side of the interface. Given that the carrier diffusion length is larger for electrons than holes, the process exhibits higher IPCE values but a slower photocurrent rise time due to the electron diffusion process.
It is interesting to consider how this photoelectrochemical behaviour can translate into a photovoltaic device. Based on the spectral response of anatase (band gap 3.2 eV), a short circuit photocurrent of 1.34 mA under AM 1.5G illumination could be predicted assuming 100% quantum efficiency. This would result in a maximum power conversion efficiency of 0.82% based on an open circuit voltage of 600 mV (a value close to the entire ideally polarisable window of the ITIES in Fig. 1b). Given the fact that this approach is rather versatile, this theoretical efficiency can be significantly improved by incorporating photostable materials with more appropriate band gaps. There are obvious mechanical limitations associated with fluid PV active layers, although gelification of the electrolytes can solve some of the limitations at room temperature.30 More challenging issues are related to designing reversible redox probes in the organic and aqueous electrolyte acting as charge relays. These redox species should be highly concentrated to minimise the formation of diffusion layers, while exhibiting negligible partition coefficient and high degree of optical transparency. The latter two conditions also apply to the supporting electrolytes.
Conclusions
We have demonstrated for the first time that IPCE values above 75% can be obtained from a thin layer of photoactive materials assembled at polarisable liquid/liquid interfaces in the presence of redox species in solution. It is interesting to establish a parallel with mesoporous photoelectrodes commonly used as photoanodes in dye-sensitised solar cells and solar fuel generation. In such systems, the carrier diffusion length is required to be of the same order as the film thickness to ensure efficient charge collection and high IPCE values. This boundary condition is not required in our approach. In fact, the efficiency of the process improves the faster the carriers are extracted from the nanostructured film (e.g. electron capture by dissolved oxygen). Another fundamental aspect unique to our approach is that an interfacial potential, which can be externally biased, can operate in the photoelectrochemical responses. Consequently, there is an interplay of different length-scales associated with the mesoscopic TiO2 film and the molecularly sharp polarisable liquid/liquid boundary. We believe these interfaces offer a unique platform for examining a range of nanostructured materials without ohmic contacts, as well as studying complex photocatalytic processes in the presence of tunable potential bias.Acknowledgements
The authors acknowledge discussions and fruitful collaboration with Andrew Rodgers, Peter Toth and Robert Dryfe from the University of Manchester. We would also like to thank Duncan Tarling (University of Bristol) for the help in designing and making of the photoelectrochemical cells. DP, DT and DJF acknowledge the funding from EPSRC (grants EP/K007025/1 and EP/L017792/1). KB acknowledges the PhD studentship supported by the Bristol Centre for Functional Nanomaterials (EP/L016648/1). The authors are also grateful to Jo Humphrey (School of Chemistry) for her support with NMR measurements.Notes and references
- B. C. O'Regan and J. R. Durrant, Acc. Chem. Res., 2009, 42, 1799–1808 CrossRef PubMed.
- A. Hagfeldt and M. Gratzel, Chem. Rev., 1995, 95, 49–68 CrossRef CAS.
- B. Oregan and M. Gratzel, Nature, 1991, 353, 737–740 CrossRef CAS.
- P. V. Kamat, J. Phys. Chem. C, 2008, 112, 18737–18753 CAS.
- F. Bella, C. Gerbaldi, C. Barolo and M. Graetzel, Chem. Soc. Rev., 2015, 44, 3431–3473 RSC.
- A. Listorti, B. O'Regan and J. R. Durrant, Chem. Mater., 2011, 23, 3381–3399 CrossRef CAS.
- L. Peter, Acc. Chem. Res., 2009, 42, 1839–1847 CrossRef CAS PubMed.
- M. A. Green, A. Ho-Baillie and H. J. Snaith, Nat. Photonics, 2014, 8, 506–514 CrossRef CAS.
- S. Kazim, M. K. Nazeeruddin, M. Graetzel and S. Ahmad, Angew. Chem., Int. Ed., 2014, 53, 2812–2824 CrossRef CAS PubMed.
- D. Cahen, G. Hodes, M. Gratzel, J. F. Guillemoles and I. Riess, J. Phys. Chem. B, 2000, 104, 2053–2059 CrossRef CAS.
- J. Bisquert, D. Cahen, G. Hodes, S. Ruhle and A. Zaban, J. Phys. Chem. B, 2004, 108, 8106–8118 CrossRef CAS.
- A. Hagfeldt and M. Gratzel, Acc. Chem. Res., 2000, 33, 269–277 CrossRef CAS PubMed.
- X. Y. Yu, M. S. Prevot, N. Guijarro and K. Sivula, Nat. Commun., 2015, 6, 7596 CrossRef PubMed.
- J. B. Edel, A. A. Kornyshev and M. Urbakh, ACS Nano, 2013, 7, 9526–9532 CrossRef CAS PubMed.
- H. Mehl, M. M. Oliveira and A. J. Gorgatti Zarbin, J. Colloid Interface Sci., 2015, 438, 29–38 CrossRef CAS PubMed.
- R. A. W. Dryfe, Phys. Chem. Chem. Phys., 2006, 8, 1869–1883 RSC.
- A. N. J. Rodgers, S. G. Booth and R. A. W. Dryfe, Electrochem. Commun., 2014, 47, 17–20 CrossRef CAS.
- X. J. Bian, M. D. Scanlon, S. N. Wang, L. Liao, Y. Tang, B. H. Liu and H. H. Girault, Chem. Sci., 2013, 4, 3432–3441 RSC.
- P. Ge, A. J. Olaya, M. D. Scanlon, I. Hatay Patir, H. Vrubel and H. H. Girault, ChemPhysChem, 2013, 14, 2308–2316 CrossRef CAS PubMed.
- B. Su, D. J. Fermin, J. P. Abid, N. Eugster and H. H. Girault, J. Electroanal. Chem., 2005, 583, 241–247 CrossRef CAS.
- D. J. Fermin, H. Jensen, J. E. Moser and H. H. Girault, ChemPhysChem, 2003, 4, 85–89 CrossRef CAS PubMed.
- D. J. Fermin, Z. F. Ding, H. D. Duong, P. F. Brevet and H. H. Girault, J. Phys. Chem. B, 1998, 102, 10334–10341 CrossRef CAS.
- D. J. Fermin, H. D. Duong, Z. F. Ding, P. F. Brevet and H. H. Girault, Phys. Chem. Chem. Phys., 1999, 1, 1461–1467 RSC.
- V. J. Cunnane, G. Geblewicz and D. J. Schiffrin, Electrochim. Acta, 1995, 40, 3005–3014 CrossRef CAS.
- D. Plana and D. J. Fermín, J. Electroanal. Chem., 2015 DOI:10.1016/j.jelechem.2015.09.030.
- Z. Samec, N. Eugster, D. J. Fermin and H. H. Girault, J. Electroanal. Chem., 2005, 577, 323–337 CrossRef CAS.
- Q. Zhang, V. Celorrio, K. Bradley, F. Eisner, D. Cherns, W. Yan and D. J. Fermin, J. Phys. Chem. C, 2014, 118, 18207–18213 CAS.
- H. Jensen, J. J. Kakkassery, H. Nagatani, D. J. Fermin and H. H. Girault, J. Am. Chem. Soc., 2000, 122, 10943–10948 CrossRef CAS.
- H. Jensen, D. J. Fermin, J. E. Moser and H. H. Girault, J. Phys. Chem. B, 2002, 106, 10908–10914 CrossRef CAS.
- D. Ibanez, D. Plana, A. Heras, D. J. Fermin and A. Colina, Electrochem. Commun., 2015, 54, 14–17 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Photoelectrochemical cell compositions and details of LED illumination. See DOI: 10.1039/c6cp02231f |
| ‡ Current address: Lennard-Jones Laboratories, School of Physical and Geographical Sciences, Keele University, Keele, Staffordshire ST5 5BG. |
| This journal is © the Owner Societies 2016 |